
Genetic Models of Apoptosis-Induced Proliferation Decipher Activation of JNK and Identify a Requirement of EGFR Signaling for Tissue Regenerative Responses in
Recent work in several model organisms has revealed that apoptotic cells are able to stimulate neighboring surviving cells to undergo additional proliferation, a phenomenon termed apoptosis-induced proliferation. This process depends critically on apoptotic caspases such as Dronc, the Caspase-9 ortholog in Drosophila, and may have important implications for tumorigenesis. While it is known that Dronc can induce the activity of Jun N-terminal kinase (JNK) for apoptosis-induced proliferation, the mechanistic details of this activation are largely unknown. It is also controversial if JNK activity occurs in dying or in surviving cells. Signaling molecules of the Wnt and BMP families have been implicated in apoptosis-induced proliferation, but it is unclear if they are the only ones. To address these questions, we have developed an efficient assay for screening and identification of genes that regulate or mediate apoptosis-induced proliferation. We have identified a subset of genes acting upstream of JNK activity including Rho1. We also demonstrate that JNK activation occurs both in apoptotic cells as well as in neighboring surviving cells. In a genetic screen, we identified signaling by the EGFR pathway as important for apoptosis-induced proliferation acting downstream of JNK signaling. These data underscore the importance of genetic screening and promise an improved understanding of the mechanisms of apoptosis-induced proliferation.
Published in the journal:
. PLoS Genet 10(1): e32767. doi:10.1371/journal.pgen.1004131
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004131
Summary
Recent work in several model organisms has revealed that apoptotic cells are able to stimulate neighboring surviving cells to undergo additional proliferation, a phenomenon termed apoptosis-induced proliferation. This process depends critically on apoptotic caspases such as Dronc, the Caspase-9 ortholog in Drosophila, and may have important implications for tumorigenesis. While it is known that Dronc can induce the activity of Jun N-terminal kinase (JNK) for apoptosis-induced proliferation, the mechanistic details of this activation are largely unknown. It is also controversial if JNK activity occurs in dying or in surviving cells. Signaling molecules of the Wnt and BMP families have been implicated in apoptosis-induced proliferation, but it is unclear if they are the only ones. To address these questions, we have developed an efficient assay for screening and identification of genes that regulate or mediate apoptosis-induced proliferation. We have identified a subset of genes acting upstream of JNK activity including Rho1. We also demonstrate that JNK activation occurs both in apoptotic cells as well as in neighboring surviving cells. In a genetic screen, we identified signaling by the EGFR pathway as important for apoptosis-induced proliferation acting downstream of JNK signaling. These data underscore the importance of genetic screening and promise an improved understanding of the mechanisms of apoptosis-induced proliferation.
Introduction
Apoptosis is the major form of programmed cell death. It is used during development and under stress conditions to remove excess, unwanted or damaged cells. Deregulated apoptosis can give rise to malignancies including cancer and neurodegeneration [1]. A central step for the execution of apoptosis is the activation of caspases, a family of cysteine-proteases that are ubiquitously expressed as inactive zymogens [2]. There are two different types of caspases. Initiator caspases are activated by incorporation into multimeric complexes such as the apoptosome [3] in response to developmental signals, cellular stress and injury. The initiator caspase complex cleaves and activates effector caspases which then proteolytically process a large number of cellular proteins inducing the death of the cell.
Caspases are very well conserved in the animal kingdom. Of the seven caspases in Drosophila, only the initiator caspase Dronc and the two effector caspases DrICE and Dcp-1 have been implicated in apoptosis in imaginal discs [4]–[12]. Caspases are negatively regulated by inhibitor of apoptosis proteins (IAP) which directly bind to processed caspases and inhibit their activity [13]. Drosophila IAP1 (Diap1) binds to and inhibits Dronc, DrICE and Dcp-1 [14], [15]. In cells committed to die, IAP-antagonists such as Reaper, Hid and Grim [16]–[18] promote ubiquitin-mediated degradation of Diap1, thus releasing Dronc, DrICE and Dcp-1 from Diap1 inhibition [19]–[23]. Dronc associates with the scaffolding protein Ark (Apaf-1 related killer) to form the apoptosome which triggers activation of DrICE and Dcp-1.
Developing organisms have the ability to compensate for massive apoptotic cell loss by inducing compensatory proliferation. For example, developing Drosophila imaginal discs can form a normal-sized and patterned organ even after more than 50% of their cells have been killed by X-ray treatment due to compensatory proliferation [24]. Surprisingly, work in Drosophila, and later in hydra, Xenopus, planarians, newt and mice, has revealed that apoptotic caspases may be the driving force for compensatory proliferation in apoptotic tissue [12], [25]–[35] (reviewed in [36]–[38]). Because this regenerative proliferation requires apoptotic caspases, it has been termed Apoptosis-induced Proliferation, henceforth referred to as AiP [38], [39].
There are two commonly used experimental models that study AiP in larval imaginal discs, usually wing and eye discs in Drosophila. The first type of model takes advantage of the fact that another caspase inhibitor, the P35 protein from Baculovirus, specifically inhibits the effector caspases DrICE and Dcp-1, but not the initiator caspase Dronc [14], [40], [41]. Therefore, induction of apoptosis in p35-expressing cells triggers the apoptotic pathway down to Dronc, but cannot execute cell death because of inhibition of effector caspases by P35. These cells are referred to as ‘undead’ cells. Consequently, Dronc is functional in ‘undead’ tissues and can fulfill non-apoptotic roles including AiP which triggers overgrowth [12], [25]–[28] which may be relevant for tumorigenesis. More, recently, P35-independent models of AiP have been described [42]–[44]. In these models, apoptosis is temporally induced followed by analysis of the events leading to replacement of the dying tissue. Because they mimic the conditions of normal regenerative growth, we referred to them as ‘genuine’ AiP models [45].
Much of what we know about AiP came from studies of ‘undead’ cells. In ‘undead’ cells, Dronc activates p53 and the stress-kinase JNK, encoded by basket (bsk) in Drosophila [12], [25]–[27], [46], [47]. JNK activity is both necessary and sufficient to induce AiP, and it may do this by expression of the Wnt family members wingless (wg) and the TGFβ/BMP-family member decapentaplegic (dpp), both of which are potent mitogens [26], [27], [42], [43], [48]–[51]. There are similarities and differences between the ‘undead’ and ‘genuine’ models. Both models involve JNK signaling, but the location of JNK activity appears to be different. While it is believed that under p35-expressing conditions, JNK activity occurs only in ‘undead’ cells [27], this is less clear in the ‘genuine’AiP model. Initially, it was reported that JNK is activated only in neighboring surviving cells [43]. More recently, it was shown that JNK is activated in both apoptotic and neighboring, surviving cells [44]. The role of Wg and Dpp in both models is also unclear. wg is not induced in all ‘undead’ cells and concern has been raised about the involvement of wg and dpp in ‘genuine’ AiP [26], [44], [46] suggesting that other signaling pathways are also critical for AiP.
There are many other open questions in the field. For example, although it is well established that Dronc can stimulate JNK activity, the molecular mechanism of this interaction is not known. Furthermore, while JNK is best characterized for its ability to induce apoptosis [52], it is not always known how JNK induces proliferation [50], [53]. For example, while in wing imaginal discs, JNK stimulates proliferation through activation of Yorkie, the downstream target of the Hippo growth control pathway, this does not appear to be a mechanism in eye imaginal discs [54], [55], the preferred model of this study (see below). This question is also relevant for understanding of tumorigenesis, as for example death receptor signaling by Fas (CD95) can promote tumor growth through JNK-induced proliferation [56]. These considerations stress the necessity of a convenient genetic screening system to identify the genes and mechanisms involved in AiP.
Here, we present and test the feasibility of ‘undead’ and ‘genuine’ genetic models of AiP in eye imaginal discs. We identify additional components in the JNK pathway that mediate the activation of JNK by Dronc. We show that JNK activation occurs in dying cells as well as in neighboring surviving cells depending on the conditions used. We report the results of a pilot screen using the ‘undead’ AiP model that led to the identification of Spi/EGFR signaling as essential component for AiP. Finally, we demonstrate that Spi is at least partially required for regeneration in a ‘genuine’ AiP model of the eye disc.
Results
The ey>hid-p35 model
eyeless (ey) is a regulatory gene for eye development and is expressed during the growth phase of eye imaginal discs [57]. With the move of the morphogenetic furrow (MF) in 3rd instar larvae, ey expression ceases in and posterior to the MF [57]. Therefore, co-expression of hid and p35 during the growth phase of the eye disc using ey-Gal4 (referred to as ey>hid-p35) may provide a convenient model to induce AiP. Correspondingly, in eye imaginal discs, the anterior portion of the eye disc is overgrown compared to controls forming an expanded head capsule due to increased cell proliferation [29] (Figure 1A,B,C,D). Additional ocelli and bristles are observed (Figure 1D, arrow). The anterior overgrowth is at the expense of posterior tissue (Figure 1A,B) which specifies the retina. As a result, eyes are smaller than wild-type and often absent (Figure 1E,F). In eye discs, we use ELAV labeling which labels photoreceptor neurons, as a marker to assess the extent of anterior overgrowth and distortion of the eye disc (Figure 1B,G). We refer to these phenotypes as AiP phenotypes. The small eye tissue is likely due to the expansion of Wg expression anterior to the MF (Figure 1G,H) which is an inhibitor of MF progression [58]. We also observed anterior expansion of dpp-lacZ expression [29]. Finally, expression of the JNK marker puc-lacZ and TRE-dsRed [59] are strongly expanded anterior to the MF (Figure 1I,J; Suppl. Figure S1). Therefore, known markers of AiP are induced in the ey>hid-p35 model which therefore may represent a convenient AiP model for genetic screening.

The ey>hid-p35 model requires the apoptosome components dronc and ark, but is independent of effector caspases
To test the feasibility of the ey>hid-p35 model for genetic screening, we first examined if mutants and RNA interference (RNAi) of caspases and ark genetically modify the AiP phenotype. Heterozygosity of dronc and dronc RNAi strongly suppressed the AiP phenotype (Figure 2A,B). Under these conditions, more than 95% of the flies display completely normal eye and head morphology. Heterozygosity of the apoptosome component ark also suppresses the AiP phenotype to a similar extend (Figure 2C). Therefore, these data demonstrate that the ey>hid-p35 model is sensitive to genetic alterations and extend previous findings that AiP not only requires dronc, but also ark, i.e. a functional apoptosome.
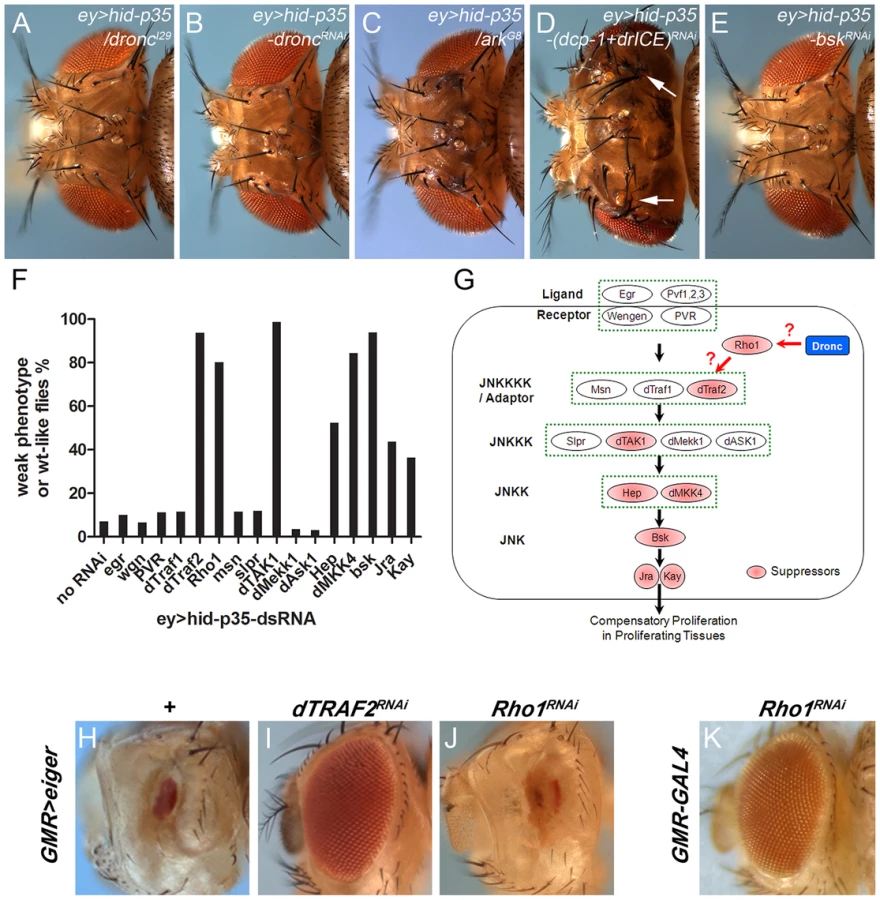
Because this type of AiP is dependent on effector caspase inhibition by P35, it was inferred that it does not require effector caspases [25], [27], [28]. However, it was recently suggested that despite P35 inhibition, effector caspases may still be active at low levels in ‘undead’ cells [60]. This low level effector caspase activity may be insufficient to induce apoptosis, but sufficient to trigger non-apoptotic functions such as invasive behavior of ‘undead’ cells [60]. To test this possibility for AiP, we further reduced DrICE and Dcp-1 activity by double RNAi due to the redundancy of these two effector caspases [8]. However, in contrast to the invasive behavior of ‘undead’ cells [60], the AiP phenotype was not suppressed by dcp-1;drICE double RNAi (Figure 2D). The RNAi stocks used are functional as dcp-1;drICE double RNAi suppresses hid activity in a different apoptotic model, GMR-hid (Suppl. Figure S2). In summary, the overgrowth of the ey>hid-p35 model is dependent on the apoptosome components Dronc and Ark, but independent of effector caspases.
Identification of JNK pathway components involved in AiP
It is unknown how the apoptosome induces JNK activity for AiP. To obtain further insight into this question, we tested components of the JNK pathway in a pilot RNAi screen for modification of the ey>hid-p35 model. As expected, RNAi targeting bsk, the JNK ortholog in Drosophila, completely suppresses the AiP phenotypes in more than 90% of the flies (Figure 2E,F,G). Downstream of JNK, RNAi knockdown of the components of the AP1 transcription factor, jun-related antigen (jra) and the Fos ortholog kayak (kay), also suppressed the ey>hid-p35 AiP phenotypes, although to a lesser extent (Figure 2F,G) suggesting that they are at least partially required for AiP.
To identify upstream components in the JNK pathway involved in AiP, we tested RNAi lines targeting all known components in the JNK pathway [52]. Interestingly, only a subset of them were found to suppress the AiP phenotypes (Figure 2F,G). This includes the JNKKK dTak1 and the JNKKs hemipterous (hep) and MKK4 (Figure 2F,G). The non-redundant functions of hep and MKK4 for AiP is puzzling, but has been previously reported in different contexts [61], [62]. Further upstream in the JNK signaling pathway, we only identified Traf2 (also known as Traf6) as AiP suppressor (Figure 2F,G). Another regulator of JNK signaling, the small GTPase Rho1 [63]–[66], was also identified as AiP suppressor. In contrast, the two ligand/receptor systems known to activate JNK, Eiger/Wengen and Pvf/PVR, do not suppress AiP (Figure 2F,G). The RNAi lines against these genes are functional as shown in Suppl. Figure S3 and in [67]–[69].
Theoretically, it is possible that the suppression of AiP by these RNAi transgenes is an indirect result of suppression of apoptosis, as observed in the case of dronc and ark mutants or RNAi (Figure 2B,C; Suppl. Figure S4B). To exclude this possibility, we labeled ey>hid-p35 eye imaginal discs expressing these RNAi constructs with cleaved Caspase-3 (Cas3*) antibody, a marker of Dronc activity [70], and ELAV antibody to evaluate rescue of disc morphology. Despite the rescue of disc morphology, Cas3* labeling is not significantly suppressed by these RNAi constructs (Suppl. Figure S4C–H) suggesting that the suppression of the AiP phenotype by reducing JNK activity is not due to suppression of caspase activity.
Because Rho1 is the least well characterized regulatory component in the JNK pathway, we examined the effect of Rho1 knockdown on JNK activity in the AiP model. Loss of Rho1 suppresses puc-lacZ in ey>hid-p35 eye discs (Suppl. Figure S5A,B). Rho1 RNAi also suppresses the AiP marker Wg (Suppl. Figure S5C,D). These data show that Rho1 acts genetically upstream of JNK in the AiP model consistent with previous reports [63]–[66].
To further place Rho1 into the AiP pathway and to relate it to Traf2, we examined the ability of Rho1 and Traf2 to suppress GMR-eiger, a known inducer of JNK activity causing a strong eye ablation phenotype (Figure 2H) [71], [72]. Interestingly, while Traf2 knockdown effectively suppresses GMR-eiger as reported [73], Rho1 RNAi does not (Figure 2I,J). It is possible that Rho1 RNAi disrupts eye development by itself and that may be the reason for the failure to suppress GMR-eiger. However, Rho1 RNAi does not disrupt eye development (Figure 2K). These observations suggest that the role of Rho1 for JNK activation is independent of Eiger which is also consistent with the observation that Eiger knockdown does not suppress AiP (Figure 2F,G). Furthermore, these data raise the possibility that Traf2 serves as an integration point of both Eiger signaling and AiP for JNK activation. For these reasons, we place Rho1 upstream of Traf2 in the AiP pathway (Figure 2G), but there may also be other ways by which Rho1 controls JNK activation.
Activation of JNK signaling in ‘undead’/dying cells and neighboring, surviving cells
The lack of a requirement of Eiger/Wengen and Pvf/PVR in our AiP model (Figure 2F,G) may suggest that activation of JNK occurs in dying cells. However, conflicting data have been reported about the location of JNK activity in various AiP models. Initially, JNK signaling was observed in ‘undead’ cells [27]. In a p35-independent regeneration model, it was reported that JNK signaling occurs only in neighboring surviving cells [43]. More recently, JNK activity was reported to be both in dying and neighboring surviving cells [44]. While there are experimental differences between these studies, none of them used a mosaic approach to determine the location of JNK activation. Therefore, to clarify this issue, we re-examined both ‘undead’ and ‘genuine’ AiP models for location of JNK activity.
In a mosaic ‘undead’ model, we expressed hid and p35 in clones in eye and wing discs using a FLP-out approach and analyzed puc-lacZ expression as JNK reporter. GFP was used to mark hid/p35-expressing clones. Using this approach, puc-lacZ is predominantly expressed in hid/p35 expressing cells (Figure 3A,B; arrows). However, we also noted a few examples where puc-lacZ was expressed in GFP− tissue (Figure 3A,B; arrowheads). These observations suggest that JNK activation occurs largely in ‘undead’ cells, but also in neighboring, normal cells.

To address this question in a ‘genuine’ (p35-independent) AiP model, we repeated the experiments by Bergantinos et al. (2010) [43] in wing imaginal discs. These authors reported JNK activity in neighboring surviving cells only. We induced hid in a temporally and spatially controlled manner using ptc-Gal4 and tub-Gal80ts (ptcts>hid) by temperature shifts for various times. In control experiments, just expressing GFP in the ptc domain does not affect the puc-lacZ pattern (Figure 3C,C′″). However, when hid expression was induced, depending on the conditions, different results were obtained regarding the location of JNK activity. In response to a short pulse (6 hours) of hid expression followed by a 6 hours recovery period (ts6hR6h), an elevation of puc-lacZ activity was detected in dying cells and neighboring, surviving cells (Figure 3E″, E′″; dying cells containing puc-lacZ are highlighted by arrows, while surviving cells are marked by arrowheads). This JNK activity was induced during the recovery period, because immediately after hid induction (ts6hR0h), no alteration of puc-lacZ expression was detected (Figure 3D′″). However, when hid expression was induced for a long period (16 hours) followed by 6 hours recovery (ts16hRh), puc-lacZ was strongly down-regulated in dying cells, likely as a result of apoptosis in these cells. Nevertheless, upregulation of puc-lacZ was detected in neighboring surviving cells (Figure 3F″, arrowheads). This result is consistent with Bergantinos et al. (2010) [43]. However, the upregulation of puc-lacZ still occurred in GFP+ cells (Figure 3F″″,F′″″), i.e. in the ptc domain which had been exposed to hid 6 hours earlier, but have survived for unknown reasons. Similar results were observed in Figure 3E: the surviving cells inducing puc-lacZ are located in the GFP+ region, i.e. in the ptc domain (Figure 3E″″,E′″″; arrowheads). Thus, it is not clear whether JNK activity in surviving cells is induced autonomously in response to hid expression, or by a signaling event from the dying Cas3*-positive cells. In any case, these data show that both in ‘undead’ and ‘genuine’ AiP models, JNK activity can be detected in ‘undead’/dying cells as well as in neighboring, surviving cells.
Identification of spi as AiP suppressor
A systematic mutagenesis screen for genes involved in AiP has not been performed to date due to absence of a convenient screening assay. However, the data presented in Figure 2 demonstrate that suppression of ey>hid-p35-induced AiP provides a convenient assay for genetic screening. Therefore, as proof of principle, we screened a total of 106 chromosomal deficiencies deleting segments on the left arm of chromosome 2 (2L) for modification of the AiP phenotype and identified four chromosomal segments as dominant AiP suppressors and seven deficiencies as dominant AiP enhancers (Table 1; Suppl. Table S1), validating the deficiency approach. Enhancers display an even stronger AiP phenotype with severely overgrown head cuticle and strong semi-lethality.

To identify the genes in the deficiencies that dominantly cause the suppression of AiP, we tested available mutants and UAS-RNAi stocks against all genes that map to these deficiencies. This approach has been completed for Df(2L)ED1303 (Table 1) and led to the identification of spitz (spi) as a potential regulator of AiP (compare Figure 4C–F,I with Figure 4A,B). spi encodes the EGF ortholog in Drosophila [74]. Therefore, our deficiency screen raises the hypothesis that the EGFR pathway regulates AiP. Consistently, heterozygosity of Egfr suppresses the ey>hid-p35 phenotype in eye discs (compare Figure 4G–I with Figure 4A,B). We also found that Egfr RNAi suppresses an AiP model in wing imaginal discs (nub>hid-p35) (Suppl. Figure S6). Downstream of EGFR, mutant alleles of the Drosophila orthologs of Ras (Dras) and MAPK (rolled (rl)) act as dominant suppressors of ey>hid-p35 (Figure 4I) suggesting that MAPK activity is required for AiP. These data imply that EGFR/Ras/MAPK signaling is essential for AiP in both eye and wing discs. These findings are exciting giving the controversy of the role of Wg and Dpp for AiP (see Introduction) [26], [46].

To further characterize the involvement of Spi/EGFR signaling for AiP, we took advantage of the spi01068 allele which is an enhancer trap insertion of lacZ into the spi gene (spi-lacZ) and can serve as a reporter for spi expression [75]. This analysis is complicated by the fact that this spi allele itself is a dominant suppressor of AiP: about 75% of the ey>hid-p35/spi01068 flies show a weak AiP phenotype, while 25% are not suppressed and still show a moderately strong AiP phenotype (Figure 4E,F,I). Consistently, in about 25% of ey>hid-p35 eye discs (n = 28), we observed a strong induction of β-Gal labeling compared to control discs (Figure 5A,B). This percentage corresponds to the number of heterozygous spi01068 flies which display a moderately strong AiP phenotype (Figure 4I). The remaining 75% of ey>hid-p35 eye discs heterozygous for spi01068 show a normalization of disc morphology as visualized by ELAV labeling and spi-lacZ expression (Figure 5C). In addition, we found that a target gene of the EGFR pathway, kekkon-1(kek) [76] is induced during AiP (Figure 5D,E,F).

To determine the position of Spi/EGFR signaling in the AiP pathway, we performed epistasis experiments between spi and bsk. Heterozygosity of spi1 dominantly suppresses the adult AiP phenotype of ey>hid-p35 (Figure 4C,D,I). This suppression can also be visualized by the normalization of the ELAV pattern in ey>hid-p35 eye imaginal discs (Figure 5G, compare to Figure 1J). However, despite the normalization of the ELAV pattern, puc-lacZ expression is not reduced in this genetic background (Figure 5G,G′) suggesting that spi acts genetically downstream of bsk. This is further confirmed by the reciprocal experiment in which bsk RNAi completely normalizes the spi-lacZ pattern in ey>hid-p35 background (Figure 5H). bsk RNAi also normalizes the kek-lacZ pattern in ey>hid-p35 background (Figure 5I). These observations suggest that Spi/EGFR signaling acts genetically downstream of bsk activity. Because Spi is a secreted signaling molecule, these findings may imply that EGFR activation occurs in cells adjacent to apoptotic, JNK-activating cells. This assumption is directly confirmed by the observation that kek-lacZ activity, a downstream marker of EGFR signaling, and Cas3* labeling as apoptotic marker do not overlap (Figure 5F, arrows). In summary, these data imply that spi expression occurs downstream of Bsk/JNK activity and that EGFR signaling acts in signal-receiving, proliferating cells.
Characterization of ‘genuine’ AiP in the eye imaginal disc: the DEts>hid model
Finally, we tested if genes identified in the ‘undead’ (P35-dependent) AiP model are also involved in ‘genuine’ (P35-independent) regeneration in the eye disc. To accomplish this we used a similar approach as previously described in wing discs [42]–[44]. hid expression was spatially restricted to the dorsal half of the eye disc by dorsal eye-Gal4 (DE-Gal4) [77] and controlled by Gal80ts [78] by a transient temperature shift (ts) to 30°C for 12 hours (Figure 6E). We refer to this system as DEts>hid. This model also induces GFP to label hid-expressing cells. Before and after the temperature shift, animals were incubated at 18°C (Figure 6E) to inhibit Gal4 activity and therefore hid and GFP expression. Note that although GFP is expressed only during the 30°C pulse, it is a rather stable protein and can be detected in control discs 72 h later (Figure 6D).

In experimental discs immediately after the 30°C pulse (recovery 0 hours – R0 h), a strong apoptotic response is detectable (Figure 6A′) which causes tissue loss and disruption of the bilateral symmetry of the disc 24 hours later (R24 h). In extreme cases, this treatment can result in ablation of the entire dorsal half (Figure 6B, asterisk), but usually some dorsal tissue remains. At that time, many cells are still Cas3*-positive (Figure 6B). 72 hours after the temperature shift (R72 h), the disc has fully recovered in shape and also has a normal photoreceptor pattern as judged by ELAV labeling (Figure 6C). Cas3* activity is no longer detectable. The recovery is the result of increased proliferation in the dorsal half of the eye disc (compare Figure 6G″ to Figure 6F″; quantified in Figure 6H). The reduction of the GFP signal in the dorsal part (Figure 6C,G′) compared to the control disc (Figure 6D) suggests that most of the GFP+ cells have died and have been replaced by new, GFP−, cells.
Interestingly, a group of apoptotic cells appears to migrate out of the dorsal half into the center of the disc (Figure 6B; arrow). At R72 h, only these cells still show strong GFP+-labeling (Figure 6C; arrow). This ‘escape’ response of these ‘genuine’ apoptotic cells is reminiscent of the invasive behavior of ‘undead’ cells in wing discs which move out of the ‘undead’ domain [60]. What makes these cells move is unknown, but an interesting avenue for further research in the future.
Requirement of bsk and spi for regeneration in the ‘genuine’ AiP model DEts>hid
Because JNK activity is essential for ‘undead’ AiP (Figure 2E), we examined a requirement of bsk in the ‘genuine’ (P35-independent) DEts>hid model. First, we examined if JNK activity is induced in the DEts>hid model. Consistent with the ‘genuine’ AiP models in the wing [42]–[44], this was indeed observed. TRE-dsRed as marker of JNK activity [59] peaked at 6 h after recovery (R6 h) and is still detectable at R12 h (Suppl. Figure S7B,C). It is mostly gone after 24 h recovery (Suppl. Figure S7D). TRE-dsRed is confined to the GFP+ area, i.e. in the death domain (Suppl. Figure S7B,C).
Second, we determined if bsk is genetically required for tissue regeneration in the DEts>hid model. We used the photoreceptor pattern (ELAV) as a marker to reveal disc outline and thus assess the degree of regeneration. Control discs (DEts>hid) at R72 h had completely regenerated (Figure 7A; n = 30). However, if bsk was inactivated by RNAi during the apoptosis-inducing phase (Figure 6E), about 35% (9 of 25 discs) of the discs show incomplete regeneration (Figure 7B). The incomplete regeneration after bsk RNAi is weak, presumably because the 12 h down-regulation of bsk during the temperature shift is not sufficient to completely remove Bsk activity. It is also possible that Bsk is resynthesized quickly during the recovery period. Nevertheless, the incomplete regeneration after bsk RNAi suggests that Bsk is at least partially required for tissue regeneration after DEts>hid-induced tissue loss.

Next, we examined whether Spi/EGFR signaling is activated in the DEts>hid model. spi-lacZ expression is induced in the ablated GFP-expressing dorsal domain of the disc compared to controls (Figure 7F,F′,G,G′). kek-lacZ as EGFR signaling marker is also strongly induced in the dorsal domain compared to controls (Figure 7H,H′,I,I′; arrow).
To determine if spi is genetically required in the DEts>hid regeneration assay, we inactivated it by RNAi during the 30°C temperature shift, following the protocol in Figure 6E. In a control experiment, because spi is required for photoreceptor differentiation posterior to the morphogenetic furrow [79], [80], we tested if a 12 h spi RNAi treatment followed by 72 h recovery (R72 h) affects normal photoreceptor differentiation. However, eye discs treated in this way have a normal ELAV pattern (Figure 7C,C′; n = 20). After this control experiment, we tested for a genetic requirement of spi for regeneration of lost tissue due to hid expression. Strikingly, the regeneration response as judged by ELAV labeling was partially impaired when spi was inactivated by RNAi during hid induction (Figure 7D,D′; arrow). All experimental discs (n = 30) showed incomplete regeneration. The regeneration is only weakly affected, likely because spi is inactivated by RNAi only during the 30°C pulse during hid expression (Figure 6E) and is likely restored soon after reducing the temperature to 18°C. Nevertheless, in a heterozygous spi condition, the discs also incompletely regenerated after DEts>hid treatment (Figure 7E,E′; N = 20). In summary, because spi RNAi and spi heterozygosity cause a partial failure to regenerate, these data imply a requirement of spi for regeneration in the ‘genuine’ DEts>hid AiP model. Furthermore, these data support the notion that genetic screening using the simpler ‘undead’ AiP model can lead to identification of genes that may also have important roles for regeneration in ‘genuine’ AiP.
Discussion
Apoptosis-induced proliferation (AiP) appears to be a mechanism by which developing organisms replace dying cells under stress conditions and initiate regenerative responses (reviewed by [37], [45]). In this paper, we described two AiP models in the developing Drosophila eye. The ‘undead’ ey>hid-p35 model generates a hyperplastic overgrowth phenotype. To date this is the only known phenotype that provides a robust and convenient assay for genetic screening and identification of novel regulators of AiP. In contrast, we have not identified a similar robust and convenient phenotype that would allow direct screening for genes involved in ‘genuine’ (p35-independent) AiP and regeneration. Nevertheless, we developed the DEts>hid model to verify genes identified in the ‘undead’ screen as being involved in ‘genuine’ AiP and regeneration.
Although the use of p35 to keep dying cells in an ‘undead’ condition may be considered as unphysiological and artificial, to date all genes identified under p35-expressing conditions such as JNK, Wg and Spi, were also found to be involved in AiP in p35-independent models [42], [43] (this study). Furthermore, cancer cells may resemble ‘undead’ cells. They often initiate, but cannot execute the apoptotic program due to genetic loss or inactivation of effector caspases or other apoptotic components [81]–[84]. Such ‘undead’ cancer cells may contribute to tumor growth. Therefore, our p35-expressing AiP model could provide insights into new regulators of AiP as well as how impaired apoptosis may promote tumor growth.
Apoptotic caspases play a critical role for AiP. In Drosophila, the initiator caspase Dronc is required for activation of JNK activity which triggers AiP. However, it is unknown how Dronc activates JNK for AiP. Using RNAi, a specific subset of components in the JNK pathway were identified as required for AiP. The most upstream genes in the JNK pathway are Rho1 and Traf2. Traf2 appears to be an integration point for Eiger - and AiP-induced JNK activation, the latter one being mediated through Rho1 (Figure 2G). However, it is unknown how Dronc triggers Rho1 activation. It is unlikely that Dronc proteolytically cleaves Rho1 for two reasons. First, Rho1 does not contain a putative Dronc cleavage site [40], [85]. Second, a proteolytic cleavage is likely to destroy Rho1; however, our genetic analysis implies that Rho1 function is required for AiP (Figure 2, Suppl. Figure S5). Therefore, it remains unknown how Dronc triggers Rho1 and thus JNK activation.
Interestingly, extracellular signaling pathways (Eiger/Wengen and Pvf/PVR) known to activate JNK [52] did not score as suppressors of AiP, suggesting that Dronc may autonomously activate JNK activity. This is also consistent with our observation that JNK activity occurs largely in hid - and p35-expressing clones (Figure 3). Nevertheless, it is also possible that a third extracellular signal is generated by ‘undead’ cells in a Dronc-dependent manner that triggers JNK activity in an autocrine and/or paracrine manner. The observation that in both ‘undead’ and ‘genuine’ AiP models JNK activity is also detectable in neighboring surviving cells (Figure 3) may support such a mechanism. Further work is necessary to reveal the exact mode of JNK activation by ‘undead’/dying cells.
In the ‘genuine’ (p35-independent) AiP model (ptcts>hid), JNK activity is detectable in both dying and surviving cells. However, the surviving cells with increased JNK activity are also present in the ptc domain (Figure 3E,F) which was exposed to hid expression during the temperature shift. JNK activity is also restricted to the death domain in the DEts>hid model (Suppl. Figure S7). Therefore, it is unclear whether a signaling mechanism from dying cells induces JNK activity in surviving cells, or whether the previous hid induction accounts for the JNK activity in surviving cells. It is also unclear how these cells survive. Even after a 16 h pulse of hid induction causing a strong apoptotic response in a large fraction of cells in the ptc domain, some cells survive (Figure 3F). They may receive survival signals from cells outside of the ptc domain, but that needs to be determined. These are interesting questions to be addressed in the future.
We have tested signaling pathways known to be involved in growth control for modification of the ey>hid-p35 model. One example is the Hippo/Warts/Yorkie pathway [86], [87]. However, neither mutants of this pathway nor transcriptional reporters (ex-lacZ) scored positive in the ey>hid-p35 model (data not shown). Therefore, at least in the eye disc, not every pathway involved in growth control is also involved in AiP. These observations stressed the necessity to perform unbiased genetic screens aimed at identifying the genes and mechanisms involved in AiP.
Therefore, we performed a pilot screen for modifiers of the ey>hid-p35 AiP model using deficiencies of chromosome arm 2L. We identified four deficiencies as suppressors and three as enhancers (Table 1). Identification of AiP enhancers implies that there is also negative regulation of AiP. In one case we identified spi, encoding the Drosophila EGF ortholog, as a suppressor of AiP suggesting an involvement of EGFR signaling for AiP. This is further confirmed by the strong transcriptional induction of spi and an EGFR target gene, kekkon, in our AiP model. We also found that EGFR signaling is involved in an ‘undead’ AiP model in the wing and – more importantly – in the ‘genuine’ DEts>hid regeneration model in the eye. The latter finding is crucial as it demonstrates that genes identified in the ‘undead’ screen may be relevant players for ‘genuine’ regeneration in response to apoptotic tissue loss. An involvement of EGFR and MAPK for regeneration is not unprecedented. It was previously shown that EGF is one of a few signals that stimulate hepatocyte proliferation during liver regeneration in mammals [88], [89]. In the Hydra regeneration model, apoptosis-induced proliferation depends on MAPK activation [90]. Therefore, these findings and considerations validate our screening approach using the ‘undead’ AiP model.
Identification of Spi/EGFR signaling as suppressor of AiP was unexpected because EGFR signaling negatively regulates the apoptotic activity of hid [91], [92]. Thus, by reducing EGFR activity, hid has increased apoptotic activity which is expected to induce even more AiP. Therefore, the AiP phenotype should be enhanced by heterozygosity of EGFR pathway components. However, the identification of spi, Egfr, Dras and rl as suppressor of AiP suggests that EGFR signaling is also required for AiP. One possibility to explain these two opposing functions of EGFR (negative regulation of hid and positive requirement for AiP) may be the exclusive appearance of Cas3*-positive areas and areas with EGFR activity (Figure 5F). Accordingly, while the Spi signal is generated in Cas3*-positive, apoptotic areas, it signals to neighboring Cas3*-negative, surviving areas to inactivate Hid and promote proliferation.
The identification of Spi/EGFR signaling may help to resolve a controversy about the signaling pathways involved in AiP. The exact roles of Wg and Dpp for AiP are unclear [26], [44], [46] and signaling by the EGFR pathway may contribute to the proliferative response in AiP.
Recently, a genetic screen has been reported aimed at identification and characterization of genes required for compensatory growth [93]. These authors induced apoptosis conditionally using a temperature-sensitive cell lethal mutant (sec5ts). Under normal conditions, the ablated tissue is replaced by new tissue due to compensatory proliferation. The authors then screened for mutants that fail to renew the lost tissue [93]. This was done in a clonal screen for chromosome arm 2L, the same chromosome arm we screened in the deficiency screen in our model. However, the genes identified in the sec5ts screen [93] do not map to the deficiencies that we have identified in our analysis (Table 1). Because it is unknown if sec5ts–induced compensatory proliferation requires caspase activity in apoptotic cells [93], it is not clear if this is a model of apoptosis-induced proliferation.
In summary, we have developed and tested the feasibility of the ey>hid-p35 model for genetic screening. We are confident that this model will close gaps in our understanding of AiP regulation under p35-expressing conditions and in p35-independent regeneration. Finally, it will have implications for the understanding of tumorigenesis by ‘undead’ as well as ‘genuine’ apoptotic tumor cells [94].
Materials and Methods
Fly stocks and genetics
The following mutants and transgenic stocks were used: droncI29; arkG8; spi1; spi01068; Egfrf2; rasΔC40b, rl10a, ey-Gal4; ptc-Gal4; DE-Gal4; tub-Gal80ts; UAS-p35; UAS-hid; UAS-GFP; wg-lacZ; puc-lacZ; kek1-lacZ; spi-lacZ = spi01068; TRE-dsRed; GMR-hid; GMR-Gal4 UAS-egr. UAS-based RNAi stocks of the following genes were obtained from various stock centers (VDRC, Bloomington and NIG) and were tested for suppression of AiP: dronc, dcp-1, drICE; bsk, egr, wgn, PVR, dTraf1, dTraf2, Rho1, msn, slpr, Tak1, dMekk1, dAsk1, hep, dMkk4, Jra, kay, spi, Egfr. The exact genotype of ey>hid-p35 is UAS-hid; ey-Gal4 UAS-p35/CyO,tub-Gal80. Expression of tub-Gal80 in this stock suppresses the semi-lethality associated with ey-induced expression of hid and p35.
Mosaic analysis
Larvae of the following genotype were heat shocked for 15min at 37°C, raised at room temperature for 48 h before they were analyzed at the late 3rd instar larval stage. Genotype: hs-FLP/UAS-hid; UAS-p35/act>y+>Gal4 UAS-GFP; puc-lacZ/+.
Tissue ablation using ptcts>hid and DEts>hid
Larvae of genotype UAS-hid/+; ptc-Gal4 tub-Gal80ts/+; UAS-GFP/+ (Figure 3) and UAS-hid/+; UAS-GFP/+; DE-Gal4 tub-Gal80ts/+ (Figure 6) were raised at 18°C. hid expression was induced by temporal temperature shift to 30°C for the indicated amount of time (Figure 3) or for 12 hours (Figure 6E). After the indicated recovery periods at 18°C, discs were dissected and analyzed as indicated in the panels.
PH3 labelling and statistics in DEts>hid model
Two rounds of experiments (ts12hR24h, at least 20 discs were analyzed each round) were done for both DEts>GFP (control) and DEts>hid. Increase of PH3-positive cells in dorsal eye disc portions of DEts>hid animals are consistently observed. PH3-positive cell numbers were counted in dorsal (GFP+) and ventral eye disc portions in selected discs. Size of the dorsal (GFP+) and ventral eye disc portions were measured through the “histogram” function in Adobe Photoshop CS. To compare the density of PH3+ cells in each disc portion, number of PH3+ cells were divided by size (in pixels) of the corresponding tissue which is used to calculate the number of cells in 100,000 pixels (density). Such normalized density of PH3+ cells in various portions of eye discs (mean ± SD) were used for the statistical chart. PH3+ cell numbers in 100,000 pixels is on average 88 in DEts>hid dorsal eye discs compared to 62 in the control dorsal discs. Their statistical significance was evaluated through a two-tailed, unpaired Student's t-Tests (P<0.04). In contrast, the number of PH3+ cells are comparable in ventral disc portions of each genotype suggesting that increased proliferation mostly occurred in the dorsal part of the disc (at least at the time point of R24 h).
Immunohistochemistry
Imaginal discs were dissected from late 3rd instar larvae and stained using standard protocols. Antibodies to the following primary antigens were used: PH3 (Upstate), anti-cleaved Caspase-3 (Cell Signaling), β-GAL (Promega), ELAV and Wg (DHSB). Secondary antibodies were donkey Fab fragments from Jackson ImmunoResearch. Images were taken with either a Zeiss AxioImager or a confocal microscope.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. ThompsonCB (1995) Apoptosis in the pathogenesis and treatment of disease. Science 267 : 1456–1462.
2. KumarS (2007) Caspase function in programmed cell death. Cell Death Differ 14 : 32–43.
3. BaoQ, ShiY (2007) Apoptosome: a platform for the activation of initiator caspases. Cell Death Differ 14 : 56–65.
4. DorstynL, ColussiPA, QuinnLM, RichardsonH, KumarS (1999) DRONC, an ecdysone-inducible Drosophila caspase. Proc Natl Acad Sci U S A 96 : 4307–4312.
5. FraserAG, McCarthyNJ, EvanGI (1997) drICE is an essential caspase required for apoptotic activity in Drosophila cells. Embo J 16 : 6192–6199.
6. SongZ, McCallK, StellerH (1997) DCP-1, a Drosophila cell death protease essential for development. Science 275 : 536–540.
7. XuD, LiY, ArcaroM, LackeyM, BergmannA (2005) The CARD-carrying caspase Dronc is essential for most, but not all, developmental cell death in Drosophila. Development 132 : 2125–2134.
8. XuD, WangY, WilleckeR, ChenZ, DingT, et al. (2006) The effector caspases drICE and dcp-1 have partially overlapping functions in the apoptotic pathway in Drosophila. Cell Death Differ 13 : 1697–1706.
9. ChewSK, AkdemirF, ChenP, LuWJ, MillsK, et al. (2004) The apical caspase dronc governs programmed and unprogrammed cell death in Drosophila. Dev Cell 7 : 897–907.
10. DaishTJ, MillsK, KumarS (2004) Drosophila caspase DRONC is required for specific developmental cell death pathways and stress-induced apoptosis. Dev Cell 7 : 909–915.
11. MuroI, BerryDL, HuhJR, ChenCH, HuangH, et al. (2006) The Drosophila caspase Ice is important for many apoptotic cell deaths and for spermatid individualization, a nonapoptotic process. Development 133 : 3305–3315.
12. KondoS, Senoo-MatsudaN, HiromiY, MiuraM (2006) DRONC coordinates cell death and compensatory proliferation. Mol Cell Biol 26 : 7258–7268.
13. VauxDL, SilkeJ (2005) IAPs, RINGs and ubiquitylation. Nat Rev Mol Cell Biol 6 : 287–297.
14. MeierP, SilkeJ, LeeversSJ, EvanGI (2000) The Drosophila caspase DRONC is regulated by DIAP1. Embo J 19 : 598–611.
15. ZachariouA, TenevT, GoyalL, AgapiteJ, StellerH, et al. (2003) IAP-antagonists exhibit non-redundant modes of action through differential DIAP1 binding. Embo J 22 : 6642–6652.
16. WhiteK, GretherME, AbramsJM, YoungL, FarrellK, et al. (1994) Genetic control of programmed cell death in Drosophila. Science 264 : 677–683.
17. GretherME, AbramsJM, AgapiteJ, WhiteK, StellerH (1995) The head involution defective gene of Drosophila melanogaster functions in programmed cell death. Genes Dev 9 : 1694–1708.
18. ChenP, NordstromW, GishB, AbramsJM (1996) grim, a novel cell death gene in Drosophila. Genes Dev 10 : 1773–1782.
19. HaysR, WicklineL, CaganR (2002) Morgue mediates apoptosis in the Drosophila melanogaster retina by promoting degradation of DIAP1. Nat Cell Biol 4 : 425–431.
20. HolleyCL, OlsonMR, Colon-RamosDA, KornbluthS (2002) Reaper eliminates IAP proteins through stimulated IAP degradation and generalized translational inhibition. Nat Cell Biol 4 : 439–444.
21. RyooHD, BergmannA, GonenH, CiechanoverA, StellerH (2002) Regulation of Drosophila IAP1 degradation and apoptosis by reaper and ubcD1. Nat Cell Biol 4 : 432–438.
22. WingJP, SchreaderBA, YokokuraT, WangY, AndrewsPS, et al. (2002) Drosophila Morgue is an F box/ubiquitin conjugase domain protein important for grim-reaper mediated apoptosis. Nat Cell Biol 4 : 451–456.
23. YooSJ, HuhJR, MuroI, YuH, WangL, et al. (2002) Hid, Rpr and Grim negatively regulate DIAP1 levels through distinct mechanisms. Nat Cell Biol 4 : 416–424.
24. HaynieJL, BryantPJ (1977) The effects of X-rays on the proliferation dynamics of cells in the imaginal disc of Drosophila melanogaster. Rouxs Arch Dev Biol 183 : 85–100.
25. Perez-GarijoA, MartinFA, MorataG (2004) Caspase inhibition during apoptosis causes abnormal signalling and developmental aberrations in Drosophila. Development 131 : 5591–5598.
26. Perez-GarijoA, ShlevkovE, MorataG (2009) The role of Dpp and Wg in compensatory proliferation and in the formation of hyperplastic overgrowths caused by apoptotic cells in the Drosophila wing disc. Development 136 : 1169–1177.
27. RyooHD, GorencT, StellerH (2004) Apoptotic cells can induce compensatory cell proliferation through the JNK and the Wingless signaling pathways. Dev Cell 7 : 491–501.
28. HuhJR, GuoM, HayBA (2004) Compensatory proliferation induced by cell death in the Drosophila wing disc requires activity of the apical cell death caspase Dronc in a nonapoptotic role. Curr Biol 14 : 1262–1266.
29. FanY, BergmannA (2008) Distinct mechanisms of apoptosis-induced compensatory proliferation in proliferating and differentiating tissues in the Drosophila eye. Dev Cell 14 : 399–410.
30. CheraS, GhilaL, DobretzK, WengerY, BauerC, et al. (2009) Apoptotic cells provide an unexpected source of Wnt3 signaling to drive hydra head regeneration. Dev Cell 17 : 279–289.
31. TsengAS, AdamsDS, QiuD, KoustubhanP, LevinM (2007) Apoptosis is required during early stages of tail regeneration in Xenopus laevis. Dev Biol 301 : 62–69.
32. PellettieriJ, FitzgeraldP, WatanabeS, MancusoJ, GreenDR, et al. (2010) Cell death and tissue remodeling in planarian regeneration. Dev Biol 338 : 76–85.
33. PellettieriJ, Sanchez AlvaradoA (2007) Cell turnover and adult tissue homeostasis: from humans to planarians. Annu Rev Genet 41 : 83–105.
34. VlaskalinT, WongCJ, TsilfidisC (2004) Growth and apoptosis during larval forelimb development and adult forelimb regeneration in the newt (Notophthalmus viridescens). Dev Genes Evol 214 : 423–431.
35. LiF, HuangQ, ChenJ, PengY, RoopDR, et al. (2010) Apoptotic cells activate the “phoenix rising” pathway to promote wound healing and tissue regeneration. Sci Signal 3: ra13.
36. FanY, BergmannA (2008) Apoptosis-induced compensatory proliferation. The Cell is dead. Long live the Cell! Trends Cell Biol 18 : 467–473.
37. BergmannA, StellerH (2010) Apoptosis, stem cells, and tissue regeneration. Science Signaling 3: re8.
38. RyooHD, BergmannA (2012) The role of apoptosis-induced proliferation for regeneration and cancer. Cold Spring Harb Perspect Biol 4: a008797.
39. MollereauB, Perez-GarijoA, BergmannA, MiuraM, GerlitzO, et al. (2013) Compensatory proliferation and apoptosis-induced proliferation: A need for clarification. Cell Death Differ 20 (1) 181.
40. HawkinsCJ, YooSJ, PetersonEP, WangSL, VernooySY, et al. (2000) The Drosophila caspase DRONC cleaves following glutamate or aspartate and is regulated by DIAP1, HID, and GRIM. J Biol Chem 275 : 27084–27093.
41. ClemRJ, FechheimerM, MillerLK (1991) Prevention of apoptosis by a baculovirus gene during infection of insect cells. Science 254 : 1388–1390.
42. Smith-BoltonRK, WorleyMI, KandaH, HariharanIK (2009) Regenerative growth in Drosophila imaginal discs is regulated by Wingless and Myc. Dev Cell 16 : 797–809.
43. BergantinosC, CorominasM, SerrasF (2010) Cell death-induced regeneration in wing imaginal discs requires JNK signalling. Development 137 : 1169–1179.
44. HerreraSC, MartinR, MorataG (2013) Tissue homeostasis in the wing disc of Drosophila melanogaster: immediate response to massive damage during development. PLoS Genet 9: e1003446.
45. Ryoo HD, Bergmann A (2012) The role of apoptosis-induced proliferation for regeneration and cancer. In: Cell Survival and Cell Death EH Baehrecke, DR Green, S Kornbluth and G Salvesen Eds, Cold Spring Harbor Press, Cold Spring Harbor, NY
46. WellsBS, YoshidaE, JohnstonLA (2006) Compensatory proliferation in Drosophila imaginal discs requires Dronc-dependent p53 activity. Curr Biol 16 : 1606–1615.
47. WellsBS, JohnstonLA (2012) Maintenance of imaginal disc plasticity and regenerative potential in Drosophila by p53. Dev Biol 361 : 263–276.
48. ShlevkovE, MorataG (2012) A dp53/JNK-dependant feedback amplification loop is essential for the apoptotic response to stress in Drosophila. Cell Death Differ 19 : 451–460.
49. SuissaY, ZivO, DinurT, AramaE, GerlitzO (2011) The NAB-Brk signal bifurcates at JNK to independently induce apoptosis and compensatory proliferation. J Biol Chem 286 : 15556–15564.
50. UhlirovaM, JasperH, BohmannD (2005) Non-cell-autonomous induction of tissue overgrowth by JNK/Ras cooperation in a Drosophila tumor model. Proc Natl Acad Sci U S A 102 : 13123–13128.
51. WarnerSJ, YashiroH, LongmoreGD (2010) The Cdc42/Par6/aPKC polarity complex regulates apoptosis-induced compensatory proliferation in epithelia. Curr Biol 20 : 677–686.
52. IgakiT (2009) Correcting developmental errors by apoptosis: lessons from Drosophila JNK signaling. Apoptosis 14 : 1021–1028.
53. WestonCR, DavisRJ (2007) The JNK signal transduction pathway. Curr Opin Cell Biol 19 : 142–149.
54. SunG, IrvineKD (2011) Regulation of Hippo signaling by Jun kinase signaling during compensatory cell proliferation and regeneration, and in neoplastic tumors. Dev Biol 350 : 139–151.
55. SunG, IrvineKD (2013) Ajuba family proteins link JNK to Hippo signaling. Sci Signal 6: ra81.
56. ChenL, ParkSM, TumanovAV, HauA, SawadaK, et al. (2010) CD95 promotes tumour growth. Nature 465 : 492–496.
57. HalderG, CallaertsP, FlisterS, WalldorfU, KloterU, et al. (1998) Eyeless initiates the expression of both sine oculis and eyes absent during Drosophila compound eye development. Development 125 : 2181–2191.
58. LegentK, TreismanJE (2008) Wingless signaling in Drosophila eye development. Methods Mol Biol 469 : 141–161.
59. ChatterjeeN, BohmannD (2012) A versatile PhiC31 based reporter system for measuring AP-1 and Nrf2 signaling in Drosophila and in tissue culture. PLoS One 7: e34063.
60. RudrapatnaVA, BangiE, CaganRL (2013) Caspase signalling in the absence of apoptosis drives Jnk-dependent invasion. EMBO Rep 14 : 172–177.
61. BoutrosM, AgaisseH, PerrimonN (2002) Sequential activation of signaling pathways during innate immune responses in Drosophila. Dev Cell 3 : 711–722.
62. GeukingP, NarasimamurthyR, LemaitreB, BaslerK, LeulierF (2009) A non-redundant role for Drosophila Mkk4 and hemipterous/Mkk7 in TAK1-mediated activation of JNK. PLoS One 4: e7709.
63. NeischAL, SpeckO, StronachB, FehonRG (2010) Rho1 regulates apoptosis via activation of the JNK signaling pathway at the plasma membrane. J Cell Biol 189 : 311–323.
64. BrumbyAM, GouldingKR, SchlosserT, LoiS, GaleaR, et al. (2011) Identification of novel Ras-cooperating oncogenes in Drosophila melanogaster: a RhoGEF/Rho-family/JNK pathway is a central driver of tumorigenesis. Genetics 188 : 105–125.
65. KhooP, AllanK, WilloughbyL, BrumbyAM, RichardsonHE (2013) In Drosophila, RhoGEF2 cooperates with activated Ras in tumorigenesis through a pathway involving Rho1-Rok-Myosin-II and JNK signalling. Dis Model Mech 6 : 661–678.
66. RudrapatnaVA, BangiE, CaganRL (2013) A Jnk-Rho-Actin remodeling positive feedback network directs Src-driven invasion. Oncogene [epub ahead of print].
67. WuY, BrockAR, WangY, FujitaniK, UedaR, et al. (2009) A blood-borne PDGF/VEGF-like ligand initiates wound-induced epidermal cell migration in Drosophila larvae. Curr Biol 19 : 1473–1477.
68. BabcockDT, LandryC, GalkoMJ (2009) Cytokine signaling mediates UV-induced nociceptive sensitization in Drosophila larvae. Curr Biol 19 : 799–806.
69. LeschC, JoJ, WuY, FishGS, GalkoMJ (2010) A targeted UAS-RNAi screen in Drosophila larvae identifies wound closure genes regulating distinct cellular processes. Genetics 186 : 943–957.
70. FanY, BergmannA (2010) The cleaved-Caspase-3 antibody is a marker of Caspase-9-like DRONC activity in Drosophila. Cell Death Differ 17 : 534–539.
71. MorenoE, YanM, BaslerK (2002) Evolution of TNF signaling mechanisms: JNK-dependent apoptosis triggered by Eiger, the Drosophila homolog of the TNF superfamily. Curr Biol 12 : 1263–1268.
72. IgakiT, KandaH, Yamamoto-GotoY, KanukaH, KuranagaE, et al. (2002) Eiger, a TNF superfamily ligand that triggers the Drosophila JNK pathway. Embo J 21 : 3009–3018.
73. XueL, IgakiT, KuranagaE, KandaH, MiuraM, et al. (2007) Tumor suppressor CYLD regulates JNK-induced cell death in Drosophila. Dev Cell 13 : 446–454.
74. RutledgeBJ, ZhangK, BierE, JanYN, PerrimonN (1992) The Drosophila spitz gene encodes a putative EGF-like growth factor involved in dorsal-ventral axis formation and neurogenesis. Genes Dev 6 : 1503–1517.
75. SpradlingAC, SternD, BeatonA, RhemEJ, LavertyT, et al. (1999) The Berkeley Drosophila Genome Project gene disruption project: Single P-element insertions mutating 25% of vital Drosophila genes. Genetics 153 : 135–177.
76. MusacchioM, PerrimonN (1996) The Drosophila kekkon genes: novel members of both the leucine-rich repeat and immunoglobulin superfamilies expressed in the CNS. Dev Biol 178 : 63–76.
77. MorrisonCM, HalderG (2010) Characterization of a dorsal-eye Gal4 Line in Drosophila. Genesis 48 : 3–7.
78. McGuireSE, LePT, OsbornAJ, MatsumotoK, DavisRL (2003) Spatiotemporal rescue of memory dysfunction in Drosophila. Science 302 : 1765–1768.
79. TioM, MaC, MosesK (1994) spitz, a Drosophila homolog of transforming growth factor-alpha, is required in the founding photoreceptor cells of the compound eye facets. Mech Dev 48 : 13–23.
80. FreemanM (1994) The spitz gene is required for photoreceptor determination in the Drosophila eye where it interacts with the EGF receptor. Mech Dev 48 : 25–33.
81. KurokawaH, NishioK, FukumotoH, TomonariA, SuzukiT, et al. (1999) Alteration of caspase-3 (CPP32/Yama/apopain) in wild-type MCF-7, breast cancer cells. Oncol Rep 6 : 33–37.
82. DevarajanE, SahinAA, ChenJS, KrishnamurthyRR, AggarwalN, et al. (2002) Down-regulation of caspase 3 in breast cancer: a possible mechanism for chemoresistance. Oncogene 21 : 8843–8851.
83. IolasconA, BorrielloA, GiordaniL, CucciollaV, MorettiA, et al. (2003) Caspase 3 and 8 deficiency in human neuroblastoma. Cancer Genet Cytogenet 146 : 41–47.
84. GhavamiS, HashemiM, AndeSR, YeganehB, XiaoW, et al. (2009) Apoptosis and cancer: mutations within caspase genes. J Med Genet 46 : 497–510.
85. SnipasSJ, DragM, StennickeHR, SalvesenGS (2008) Activation mechanism and substrate specificity of the Drosophila initiator caspase DRONC. Cell Death Differ 15 : 938–945.
86. IrvineKD (2012) Integration of intercellular signaling through the Hippo pathway. Semin Cell Dev Biol 23 (7) 812–7.
87. HalderG, JohnsonRL (2011) Hippo signaling: growth control and beyond. Development 138 : 9–22.
88. LiQ, LiuDW, ZhangLM, ZhuB, HeYT, et al. (2005) Effects of augmentation of liver regeneration recombinant plasmid on rat hepatic fibrosis. World J Gastroenterol 11 : 2438–2443.
89. PawlowskiR, JuraJ (2006) ALR and liver regeneration. Mol Cell Biochem 288 : 159–169.
90. CheraS, GhilaL, WengerY, GalliotB (2011) Injury-induced activation of the MAPK/CREB pathway triggers apoptosis-induced compensatory proliferation in hydra head regeneration. Dev Growth Differ 53 : 186–201.
91. BergmannA, AgapiteJ, McCallK, StellerH (1998) The Drosophila gene hid is a direct molecular target of Ras-dependent survival signaling. Cell 95 : 331–341.
92. KuradaP, WhiteK (1998) Ras promotes cell survival in Drosophila by downregulating hid expression. Cell 95 : 319–329.
93. GerholdAR, RichterDJ, YuAS, HariharanIK (2011) Identification and characterization of genes required for compensatory growth in Drosophila. Genetics 189 : 1309–1326.
94. HuangQ, LiF, LiuX, LiW, ShiW, et al. (2011) Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat Med 17 : 860–866.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 1
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- GATA6 Is a Crucial Regulator of Shh in the Limb Bud
- Large Inverted Duplications in the Human Genome Form via a Fold-Back Mechanism
- Differential Effects of Collagen Prolyl 3-Hydroxylation on Skeletal Tissues
- Affects Plant Architecture by Regulating Local Auxin Biosynthesis
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy