
Meta-Analysis Identifies Gene-by-Environment Interactions as Demonstrated in a Study of 4,965 Mice
Identifying environmentally-specific genetic effects is a key challenge in understanding the structure of complex traits. Model organisms play a crucial role in the identification of such gene-by-environment interactions, as a result of the unique ability to observe genetically similar individuals across multiple distinct environments. Many model organism studies examine the same traits but under varying environmental conditions. For example, knock-out or diet-controlled studies are often used to examine cholesterol in mice. These studies, when examined in aggregate, provide an opportunity to identify genomic loci exhibiting environmentally-dependent effects. However, the straightforward application of traditional methodologies to aggregate separate studies suffers from several problems. First, environmental conditions are often variable and do not fit the standard univariate model for interactions. Additionally, applying a multivariate model results in increased degrees of freedom and low statistical power. In this paper, we jointly analyze multiple studies with varying environmental conditions using a meta-analytic approach based on a random effects model to identify loci involved in gene-by-environment interactions. Our approach is motivated by the observation that methods for discovering gene-by-environment interactions are closely related to random effects models for meta-analysis. We show that interactions can be interpreted as heterogeneity and can be detected without utilizing the traditional uni - or multi-variate approaches for discovery of gene-by-environment interactions. We apply our new method to combine 17 mouse studies containing in aggregate 4,965 distinct animals. We identify 26 significant loci involved in High-density lipoprotein (HDL) cholesterol, many of which are consistent with previous findings. Several of these loci show significant evidence of involvement in gene-by-environment interactions. An additional advantage of our meta-analysis approach is that our combined study has significantly higher power and improved resolution compared to any single study thus explaining the large number of loci discovered in the combined study.
Published in the journal:
. PLoS Genet 10(1): e32767. doi:10.1371/journal.pgen.1004022
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004022
Summary
Identifying environmentally-specific genetic effects is a key challenge in understanding the structure of complex traits. Model organisms play a crucial role in the identification of such gene-by-environment interactions, as a result of the unique ability to observe genetically similar individuals across multiple distinct environments. Many model organism studies examine the same traits but under varying environmental conditions. For example, knock-out or diet-controlled studies are often used to examine cholesterol in mice. These studies, when examined in aggregate, provide an opportunity to identify genomic loci exhibiting environmentally-dependent effects. However, the straightforward application of traditional methodologies to aggregate separate studies suffers from several problems. First, environmental conditions are often variable and do not fit the standard univariate model for interactions. Additionally, applying a multivariate model results in increased degrees of freedom and low statistical power. In this paper, we jointly analyze multiple studies with varying environmental conditions using a meta-analytic approach based on a random effects model to identify loci involved in gene-by-environment interactions. Our approach is motivated by the observation that methods for discovering gene-by-environment interactions are closely related to random effects models for meta-analysis. We show that interactions can be interpreted as heterogeneity and can be detected without utilizing the traditional uni - or multi-variate approaches for discovery of gene-by-environment interactions. We apply our new method to combine 17 mouse studies containing in aggregate 4,965 distinct animals. We identify 26 significant loci involved in High-density lipoprotein (HDL) cholesterol, many of which are consistent with previous findings. Several of these loci show significant evidence of involvement in gene-by-environment interactions. An additional advantage of our meta-analysis approach is that our combined study has significantly higher power and improved resolution compared to any single study thus explaining the large number of loci discovered in the combined study.
Introduction
Identifying environmentally specific genetic effects is a key challenge in understanding the structure of complex traits. In humans, gene-by-environment (GxE) interactions have been widely discussed [1]–[12] yet only a few have been replicated. One reason for this discrepancy is the inability to accurately control for environmental conditions in humans as well as the inability to observe the same individuals in multiple distinct environments. Model organisms do not share such difficulties and for this reason can play a crucial role in the identification of gene-by-environment interactions. For example, in many mouse genetic studies the same traits are examined under different environmental conditions. Specifically, knock-out or diet-controlled mice are often utilized in the study of cholesterol levels. The availability of these studies presents a unique opportunity to identify genomic loci involved in gene-by-environment interactions as well as those loci involved in the trait independent of the environment.
In order to utilize genetic studies in model organisms to identify gene-by-environment interactions, one needs to directly compare the effects of genetic variations in studies conducted under different conditions. This practice is complicated for a number of reasons, when combining more than two studies. First, environmental conditions are often variable across studies and do not fit to the standard univariate model for interactions. For example, in one study, cholesterol may be examined under different diet conditions (eg. low fat and high fat) and then in another study cholesterol is examined using gene knockouts. In this case, it is not straightforward to analyze these studies in aggregate using a single variable to represent the environmental condition. Applying a multivariate model, one in which the environment is represented using multiple environmental variables, results in increased degrees of freedom and low statistical power. Second, model organisms such as the mouse exhibit a large degree of population structure. Population structure is well-known for causing false positives and spurious associations [13], [14] in association analysis and can be expected to complicate the ability to combine separate studies.
In this paper, we propose a random-effects based meta-analytic approach to combine multiple studies conducted under varying environmental conditions and show that this approach can be used to identify both genomic loci involved in gene-by-environment interactions as well as those loci involved in the trait independent of the environment. By making the connection between gene-by-environment interactions and random effects model meta-analysis, we show that interactions can be interpreted as heterogeneity and detected without requiring uni - or multi-variate models. We also define an approach for correcting population structure in the random effects model meta-analysis, extending the methods developed for fixed effects model meta-analysis [15]. We show that this method enables the analyses of large scale meta-analyses with dozens of heterogeneous studies and leads to dramatic increases in power. We demonstrate that insights regarding gene-by-environment interactions are obtained by examining the differences in effect sizes among studies facilitated by the recently developed m-value statistic [16], which allows us to distinguish between studies having an effect and studies not having an effect at a given locus.
We applied our approach, which we refer to as Meta-GxE, to combine 17 mouse High-density lipoprotein (HDL) studies containing 4,965 distinct animals. To our knowledge, this is the largest mouse genome-wide association study conducted to date. The environmental factors of the 17 studies vary greatly and include various diet conditions, knock-outs, different ages and mutant animals. By applying our method, we have identified 26 significant loci. Consistent with the experience of meta-analysis in human studies, our combined study finds many loci which were not discovered in any of the individual studies. Among the 26, 24 loci have been previously implicated in having an effect on HDL cholesterol or closely related lipid levels in the blood, while 2 loci are novel findings. In addition, our study provides insights into genetic effects on several disease loci and their relationship between environment and sex. For example, we identified 3 loci (Chr10 : 21399819, Chr19 : 3319089, ChrX:151384614), where female mice show a more significant effect on HDL phenotypes than male mice. We also identified 7 loci (Chr1 : 171199523, Chr8 : 46903188, Chr8 : 64150094, Chr8 : 84073148, Chr10 : 90146088, Chr11 : 69906552, Chr15 : 21194226) where male mice show a more significant effect on HDL than female mice. In addition, many of the loci show strong gene-by-environment interactions. Using additional information describing the studies and our predictions of which studies do and do not contain an effect, we gain insights into the interaction. For example, locus on chromosome 8 (Chr8 : 84073148) shows a strong sex by mutation-driven LDL level interaction, which affects HDL cholesterol levels.
Part of the reason for our success in identifying a large number of loci is that our study combined multiple mouse genetic studies many of which use very different mapping strategies. Over the past few years, many new strategies have been proposed beyond the traditional F2 cross [17] which include the hybrid mouse diversity panel (HMDP) [18], [19], heterogeneous outbred stocks [20], commercially available outbred mice [21], and the collaborative cross [22]. In our current study, we are combining several HMDP studies with several F2 cross studies and benefit from the statistical power and resolution advantages of this combination [15]. The methodology presented here can serve as a roadmap for both performing and planning large scale meta-analysis combining the advantages of many different mapping strategies. Meta-GxE is publicly available at http://genetics.cs.ucla.edu/metagxe/.
Results
Discovering environmentally-specific loci using meta-analysis
The Meta-GxE strategy uses a meta-analytic approach to identify gene-by-environment interactions by combining studies that collect the same phenotype under different conditions. Our method consists of four steps. First, we apply a random effects model meta-analysis (RE) to identify loci associated with a trait considering all of the studies together. The RE method explicitly models the fact that loci may have different effects in different studies due to gene-by-environment interactions. Second, we apply a heterogeneity test to identify loci with significant gene-by-environment interactions. Third, we compute the m-value of each study to identify in which studies a given variant has an effect and in which it does not. Forth, we visualize the result through a forest plot and PM-plot to understand the underlying nature of gene-by-environment interactions.
We illustrate our methodology by examining a well-known region on mouse chromosome 1 harboring the Apoa2 gene, which is known to be strongly associated with HDL cholesterol [23]. Figure 1 shows the results of applying our method to this locus. We first compute the effect size and its standard deviation for each of the 17 studies. These results are shown as a forest plot in Figure 1 (a). Second we compute the P-value for each individual study also shown in Figure 1 (a). If we were to follow traditional methodology and evaluate each study separately, we would declare an effect present in a study if the P-value exceeds a predefined genome-wide significance threshold (P ). In this case, we would only identify the locus as associated in a single study, HMDP-chow(M) (P = ). On the other hand, in our approach, we combine all studies to compute a single P-value for each locus taking into account heterogeneity between studies. This approach leads to increased power over the simple approach considering each study separately. The combined meta P-value for the Apoa2 locus is very significant (), which is consistent with the fact that the largest individual study only has 749 animals compared to 4,965 in our combined study.
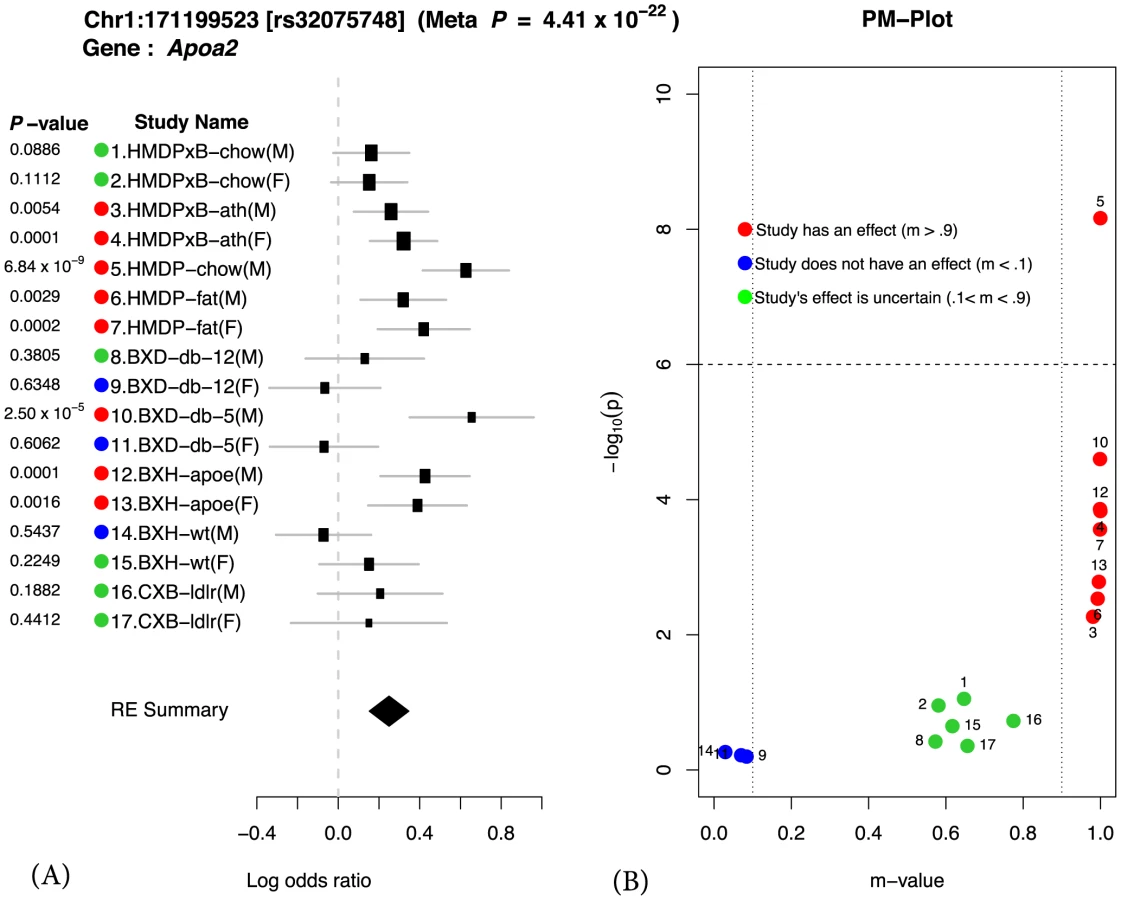
In order to evaluate how significantly different the effect sizes of the locus are between studies, we apply a heterogeneity test. The statistical test is based on Cochran's Q test [24], [25], which is a non-parametric test for testing if studies have the same effect or not. In this locus, the effect sizes are clearly different and not surprisingly the P-value of the heterogeneity test is significant (). This provides strong statistical evidence of a gene-by-environment interaction at the locus. Below we more formally describe how heterogeneity in effect size at a given locus can be interpreted as gene-by-environment interaction.
If a variant is significant in the meta-analytic testing procedure, then this implies that the variant has an effect on the phenotype in one or more studies. Examining in which subset of the studies an effect is present and comparing to the environmental conditions of the studies can provide clues to the nature of gene-by-environment interactions at the locus. However, the presence of the effect may not be reflected in the study-specific P-value due to a lack of statistical power. Therefore, it is difficult to distinguish only by a P-value if an effect is absent in a particular study due to a gene-by-environment interaction at the locus or a lack of power. In order to identify which studies have effects, we utilize a statistic called the m-value [16], which estimates the posterior probability of an effect being present in a study given the observations from all other studies. We visualize the results through a PM-plot, in which P-values are simultaneously visualized with the m-values at each tested locus. These plots allow us to identify in which studies a given variant has an effect and in which it does not. M-values for a given variant have the following interpretation: a study with a small m-value() is predicted not to be affected by the variant, while a study with a large m-value() is predicted to be affected by the variant.
The PM-plot for the Apoa2 locus is shown in Figure 1 (b). If we only look at the separate study P-values (y-axis), we can conclude that this locus only has an effect in HMDP-chow(M). However, if we look at m-value (x-axis), then we find 8 studies (HMDPxB-ath(M), HMDPxB-ath(F), HMDP-chow(M), HMDP-fat(M), HMDP-fat(F), BxD-db-5(M), BxH-apoe(M), BxH-apoe(F)), where we predict that the variation has an effect, while in 3 studies (BxD-db-12(F), BxD-db-5(F), BxH-wt(M)) we predict there is no effect. The predictions for the remaining 6 studies are ambiguous.
From Figure 1, we observe that differences in effect sizes among the studies are remarkably consistent when considering the environmental factors of each study as described in Table 1. For example, when comparing study 1–4, the effect size of the locus decreases in both the male and female HMDPxB studies in the chow diet (chow study) relative to the fat diet (ath study). Thus we can see that when the mice have Leiden/CETP transgene, which cause high total cholesterol level and high LDL cholesterol level, effect size of this locus on HDL cholesterol level in blood is affected by the fat level of diet. Similarly, when comparing study 12–15, the knockout of the Apoe gene affects the effect sizes for both male and female BxH crosses. However, in the BxD cross (study 8–11), where each animal is homozygous for a mutation causing a deficiency of the leptin receptor, the effect of the locus is very strong in the young male animals, while as animals get older and become fatter, the effect becomes weaker. However in the case of female mice, the effect of the locus is nearly absent at both 5 and 12 weeks of age. Thus we can see that sex plays an important role in affecting HDL when the leptin receptor activity is deficient. We note that there are many genes in this locus and the genetic mechanism of interactions may involve genes other than Apoa2. Despite this caveat, the results of Meta-GxE at this locus provides insights into the nature of GxE and can provide a starting point for further investigation.

We note that an alternate explanation for differences in effect sizes between studies is the presence of gene-by-gene interactions and differences in the genetic backgrounds of the studies. While this is a possible explanation for differences in effect sizes between the different crosses and the HMDP studies, in Figure 1, we see many differences in effect sizes among studies with the same genetic background. Thus gene-by-gene interactions can only partially explain the differences in observed effect sizes.
The connection between random effects meta-analysis and gene-by-environment interactions
Gene-by-environment interactions, random effects meta-analysis and heterogeneity testing are closely related. Suppose we have studies each conducted under different environmental conditions. We define the following linear model, where is the observed phenotype for study , is the phenotype mean for study , is the genetic effect on the phenotype for study , is the genotype, and is the residual error.

Since each environment is different, the effect size is partially determined by environmentally-specific factors and partially determined by factors common to all studies. Given that we can decompose the effect into environment-independent and environment-dependent factors. Then we define the following linear model, where is the environment-independent genetic effect and is the environment-dependent genetic effect for study .

In order to test for the presence of an effect shared across environments, we test the null hypothesis and to test for the presence of a gene-by-environment interaction, we test the hypothesis that .
In the random effects meta-analysis, we assume that the effect size is sampled from a normal distribution with mean and variance , denoted . Under this assumption, we test the null hypothesis and , in order to obtain a study-wide P-value. Additionally, we perform a heterogeneity test to test the null hypothesis versus the alternative hypothesis . We posit that by conducting hypotheses tests in the meta-analysis framework, we are simultaneously testing for the presence of environmentally-independent and environmentally-specific effects and that by applying heterogeneity testing we are testing for only environmentally-specific effects.
Consider that in the meta-analysis framework is analogous to and the variation () around is analogous to variation among s. In the random effects meta analysis testing framework we are testing if and . This is equivalent to testing both environmentally-independent () and environmentally-dependent () effects simultaneously. In heterogeneity testing, we test the null hypothesis versus the alternative hypothesis . When the environmentally-dependent effect () is 0 it means that and thus . When , we expect that will vary around , so that we do not expect that . Since the variation () of around is analogous to the variable , heterogeneity testing in the meta-analysis framework is approximately equivalent to testing for environmentally-specific effects.
Gene-by-environment interactions are prevalent in mouse association studies
The presence of heterogeneity in the effect size at causal genetic loci due to gene-by-environment interactions is naturally expected in mouse genetic studies when combining studies with varying environmental conditions. One extreme example comes from a knock-out experiment. If the knocked-out gene is causal for a particular trait, then we can expect that the gene would have no effect on a knock-out mouse, while the gene would have an effect on the wild type mouse. This is a binary form of heterogeneity. In a less extreme form of heterogeneity, the effect of a given gene may be affected by an environmental factor which varies in different mice – ranging from small effects to large effects.
To see the relationship between significance of the association and gene-by-environment interactions, we compute and compare this P-value for each SNP from the 17 studies using the random effects meta-analysis to a measure of heterogeneity. Heterogeneity can be assessed by statistic, which describes the percentage of variation across studies that is due to heterogeneity rather than chance [26].
Figure 2 compares statistic with the meta-analysis P-value for each SNP. In this figure, we see that is uniformly distributed for the non-significant SNPs (blue dots), while it is right skewed for significant SNPs (red dots), indicating that more significant SNPs have a greater potential for exhibiting heterogeneity in effect. Since heterogeneity in this case can be interpreted as representing gene-by-environment interactions, as heterogeneity is induced by differences in the environment, we see that the presence of a GxE interaction confers higher power to detect an association.

Power of meta-analysis for detecting gene-by-environment interactions
The power to identify both gene-by-environment and main effects in a meta-analysis of mouse studies depends on both the main effect size and the amount of heterogeneity. We performed simulations using the genotypes of the 17 mouse studies analyzed in this paper. We simulated a range of main effect (mean effect) sizes and a range of gene-by-environment effects. We are simulating the realistic scenario in which we do not know exactly the set of covariates which are responsible for the gene-by-environment effects. We simulated gene-by-environment effects by drawing the effect in each study from a distribution with a mean given by the main effect size and a variance controlling the magnitude of gene-by-environment interactions. If this variance is small, then all of the studies have close to the same effect size and there are few gene-by-environment effects. If the variance is high, then there are strong gene-by-environment effects. Figure 3 shows the results of our simulations. 1000 simulated phenotypes were generated for each mean and variance pair. Statistical power is estimated by computing the proportion of the datasets in which a simulated effect is detected. We observe that the power is high for a wide range of main effect sizes and gene-by-environment effect sizes which is explained by the large sample size of the study. We also observe that even for small main effects, if there are strong gene-by-environment effects, we can still identify the locus. This is because in this case a subset of the studies will have strong effect sizes due to gene-by-environment effects.

Our approach is not the only way to analyze a meta-analysis study. We compare the power to two other meta-analytic approaches. The first is the traditional meta-analysis strategy which uses a fixed effects model (FE) in which all of the effect sizes across studies are assumed to be the same. We utilize an extension of the fixed effects model which corrects for population structure [15]. A second alternate strategy is to simply apply the heterogeneity test (HE), which in our framework is only applied to loci first identified using random effects meta-analysis. The HE test follows the intuition that loci with high heterogeneity will harbor gene-by-environment interactions. For the purposes of the comparison we refer to Meta-GxE as the random effects (RE) model.
The level of gene-by-environment interactions can be simulated by changing both the environment-dependent and environment-independent effect simultaneously, when simulating the phenotype. Figure 4 (a)–(c) shows the power of the three approaches (RE, FE, HE) respectively when we vary the mean and variance of the effect size distribution we sampled from. In this simulation study, mean effect represents shared effect and variance of the effect size represents interaction effect. As expected, RE has high power in cases where the shared effect or the interaction effect is large. FE has high power when the shared effect is large and the HE test has high power when the interaction effect is large. Figure 4 (d) shows the heatmap which is colored with the color of highest powered approach. FE is most powerful at the top-left region, HE is most powerful at the bottom-right region, while RE is most powerful for a majority of the simulations. In the Text S1, we show through simulations that our methodology outperforms the alternative fixed effects and heterogeneity testing approaches when the effect is present in a subset of the studies, which is another possible interaction model we can assume. We also show in the Text S1 that our approach is more powerful than the traditional uni - or multi-variate gene-by-environment association approach which assumes knowledge of the covariates involved in gene-by-environment interactions. For the traditional uni - or multi-variate approach, required knowledge includes kinds of variable (e.g. sex, age, gene knockouts) and encoding of the variables (e.g. binary values, continuous values). In the Text S1, we also show the our proposed approach controls the false positive rate.?

Application to 17 mouse HDL studies
We applied Meta-GxE to 17 mouse genetic studies conducted under various environmental conditions where each study measured HDL cholesterol. Table 1 summarizes each study. More details are provided in the Materials and Methods section and in Text S1. We analyzed all 17 studies together and we also analyzed the 9 male and 8 female studies separately. Some significant associations are shared and some associations are specific to males and females.
The Manhattan plots in Figure 5 show the meta-GxE result when applied to the 17 studies, 9 male only studies and 8 female only studies. Table 2 summarizes 26 significant peaks (P) showing the P-values obtained by applying meta-GxE to the male only studies (9 studies), the female only studies (8 studies) and the male+female studies (17 studies). For each significant locus, we computed m-values, interpreted as the posterior probability of having an effect on the phenotype and report the number of studies with an effect (E), the number of studies with ambiguous effect size (A) and the number of studies without an effect (N). We also report the number of individual studies where the locus was significant (P). As seen in the table, many of the loci were not significant in any of the individual studies and would not have been discovered without combining the studies. We note that we use a more stringent genome wide threshold of P than was used in the original studies. The Genes in Region and Gene Refs columns contain the gene names near the locus previously known to affect HDL cholesterol level or closely related lipid level in the blood and associated literature citations.


Among the 26 loci that we identified by applying Meta-GxE, 24 loci are near the genes (mostly genes are located within 1MB of the peak) known to affect HDL or closely related lipid level in the blood, while 3 loci are novel.
For example, we identified 3 loci (Chr10 : 21399819, Chr19 : 3319089, ChrX:151384614) female mice show a more significant effect on HDL phenotypes than male mice. We also identified 7 loci (Chr1 : 171199523, Chr8 : 46903188, Chr8 : 64150094, Chr8 : 84073148, Chr10 : 90146088, Chr11 : 69906552, Chr15 : 21194226) where male mice show a more significant effect on HDL than female mice.
Interestingly, we observed that in 3 loci (Chr10 : 21399819, Chr19 : 3319089, ChrX:151384614), female mice are more highly affected, while in 7 loci (Chr1 : 171199523, Chr8 : 46903188, Chr8 : 64150094, Chr8 : 84073148, Chr10 : 90146088, Chr11 : 69906552, Chr15 : 21194226) male mice are more highly affected. Among 26 loci, many show a significant heterogeneity in effect sizes between the 17 studies, which we interpret as gene-by-environment interactions.
One interesting example showing strong gene-by-environment interaction is a locus in Chr8 : 84073148. This locus is located near the gene , which is known to affect the abnormal lipid levels in blood [27]. Figure 6 shows the forest plot and PM-plot for this locus. If we look at the forest plot of the locus in Figure 6, we can easily see that there are two groups: 12 studies with an effect (red dots) and 5 studies with an ambiguous prediction of the existence of an effect (green dots). Interestingly, the log odds ratios of effect size for the 12 studies with an effect is about the same (around 0.2). The common characteristic in 4 of the 5 studies (HMDPxB-chow(F), HMDPxB-ath(F), BXH-apoe(F), CXB-ldlr(F)) is that they are female mice with high LDL levels in the blood. In addition, in all 4 cases, these high LDL levels are caused by mutant genes. Mice in HMDPxB-chow and HMDPxB-ath studies have transgenes for both Apoe Leiden and for human Cholesterol Ester Transfer Protein (CETP), while mice in the BXH-apoe and CXB-ldlr studies carried knockouts of the genes for Apoe and LDL receptor, respectively. This is a strong evidence that there is an interaction between sex×mutation-driven LDL levels through this locus (Chr8 : 84073148) when affecting HDL levels in mice.

Figures S5, S6, S7, S8, S9, S10, S11, S12, S13, S14, S15, S16, S17, S18, S19, S20, S21, S22, S23, S24, S25, S26, S27, S28, S29, S30 show the forest plots and PM-plots for each locus, which show information such as effect sizes, the direction of the effect, which study has an effect and which study does not have an effect for each of 17 studies at the given locus.
Discussion
In this paper, we present a new meta-analysis approach for discovering gene-by-environment interactions that can be applied to a large number of heterogeneous studies each conducted in different environments and with animals from different genetic backgrounds. We show the practical utility of the proposed method by applying it to 17 mouse HDL studies containing 4,965 mice, and we successfully identify many known loci involved in HDL. Consistent with the results of meta-analysis in human studies, our combined study finds many loci which were not discovered in any of the individual studies.
A point of emphasis is that in our study design, in each of the combined studies, all of the individuals in the study are subject to only a single environment. This is distinct from other approaches for discovery of gene-by-environment interactions using meta-analysis such as those described in [28]. In these approaches, in each of the combined studies, the individuals in the study are subject to multiple environments and information on each individual's environment is collected. Gene-by-environment statistics are then computed in each study and then combined in the meta-analysis. In our study design, we compute main effect sizes for each SNP and then look for variants where the effect sizes are different suggesting the presence of a gene-by-environment interaction.
In our meta-analysis approach, we assume that we do not have any prior knowledge of the effect size in any particular study. However one might incorporate prior knowledge of the specific environmental effects. In some cases, one might know that some of the studies have similar effect sizes as compared to others. Or the prior knowledge might suggest that one specific study needs to be eliminated in the meta analysis. If we utilize such prior knowledge, we may be able to achieve even higher statistical power.
In this paper we have addressed how to perform meta-analysis when the studies have different genetic structures, building off the results of our previous study [15]. While in this paper we combine 7 HMDP studies with 10 genetic crosses, the approach in principle can be used to combine any variety of study types. Recently, several strategies for mouse genome-wide association mapping have been proposed [29] [17]. These include HMDP [18], collaborative cross [30] and outbredstock [21] [17]. The approach presented here can be utilized to combine these different kinds of studies and is a roadmap for integrating the results of different strategies for mouse GWAS.
Although we have focused on explaining heterogeneity by gene-by-environment interaction, it is possible that the differences in effect sizes can be caused by gene-by-gene interactions on different genetic backgrounds, where the interacting variants differ in frequency in the different studies. While gene-by-gene interactions certainly contribute to locus heterogeneity, we predict that, in combining studies with similar genetic structures, locus heterogeneity more likely arises from gene-by-environment interactions. In any case, determining whether or not these heterogeneous loci are environment-driven or interaction-driven is an important and interesting direction for future study.
Materials and Methods
Standard study design for testing gene-by-environment interactions
In the model organism studies for which we can control the environment, the standard study design for testing gene-by-environment interactions is to combine multiple cohorts whose environments are known. The environmental value that we vary is typically a quantitative measure that we can model with a single random variable. Thus, the standard univariate linear model can be applied

Multivariate interactions model
For more complicated scenarios where the different environments can not be modeled with a single variable, a straightforward extension of the standard univariate interactions model is the multivariate model. Suppose that there are k different possible environments and the information on the environments of each individual are captured by a matrix D which has k columns where each column corresponds to one environment. Then, the standard multivariate interactions model will be


Standard meta-analysis approach
Before we describe the relationship between gene-by-environment interactions and meta-analysis, we first describe the standard fixed effects and random effects meta-analysis in details.
Fixed effects model meta analysis
In standard meta-analysis, we have studies. In each of the studies, we estimate the effect size of interest. Suppose that we estimate the genetic effect in study ,

We can obtain the estimates of and its variance . In the fixed effects model meta-analysis, we assume that the underlying effect sizes are the same as (). The best estimate of is the inverse-variance weighted effect size,

Testing heterogeneity
The phenomenon that the underlying effect sizes differ between studies is called heterogeneity. The presence of heterogeneity is tested using the Cochran's Q test [24], [25]. Cochran's Q test is a non-parametric test for testing if N studies have the same effect or not. Particularly it tests the null hypothesis versus the alternative hypothesis . Cochran's Q statistic can be calculated as the weighted sum of squared differences between individual study effects and the pooled effect across studies.

Random effects model meta analysis
Under the random effects model meta-analysis, we explicitly model heterogeneity by assuming a hierarchical model. We assume that the effect size of each study is a random variable sampled from a distribution with amean and variance ,

Similarly to the traditional random effects model [24], we use the likelihood ratio framework considering each statistic as a single observation. Since we assume no heterogeneity under the null, and under the null hypothesis. The likelihoods are then



Relation between gene-by-environment interactions and meta-analysis
Here we explain more about the relationship between gene-by-environment interactions and meta-analysis based on the explanation in Results section. If we do not consider the interactions, it has been already known that the fixed effects model meta-analysis is approximately equivalent to the linear model of combined cohorts [35]. That is, the fixed effects model equation (5) gives approximately equivalent results to the combined linear model


Similarly, by considering interactions, we extend this argument to show the relationship between gene-by-environment interactions and meta-analysis. We consider the relationship between equation (3) and equation (4). For simplicity of the notation, we consider the case where the matrix D is defined in such a way that each individual is only in one environment such that the D matrix is equivalent to the matrix A described above. If we assume the constant error variance assumption, we establish the following relationship,

Suppose that there are no interactions (null hypothesis of interaction testing). Then, for each study . Thus, the effect size of meta-analysis is equivalent to , the genetic effects that are invariant across studies. Therefore, (null hypothesis of heterogeneity testing). On the other hand, suppose that (null hypothesis of heterogeneity testing). Naturally, for all studies (null hypothesis of interaction testing). This shows that the null hypothesis of the interactions test in the model (3) and the null hypothesis of the heterogeneity test in meta-analysis are equivalent. As a result, we can utilize meta-analytic heterogeneity testing to detect interactions.
Using reasoning, it is straightforward to show that we can utilize the random effects model meta-analysis method to detect the mean effect and the interaction effect at the same time, which can be powerful for identifying loci bearing both kinds of effects.
Controlling for population structure within studies
Model organism such as the mouse are well-known to exhibit population structure or cryptic relatedness [36], [37], where genetic similarities between individuals both inhibit the ability to find true associations and cause the appearance of a large number of false or spurious associations. Mixed effects models are often used in order to correct this problem [38]–[42]. Methods employing a mixed effects correction account for the genetic similarity between individuals with the introduction of a random variable into the traditional linear model.

In the model in equation (9), the random variable represents the vector of genetic contributions to the phenotype for individuals in population . This random variable is assumed to follow a normal distribution with , where is the kinship coefficient matrix for population . With this assumption, the total variance of is given by . A z-score statistic is derived for the test by noting the distribution of the estimate of . In order to avoid complicated notation, we introduce a more basic matrix form of the model in equation (9), shown in equation (10).

In equation (10), is a matrix with the first column being a vector of 1 s representing the global mean and the second vector is the vector and is a coefficient vector containing the mean and genotype effect . We note that this form also easily extends to models with multiple covariates. The maximum likelihood estimate for in population is given by which follows a normal distribution with a mean equal to the true and variance . The estimates of the effect size and standard error of the () are then given in equation (11) and equation (12), where is a vector used to select the appropriate entry in the vector .


Meta-analysis of studies with population structure
When we test gene-by-environment interactions with meta analysis approaches, one important step is correcting for population structure. This can be achieved by correcting for population structure within each study first as described above. For example, consider the random effects model meta-analysis method that we primarily focus on. We employ population structure control, using (11) and (12). Then the likelihood ratio test statistic will be

Identifying studies with an effect
After identifying loci exhibiting interaction effects, we employ the meta-analysis interpretation framework that we recently developed. The m-value [16] is the posterior probability that the effect exists in each study. Suppose we have number of studies we want to combine. Let be the vector of estimated effect sizes and be the vector of estimated variance of effect sizes. We assume that the effect size follows the normal distribution.









The m-values have the following interpretations: small m-values(0.1) represent a study that is predicted to not have an effect, large m-values(0.9) represent a study that is predicted to have an effect, otherwise it is ambiguous to make a prediction. It was previously reported that m-values can accurately distinguish studies having an effect from the studies not having an effect [16]. For interpreting and understanding the result of the meta-analysis, it is informative to look at the P-value and m-value at the same time. We propose to apply the PM-plot framework [16], which plots the P-values and m-values of each study together in two dimensions. Figure 1 (b) shows one example of a PM-plot. In this example, studies with an m-value less than are interpreted as studies not having an effect while studies with an m-value greater than are interpreted as studies having an effect. For studies with an m-value between and , we cannot make a decision. One reason that studies are ambiguous () is that they are underpowered due to small sample size. If the sample size increases, the study can be drawn to either the left or the right side.
Supporting Information
Zdroje
1. GerkeJ, LorenzK, RamnarineS, CohenB (2010) Gene-environment interactions at nu - cleotide resolution. PLoS Genet 6: e1001144.
2. MureaM, MaL, FreedmanBI (2012) Genetic and environmental factors associated with type 2 diabetes and diabetic vascular complications. Rev Diabet Stud 9 : 6–22.
3. SmithEN, KruglyakL (2008) Gene-environment interaction in yeast gene expression. PLoS Biol 6: e83.
4. TalmudPJ (2007) Gene-environment interaction and its impact on coronary heart disease risk. Nutr Metab Cardiovasc Dis 17 : 148–52.
5. ForsythJK, EllmanLM, TanskanenA, MustonenU, HuttunenMO, et al. (2012) Genetic risk for schizophrenia, obstetric complications, and adolescent school outcome: Evidence for gene-environment interaction. Schizophr Bull 39 : 1067–76.
6. OrozcoLD, BennettBJ, FarberCR, GhazalpourA, PanC, et al. (2012) Unraveling inammatory responses using systems genetics and gene-environment interactions in macrophages. Cell 151 : 658–70.
7. DaiX, WuC, HeY, GuiL, ZhouL, et al. (2013) A genome-wide association study for serum bilirubin levels and gene-environment interaction in a chinese population. Genet Epidemiol 37 : 293–300.
8. PatelCJ, ChenR, KodamaK, IoannidisJPA, ButteAJ (2013) Systematic identification of interaction effects between genome - and environment-wide associations in type 2 diabetes mellitus. Hum Genet 132 : 495–508.
9. WuC, KraftP, ZhaiK, ChangJ, WangZ, et al. (2012) Genome-wide association analyses of esophageal squamous cell carcinoma in chinese identify multiple susceptibility loci and gene-environment interactions. Nat Genet 44 : 1090–7.
10. MaJ, XiaoF, XiongM, AndrewAS, BrennerH, et al. (2012) Natural and orthogonal interaction framework for modeling gene-environment interactions with application to lung cancer. Hum Hered 73 : 185–94.
11. GaoJ, NallsMA, ShiM, JoubertBR, HernandezDG, et al. (2012) An exploratory analysis on gene-environment interactions for parkinson disease. Neurobiol Aging 33 : 2528.e1–6.
12. WeiS, WangLEE, McHughMK, HanY, XiongM, et al. (2012) Genome-wide gene - environment interaction analysis for asbestos exposure in lung cancer susceptibility. Carcinogenesis 33 : 1531–7.
13. PriceAL, PattersonNJ, PlengeRM, WeinblattME, ShadickNA, et al. (2006) Principal components analysis corrects for stratification in genome-wide association studies. Nature Genetics 38 : 904.
14. DevlinB, RoederK, WassermanL (2001) Genomic control, a new approach to genetic - based association studies. Theor Popul Biol 60 : 155–66.
15. FurlotteNA, KangEY, Van NasA, FarberCR, LusisAJ, et al. (2012) Increasing asso - ciation mapping power and resolution in mouse genetic studies through the use of meta - analysis for structured populations. Genetics 191 : 959–67.
16. HanB, EskinE (2012) Interpreting meta-analyses of genome-wide association studies. PLoS Genet 8: e1002555.
17. FlintJ, EskinE (2012) Genome-wide association studies in mice. Nature Reviews Genetics 13 : 807.
18. BennettBJ, FarberCR, OrozcoL, KangHM, GhazalpourA, et al. (2010) A high - resolution association mapping panel for the dissection of complex traits in mice. Genome Res 20 : 281–90.
19. GhazalpourA, RauCD, FarberCR, BennettBJ, OrozcoLD, et al. (2012) Hybrid mouse diversity panel: a panel of inbred mouse strains suitable for analysis of complex genetic traits. Mamm Genome 23 : 680–92.
20. ValdarW, SolbergLC, GauguierD, BurnettS, KlenermanP, et al. (2006) Genome-wide genetic association of complex traits in heterogeneous stock mice. Nat Genet 38 : 879–87.
21. YalcinB, NicodJ, BhomraA, DavidsonS, CleakJ, et al. (2010) Commercially available outbred mice for genome-wide association studies. PLoS Genet 6: e1001085.
22. AylorDL, ValdarW, Foulds-MathesW, BuusRJ, VerdugoRA, et al. (2011) Genetic analysis of complex traits in the emerging collaborative cross. Genome Res 21 : 1213–22.
23. WardenCH, HedrickCC, QiaoJH, CastellaniLW, LusisAJ (1993) Atherosclerosis in transgenic mice overexpressing apolipoprotein a-ii. Science 261 : 469–72.
24. DerSimonianR, LairdN (1986) Meta-analysis in clinical trials. Control Clin Trials 7 : 177–88.
25. Cochran WG (2009) The Combination of Estimates from Different Experiments. doi: 10.2307/3001666.
26. HigginsJPT, ThompsonSG (2002) Quantifying heterogeneity in a meta-analysis. Stat Med 21 : 1539–58.
27. NiswenderCM, WillisBS, WallenA, SweetIR, JettonTL, et al. (2005) Cre recombinase - dependent expression of a constitutively active mutant allele of the catalytic subunit of protein kinase a. Genesis 43 : 109–19.
28. ManningAK, LaValleyM, LiuCTT, RiceK, AnP, et al. (2011) Meta-analysis of gene - environment interaction: joint estimation of snp and snp×environment regression coeffi - cients. Genetic Epidemiology 35 : 11.
29. KirbyA, KangHM, WadeCM, CotsapasC, KostemE, et al. (2010) Fine mapping in 94 inbred mouse strains using a high-density haplotype resource. Genetics 185 : 1081–95.
30. ChurchillGA, AireyDC, AllayeeH, AngelJM, AttieAD, et al. (2004) The collaborative cross, a community resource for the genetic analysis of complex traits. Nat Genet 36 : 1133–7.
31. IoannidisJPA, PatsopoulosNA, EvangelouE (2007) Heterogeneity in meta-analyses of genome-wide association investigations. PLoS One 2: e841.
32. IoannidisJPA, PatsopoulosNA, EvangelouE (2007) Uncertainty in heterogeneity esti - mates in meta-analyses. BMJ 335 : 914–6.
33. HanB, EskinE (2011) Random-effects model aimed at discovering associations in meta - analysis of genome-wide association studies. Am J Hum Genet 88 : 586–98.
34. HardyRJ, ThompsonSG (1996) A likelihood approach to meta-analysis with random effects. Stat Med 15 : 619–29.
35. LinDYY, SullivanPF (2009) Meta-analysis of genome-wide association studies with over - lapping subjects. Am J Hum Genet 85 : 862–72.
36. DevlinB, RoederK, BacanuSA (2001) Unbiased methods for population-based association studies. Genet Epidemiol 21 : 273–84.
37. VoightBF, PritchardJK (2005) Confounding from cryptic relatedness in case-control association studies. PLoS Genet 1: e32.
38. LangeK (2002) Mathematical and statistical methods for genetic analysis. Springer Verlag
39. YuJ, PressoirG, BriggsWH, Vroh BiI, YamasakiM, et al. (2006) A unified mixed - model method for association mapping that accounts for multiple levels of relatedness. Nat Genet 38 : 203–8.
40. KangHM, ZaitlenNA, WadeCM, KirbyA, HeckermanD, et al. (2008) Efficient control of population structure in model organism association mapping. Genetics 178 : 1709.
41. LippertC, QuonG, KangEY, KadieCM, ListgartenJ, et al. (2013) The benefits of selecting phenotype-specific variants for applications of mixed models in genomics. Scientific Reports 3 : 1815.
42. ListgartenJ, LippertC, KangEY, XiangJ, KadieCM, et al. (2013) A powerful and efficient set test for genetic markers that handles confounders. Bioinformatics 29 : 1526–1533.
43. StephensM, BaldingDJ (2009) Bayesian statistical methods for genetic association stud - ies. Nat Rev Genet 10 : 681–90.
44. ParksBW, NamE, OrgE, KostemE, NorheimF, et al. (2013) Genetic control of obesity and gut microbiota composition in response to high-fat, high-sucrose diet in mice. Cell Metab 17 : 141–52.
45. DavisRC, van NasA, CastellaniLW, ZhaoY, ZhouZ, et al. (2012) Systems genetics of susceptibility to obesity-induced diabetes in mice. Physiol Genomics 44 : 1–13.
46. WangS, YehyaN, SchadtE, WangH, DrakeT, et al. (2006) Genetic and genomic analysis of a fat mass trait with complex inheritance reveals marked sex specificity. PLoS genetics 2: e15.
47. van NasA, Ingram-DrakeL, SinsheimerJS, WangSS, SchadtEE, et al. (2010) Expression quantitative trait loci: replication, tissue - and sex-specificity in mice. Genetics 185 : 1059–68.
48. van den MaagdenbergAM, HofkerMH, KrimpenfortPJ, de BruijnI, van VlijmenB, et al. (1993) Transgenic mice carrying the apolipoprotein e3-leiden gene exhibit hyperlipopro - teinemia. J Biol Chem 268 : 10540–5.
49. JiangXC, AgellonLB, WalshA, BreslowJL, TallA (1992) Dietary cholesterol increases transcription of the human cholesteryl ester transfer protein gene in transgenic mice. dependence on natural anking sequences. J Clin Invest 90 : 1290–5.
50. TakasugaS, HorieY, SasakiJ, Sun-WadaGHH, KawamuraN, et al. (2013) Critical roles of type iii phosphatidylinositol phosphate kinase in murine embryonic visceral endoderm and adult intestine. Proc Natl Acad Sci U S A 110 : 1726–31.
51. HeC, BassikMC, MoresiV, SunK, WeiY, et al. (2012) Exercise-induced bcl2-regulated autophagy is required for muscle glucose homeostasis. Nature 481 : 511–5.
52. PlumpAS, AzrolanN, OdakaH, WuL, JiangX, et al. (1997) Apoa-i knockout mice: characterization of hdl metabolism in homozygotes and identification of a post-rna mechanism of apoa-i up-regulation in heterozygotes. J Lipid Res 38 : 1033–47.
53. LiegelR, ChangB, DubielzigR, SidjaninDJ (2011) Blind sterile 2 (bs2), a hypomorphic mutation in agps, results in cataracts and male sterility in mice. Mol Genet Metab 103 : 51–9.
54. HofmannJJ, ZoveinAC, KohH, RadtkeF, WeinmasterG, et al. (2010) Jagged1 in the portal vein mesenchyme regulates intrahepatic bile duct development: insights into alagille syndrome. Development 137 : 4061–72.
55. FareseRV, SajanMP, YangH, LiP, MastoridesS, et al. (2007) Muscle-specific knockout of pkc-lambda impairs glucose transport and induces metabolic and diabetic syndromes. J Clin Invest 117 : 2289–301.
56. QiaoJH, TripathiJ, MishraNK, CaiY, TripathiS, et al. (1997) Role of macrophage colony-stimulating factor in atherosclerosis: studies of osteopetrotic mice. Am J Pathol 150 : 1687–99.
57. StanfordKI, WangL, CastagnolaJ, SongD, BishopJR, et al. (2010) Heparan sulfate 2-o-sulfotransferase is required for triglyceride-rich lipoprotein clearance. J Biol Chem 285 : 286–94.
58. MorganH, BeckT, BlakeA, GatesH, AdamsN, et al. (2010) Europhenome: a repository for high-throughput mouse phenotyping data. Nucleic Acids Res 38: D577–85.
59. LeshanRL, Greenwald-YarnellM, PattersonCM, GonzalezIE, MyersMG (2012) Leptin action through hypothalamic nitric oxide synthase-1-expressing neurons controls energy balance. Nat Med 18 : 820–3.
60. NakataS, TsutsuiM, ShimokawaH, SudaO, MorishitaT, et al. (2008) Spontaneous myocardial infarction in mice lacking all nitric oxide synthase isoforms. Circulation 117 : 2211–23.
61. LiLO, EllisJM, PaichHA, WangS, GongN, et al. (2009) Liver-specific loss of long chain acyl-coa synthetase-1 decreases triacylglycerol synthesis and beta-oxidation and alters phospholipid fatty acid composition. J Biol Chem 284 : 27816–26.
62. NishinaPM, NaggertJK, VerstuyftJ, PaigenB (1994) Atherosclerosis in genetically obese mice: the mutants obese, diabetes, fat, tubby, and lethal yellow. Metabolism 43 : 554–8.
63. CiraoloE, IezziM, MaroneR, MarengoS, CurcioC, et al. (2008) Phosphoinositide 3 - kinase p110beta activity: key role in metabolism and mammary gland cancer but not development. Sci Signal 1: ra3.
64. GuptaS, PabloAM, JiangXc, WangN, TallAR, et al. (1997) Ifn-gamma potentiates atherosclerosis in apoe knock-out mice. J Clin Invest 99 : 2752–61.
65. KimI, AhnSHH, InagakiT, ChoiM, ItoS, et al. (2007) Differential regulation of bile acid homeostasis by the farnesoid x receptor in liver and intestine. J Lipid Res 48 : 2664–72.
66. WiedmerT, ZhaoJ, LiL, ZhouQ, HevenerA, et al. (2004) Adiposity, dyslipidemia, and insulin resistance in mice with targeted deletion of phospholipid scramblase 3 (plscr3). Proc Natl Acad Sci U S A 101 : 13296–301.
67. FanCY, PanJ, ChuR, LeeD, KluckmanKD, et al. (1996) Hepatocellular and hepatic peroxisomal alterations in mice with a disrupted peroxisomal fatty acyl-coenzyme a oxidase gene. J Biol Chem 271 : 24698–710.
68. SainsburyA, BaldockPA, SchwarzerC, UenoN, EnriquezRF, et al. (2003) Synergistic effects of y2 and y4 receptors on adiposity and bone mass revealed in double knockout mice. Mol Cell Biol 23 : 5225–33.
69. EdmondsonAC, BraundPS, StylianouIM, KheraAV, NelsonCP, et al. (2011) Dense genotyping of candidate gene loci identifies variants associated with high-density lipoprotein cholesterol. Circ Cardiovasc Genet 4 : 145–55.
70. ForestiO, RuggianoA, Hannibal-BachHK, EjsingCS, CarvalhoP (2013) Sterol home - ostasis requires regulated degradation of squalene monooxygenase by the ubiquitin ligase doa10/teb4. eLife 2: e00953.
71. LiuSPP, LiYSS, ChenYJJ, ChiangEPP, LiAFY, et al. (2007) Glycine n - methyltransferase-/ - mice develop chronic hepatitis and glycogen storage disease in the liver. Hepatology 46 : 1413–25.
72. BourreJM, Cl_ementM, G_erardD, Chaudi_ereJ (1989) Alterations of cholesterol synthe - sis precursors (7-dehydrocholesterol, 7-dehydrodesmosterol, desmosterol) in dysmyelinat - ing neurological mutant mouse (quaking, shiverer and trembler) in the pns and the cns. Biochim Biophys Acta 1004 : 387–90.
73. FujinoT, AsabaH, KangMJJ, IkedaY, SoneH, et al. (2003) Low-density lipoprotein receptor-related protein 5 (lrp5) is essential for normal cholesterol metabolism and glucose - induced insulin secretion. Proc Natl Acad Sci U S A 100 : 229–34.
74. KawaharaY, GrimbergA, TeegardenS, MombereauC, LiuS, et al. (2008) Dysregulated editing of serotonin 2c receptor mrnas results in energy dissipation and loss of fat mass. J Neurosci 28 : 12834–44.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 1
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- GATA6 Is a Crucial Regulator of Shh in the Limb Bud
- Large Inverted Duplications in the Human Genome Form via a Fold-Back Mechanism
- Differential Effects of Collagen Prolyl 3-Hydroxylation on Skeletal Tissues
- Affects Plant Architecture by Regulating Local Auxin Biosynthesis
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy