
The Candidate Splicing Factor Sfswap Regulates Growth and Patterning of Inner Ear Sensory Organs
The Notch signaling pathway is thought to regulate multiple stages of inner ear development. Mutations in the Notch signaling pathway cause disruptions in the number and arrangement of hair cells and supporting cells in sensory regions of the ear. In this study we identify an insertional mutation in the mouse Sfswap gene, a putative splicing factor, that results in mice with vestibular and cochlear defects that are consistent with disrupted Notch signaling. Homozygous Sfswap mutants display hyperactivity and circling behavior consistent with vestibular defects, and significantly impaired hearing. The cochlea of newborn Sfswap mutant mice shows a significant reduction in outer hair cells and supporting cells and ectopic inner hair cells. This phenotype most closely resembles that seen in hypomorphic alleles of the Notch ligand Jagged1 (Jag1). We show that Jag1; Sfswap compound mutants have inner ear defects that are more severe than expected from simple additive effects of the single mutants, indicating a genetic interaction between Sfswap and Jag1. In addition, expression of genes involved in Notch signaling in the inner ear are reduced in Sfswap mutants. There is increased interest in how splicing affects inner ear development and function. Our work is one of the first studies to suggest that a putative splicing factor has specific effects on Notch signaling pathway members and inner ear development.
Published in the journal:
. PLoS Genet 10(1): e32767. doi:10.1371/journal.pgen.1004055
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004055
Summary
The Notch signaling pathway is thought to regulate multiple stages of inner ear development. Mutations in the Notch signaling pathway cause disruptions in the number and arrangement of hair cells and supporting cells in sensory regions of the ear. In this study we identify an insertional mutation in the mouse Sfswap gene, a putative splicing factor, that results in mice with vestibular and cochlear defects that are consistent with disrupted Notch signaling. Homozygous Sfswap mutants display hyperactivity and circling behavior consistent with vestibular defects, and significantly impaired hearing. The cochlea of newborn Sfswap mutant mice shows a significant reduction in outer hair cells and supporting cells and ectopic inner hair cells. This phenotype most closely resembles that seen in hypomorphic alleles of the Notch ligand Jagged1 (Jag1). We show that Jag1; Sfswap compound mutants have inner ear defects that are more severe than expected from simple additive effects of the single mutants, indicating a genetic interaction between Sfswap and Jag1. In addition, expression of genes involved in Notch signaling in the inner ear are reduced in Sfswap mutants. There is increased interest in how splicing affects inner ear development and function. Our work is one of the first studies to suggest that a putative splicing factor has specific effects on Notch signaling pathway members and inner ear development.
Introduction
The organ of Corti is an excellent system to study mechanisms of cell patterning due to its highly organized array of sensory cells. It contains one row of inner hair cells, three rows of outer hair cells and several classes of specialized supporting cells, including pillar and Deiters' cells. The signals responsible for this intricate and fine-grained cellular pattern are beginning to be understood, and include the Notch signaling pathway. The Notch1 receptor is expressed in supporting cells, while the Notch ligands Jagged2 (Jag2), Delta1 and Delta3 are expressed in hair cells after they differentiate from prosensory precursors [1], [2], [3], [4]. Supernumerary inner and outer hair cells are generated at the expense of supporting cells in the absence of Notch1, Jag2 or Delta1 [5], [6], [7], [8], [9]. In addition, mutations in members of the Hes and Hey family of downstream Notch effectors also cause an increase in hair cell numbers at the expense of supporting cells, with mutations of multiple Hes/Hey family members causing progressively more severe phenotypes [10], [11]. These studies suggest that lateral inhibition mediated by Notch signaling acts to regulate and maintain the correct proportion of hair cells and supporting cells in inner ear sensory organs.
The Notch ligand, Jagged1 (Jag1) is expressed in all sensory organs of the inner ear prior to the onset of hair cell differentiation [2], [3]. In the developing mouse cochlea, Jag1 is expressed broadly at first, and then becomes excluded from the prosensory domain and restricted to Kölliker's organ by E13.5 [12]. As prosensory progenitors in the cochlea differentiate into hair cells and supporting cells, Jag1 is down-regulated from Kölliker's organ and is expressed with Notch1 in supporting cells [2], [3]. Although several hypotheses have been proposed for the mechanism of Jag1 function in the developing cochlea, the precise role of this gene is still poorly understood. Conditional inactivation of Jag1 in the developing inner ear leads to a severely disrupted organ of Corti [6], [13]. Sensory cells are entirely absent from the basal region of the Jag1 conditional mutant cochlea, whereas two rows of inner hair cells but no outer hair cells are observed in the apical region of the cochlea [6], [13]. Jag1 mutant heterozygotes generated by ENU mutagenesis show a milder phenotype; they lack some cells in the third row of outer hair cells and display ectopic inner hair cells [14], [15].
As part of a study to determine whether self-inactivating (SIN) lentiviruses can be used for efficient insertional mutagenesis in transgenic mice, we used a tyrosinase-expressing lentiviral vector to infect pre-implantation albino (FVB/N) mouse embryos by subzonal injection. Tyrosinase expression rescues albinism and provides a visible, dosage-sensitive, reporter for different integration sites. Transgenic founder (F0) mice were bred to establish families with single lentiviral integration sites and the mice were then inbred and assayed for evidence of insertional mutations. In one family (OVE2267B), homozygous mice displayed a robust circling behavior, suggesting inner ear defects. The lentiviral integration site in this family was mapped to the Sfswap gene. Sfswap was originally identified in Drosophila as a suppressor of the transposon-induced white-apricot mutation [16]. Sfswap encodes an RS-domain containing (SR-Like) protein that is a putative splicing factor. RS-domain containing proteins are known to regulate many aspects of RNA processing, including splicing, transcript elongation, transcript stability, nuclear export and miRNA cleavage as well as genome stability, (reviewed in [17]). In Drosophila, Sfswap regulates splicing of several genes, including Sfswap itself [18], [19], [20]. In vitro evidence suggests that Sfswap is involved in RNA processing in mammals as well by promoting fully spliced transcripts [21], [22], [23]. It is unclear whether Sfswap regulates other aspects of RNA processing, however some evidence in Drosophila suggests Sfswap may influence transcript stability [18], [24]. Our Sfswap mouse mutants have hearing loss, circling behavior, and show cochlear defects that are remarkably similar to those seen in Jag1 hypomorphic mutants - they show reduced numbers of outer hair cells and their associated supporting cells and increased numbers of inner hair cells. Compound mutants of Sfswap and Jag1 have a more pronounced cochlear phenotype and have truncations of their semicircular canals, suggestive of a genetic interaction. Moreover, we show that levels of expression of a number of genes involved in Notch signaling in the inner ear, such as Hey1, Neurl1, Numb, and MamlD1, are also affected in the Sfswap mutants. Our results suggest that Sfswap is necessary for the proper development and patterning of sensory structures of the inner ear and shows a genetic interaction with Jagged1 which may be mediated by a reduction in several genes involved in Notch signaling.
Results
Generation and identification of the SfswapTg allele
We conducted a random insertional mutagenesis study using a tyrosinase-tagged lentiviral vector (Figure 1A) to infect pre-implantation albino (FVB/N) mouse embryos. Lentiviral infection provides a number of distinct advantages for insertional mutagenesis. Lentiviral integration sites are scattered throughout the genome, are single copy, and are well-defined since integration is catalyzed by the lentiviral integrase. Since the injection needle is not inserted into the pronucleus of the embryos, the genomic DNA is not mechanically damaged and the yield of transgenic newborns is much higher. The tyrosinase minigene rescues albinism and provides a dosage-dependent, visible, reporter gene. Greater than 85% of the newborn mice from infected embryos were pigmented, verifying efficient transgenesis and effective expression of the reporter gene (data not shown). F0 mice were bred to FVB/N partners to generate F1 offspring and the F1 mice were again bred to albino mice to establish families with a single lentiviral insertion site. Mice were then inbred and assayed for evidence of insertional mutations. Eighty unique mutant phenotypes were identified (data not shown). This manuscript describes the characterization of the insertional mutation in family OVE2267B. Using inverse PCR, the integration site in this family was amplified, sequenced, and shown to be located in the fourth intron of Sfswap, 115 bases 5′ of exon 5. The mutation was labeled SfswapTg(Tyr)2267BOve (MGI ID: 5287267), and will be referred to throughout this study as SfswapTg or Tg. The tyrosinase minigene allows for identification of genotype by coat color when transgenics are maintained in an albino background [25]. Homozygote Tg mice are more darkly pigmented than heterozygotes and non-transgenic mice are albino. We back-crossed SfswapTg mice onto the FVB/N background for more than 10 generations. Tyrosinase expression and the inner ear phenotypes in homozygous mutants correlated 100% with the insertion in Sfswap as assayed by PCR. Additionally, Southern blots showed a single lentiviral integration site (data not shown). Northern blot analysis of Sfswap revealed that the wild-type Sfswap transcript was significantly reduced in SfswapTg/Tg mice (Figure 1B). In its place we observed an accumulation of Sfswap RNA migrating at an abnormal size (greater than 10 Kb). This RNA is most likely unspliced or incompletely spliced Sfswap mRNA, suggesting a hypomorphic allele. Reverse transcriptase PCR (RT-PCR), followed by sequencing, reveals that some of the Sfswap mRNA produced in SfswapTg/Tg mice includes sequences from the lentiviral insert and exclusion of exons surrounding the insert (data not shown). These abnormal RNAs are not found in mice heterozygous for the transgene, suggesting that SfswapTg is likely to be a recessive allele. Previous studies have demonstrated that Sfswap can regulate the splicing of its own transcript [18], [21], [22]. Disruption of Sfswap function by the mutation may explain why the Sfswap transcript is aberrantly spliced in homozygotes but not heterozygotes.

We initially identified SfswapTg/Tg mutants in our screen because they displayed a significant circling behavior (Figure 1C, Video S1). In a 30-minute open field assay, Tg/Tg mice circle almost five times more than wild-type or heterozygous littermates. SfswapTg/Tg mice are also almost twice as active as littermates (Figure 1D). Since circling behavior is often associated with vestibular dysfunction, we tested SfswapTg/Tg mice for balance defects. We found that Tg/Tg mice curl and grasp their feet in a tail hang assay, circle and tumble in a forced swim test, and have a delay in righting behavior (data not shown), all indicative of vestibular defects [26]. SfswapTg/Tg mutants on the FVB/N background are about 23% smaller than wild-type (WT) littermates at 8 weeks (WT = 29+/−1.11 g, Tg/Tg = 22.28+/−0.99 g p = 0.001), and this size difference is detectable at birth (WT = 1.39+/−0.045 g, Tg/Tg = 1.22+/−0.056 g, p = 0.008). Tg/Tg animals mate at very low frequency in the FVB background, but when crossed to a C57Bl/6 background, the circling behavior ceases and homozygous mice are able to mate more successfully.
Balance defects are often associated with hearing deficits in mice. To test if Sfswap mutants also have hearing defects we measured auditory-evoked brainstem responses (ABR) and found that Tg/Tg mice have an average 18 dB increase in threshold to elicit an auditory response, indicating a moderate degree of hearing loss (Figure 1E). Furthermore, ABR waveforms to auditory stimuli presented at intensities above threshold in SfswapTg/Tg mice have a qualitatively normal shape but smaller peak-to-peak amplitudes (Figure S1). This suggests that the auditory pathway is intact in Sfswap mice, but that fewer neurons are being stimulated to suprathreshold stimuli compared to wild-type mice. We next performed distortion product otoacoustic emissions assays (DPOAE) to identify if the hearing loss is at the level of hair cells. Tg/Tg mutants display an average 15 dB increase in DPOAE thresholds (Figure 1F), suggesting that outer hair cell defects may contribute to the hearing deficits [27]. To test how hearing loss affects behavior, we tested auditory startle responses. In this assay, mice are first acclimated to 70 dB white noise and their movement in response to subsequent varying sound pressures is recorded. As previously reported [28], the magnitude of the startle response increases rapidly between 100–120 dB in wild type mice (Figure 1G). We found that the startle responses in Tg/Tg mice are significantly attenuated compared to wild type. The maximum response we observe in Tg/Tg mice at 118 dB resembles those seen in wild type at sound pressures between 102–106 dB. This reduction in the threshold required to elicit a startle response resembles the increases we observed in ABR and DPOAE thresholds.
RNA in situ hybridization reveals that Sfswap is expressed in the inner ear and brain at E10.5 (Figure 2A). Sfswap is expressed broadly at low levels throughout the E10.5 embryo. Sfswap is expressed broadly and uniformly in the cochlea and surrounding mesenchyme as early as E13.5 in wild-type embryos (Figure 2B, C). Expression of Sfswap persists through birth and is maintained broadly in the cochlea, spiral ganglion, cristae, utricle, and saccule, but is reduced in the surrounding tissues (Figure 2D, E, F, G).

SfswapTg/Tg mutants exhibit defects in hair cells and supporting cells in the cochlea
To study the effects of the SfswapTg mutation on inner ear morphology, we performed paint fills of the inner ear at E13.5–16.5. We found no defects in semicircular canal structure, and all components of the inner ear appear to be present. However, the cochlea is shorter, and the organs of the vestibular labyrinths are reduced in size (Figure 3A). Measurement of the flat-mounted cochlear preparations reveals a 38% reduction in the length of the cochlea (WT = 5058.5+/−201.45 µm Tg/Tg = 3135.9+/−221.92 µm p = 5×10−6). We stained surface preparations of newborn Tg/Tg cochleas with fluorescently-labeled phalloidin to detect actin in stereocilia and found regions of the mutant cochlea are missing the third row of outer hair cells and regions that have ectopic inner hair cells (Figure 3B, C). We quantified the number of hair cells per 200 µm and found there are significantly fewer third row outer hair cells throughout the length of the cochlea and significantly more inner hair cells at the apex (Figure 3C, Table 1). Although inner hair cells are locally increased, we found a significant decrease in total numbers of both inner and outer hair cells (37.5% and 50% respectively; Figure 3D).


To determine if there are also defects in supporting cells in SfswapTg/Tg cochleas, we examined expression of the transcription factor Prox1 on whole mount and sectioned inner ears to reveal pillar cells and Deiters' cells (Figure 4A, B) [29]. We found that in regions of the cochlea where outer hair cells are missing, a row of Prox1+ supporting cells are typically missing as well. Based on morphology and location, the missing supporting cell is likely a Deiters' cell or the outer pillar cell. In addition, we examined expression of the low affinity NGF receptor p75 that marks pillar cell apical processes (Figure 4C) and β-tectorin, which marks pillar cells and the greater epithelial ridge (Figure 4D). We found that in the mid-regions of the mutant cochlea individual pillar cell pairs are often absent, and replaced with an ectopic cell that is either a hair cell or an undifferentiated cell. β-tectorin is also absent in some regions of the mutant cochlea in a location corresponding to pillar cells, suggesting that the supporting cell loss is due at least in part to missing pillar cells. We also found that the hair cell marker parvalbumin is occasionally ectopically expressed in the Hensen's cell region of Tg/Tg mutants (Figure 4B). These ectopic parvalbumin-positive cells did not express other hair cell markers such as Myosin VI or show hair bundles with phalloidin staining, suggesting these ectopic cells are not bona fide hair cells, but rather represent mis-expression of at least one hair cell marker.

To determine if the cochlear defects we observed in Tg/Tg mice were due to gross abnormalities in the formation of the prosensory domain which gives rise to the organ of Corti, we examined mutant and wild-type animals for expression of the prosensory domain markers Sox2, and p27kip1 by antibody staining, and Hey2 by in situ hybridization. We used Jag1 antibodies and an in situ probe for Bmp4 to reveal the greater epithelial ridge and outer sulcus, respectively, which form a boundary with the prosensory domain. We did not observe any significant differences in the size of the prosensory domain, nor of the regions of the cochlea that border the prosensory domain in SfswapTg/Tg (Figure S2).
SfswapTg/Tg mutant mice have severe vestibular organ defects
As described above, SfswapTg/Tg mice were initially identified as exhibiting circling behavior indicative of a vestibular defect. To analyze cellular defects that might cause this behavior, we stained flat mounts of the cristae and maculae of newborn mice with fluorescently labeled phalloidin to detect actin in stereociliary bundles, and measured the area of the sensory structures. Anterior and horizontal canal cristae remained attached to the utricle during dissection so that they could be unambiguously identified based on location. All semicircular canal cristae and the maculae of the utricle and saccule are smaller in Tg/Tg mutants than in wild-type mice (Figure 5A, B, C). Strikingly, the saccule is reduced to 25% of wild-type size. In addition, the anterior semicircular canal cristae exhibited smaller or absent eminentia cruciata in 85% of ears examined. The defects observed in vestibular organs likely contribute to the circling behavior of Sfswap mutant mice. We further analyzed the source of this reduction by examining early known markers of the vestibular cristae and maculae, Bmp4 and Lunatic Fringe (Lfng), respectively [30]. We found that the anterior stripe of Bmp4 expression, which corresponds to the presumptive horizontal and anterior canal cristae, is reduced in mutants at E10.5. The posterior spot, which will develop into the posterior crista, is small or indiscernible at this age (Figure 5D). Similarly, Lfng expression, which marks the future utricle, saccule, and neurogenic domain, is severely reduced in mutants at E10.5 (Figure 5D). These data suggest that the size reduction in the maculae and cristae in SfswapTg/Tg mice may represent an early developmental defect.

Finally, we tested for defects in mechanotransduction in the hair cells of SfswapTg/Tg mutant pups by intraperitoneal injection of the dye AM1-43, which can be taken up through mechanotransduction channels [31]. We found no differences in AM1-43 uptake in maculae, cristae, or cochleas between mutant and control mice (Figure S3). This data combined with the ABR and DPOAE data suggest that the remaining hair cells in SfswapTg/Tg mutant mice are likely to be functional.
Sfswap interacts genetically with Jag1
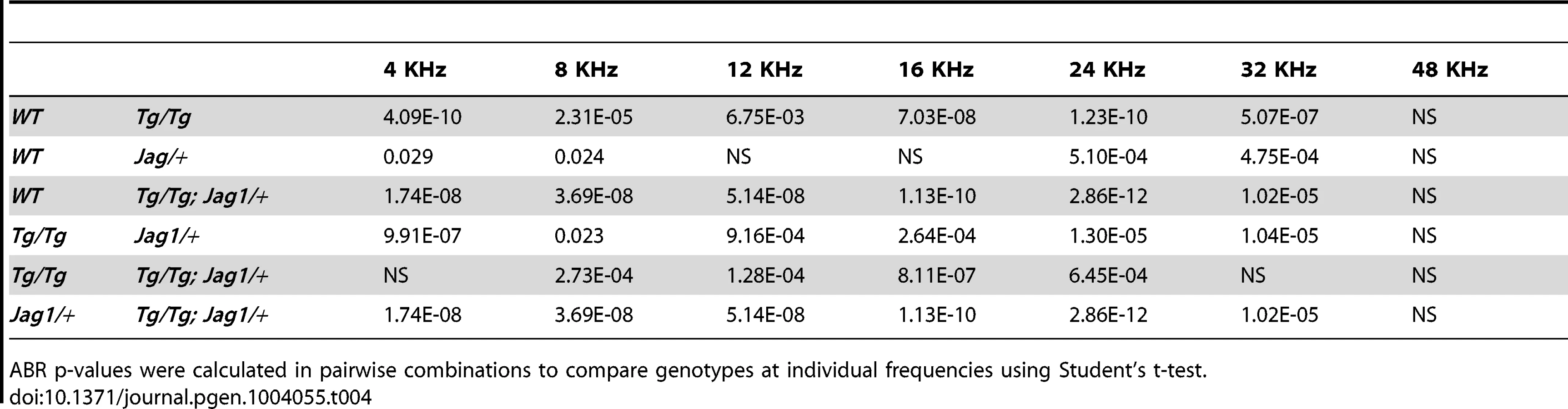
SfswapTg/Tg mutants have a cochlear phenotype that is strikingly similar to that of Jag1 heterozygotes and point mutants. Jag1 heterozygous mutants display loss of the third row of outer hair cells, ectopic or extra inner hair cells, reduced expression of Bmp4 and Lfng at 10.5, disruptions in canal cristae and utricular macula, and small body size [14], [15], [32]. These defects can vary according to the genetic background [33]. We found that Jag1/+ mutants also have fewer supporting cells in a pattern similar to SfswapTg/Tg mutants (data not shown). These similarities lead us to hypothesize that Sfswap and Jag1 function in the same genetic pathway. To test this, we crossed Jag1 knockout mice (B6;129S-Jag1tm1Grid/J referred to hereafter as Jag1+/−) maintained on a C57BL/6 background, with our SfswapTg/+ mutants raised in an FVB/N background to obtain SfswapTg/+; Jag1+/− F1 progeny. These were then crossed again to SfswapTg/+ mice on an FVB/N background to generate SfswapTg/Tg; Jag1+/ − mutants. Most of these compound mutants (10/14) exhibited semicircular canal truncations, but neither SfswapTg/Tg nor Jag1+/− mutants have canal truncations on this genetic background (Figure 6A). Jag1+/− mutants are known to have variations in semicircular canal defects depending on the genetic background [33]. To test if this is the case for the FVB/N background on which our SfswapTg mice were maintained, Jag1+/− mice were crossed one and two generations to wild-type FVB/N mice. Surprisingly, one generation was enough to completely suppress the canal truncations (Figure 6B). This indicates that there is a strong suppressor of canal truncations in the FVB/N background, and the canal truncations found in SfswapTg/Tg; Jag1+/− mice are therefore strongly indicative of an interaction between these two genes. To further test this hypothesis, we stained cochlear flat mounts from single and compound mutants with fluorescently labeled phalloidin to visualize stereocilia. We found that SfswapTg/Tg; Jag1+/− mutants have a more pronounced cochlear phenotype than either single mutant alone, showing a loss of outer hair cells extending into the second and first rows, increased inner hair cells throughout the length of the cochlea and the addition of a fourth row of outer hair cells in the apex (Figure 6C). In addition, compound mutant cochleas are significantly shorter than either Sfswap or Jag1 mutant cochleas (Figure 6D, Table 2). Quantitatively, the number of outer hair cells in the mid-turn and base of the cochlea are significantly fewer in the compound mutant than either mutant alone (Figure 6E, Table 2). For audiological measurements, we out-crossed the Jag1+/− mice to the FVB background for an additional three generations. These Jag1+/− mice were then crossed to SfswapTg/+ mice to generate compound heterozygotes that were then intercrossed to produce SfswapTg/Tg; Jag1+/−, Jag1+/− and SfswapTg/Tg mice. ABR and DPOAE measurements of compound mutants also showed increased threshold compared to those of Tg/Tg or Jag1 mutants (Figure 6F, G; Tables 3–6). Interestingly, we find that Jag1+/− mice have a significant increase compared to WT ABR and DPOAE thresholds; this difference is evident at high frequencies. However, compound mutants also have increased thresholds compared to Tg/Tg at frequencies below 20 kHz, a range that is only mildly affected in Jag1+/− mice. The defects found in SfswapTg/Tg; Jag1+/− cochlear hair cells, semicircular canals, and auditory responses are greater than expected for a simple additive effect, implying a synergistic relationship between Jag1 and Sfswap in inner ear development.





To further analyze the mechanism for Sfswap's interaction with Jag1, we analyzed expression of Jag1 in cochlear whole mounts and at the otocyst stage, but found no differences in expression (Figure 7A, B). We next examined expression of the downstream target Hey1 at E10.5 and found a significant reduction in expression (Figure 7C), despite normal Jag1 expression. To determine if transcripts for genes in the Notch signaling pathway are incorrectly spliced in Tg/Tg mutants, we performed RT-PCR of splice junctions for Notch pathway genes using inner ear mRNA. We analyzed Jag1, Notch1, Hes1, Hes5, Hey1, Hey2, HeyL, Mfng, Lfng, Rbpj, Delta1, Numb, NumbL, Maml1, Maml2, Maml3, MamlD1, Neuralized1A (Neurl1A), and Sfswap and found no differences in splicing in any gene except for Sfswap (Figure 7D and data not shown). We found slight reductions in levels of Neurl1A and Numb mRNA and significant reductions in MamlD1 mRNA visible by RT-PCR, all of which could potentially contribute to the Jagged1-like phenotype of SfswapTg/Tg mice. To test for splice differences in these genes that would not be detectible by RT-PCR, we performed Northern blots using brain RNA. However, we found no detectable differences in splicing in Sfswap mutants in Jag1, Neurl1A, MamlD1, or Numb (Figure S4).

Discussion
Using a lentivirus-based insertional mutagenesis strategy, we have identified the putative splicing factor Sfswap as an essential gene for inner ear development. SfswapTg/Tg mutants exhibit circling and balance dysfunction associated with vestibular defects and also have a moderate (20–25 dB) hearing loss. The mutants exhibit a partial loss of the third row of outer hair cells and show ectopic inner hair cells, together with some missing pillar cells. All organs in the vestibular system are smaller than their wild-type counterparts, particularly the utricle and saccule, which are less than 50% of the size of wild-type organs. Our data are consistent with Sfswap regulating aspects of Jagged1 signaling. First, the cochlear phenotype of SfswapTg/Tg mice is strikingly similar to that seen in Jag1 heterozygous point mutants [14], [15]. Second, SfswapTg/Tg mice have otocyst patterning defects similar to those seen in Jag1 mutants [32]. Finally, we show that our Sfswap mutant allele exacerbates the phenotype of Jag1 heterozygous mice. We discuss these phenotypes in more detail below.
SfswapTg/Tg mutants have reduced levels of the early sensory markers Bmp4, Lfng, and Hey1 and significantly smaller sensory organs. Jag1 conditional mutants similarly have severely reduced vestibular structures [13]. Jag1 mutants also show changes in expression of sensory markers such as Bmp4, Sox2, Lfng and Hey1 at E10.25 [32]. Vestibular structures are also reduced in Bmp4 conditional or hypomorphic mutants [34], particularly in the horizontal lateral canal. We suggest the reduction of Bmp4 expression in our mutants may be due to defects in Jag1 signaling and leads to the smaller vestibular structures and reduced eminentia cruciata of the anterior crista that we observe in SfswapTg/Tg mutants.
The unique combination of loss of outer hair cells and ectopic inner hair cells that we observe in the cochleas of SfswapTg/Tg mutants has only been observed previously in heterozygous knockout or point mutants of Jag1 [14], [15]. To date, there is no definitive molecular explanation for this unique phenotype. Kiernan and colleagues [14] suggested that Jag1 may have two roles, an early one in which Jag1 helps specify sensory organs, and a later one in which Jag1 helps define hair cell and supporting cell identity through lateral inhibition. We did not see significant changes in the size of the cochlear prosensory domain in SfswapTg/Tg mutants, although if there are small changes in the prosensory domain, they would be difficult to discern using the markers and techniques currently available. Ectopic rows of inner hair cells can be seen in Jag1 heterozygotes and in apical regions of the Jag1 conditional homozygote cochlea [6], [13]. In SfswapTg/Tg mutants, we also observe an extra row of inner hair cells in the apical region of the cochlea. Since Jag1 is expressed at the boundary of the greater epithelial ridge and the prosensory domain where the first inner hair cells form, it is possible that a partial loss of Jag1-Notch signaling leads to the inappropriate formation of extra inner hair cells in Jag1 and SfswapTg/Tg mutants.
Although our data support a genetic interaction between Sfswap and Jag1, we currently have no evidence for a direct interaction. It is possible that Jag1 mRNA splicing or stability is regulated by Sfswap. However, we did not detect significant differences in the levels or splicing pattern of Jag1 transcripts, and we have not detected significant differences in Jag1 protein expression in the cochlea by immunofluorescence. Since the cochlear phenotype of Jag1 mutants can vary according to both the type of mutation (point mutation versus null) and the genetic background on which mutants are maintained [13], [14], [15], [33], it is clear that hair cell patterning in the cochlea is exquisitely sensitive to levels of Jag1. It is possible that SfswapTg/Tg mice are causing small changes in Jag1 expression below the limits of detection that are sufficient to affect cochlear patterning. Alternatively, it is possible that Sfswap is regulating the splicing or stability of modifiers of the Jag1 locus. We screened a variety of Notch pathway genes for possible expression changes in Sfswap mutant cochleas. Several genes showed significant changes by RT-PCR including the mouse neuralized homologue Neurl1A, the Notch transcriptional co-activator MamlD1, and Numb (Figure 7D). Neurl1A is an E3 ubiquitin ligase that has been shown to regulate turnover and endocytosis of Jag1 in vitro [35]. It is therefore possible that small changes in Neurl1A activity may be sufficient to disrupt hair cell patterning. Similarly, Numb has been shown to regulate Notch signaling through endocytosis and ubiquitinization of the intracellular domain of Notch [36], [37]. Interestingly, Numb has recently been shown to be broadly expressed in the rat cochlea during development and over-expression can modulate levels of Atoh1, suggesting a possible role in hair cell development [38]. Mastermind proteins are transcriptional co-activators that are essential for Notch signaling [39]. It is possible that a reduction in MamlD1 can affect Jagged1-Notch signaling through a reduction in canonical or non-canonical Notch signaling [40], [41]. It is also possible that the simultaneous reduction in two or more of these proteins results in the phenotypes seen in the Sfswap mutants.
Sfswap was discovered in Drosophila as a suppressor of the transposon-induced white-apricot mutation [16]. White-apricot mutants have a transposon insertion that disrupts the white transcript by splice inclusion and this disruption is partially suppressed by mutation of Sfswap through selective exclusion of the transposon [42], [43], [44]. Beyond this system, no evidence has been identified for the in vivo function or targets of Sfswap in Drosophila. In mice, in vitro studies have identified Fibronectin and CD45 as putative splice targets of Sfswap [21], although in vivo targets have yet to be confirmed. In humans, the nonsyndromic autosomal dominant deafness locus DFNA41 contains at least 100 genes, including SFSWAP [45]. The pathological mutation in this region has yet to be identified, making SFSWAP a potential candidate for future gene sequencing efforts.
Many genes undergo complex splicing patterns in the inner ear. These include several genes that are involved in ion transport including Atp2b [46], Kcnq4 [47], [48], Cav1.3 [49], BK channels [50], [51], [52], and P2X2 [53]. Spliced isoforms of these channels have been proposed to be involved in electrical differences in tonotopy, electromotility of outer hair cells, and differences between sensory and neuronal cells. Alternative splicing has been found to affect protein targeting [49], [54], [55], electrical properties channels [56], [57], [58], [59], cell viability [60], [61], and stereociliary organization [62] in the inner ear. There are also several examples of mutations in distinct alternative spliced isoforms that cause different pathologies in the ear. For example, loss of one isoform of PCDH15 results in stereociliary defects, while mutations in the other two isoforms of this gene have no significant effect on hair cells [62]. Similarly, mutations in harmonin are typically associated with Usher Syndrome 1C, whereas a mutation in an alternative exon results in non-syndromic deafness at the DFNB18 locus with normal vision [63]. Despite the significance of alternative splicing in the inner ear, only one splicing factor has been identified that functions in the inner ear [61]. Srrm4 is an SR-like protein that has recently been identified to be mutated in the Bronx waltzer (bv) mouse. Mutation at this locus results in degeneration of inner hair cells after E17.5 [64]. Gene ontology analysis of exons regulated by Srrm4 suggests this factor regulates splicing of genes involved in synaptic transmission and the secretory pathway. We have now identified a second SR-like protein, Sfswap, which has distinct and highly specific effects on patterning of the inner ear. Our work provides the first evidence that a putative splicing factor is necessary for establishing the size of sensory organs in the ear and for proper patterning of mechanosensory hair cells in the organ of Corti.
Materials and Methods
Generation of Sfswap transgenic mice
A detailed account of the generation of Sfswap transgenic mice by lentiviral insertional mutagenesis with a tyrosinase minigene is given in Text S1.
RNA analysis
For RT-PCR, RNA was isolated from E15.5 inner ears using the Ambion PureLink RNA mini kit (12183018A) according to the manufacturer's instructions. cDNA was generated using the Superscript III First Strand system (Invitrogen 18080-051) according to manufacturer's instructions. RT-PCRs for exon splicing events were performed using exon tiling primers (Tables S1 and S2). See Text S1 for Northern blot protocol.
Probe generation
Plasmid probes for in situ hybridizations and Northern blots were obtained from Brigid Hogan (Bmp4),Gerry Weinmaster (Jag1), Elias Pavlopoulos (Neurl1A), Yutaka Hata (Numb, Addgene Plasmid 37012 [65]) or cloned using TOPO 2.1 vector (Life Technologies). PCR primers used to clone probes are as follows: Sfswap exons 1–4: F-GCTGTGTTGAAGTTGCGAAG and, R-CATCAGACGGGACGCTTAAT; Sfswap exons 15–18: F-AAAGGACCCGTTCCAGAAGT and R-CCACTGACTGACCCAGGAGT; beta-Actin: F - TGTTACCAACTGGGACGACA and R - AAGGAAGGCTGGAAAAGAGC; MamlD1 F-TCCATTTCCCATCTCCTCAG and R - AGCCTTCCAAAAGCTCTTCC.
Some in situ probe templates were generated through direct PCR with the addition of a T7 promoter (T7 sequence is underlined). These include Lfng: F-GTTCCGCTCTGTCCATTGC R - GGATCCTAATACGACTCACTATAGGGAGCCCACTATGGGCGACTTTC and Hey1: F-AGACCTTGGGGGACAGAGAT and R-GGATCCTAATACGACTCACTATAGGGAGAACGGTGAAATCCGTGAGAC.
Histology and in situ hybridization
For cryosections, heads were fixed in 4% paraformaldehyde overnight at 4 degrees. Heads were then washed briefly in PBS, immersed in 30% sucrose overnight, equilibrated in OCT, then embedded in OCT. Sections were taken between 8 and 14 µm. For P0 and older whole mounts, heads were fixed overnight in 4% paraformaldehyde. Cochleas, cristae, and maculae were then dissected out and processed. For immunohistochemistry, some tissue was first boiled for 10 minutes in 10 mM citric acid for antigen retrieval. All samples were washed 3 times in PBS, then 30 minutes in blocking buffer (PBS with 10% goat serum and 0.1% Triton-X or Tween-20). Tissue was then stained overnight in primary antibody diluted in blocking buffer. Tissue was then washed 3 times in PBS then incubated for 2 hours with the appropriate secondary antibody at 1∶400 dilution in blocking buffer. In tissues where Topro3 (1∶10,000, Invitrogen) or phalloidin (1∶200, Invitrogen) were used, they were added to the secondary antibody cocktail. Tissue was then washed 3 times in PBS and mounted in either Vectashield (Vector Labs) or Prolong Gold+Antifade (Invitrogen).
Antibodies used: Myo6 (Proteus), Prox1 (Millipore Bioscience Research Reagents), Parv (Sigma), Jag1 (Santa Cruz Biotechnology), p27Kip1 (Neomarker), p75 (Advanced Targeting Systems), Alexa 488 anti mouse and rabbit (Invitrogen), Cy5 anti mouse and rabbit (Jackson Immunolabs). In situ hybridization was performed as previously described [66], [67]. Digoxigenin labeled riboprobes were synthesized according to standard protocols [68].
Imaging and measurements
Images were taken using a Zeiss LSM 510 confocal microscope, a Zeiss Axiophot microscope, or a Zeiss dissecting scope. Images were processed using Axiovision software or ImageJ, then further processed in Adobe Photoshop. Inner and outer hair cells were counted in P0 cochleas per 200 µm using Axiovision software. Cochlea lengths were measured at P0 using Axiovision software. Vestibular areas were measured on P0 vestibular flat mounts using ImageJ.
Paint fills
Paint fills were performed as previously described [69], [70]. In brief, E13.5–E16.5 heads were fixed in Bodian's fix. Heads were dehydrated in an ethanol series, and then cleared with methyl salicylate. Inner ears were filled with white gloss paint in methyl salicylate using a Picospritzer III pressure injector (General Valve Corporation) and stored and photographed in methyl salicylate.
Behavioral analysis and audiological measurements
Open field activity was measured using the Versamax System for automated activity recording. Mice were acclimated for at least 30 minutes to the testing room illuminated to 400 lux light and with 60 dB white noise before testing. The open field is a 40×40×30 cm Plexiglas arena. Tests were performed for 30 minutes during which time horizontal and vertical activity were measured via beam breaks. Chambers were cleaned with alcohol before and after each run to minimize interfering odors. Pre-pulse inhibition and startle responses were measured using a San Diego Instruments system. Mice were acclimated for at least 30 minutes in a separate nearby room before testing. Mice were placed in cylindrical restraint tube in a sound-attenuating chamber to minimize movement of the mouse and interfering noise. Mice were acclimated in the testing chamber to 70 dB white noise for 5 minutes before test pulses were delivered. Mice were then presented with 6 rounds of 8 test types in a pseudorandom order with 10–20 seconds between trials. These consist of no stimulus, startle at 120 dB for 40 ms, pre-pulse trials (74, 78, and 82 dB for 20 ms) and pre-pulse inhibition trials of each sound 100 ms prior to a 120 dB startle. Responses were recorded every 1 ms for 65 ms following the stimulus. Tg/Tg mice fail to present a threshold level startle response, so percent pre-pulse inhibition could not be calculated and only startle responses are reported. ABR and DPOAE were performed as previously described [71].
Statistics
Significance was measured using students T-Test or ANOVA with repeated measures where appropriate using SPSS software. Graphs were generated using Prism.
Ethics statement
All animal experiments in this study were carried out in accordance with the Institutional Animal Care and Use Committee at Baylor College of Medicine.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. LewisAK, FrantzGD, CarpenterDA, de SauvageFJ, GaoWQ (1998) Distinct expression patterns of notch family receptors and ligands during development of the mammalian inner ear. Mech Dev 78 : 159–163.
2. MurataJ, TokunagaA, OkanoH, KuboT (2006) Mapping of notch activation during cochlear development in mice: implications for determination of prosensory domain and cell fate diversification. J Comp Neurol 497 : 502–518.
3. MorrisonA, HodgettsC, GosslerA, Hrabe de AngelisM, LewisJ (1999) Expression of Delta1 and Serrate1 (Jagged1) in the mouse inner ear. Mech Dev 84 : 169–172.
4. HartmanBH, HayashiT, NelsonBR, Bermingham-McDonoghO, RehTA (2007) Dll3 is expressed in developing hair cells in the mammalian cochlea. Dev Dyn 236 : 2875–2883.
5. KiernanAE, CordesR, KopanR, GosslerA, GridleyT (2005) The Notch ligands DLL1 and JAG2 act synergistically to regulate hair cell development in the mammalian inner ear. Development 132 : 4353–4362.
6. BrookerR, HozumiK, LewisJ (2006) Notch ligands with contrasting functions: Jagged1 and Delta1 in the mouse inner ear. Development 133 : 1277–1286.
7. LanfordPJ, LanY, JiangR, LindsellC, WeinmasterG, et al. (1999) Notch signalling pathway mediates hair cell development in mammalian cochlea. Nat Genet 21 : 289–292.
8. ZhangN, MartinGV, KelleyMW, GridleyT (2000) A mutation in the Lunatic fringe gene suppresses the effects of a Jagged2 mutation on inner hair cell development in the cochlea. Curr Biol 10 : 659–662.
9. DoetzlhoferA, BaschML, OhyamaT, GesslerM, GrovesAK, et al. (2009) Hey2 regulation by FGF provides a Notch-independent mechanism for maintaining pillar cell fate in the organ of Corti. Dev Cell 16 : 58–69.
10. TateyaT, ImayoshiI, TateyaI, ItoJ, KageyamaR (2011) Cooperative functions of Hes/Hey genes in auditory hair cell and supporting cell development. Dev Biol 352 : 329–340.
11. LiS, MarkS, Radde-GallwitzK, SchlisnerR, ChinMT, et al. (2008) Hey2 functions in parallel with Hes1 and Hes5 for mammalian auditory sensory organ development. BMC Dev Biol 8 : 20.
12. OhyamaT, BaschML, MishinaY, LyonsKM, SegilN, et al. (2010) BMP signaling is necessary for patterning the sensory and nonsensory regions of the developing mammalian cochlea. J Neurosci 30 : 15044–15051.
13. KiernanAE, XuJ, GridleyT (2006) The Notch ligand JAG1 is required for sensory progenitor development in the mammalian inner ear. PLoS Genet 2: e4.
14. KiernanAE, AhituvN, FuchsH, BallingR, AvrahamKB, et al. (2001) The Notch ligand Jagged1 is required for inner ear sensory development. Proc Natl Acad Sci U S A 98 : 3873–3878.
15. TsaiH, HardistyRE, RhodesC, KiernanAE, RobyP, et al. (2001) The mouse slalom mutant demonstrates a role for Jagged1 in neuroepithelial patterning in the organ of Corti. Hum Mol Genet 10 : 507–512.
16. GreenMM (1959) Spatial and functional properties of pseudo-alleles at the white locus in Drosophila melanogaster. Heredity 303–315.
17. TwyffelsL, GueydanC, KruysV (2011) Shuttling SR proteins: more than splicing factors. FEBS J 278 : 3246–3255.
18. ZacharZ, ChouTB, BinghamPM (1987) Evidence that a regulatory gene autoregulates splicing of its transcript. EMBO J 6 : 4105–4111.
19. ZacharZ, ChouTB, KramerJ, MimsIP, BinghamPM (1994) Analysis of autoregulation at the level of pre-mRNA splicing of the suppressor-of-white-apricot gene in Drosophila. Genetics 137 : 139–150.
20. RutledgeBJ, MortinMA, SchwarzE, Thierry-MiegD, MeselsonM (1988) Genetic interactions of modifier genes and modifiable alleles in Drosophila melanogaster. Genetics 119 : 391–397.
21. SarkissianM, WinneA, LafyatisR (1996) The mammalian homolog of suppressor-of-white-apricot regulates alternative mRNA splicing of CD45 exon 4 and fibronectin IIICS. J Biol Chem 271 : 31106–31114.
22. DenhezF, LafyatisR (1994) Conservation of regulated alternative splicing and identification of functional domains in vertebrate homologs to the Drosophila splicing regulator, suppressor-of-white-apricot. J Biol Chem 269 : 16170–16179.
23. LemaireR, WinneA, SarkissianM, LafyatisR (1999) SF2 and SRp55 regulation of CD45 exon 4 skipping during T cell activation. Eur J Immunol 29 : 823–837.
24. MountSM, GreenMM, RubinGM (1988) Partial revertants of the transposable element-associated suppressible allele white-apricot in Drosophila melanogaster: structures and responsiveness to genetic modifiers. Genetics 118 : 221–234.
25. Overbeek PA (2002) Factors affecting transgenic animal production; Pinkert C, editor. New York, NY: Elsevier Science.
26. Hardisty-HughesRE, ParkerA, BrownSD (2010) A hearing and vestibular phenotyping pipeline to identify mouse mutants with hearing impairment. Nat Protoc 5 : 177–190.
27. SalviRJ, DingD, WangJ, JiangHY (2000) A review of the effects of selective inner hair cell lesions on distortion product otoacoustic emissions, cochlear function and auditory evoked potentials. Noise Health 2 : 9–26.
28. PaylorR, CrawleyJN (1997) Inbred strain differences in prepulse inhibition of the mouse startle response. Psychopharmacology (Berl) 132 : 169–180.
29. Bermingham-McDonoghO, OesterleEC, StoneJS, HumeCR, HuynhHM, et al. (2006) Expression of Prox1 during mouse cochlear development. J Comp Neurol 496 : 172–186.
30. MorsliH, ChooD, RyanA, JohnsonR, WuDK (1998) Development of the mouse inner ear and origin of its sensory organs. J Neurosci 18 : 3327–3335.
31. MeyersJR, MacDonaldRB, DugganA, LenziD, StandaertDG, et al. (2003) Lighting up the senses: FM1-43 loading of sensory cells through nonselective ion channels. J Neurosci 23 : 4054–4065.
32. PanW, JinY, StangerB, KiernanAE (2010) Notch signaling is required for the generation of hair cells and supporting cells in the mammalian inner ear. Proc Natl Acad Sci U S A 107 : 15798–15803.
33. KiernanAE, LiR, HawesNL, ChurchillGA, GridleyT (2007) Genetic background modifies inner ear and eye phenotypes of jag1 heterozygous mice. Genetics 177 : 307–311.
34. ChangW, LinZ, KulessaH, HebertJ, HoganBL, et al. (2008) Bmp4 is essential for the formation of the vestibular apparatus that detects angular head movements. PLoS Genet 4: e1000050.
35. KoutelouE, SatoS, Tomomori-SatoC, FlorensL, SwansonSK, et al. (2008) Neuralized-like 1 (Neurl1) targeted to the plasma membrane by N-myristoylation regulates the Notch ligand Jagged1. J Biol Chem 283 : 3846–3853.
36. McGillMA, DhoSE, WeinmasterG, McGladeCJ (2009) Numb regulates post-endocytic trafficking and degradation of Notch1. J Biol Chem 284 : 26427–26438.
37. McGillMA, McGladeCJ (2003) Mammalian numb proteins promote Notch1 receptor ubiquitination and degradation of the Notch1 intracellular domain. J Biol Chem 278 : 23196–23203.
38. GaoZ, ChiFL, HuangYB, YangJM, CongN, et al. (2011) Expression of Numb and Numb-like in the development of mammalian auditory sensory epithelium. Neuroreport 22 : 49–54.
39. NamY, SlizP, SongL, AsterJC, BlacklowSC (2006) Structural basis for cooperativity in recruitment of MAML coactivators to Notch transcription complexes. Cell 124 : 973–983.
40. FukamiM, WadaY, OkadaM, KatoF, KatsumataN, et al. (2008) Mastermind-like domain-containing 1 (MAMLD1 or CXorf6) transactivates the Hes3 promoter, augments testosterone production, and contains the SF1 target sequence. J Biol Chem 283 : 5525–5532.
41. WuL, SunT, KobayashiK, GaoP, GriffinJD (2002) Identification of a family of mastermind-like transcriptional coactivators for mammalian notch receptors. Mol Cell Biol 22 : 7688–7700.
42. PirrottaV, BrocklC (1984) Transcription of the Drosophila white locus and some of its mutants. EMBO J 3 : 563–568.
43. LevisR, O'HareK, RubinGM (1984) Effects of transposable element insertions on RNA encoded by the white gene of Drosophila. Cell 38 : 471–481.
44. ZacharZ, DavisonD, GarzaD, BinghamPM (1985) A detailed developmental and structural study of the transcriptional effects of insertion of the Copia transposon into the white locus of Drosophila melanogaster. Genetics 111 : 495–515.
45. BlantonSH, LiangCY, CaiMW, PandyaA, DuLL, et al. (2002) A novel locus for autosomal dominant non-syndromic deafness (DFNA41) maps to chromosome 12q24-qter. J Med Genet 39 : 567–570.
46. ChenQ, ChuH, WuX, CuiY, ChenJ, et al. (2011) The expression of plasma membrane Ca(2+)-ATPase isoform 2 and its splice variants at sites A and C in the neonatal rat cochlea. Int J Pediatr Otorhinolaryngol 75 : 196–201.
47. BeiselKW, Rocha-SanchezSM, MorrisKA, NieL, FengF, et al. (2005) Differential expression of KCNQ4 in inner hair cells and sensory neurons is the basis of progressive high-frequency hearing loss. J Neurosci 25 : 9285–9293.
48. Rocha-SanchezSM, MorrisKA, KacharB, NicholsD, FritzschB, et al. (2007) Developmental expression of Kcnq4 in vestibular neurons and neurosensory epithelia. Brain Res 1139 : 117–125.
49. ShenY, YuD, HielH, LiaoP, YueDT, et al. (2006) Alternative splicing of the Ca(v)1.3 channel IQ domain, a molecular switch for Ca2+-dependent inactivation within auditory hair cells. J Neurosci 26 : 10690–10699.
50. RamanathanK, MichaelTH, JiangGJ, HielH, FuchsPA (1999) A molecular mechanism for electrical tuning of cochlear hair cells. Science 283 : 215–217.
51. LangerP, GrunderS, RuschA (2003) Expression of Ca2+-activated BK channel mRNA and its splice variants in the rat cochlea. J Comp Neurol 455 : 198–209.
52. SakaiY, HarveyM, SokolowskiB (2011) Identification and quantification of full-length BK channel variants in the developing mouse cochlea. J Neurosci Res 89 : 1747–1760.
53. HousleyGD, KanjhanR, RaybouldNP, GreenwoodD, SalihSG, et al. (1999) Expression of the P2X(2) receptor subunit of the ATP-gated ion channel in the cochlea: implications for sound transduction and auditory neurotransmission. J Neurosci 19 : 8377–8388.
54. HillJK, WilliamsDE, LeMasurierM, DumontRA, StrehlerEE, et al. (2006) Splice-site A choice targets plasma-membrane Ca2+-ATPase isoform 2 to hair bundles. J Neurosci 26 : 6172–6180.
55. GratiM, AggarwalN, StrehlerEE, WentholdRJ (2006) Molecular determinants for differential membrane trafficking of PMCA1 and PMCA2 in mammalian hair cells. J Cell Sci 119 : 2995–3007.
56. FicarellaR, Di LevaF, BortolozziM, OrtolanoS, DonaudyF, et al. (2007) A functional study of plasma-membrane calcium-pump isoform 2 mutants causing digenic deafness. Proc Natl Acad Sci U S A 104 : 1516–1521.
57. XuT, NieL, ZhangY, MoJ, FengW, et al. (2007) Roles of alternative splicing in the functional properties of inner ear-specific KCNQ4 channels. J Biol Chem 282 : 23899–23909.
58. ChenC, ParkerMS, BarnesAP, DeiningerP, BobbinRP (2000) Functional expression of three P2X(2) receptor splice variants from guinea pig cochlea. J Neurophysiol 83 : 1502–1509.
59. BrandleU, SpielmannsP, OsterothR, SimJ, SurprenantA, et al. (1997) Desensitization of the P2X(2) receptor controlled by alternative splicing. FEBS Lett 404 : 294–298.
60. LuikartBW, NefS, ShipmanT, ParadaLF (2003) In vivo role of truncated trkb receptors during sensory ganglion neurogenesis. Neuroscience 117 : 847–858.
61. NakanoY, JahanI, BondeG, SunX, HildebrandMS, et al. (2012) A mutation in the Srrm4 gene causes alternative splicing defects and deafness in the Bronx waltzer mouse. PLoS Genet 8: e1002966.
62. WebbSW, GrilletN, AndradeLR, XiongW, SwarthoutL, et al. (2011) Regulation of PCDH15 function in mechanosensory hair cells by alternative splicing of the cytoplasmic domain. Development 138 : 1607–1617.
63. OuyangXM, XiaXJ, VerpyE, DuLL, PandyaA, et al. (2002) Mutations in the alternatively spliced exons of USH1C cause non-syndromic recessive deafness. Hum Genet 111 : 26–30.
64. WhitlonDS, GabelC, ZhangX (1996) Cochlear inner hair cells exist transiently in the fetal Bronx Waltzer (bv/bv) mouse. J Comp Neurol 364 : 515–522.
65. KansakuA, HirabayashiS, MoriH, FujiwaraN, KawataA, et al. (2006) Ligand-of-Numb protein X is an endocytic scaffold for junctional adhesion molecule 4. Oncogene 25 : 5071–5084.
66. DavisRJ, HardingM, MoayediY, MardonG (2008) Mouse Dach1 and Dach2 are redundantly required for Mullerian duct development. Genesis 46 : 205–213.
67. HenriqueD, AdamJ, MyatA, ChitnisA, LewisJ, et al. (1995) Expression of a Delta homologue in prospective neurons in the chick. Nature 375 : 787–790.
68. SternCD (1998) Detection of multiple gene products simultaneously by in situ hybridization and immunohistochemistry in whole mounts of avian embryos. Curr Top Dev Biol 36 : 223–243.
69. KiernanAE (2006) The paintfill method as a tool for analyzing the three-dimensional structure of the inner ear. Brain Res 1091 : 270–276.
70. BaschML, OhyamaT, SegilN, GrovesAK (2011) Canonical Notch signaling is not necessary for prosensory induction in the mouse cochlea: insights from a conditional mutant of RBPjkappa. J Neurosci 31 : 8046–8058.
71. XiaA, GaoSS, YuanT, OsbornA, BressA, et al. (2010) Deficient forward transduction and enhanced reverse transduction in the alpha tectorin C1509G human hearing loss mutation. Dis Model Mech 3 : 209–223.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 1
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- GATA6 Is a Crucial Regulator of Shh in the Limb Bud
- Large Inverted Duplications in the Human Genome Form via a Fold-Back Mechanism
- Differential Effects of Collagen Prolyl 3-Hydroxylation on Skeletal Tissues
- Affects Plant Architecture by Regulating Local Auxin Biosynthesis
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy