
C/EBPα Is Required for Long-Term Self-Renewal and Lineage Priming of Hematopoietic Stem Cells and for the Maintenance of Epigenetic Configurations in Multipotent Progenitors
Transcription factors are key regulators of hematopoietic stem cells (HSCs) and act through their ability to bind DNA and impact on gene transcription. Their functions are interpreted in the complex landscape of chromatin, but current knowledge on how this is achieved is very limited. C/EBPα is an important transcriptional regulator of hematopoiesis, but its potential functions in HSCs have remained elusive. Here we report that C/EBPα serves to protect adult HSCs from apoptosis and to maintain their quiescent state. Consequently, deletion of Cebpa is associated with loss of self-renewal and HSC exhaustion. By combining gene expression analysis with genome-wide assessment of C/EBPα binding and epigenetic configurations, we show that C/EBPα acts to modulate the epigenetic states of genes belonging to molecular pathways important for HSC function. Moreover, our data suggest that C/EBPα acts as a priming factor at the HSC level where it actively promotes myeloid differentiation and counteracts lymphoid lineage choice. Taken together, our results show that C/EBPα is a key regulator of HSC biology, which influences the epigenetic landscape of HSCs in order to balance different cell fate options.
Published in the journal:
. PLoS Genet 10(1): e32767. doi:10.1371/journal.pgen.1004079
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004079
Summary
Transcription factors are key regulators of hematopoietic stem cells (HSCs) and act through their ability to bind DNA and impact on gene transcription. Their functions are interpreted in the complex landscape of chromatin, but current knowledge on how this is achieved is very limited. C/EBPα is an important transcriptional regulator of hematopoiesis, but its potential functions in HSCs have remained elusive. Here we report that C/EBPα serves to protect adult HSCs from apoptosis and to maintain their quiescent state. Consequently, deletion of Cebpa is associated with loss of self-renewal and HSC exhaustion. By combining gene expression analysis with genome-wide assessment of C/EBPα binding and epigenetic configurations, we show that C/EBPα acts to modulate the epigenetic states of genes belonging to molecular pathways important for HSC function. Moreover, our data suggest that C/EBPα acts as a priming factor at the HSC level where it actively promotes myeloid differentiation and counteracts lymphoid lineage choice. Taken together, our results show that C/EBPα is a key regulator of HSC biology, which influences the epigenetic landscape of HSCs in order to balance different cell fate options.
Introduction
Hematopoietic stem cells (HSCs) are responsible for the maintenance of a constant production of blood cells throughout life. To achieve this, HSCs have to tightly regulate their different fate options including self-renewal, proliferation, differentiation and apoptosis, as alterations in any of these may lead to HSC exhaustion, expansion or leukemia [1]. HSC fate options are controlled by a number of different pathways and are influenced both by the microenvironment and by the actions of cell-autonomous regulators such as transcription factors (TFs) and chromatin-interacting proteins [2].
Given their impact on gene expression, the influence of TFs on HSC properties has been the focus of several studies. Indeed, factors such as C-MYB, ERG, and PU.1 are all essential for preserving HSC self-renewal and their deletion have dramatic impact on hematopoietic maintenance both during fetal and adult life [3], [4], [5], [6]. Other factors, as exemplified by SOX17, are required exclusively for the maintenance of fetal HSCs, whereas GFI-1 and ETV6 only appear to play a role in an adult setting [7], [8], [9].
TF function is interpreted in a chromatin context and, accordingly, chromatin readers and writers have been shown to be important for HSC function and maintenance. Examples include the PRC1 component BMI-1 [10], [11], the maintenance DNA methyltransferase DNMT1 [12], [13] as well as the H3K4 methyltransferase MLL1 [14]. Despite the importance of both TFs and chromatin context for HSC function, our knowledge on how TF binding is interpreted within an epigenetic landscape, and how they may influence epigenetic configurations remains limited. Importantly, given their inherent developmental plasticity, stem cells have been reported to exhibit unique epigenetic signatures of which the so-called bivalent configuration is the best characterized. Work in ES cells has shown that bivalently marked genes are lowly expressed, enriched in genes involved in development/differentiation, and display active (H3K4me3) as well as repressive (H3K27me3) histone marks [15], [16]. As stem cells progress along the path of differentiation the bivalent configuration is resolved into an active or repressed state with a concomitant upregulation or downregulation, respectively, of the expression of previously marked genes [15], [16]. To what extent the bivalent signature is influenced by loss of TFs in HSCs has not been characterized.
C/EBPα is an important myeloid TF that functions not only by binding to regulatory DNA elements and directing transcription, but also through its ability to constrain proliferation by inhibiting the transcriptional activity of E2F-complexes [17], [18], [19], [20]. In the hematopoietic system loss of C/EBPα leads to a differentiation block upstream of the Granulocytic Monocytic Progenitor (GMP) accompanied by an accumulation of earlier stem and myeloid progenitor populations [17], [21].
In acute myeloid leukemia (AML), CEBPA is found mutated in approximately 10% of cases, and studies in mouse have shown that the tumor-suppressive functions of C/EBPα can be ascribed to its ability to balance the proliferation and differentiation of hematopoietic stem and progenitor cells (HSPCs) populations [18], [22]. Indeed, HSCs from mice harboring tumor-prone variants of C/EBPα displayed altered cell cycle kinetics, but how this impacts on HSC function was difficult to assess due to the leukemic transformation in these animals. Furthermore, complete loss of C/EBPα has been reported to endow fetal HSC-enriched populations with a minor competitive advantage in a transplantation setting [21] and C/EBPα has recently been shown to control the fetal-to-adult switch of HSCs [23]. However, at present we lack knowledge on the long-term self-renewal properties of C/EBPα in normal adult HSCs as well as insights into how it potentially may influence transcription and the epigenetic configuration of these cells.
In the present work we have used mouse genetics to characterize the role of C/EBPα in adult HSCs. We found that loss of Cebpa leads to HSC exhaustion, identifying C/EBPα as a crucial regulator of HSC self-renewal. To molecularly assess the function of C/EBPα in HSCs, we combined gene expression analysis with genome-wide profiling of key epigenetic marks and C/EBPα binding using Chromatin ImmunoPrecipitation coupled to sequencing (ChIP-seq). Our findings demonstrate that C/EBPα modulates key pathways associated with HSC function and differentiation, in part by affecting the epigenetic states of genes associated with some of these.
Results
Loss of C/EBPα Affects the Numbers of Immunophenotypic HSPCs
To investigate the functional importance of C/EBPα in the HSPC compartment, we generated Cebpafl/fl and Cebpafl/fl;Mx1Cre mice (hereafter termed Cebpafl/fl and CebpaΔ/Δ, respectively) and injected them with pIpC. Mice were analyzed 18–21 days later at which time point the Cebpa allele was completely recombined in CebpaΔ/Δ mice (Figure S1A). Of note, bone marrows (BM) of CebpaΔ/Δ mice had an approximately 30% reduction in cellularity compared to Cebpafl/fl littermates due to the loss of neutrophil granulocytes (Figure S1B, C).
In line with its requirement for myeloid differentiation [21] publically available gene expression profiles (http://servers.binf.ku.dk/hemaexplorer/) [24] revealed a distinct expression pattern in which Cebpa is lowly expressed in the stem cell compartment and becomes transiently upregulated during commitment to the granulocytic monocytic lineage (Figure 1A). Flow cytometry-based analyses of the myelo-erythroid progenitor compartment verified the reported block in monocytic and granulocytic differentiation at the preGM to GMP transition in CebpaΔ/Δ BMs and the concomitant increase in erythroid and megakaryocytic progenitors (Figure S1D, E) [17], [21]. Moving further upstream in the hematopoietic hierarchy, we found a prominent expansion of the MPP compartment (LSK, CD150−, CD48+) whereas the frequency of immunophenotypic HSCs (LSK, CD150+, CD48−) was unaltered. However, given the reduction in BM cellularity in CebpaΔ/Δ mice this translates into a 50% reduction in the absolute numbers of HSCs upon loss of C/EBPα (Figure 1B, C).

Loss of Cebpa Compromises HSC Activity and Leads to Their Exhaustion
The reduction of absolute HSC numbers suggests an uncharacterized function of C/EBPα in HSC biology. To test this functionally, we first transplanted BM from CebpaΔ/Δ and Cebpafl/fl mice non-competitively into lethally irradiated recipient mice. Whereas all mice receiving Cebpafl/fl BM cells survived, 4/16 of those receiving CebpaΔ/Δ BM died within 16 weeks (Figure 1D). In the remaining mice CebpaΔ/Δ donor cells contributed to the peripheral blood to a similar extent as the Cebpafl/fl control at 8 and 16 weeks post-transplantation, but exhibited a lower level of reconstitution at three weeks post-transplantation due to the requirement of C/EBPα for the production of the early reconstituting neutrophil granulocytes (Figure 1E). Importantly, we find the recipients to be completely devoid of donor-derived granulocytes at all time points (Figure 1F and data not shown) showing that Cebpa is fully recombined in the transplanted cells and that it is equally important for granulocytic differentiation in a transplantation setting. Remarkably, the frequency of CebpaΔ/Δ HSCs was reduced 20-fold in the recipient BMs 16-weeks post-transplantation, whereas the effects on other HSPC subsets were similar to those observed in non-transplanted animals (Figure 1G). To assess this further, we serially transplanted BM from primary recipients and found that only 3/17 of the secondary recipients reconstituted with CebpaΔ/Δ BM survived 16 weeks post-transplantation (Figure 1D).
Competitive BM transplantation remains the gold standard for the assessment of HSC function and we therefore transplanted CD45.2 donor cells along with equal amounts of CD45.1 competitor cells. Despite the comparable HSC frequencies in Cebpafl/fl and CebpaΔ/Δ donors, loss of Cebpa leads to a marked reduction in donor contribution both to PB and to the HSC compartment of the recipients at 18-week post-transplantation (Figure 2A–C). Importantly, the competitive disadvantage of CebpaΔ/Δ BM cells was further exacerbated upon re-transplantation of 1 million donor cells from primary recipients having equal donor contributions, and resulted in their complete failure to contribute to the HSPC compartment of secondary recipients (Figure 2D). Next, we transplanted 20 highly purified (LSK, CD150+, CD48−, CD34−) HSCs from Cebpafl/fl or CebpaΔ/Δ BM together with 200.000 unfractionated competitor BM cells. In accordance with our previous findings, CebpaΔ/Δ HSCs were markedly impaired in hematopoietic reconstitution and their contribution to the recipient HSC compartment was negligible (Figure 2E–G). Furthermore, upon re-transplantation of the BM from primary recipients, CebpaΔ/Δ donor cells were unable to contribute to hematopoietic reconstitution in secondary recipients (Figure 2H, I). Finally, by transplanting 3000 CFSE-labeled HSCs (LSK, CD150+, CD48−) and assessing their homing to hematopoietic organs we could exclude any obvious role of C/EBPα in HSC trafficking during hematopoietic reconstitution (Figure 2J).

In conclusion, loss of Cebpa has profound effects on hematopoietic reconstitution both in competitive and non-competitive settings and collectively our functional analyses demonstrate an essential role of C/EBPα in HSC self-renewal and in preventing HSC exhaustion.
Cebpa Maintains HSC Quiescence and Protects Them from Cell Death
Several reports have demonstrated that C/EBPα acts to restrict proliferation and a number of distinct mechanisms have been proposed (reviewed in [25]).
To test to what extent the anti-proliferative properties of C/EBPα may explain the loss of self-renewal in Cebpa deficient HSCs, we first analyzed the frequencies of replicating HSCs by in vivo BrdU incorporation. Surprisingly, we were unable to detect any major differences in the amounts of BrdU+ HSCs within Cebpafl/fl and CebpaΔ/Δ BMs (Figure 3A), suggesting that C/EBPα does not affect DNA synthesis in HSCs per se. In contrast, we found that loss of Cebpa lead to a marked exit from quiescence as evidenced by a reduction of CebpaΔ/Δ HSCs in G0 (Ki67 negative) and a compensatory increase of CebpaΔ/Δ HSCs in G1 (Figure 3B). Finally, we also found that the exit from quiescence was accompanied by a marked increase in CebpaΔ/Δ HSCs that underwent cell death and that this effect was restricted to immunophenotypic HSCs (Figure 3C).

We next tested the behavior of HSCs in vitro by first evaluating proliferation and cell death of single-sorted HSCs. Consistent with our in vivo analysis we found that loss of Cebpa resulted in both fewer and smaller colonies accompanied by an increase in cell death (Figure 3D, E). Secondly, we used single-cell tracking of HSCs in vitro to monitor the cell cycle length during the first three cell divisions [26]. Consistently, we detected no differences between the average cell cycle lengths by comparing Cebpafl/fl and CebpaΔ/Δ HSCs (Figure 3F). Thus both our in vivo and in vitro analysis demonstrated that C/EBPα does not regulate HSC proliferation per se but rather acts to maintain HSC quiescence and to protect HSCs from cell death.
Collectively, our phenotypic profiling of Cebpa deficient HSCs demonstrates that C/EBPα is a crucial regulator of the balance between quiescence, proliferation and cell death in HSCs. Hence, loss of C/EBPα leads to cell cycle entry, loss of HSC self-renewal and cell death.
Loss of Cebpa Affects Gene Expression Programs Associated with Stem Cell Functions and Differentiation
In order to get insights into the molecular mechanisms by which C/EBPα controls HSC self-renewal, we performed microarray-based gene expression analysis of Cebpafl/fl and CebpaΔ/Δ HSCs (LSK, CD150+, CD48−). In total, we found 110 and 261 genes to be more than 1.5x down - and upregulated, respectively, in CebpaΔ/Δ HSCs (P<0,05) (Figure 4A, B, , Figure S2A for verification by qRT-PCR). Consistent with our phenotypic analysis, gene set enrichment analysis (GSEA) demonstrated a marked negative correlation between HSC signature genes and the CebpaΔ/Δ genotype (Figure 4C). Among the gene sets correlating positively with the CebpaΔ/Δ genotype we found numerous signatures involved in quiescence/proliferation, which are likely to underlie the loss of quiescence that we observed in CebpaΔ/Δ HSCs. Similarly, we also found a positive correlation between the expression of apoptotic signatures and the CebpaΔ/Δ genotype as well as an up-regulation of gene sets involved in the DNA damage response pathway. The relevance of the latter is supported by the 10-fold increase of γH2AX (a marker of DNA damage) positive HSCs that we observe in CebpaΔ/Δ BMs (Figure 4D). Thus the increased levels in cell death and DNA damage observed upon loss of Cebpa are supported both by phenotypic and gene expression analyses.
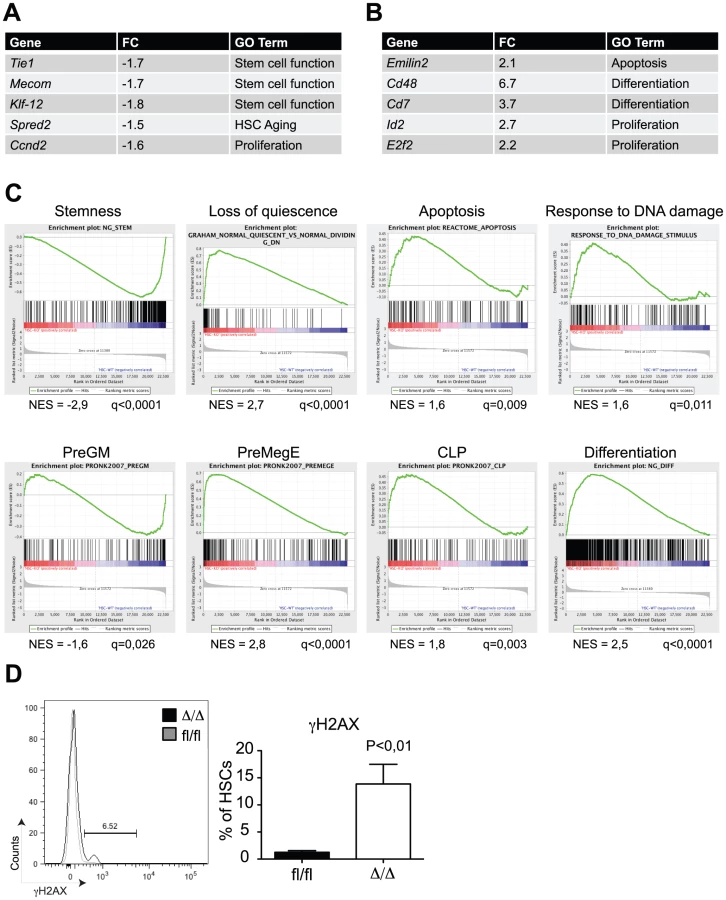
Finally, we noted that a number of gene signatures representing distinct hematopoietic lineages were selectively deregulated in CebpaΔ/Δ HSCs. Thus whereas the expression of granulocytic-monocytic gene sets were downregulated, their lymphoid and erythroid counterparts correlated positively with the CebpaΔ/Δ genotype. These findings strongly suggest that C/EBPα primes HSCs towards the granulocytic-monocytic lineage at the expense of lymphoid and erythroid differentiation and therefore plays a key role in specifying the lineage output from HSCs (Figure 4C).
Identification of C/EBPα Binding Sites in HSPCs
The gene expression analysis reported above identified numerous genes that were de-regulated upon loss of C/EBPα, however whether these are direct or indirect targets remained to be determined. To assess this we performed ChIP-seq analysis for C/EBPα binding in LSK cells, which contains a mixture of HSCs and, predominantly, MPPs.
Despite its low expression, we identified 397 high-confidence C/EBPα bound regions of which selected examples of ChIP-seq coverage are shown in Figure 5A (replicate 1) and verified by qPCR (Figure 5B, Table S2 for the full dataset and Figure S2B–D for data concerning the second replicate). In general, the C/EBPα bound regions were located closer to the transcription start site (TSS) of the nearest gene than a random set of regions, and 39% were located at proximal promoters within 5 kb from TSS (Figure 5C, ). Gratifyingly, motif search analysis in the immediate vicinity of the C/EBPα peak summit (+/−70 bps) identified C/EBP consensus motifs to be markedly overrepresented, however we also identified enrichment of RUNX, ETS and SP1 binding motifs (Figure 5D, Table S3). Interestingly, we find that PU.1 (an ETS factor) and EGR2 (reported to bind SP1 motifs [27], [28]) target genes to be downregulated and upregulated, respectively in CebpaΔ/Δ HSCs (LSK, CD150+, CD48−) (Figure 5E). This might suggest that C/EBPα activate and repress the expression of selected PU.1 and EGR2 target genes, respectively, in HSCs in order to restrict lymphoid and promote myeloid differentiation, thereby providing further support for its lineage priming function.

We next took advantage of the Genomic Regions Enrichment of Annotations Tool (GREAT), which predicts functions of cis-regulatory regions by associating proximal and distal genomic regions with putative targets [29]. Interestingly, C/EBPα associated genes were enriched in a number of gene ontology (GO) categories, the majority of which were involved in myeloid differentiation (Figure 5F, Table S4). Surprisingly, the expression of genes belonging to these GO categories was not prominently de-regulated in CebpaΔ/Δ HSCs (Figure 5G). One explanation could be that C/EBPα marks genes, which are to be expressed at latter stages of myeloid differentiation. Indeed, we find that genes belonging to the GO categories abnormal myeloid leukocyte morphology and increased granulocyte numbers, which were enriched for C/EBPα-associated genes, were upregulated during myeloid differentiation (Figure 5H). Finally, when we compare the overlap between C/EBPα bound regions in LSK cells to a similar dataset derived from GMPs, we find all regions bound by C/EBPα in LSKs to be occupied in GMPs as well. (Figure 5I, Table S5). Thus our data is consistent with a lineage priming function for C/EBPα.
Finally, in order to identify direct C/EBPα target genes in HSCs, we correlated changes in gene expression in CebpaΔ/Δ HSCs with the nearest C/EBPα bound region identified by ChIP-seq in LSK cells. In total 6/102 genes (6%) and 21/257 (8%) of the down - and upregulated genes, respectively, contained a C/EBPα bound region within 100 kb from the TSS (Table S6). Hence, the vast majority of genes displaying altered expression in CebpaΔ/Δ HSCs did not have a nearby C/EBPα binding site and are therefore unlikely to be directly regulated by C/EBPα in HSCs. The identified list of genes targeted by C/EBPα in HSCs/MPPs includes C/EBPα target genes known from other cell types such as Hmox1 and Itgal, but also new and interesting target genes such as Hmbs, Ccnd2 and Emilin2. The latter is part of a family of extracellular matrix glycoproteins and has previously been shown to induce cell death by interacting with TRAIL receptors [30], which may explain the increased cell death that we observe in CebpaΔ/Δ HSCs.
In conclusion we have identified 397 high-confidence regions bound by C/EBPα in LSK cells. C/EBPα binds to the proximal promoter and distal regulatory regions of genes involved primarily in myeloid differentiation as well as genes targeted by TFs with roles in HSC function and lymphoid differentiation. These findings suggest a role for C/EBPα in regulating lineage decisions in HSCs/MPPs.
Loss of Cebpa Affects the Epigenetic Configuration of Bivalent Genes and Pathways Involved in HSC Function
Transcriptional regulation is a complex process not only involving TFs but also the chromatin context in which their function is exerted. Chromatin structure and accessibility is influenced by post-transcriptional modifications on histone tails, which are dynamically deposited and removed by chromatin modifying enzymes and serves as recruitment platforms for other chromatin binding proteins [31], [32]. To test if loss of Cebpa affected chromatin structure we performed replicate ChIP-seq analysis for the presence of two key histone marks, H3K4me3 and H3K27me3 using CebpaΔ/Δ and Cebpafl/fl LSK cells. ChIP-seq profiles of selected genes are depicted in Figure 6 (See Figure S3A–C for replicate data).

To systematically analyze these data, we used hierarchical clustering to separate the epigenetic profiles into distinct clusters and subsequently combined these with gene expression and C/EBPα binding data (Figure 7A). From this analysis it became evident that the vast majority of genes were highly similar in terms of their expression and H3K4me3/H3K27me3 modification patterns when CebpaΔ/Δ and Cebpafl/fl LSK cells were compared (Figure 7A–C). Although some changes in histone modification between the CebpaΔ/Δ and Cebpafl/fl genotypes reached statistical significance, they were overall small and are therefore unlikely to have major phenotypic consequences. One exception was the lowly expressed cluster 6 carrying the bivalent chromatin configuration (originally identified in ES cells [33]), which is characterized by intermediate H3K4me3/high H3K27me3 at the TSS and low H3K27me3 in the gene body. This cluster displayed a specific downregulation of the H3K4me3 mark (p = 8.93×10−24) around the TSS in CebpaΔ/Δ LSKs (Figure 7D, E, Table S7) and similar to previous studies in ES cells, GO-analysis identified pathways involved in cell differentiation, regulation of cell differentiation and cell development to be enriched among the group of bivalently marked genes (Figure S4, Table S8). Strikingly, C/EBPα-bound regions were not over-represented in the bivalent cluster and the majority of genes in this cluster did not have a C/EBPα bound region within 100 kb of their TSS neither in the LSKs (Table S7) nor in the GMPs (data not shown). Thus, whereas C/EBPα is dispensable for the maintenance of the overall H3K4me3 and H3K27me3 modification patterns, it appears to affect the H3K4me3 status of the bivalent cluster through indirect mechanisms. This may in turn interfere with their expression later in differentiation, or alternatively, interfere with the ability of CebpaΔ/Δ HSPCs to initiate differentiation.

We next took a pathway-centric approach and assessed the epigenetic changes in a selection of the gene sets identified in the GSEA analysis described above. Interestingly, for the stemness signature we observed marked changes in histone modifications in the regions flanking the TSS (Figure 8A, B). Specifically, we found the H3K4me3 (p = 1.83×10−5) and H3K27me3 (p = 1.41×10−5) marks to be reduced and increased, respectively, in CebpaΔ/Δ LSK cells consistent with the changes in gene expression in HSCs. The alterations in H3K4me3 and H3K27me3 identified by ChIP-seq were verified by qPCR on selected genes as well as genes, which are part of the bivalent cluster and/or the stemness signature (Figure 8C and Figure S3D). Similar to our findings for the bivalent cluster, the majority of the genes constituting the stemness signature did not have a C/EBPα bound region within 100 kb of their TSS (Table S9), again suggesting that C/EBPα most likely affects the epigenetic marks through indirect mechanisms. For the remaining signatures we do see minor changes but none of them reached statistical significance (Figure S5).

We hypothesized that the reduced level of H3K4me3 in CebpaΔ/Δ LSK cells could, in part, be explained by altered levels of H3K4me3 readers or writers. To test this we first scrutinized the HSC gene expression data for changes in the expression of candidate genes and noted that three probesets for Tet1 were marginally downregulated (1.4-fold in all three probesets) in CebpaΔ/Δ HSCs (Table S1). TET1 belongs to the TET family of 5′-methylcytosine hydroxylases, which have emerged as important epigenetic regulators of both normal and malignant hematopoiesis [34], [35], [36]. TET1 has been shown to bind CpG-islands of TSSs marked either by H3K4me3 or the bivalent H3K4me3/H3K27me3 signature in ES cells [37],[38]. To extend these data to a hematopoietic setting we used a published TET1 dataset [37] and found that genes bound by TET1 in ES cells were enriched in the group of bivalently marked genes (cluster 6) in LSK cells (Figure 8D; p<2.2×10−16). Moreover, when TET1 occupancy was used to stratify the bivalent genes, the TET1 bound fraction (56%) showed a marked decrease in H3K4me3 levels in CebpaΔ/Δ LSKs (Figure 8E; p = 9.6×10−20). Interestingly, this effect was restricted to the bivalent genes as CebpaΔ/Δ and control LSKs showed similar H3K4me3 levels irrespectively of the binding of TET1 (Figure 8E).
Collectively, whereas C/EBPα is largely dispensable for H3K4me3 and H3K27me3 modification patterns in HSC/MPP, its loss do lead to specific changes of the epigenetic states of bivalently marked genes as well as genes associated with stem cell function.
Discussion
Proper control of gene transcription is crucial for essentially all biological processes and is orchestrated through a complex interplay between TFs, chromatin structure and epigenetic regulators. This also extends to the complex biology of HSCs as evidenced by the fact that mice deficient for TFs or epigenetic regulators are frequently associated with HSC phenotypes [2]. However, the mechanisms by which TFs act in a chromatin context to regulate transcription and facilitate epigenetic changes of HSCs are largely unknown.
In the present work we therefore analyzed the impact of deleting C/EBPα, one of the key transcriptional regulators of hematopoiesis, on HSC function, gene expression and the epigenetic status of key histone modifications. We find that conditional acute deletion of Cebpa leads to a 2-fold reduction of immunophenotypic HSCs and that Cebpa deficient HSCs were severely compromised. Furthermore, loss of C/EBPα promoted their exit from quiescence and was associated with a marked increase in cell death. These findings were supported by gene expression analyses, which revealed prominent transcriptional downregulation of genes associated with HSC function and quiescence as well as upregulation of genes associated with DNA damage and apoptosis. Among the pathways that displayed transcriptional changes upon loss of Cebpa, we find noticeable changes in the H3K4me3 and H3K27me3 marks flanking the TSS for genes involved in stem cell function (stemness signature). These genes are expressed at intermediate level (data not shown) and their epigenetic changes may reflect a drift away from an epigenetic state favoring self-renewal. In addition, whereas the overall distribution of H3K4me3 and H3K27me3 marks are largely unaltered in C/EBPα-deficient LSKs, we find that loss of C/EBPα specifically affects the H3K4me3 status of bivalent genes. Interestingly, TET1 was marginally downregulated in CebpaΔ/Δ HSCs and the H3K4me3 levels were selectively reduced in CebpaΔ/Δ LSKs at bivalent genes bound by TET1. Recently it was suggested that TET1, together with the epigenetic regulators CFP1 (a recruiter of the H3K4me3 methylase SETD1) and KDM2A (a H3K36 demethylase), work synergistically to maintain a distinct epigenetic configuration on their target genes [39]. Thus in this context, our findings suggest that downregulation of TET1 following loss of Cebpa may affect H3K4me3 status on selected genes. Whether C/EBPα affects other histone modifications such as H3K4me1 or H3K9Ac remains to be elucidated.
The importance of C/EBPα for HSC function has recently been analyzed in an adult setting. Specifically, Ye et al. [23] found that C/EBPα acts to restrict the numbers of HSCs by inhibiting their proliferation, however the effects on HSC self-renewal was not tested in a serial transplantation set-up. Furthermore, they found N-Myc to be bound by C/EBPα in c-Kit+ cells and suggested that loss of C/EBPα de-repressed N-Myc and promoted HSC expansion through an N-Myc-driven increase in proliferation. These findings contrast those reported in the present study, where we find C/EBPα to be important for the maintenance of a functional HSC pool as defined by serial transplantation and quantification of immunophenotypic HSCs. Moreover, in our HSC gene expression analysis, we do not find N-Myc to be upregulated, and C/EBPα does not bind the N-Myc promoter before the GMP state (data not shown), suggesting that C/EBPα regulates N-Myc in progenitors rather than in HSCs.
A number of methodological differences are likely to explain at least some of the differences between the findings in our study and those reported by Ye et al. First of all, whereas Ye et al. performed their analyses shortly (4–7 days after the last pIpC injection), we waited until 14–17 days after the last pIpC injection to avoid any confounding issues with the pIpC induced interferon response. Indeed, it has previously been reported that interferons influence key HSC parameters such as proliferation, apoptosis and self-renewal [40], [41], and therefore, it is likely that the HSC compartment is affected differentially 4 and 14 days after treatment with pIpC. We therefore speculate that the effect of C/EBPα loss on HSCs might evolve over time, from an initial expanding phase to a strongly compromised state ultimately resulting in increased cell death and loss of self-renewal. This would explain the increased proliferation and expansion observed early after deletion of Cebpa [23], the balanced HSC numbers three weeks after deletion, the loss of HSCs in primary recipients transplanted with C/EBPα-deficient HSCs/BM and the loss of re-population in secondary recipients.. It is recognized that HSC phenotypes can progress over time from proliferative to exhaustion especially under stress such as serial transplantations as shown for PTEN-deficiency [42], [43]. Moreover, although both studies used a conditional model that facilitates the full ablation of Cebpa, the two models are not completely identical, which could potentially explain some of the diverging results.
The functional consequences of Cebpa loss have also been assessed in other developmental settings. Thus, in a pre-leukemic setting, mutation of Cebpa leads to increased HSC proliferation but its impact on HSC function was obscured by transformation to AML [22] and in a fetal setting, E.14.5 LSK cells were reported to have a competitive advantage although their proliferative status was unaltered [21]. Together with our demonstration that loss of Cebpa has dramatic consequences for HSC self-renewal, these observations suggest that different mechanisms are governing HSC self-renewal during embryonic development, in the adult, and in a disease context. While this concept is not novel, and is supported by the selective importance of transcriptional regulators such as SOX17 [7] and BMI-1 [11] in fetal versus adult HSCs, C/EBPα is to our knowledge the first factor that impact oppositely on fetal and adult HSCs as well as being de-regulated in disease. Thus, our work highlights a key role of C/EBPα in HSC biology in different developmental contexts.
TFs often operate in combinations and our data suggest that at least some of the functional deficits of CebpaΔ/Δ HSCs may be explained through crosstalk between C/EBPα and PU.1 - two TFs, which have been reported to act both synergistically and antagonistically in progenitor cells [44], [45]. Our findings that ETS binding sites are overrepresented in C/EBPα bound regions in LSK cells and that PU.1 target genes are downregulated in CebpaΔ/Δ HSCs suggest that C/EBPα may collaborate with PU.1 to activate a transcriptional program already in HSCs. PU.1 is crucial for the development of almost all blood cells and even minor alterations in its expression level have been found to impair HSC self-renewal and lead to HSC exhaustion [46]. In this context it is tempting to speculate that C/EBPα regulates HSC self-renewal through crosstalk with PU.1 and that loss of C/EBPα reduces the output of PU.1 target genes, ultimately resulting in exhaustion of the stem cell pool.
In order to maintain hematopoietic homeostasis, HSCs have to carefully balance the choice between a number of cell fate options. Whereas early reports suggested that HSCs sampled different lineage-affiliated transcriptional programs uniformly, more recent data suggest a considerable degree of heterogeneity within the HSC compartment [47], [48], [49]. Moreover, data from cell line experiments suggest that stochastic upregulation of key regulatory molecules including lineage-instructive TFs drives differentiation along their respective lineages [50]. However, due to the technical difficulties in assessing TF binding in low-abundant cell types it is not clear whether these factors are occupying their targets already in the HSC compartment.
When analyzing the C/EBPα bound regions in the HSC/MPP-enriched LSK population we find these to be enriched for genes that were upregulated during granulocytic-monocytic differentiation. In addition, gene sets associated with myeloid differentiation are downregulated in CebpaΔ/Δ HSCs, suggesting that the sampling of myeloid transcriptional programs is in part controlled by C/EBPα in HSCs perhaps involving PU.1 crosstalk. Hence, in addition to its accepted role as a lineage-instructive factor required for the transition from preGMs to GMPs [51], our data demonstrate an additional role of C/EBPα in controlling the sampling of myeloid gene programs already in HSCs through its binding to the regulatory regions of myeloid genes.
Lineage choice may be viewed as a competition between distinct lineage-affiliated programs. Interestingly, we find that loss of Cebpa leads to upregulation of lymphoid gene expression programs in CebpaΔ/Δ HSCs, which suggests that C/EBPα may actively repress lymphoid differentiation perhaps through the inhibition of the transcriptional activities of EGR family members [52], [53]. This model is also compatible with the ability of C/EBPα to transdifferentiate lymphoid cell types into macrophages [54], [55], with the observation that a subclass of leukemia patients with silenced C/EBPα expression develops AML associated with distinct T-cell characteristics [56] and with the finding that pre-leukemic Cebpa mutant GMPs readily differentiate into T-cells [57].
In conclusion, we find that C/EBPα regulates HSC functions at several distinct levels. Not only is its loss associated with HSC exhaustion, but C/EBPα also appear to be involved in the selection of the myeloid vs. lymphoid lineage suggesting that C/EBPα primes lineage choice at the HSC level. Thus, our work highlight the complexities by which transcriptional circuitries are operating to maintain functional HSCs by balancing key cell fate options such as quiescence, cell death, self-renewal and differentiation.
Materials and Methods
Mouse Colony
Animals were housed according to institutional guidelines at the University of Copenhagen and housed according to institutional guidelines. Excision of the Cebpa allele was achieved by subjecting 10–12 weeks old Cebpafl/fl or Cebpafl/fl;Mx1Cre mice to 3 injections (day 0, 2 and 4) with 200 ul polyinosinic-polycytidylic acid (pIpC) (1.5 mg/ml in PBS, GE Healthcare) as described previously [58]. After conditional deletion of Cebpa, the mice were treated with ciprofloxacin (100 mg/L, Actavis) in the drinking water. Mice were analysed 18–21 days after first pIpC injection.
Ethics Statement
All animal work was done with approval from the Danish Animal Ethical Committee. This study was approved by the review board at the Faculty of Health Sciences, University of Copenhagen.
Flow Cytometry
BM and PB was stained with antibodies and run on a LSRII (Becton Dickinson) or FACSAria (Becton Dickinson) and analysed using the FlowJo software. Student two-tailed T-test was used to test for significance.
Transplantation Assays
All reconstitution assays were performed using the Ly-5 congenic mouse system. Briefly, whole BM or HSCs (LSK, CD150+, CD48−) from Ly-5.2 (CD45.2) donors were transplanted by tail vein injection into 10–12 week old lethally irradiated (900 cGy) Ly-5.1 (CD45.1) mice +/ − Ly-5.1 support BM cells. The recipient mice were treated with ciprofloxacin (100 mg/L, Actavis) in the drinking water. For all transplantation assays, PB was analyzed at 3–4 weeks, 8 weeks and 16–18 weeks after transplantation. The BM from the recipients was analyzed at the experimental endpoint, i.e. 16–18 weeks after transplantation. Student two-tailed T-test was used to test for significance.
Homing Assay
HSCs (LSK, CD150+, CD48−) were isolated as described above and stained with carboxyfluorescein diacetate succinimidyl ester (CFSE) (CellTrace, Invitrogen) according to manufacturer's protocol. Next, HSCs (3000/mouse) were resuspended in PBS+3% FCS and transplanted by tail vein injection into irradiated (900 cGy) Ly-5.1 (CD45.1) mice. Twelve hours later BM and spleen from recipients were isolated, enriched for c-Kit+ cells, and analyzed on a LSRII for CFSE+ cells. Student two-tailed T-test was used to test for significance.
In Vitro Cell Culture
Single HSCs (LSK, CD150+, CD48−) were sorted directly to round-bottom 96-well plates and cultured in serum-free medium (StemSpan SFEM, StemCell Technologies) supplemented with 100 ng/ml SCF and 100 ng/ml TPO (Peprotech). Each well was examined by microscopy and cell numbers were recorded every other day for a week. P-values for differences in colony size were calculated using chi-square test.
Continuous Live Imaging of HSCs
HSCs (LSK, CD150+, CD48−) were sorted and cultured in serum-free medium (StemSpan SFEM, StemCell Technologies) supplemented with SCF (100 ng/ml), IL-3 (10 ng/ml), IL-6 (10 ng/ml), TPO (100 ng/ml) (Peprotech), EPO (5 U/ml) (Promocell), penicillin (50 U), streptomycin (50 ug/ml) and Hepes (10 mM) (Gibco) in T12.5 flasks coated with fibronectin (Innovative Research). Using cell observer (Zeiss), images were acquired every 90 seconds using a VBA module controlling Zeiss AxioVision 4.8 software and analyzed using TTT software [59]. Student two-tailed T-test was used to test for significance.
Gene Expression Profiling
RNA was purified from sorted HSCs (LSK, CD150+, CD48−), amplified, labeled and hybridized to the Mouse Gene 1.0 ST GeneChip Array (Affymetrix, Santa Clara, CA, USA). Raw gene expression data are available at the Gene Expression Omnibus (GEO) online database under ID GSE42498.
ChIP-seq Analysis
ChIP-seq was performed using LSK or GMP cells from Cebpafl/fl or CebpaΔ/Δ mice. Chromatin from 100.000 or 500.000 cells was incubated with antibodies for histone marks (H3K4me3 and H3K27me3, Cell Signaling) or C/EBPα (Santa Cruz Biotechnology), respectively. The antibody-bound chromatin was captured with Protein-A sepharose beads, washed, de-cross-linked and precipitated. Precipitated DNA were mixed with 2 ng E. Coli DNA and amplified using NEB Next ChIP-seq sample prep reagent set 1 (New England Biolabs) according to manufacturer's protocol. Libraries were sequenced on an Illumina Genome Analyzer IIx or and Illumina Hiseq2000. Data was deposited in the NCBI Gene Expression Omnibus online database under ID GSE43007. See Table S10 for primers.
Bioinformatic Analyses
All reads were mapped using bowtie 0.12.7 [60] using standard parameters (Figure S3B). C/EBPα peaks were called with MACS, v. 1.4 [61] with an IgG mock sample as control and using 1*10−5 as the p-value threshold. Clusters in both positional and regular heatmaps were identified by k-means clustering using the biopython Bio Cluster module [62]. In order to identify TF motifs in C/EBPα bound regions, sequences below the peak summit +/−70 bp were analyzed.
Additional and detailed Materials and Methods are available as Supporting Information (See Text S1).
Supporting Information
{kind=link}
Zdroje
1. ZonLI (2008) Intrinsic and extrinsic control of haematopoietic stem-cell self-renewal. Nature 453 : 306–313.
2. RossiL, LinKK, BolesNC, YangL, KingKY, et al. (2012) Less is more: unveiling the functional core of hematopoietic stem cells through knockout mice. Cell Stem Cell 11 : 302–317.
3. NgAP, LoughranSJ, MetcalfD, HylandCD, de GraafCA, et al. (2011) Erg is required for self-renewal of hematopoietic stem cells during stress hematopoiesis in mice. Blood 118 : 2454–2461.
4. LoughranSJ, KruseEA, HackingDF, de GraafCA, HylandCD, et al. (2008) The transcription factor Erg is essential for definitive hematopoiesis and the function of adult hematopoietic stem cells. Nature immunology 9 : 810–819.
5. LieuYK, ReddyEP (2009) Conditional c-myb knockout in adult hematopoietic stem cells leads to loss of self-renewal due to impaired proliferation and accelerated differentiation. Proceedings of the National Academy of Sciences of the United States of America 106 : 21689–21694.
6. IwasakiH, SomozaC, ShigematsuH, DuprezEA, Iwasaki-AraiJ, et al. (2005) Distinctive and indispensable roles of PU.1 in maintenance of hematopoietic stem cells and their differentiation. Blood 106 : 1590–1600.
7. KimI, SaundersTL, MorrisonSJ (2007) Sox17 dependence distinguishes the transcriptional regulation of fetal from adult hematopoietic stem cells. Cell 130 : 470–483.
8. HockH, HamblenMJ, RookeHM, SchindlerJW, SalequeS, et al. (2004) Gfi-1 restricts proliferation and preserves functional integrity of haematopoietic stem cells. Nature 431 : 1002–1007.
9. HockH, MeadeE, MedeirosS, SchindlerJW, ValkPJ, et al. (2004) Tel/Etv6 is an essential and selective regulator of adult hematopoietic stem cell survival. Genes & development 18 : 2336–2341.
10. IwamaA, OguroH, NegishiM, KatoY, MoritaY, et al. (2004) Enhanced self-renewal of hematopoietic stem cells mediated by the polycomb gene product Bmi-1. Immunity 21 : 843–851.
11. ParkIK, QianD, KielM, BeckerMW, PihaljaM, et al. (2003) Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature 423 : 302–305.
12. BroskeAM, VockentanzL, KharaziS, HuskaMR, ManciniE, et al. (2009) DNA methylation protects hematopoietic stem cell multipotency from myeloerythroid restriction. Nature genetics 41 : 1207–1215.
13. TrowbridgeJJ, SnowJW, KimJ, OrkinSH (2009) DNA methyltransferase 1 is essential for and uniquely regulates hematopoietic stem and progenitor cells. Cell Stem Cell 5 : 442–449.
14. JudeCD, ClimerL, XuD, ArtingerE, FisherJK, et al. (2007) Unique and independent roles for MLL in adult hematopoietic stem cells and progenitors. Cell Stem Cell 1 : 324–337.
15. De GobbiM, GarrickD, LynchM, VernimmenD, HughesJR, et al. (2011) Generation of bivalent chromatin domains during cell fate decisions. Epigenetics & chromatin 4 : 9.
16. PietersenAM, van LohuizenM (2008) Stem cell regulation by polycomb repressors: postponing commitment. Current opinion in cell biology 20 : 201–207.
17. ManciniE, Sanjuan-PlaA, LucianiL, MooreS, GroverA, et al. (2012) FOG-1 and GATA-1 act sequentially to specify definitive megakaryocytic and erythroid progenitors. The EMBO journal 31 : 351–365.
18. PorseBT, BryderD, Theilgaard-MonchK, HasemannMS, AndersonK, et al. (2005) Loss of C/EBP alpha cell cycle control increases myeloid progenitor proliferation and transforms the neutrophil granulocyte lineage. J Exp Med 202 : 85–96.
19. PorseBT, PedersenTA, XuX, LindbergB, WewerUM, et al. (2001) E2F repression by C/EBPalpha is required for adipogenesis and granulopoiesis in vivo. Cell 107 : 247–258.
20. JohansenLM, IwamaA, LodieTA, SasakiK, FelsherDW, et al. (2001) c-Myc is a critical target for c/EBPalpha in granulopoiesis. Molecular and cellular biology 21 : 3789–3806.
21. ZhangP, Iwasaki-AraiJ, IwasakiH, FenyusML, DayaramT, et al. (2004) Enhancement of hematopoietic stem cell repopulating capacity and self-renewal in the absence of the transcription factor C/EBP alpha. Immunity 21 : 853–863.
22. BereshchenkoO, ManciniE, MooreS, BilbaoD, ManssonR, et al. (2009) Hematopoietic stem cell expansion precedes the generation of committed myeloid leukemia-initiating cells in C/EBPalpha mutant AML. Cancer Cell 16 : 390–400.
23. YeM, ZhangH, AmabileG, YangH, StaberPB, et al. (2013) C/EBPa controls acquisition and maintenance of adult haematopoietic stem cell quiescence. Nature cell biology 15 (4) 385–94.
24. BaggerFO, RapinN, Theilgaard-MonchK, KaczkowskiB, ThorenLA, et al. (2012) HemaExplorer: a database of mRNA expression profiles in normal and malignant haematopoiesis. Nucleic acids research 41: D1034–9.
25. JohnsonPF (2005) Molecular stop signs: regulation of cell-cycle arrest by C/EBP transcription factors. J Cell Sci 118 : 2545–2555.
26. KimuraA, RiegerMA, SimoneJM, ChenW, WickreMC, et al. (2009) The transcription factors STAT5A/B regulate GM-CSF-mediated granulopoiesis. Blood 114 : 4721–4728.
27. SwirnoffAH, MilbrandtJ (1995) DNA-binding specificity of NGFI-A and related zinc finger transcription factors. Molecular and Cellular Biology 15 : 2275–2287.
28. SeilerMP, MathewR, LiszewskiMK, SpoonerCJ, BarrK, et al. (2012) Elevated and sustained expression of the transcription factors Egr1 and Egr2 controls NKT lineage differentiation in response to TCR signaling. Nature immunology 13 : 264–271.
29. McLeanCY, BristorD, HillerM, ClarkeSL, SchaarBT, et al. (2010) GREAT improves functional interpretation of cis-regulatory regions. Nature biotechnology 28 : 495–501.
30. MongiatM, LigrestiG, MarastoniS, LorenzonE, DolianaR, et al. (2007) Regulation of the extrinsic apoptotic pathway by the extracellular matrix glycoprotein EMILIN2. Molecular and cellular biology 27 : 7176–7187.
31. KooistraSM, HelinK (2012) Molecular mechanisms and potential functions of histone demethylases. Nature reviews Molecular cell biology 13 : 297–311.
32. BannisterAJ, KouzaridesT (2011) Regulation of chromatin by histone modifications. Cell research 21 : 381–395.
33. BernsteinBE, MikkelsenTS, XieX, KamalM, HuebertDJ, et al. (2006) A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125 : 315–326.
34. ShihAH, Abdel-WahabO, PatelJP, LevineRL (2012) The role of mutations in epigenetic regulators in myeloid malignancies. Nature reviews Cancer 12 : 599–612.
35. Moran-CrusioK, ReavieL, ShihA, Abdel-WahabO, Ndiaye-LobryD, et al. (2011) Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 20 : 11–24.
36. QuivoronC, CouronneL, Della ValleV, LopezCK, PloI, et al. (2011) TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell 20 : 25–38.
37. WilliamsK, ChristensenJ, PedersenMT, JohansenJV, CloosPA, et al. (2011) TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature 473 : 343–348.
38. KohKP, YabuuchiA, RaoS, HuangY, CunniffK, et al. (2011) Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell 8 : 200–213.
39. WuH, ZhangY (2011) Tet1 and 5-hydroxymethylation: a genome-wide view in mouse embryonic stem cells. Cell Cycle 10 : 2428–2436.
40. EssersMA, OffnerS, Blanco-BoseWE, WaiblerZ, KalinkeU, et al. (2009) IFNalpha activates dormant haematopoietic stem cells in vivo. Nature 458 : 904–908.
41. SatoT, OnaiN, YoshiharaH, AraiF, SudaT, et al. (2009) Interferon regulatory factor-2 protects quiescent hematopoietic stem cells from type I interferon-dependent exhaustion. Nature medicine 15 : 696–700.
42. ZhangJ, GrindleyJC, YinT, JayasingheS, HeXC, et al. (2006) PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature 441 : 518–522.
43. YilmazOH, ValdezR, TheisenBK, GuoW, FergusonDO, et al. (2006) Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 441 : 475–482.
44. ReddyVA, IwamaA, IotzovaG, SchulzM, ElsasserA, et al. (2002) Granulocyte inducer C/EBPalpha inactivates the myeloid master regulator PU.1: possible role in lineage commitment decisions. Blood 100 : 483–490.
45. SmithLT, HohausS, GonzalezDA, DziennisSE, TenenDG (1996) PU.1 (Spi-1) and C/EBP alpha regulate the granulocyte colony-stimulating factor receptor promoter in myeloid cells. Blood 88 : 1234–1247.
46. StaberPB, ZhangP, YeM, WelnerRS, Nombela-ArrietaC, et al. (2013) Sustained PU.1 Levels Balance Cell-Cycle Regulators to Prevent Exhaustion of Adult Hematopoietic Stem Cells. Molecular cell 49 : 934–946.
47. GrafT, EnverT (2009) Forcing cells to change lineages. Nature 462 : 587–594.
48. ChallenGA, BolesNC, ChambersSM, GoodellMA (2010) Distinct hematopoietic stem cell subtypes are differentially regulated by TGF-beta1. Cell Stem Cell 6 : 265–278.
49. DykstraB, KentD, BowieM, McCaffreyL, HamiltonM, et al. (2007) Long-term propagation of distinct hematopoietic differentiation programs in vivo. Cell Stem Cell 1 : 218–229.
50. PinaC, FugazzaC, TippingAJ, BrownJ, SonejiS, et al. (2012) Inferring rules of lineage commitment in haematopoiesis. Nature cell biology 14 : 287–294.
51. NerlovC (2007) The C/EBP family of transcription factors: a paradigm for interaction between gene expression and proliferation control. Trends in cell biology 17 : 318–324.
52. LazarevicV, ZulloAJ, SchweitzerMN, StatonTL, GalloEM, et al. (2009) The gene encoding early growth response 2, a target of the transcription factor NFAT, is required for the development and maturation of natural killer T cells. Nature immunology 10 : 306–313.
53. LiS, SymondsAL, ZhuB, LiuM, RaymondMV, et al. (2011) Early growth response gene-2 (Egr-2) regulates the development of B and T cells. PLoS One 6: e18498.
54. Di TullioA, Vu ManhTP, SchubertA, CastellanoG, ManssonR, et al. (2011) CCAAT/enhancer binding protein alpha (C/EBP(alpha))-induced transdifferentiation of pre-B cells into macrophages involves no overt retrodifferentiation. Proceedings of the National Academy of Sciences of the United States of America 108 : 17016–17021.
55. XieH, YeM, FengR, GrafT (2004) Stepwise reprogramming of B cells into macrophages. Cell 117 : 663–676.
56. FigueroaME, WoutersBJ, SkrabanekL, GlassJ, LiY, et al. (2009) Genome-wide epigenetic analysis delineates a biologically distinct immature acute leukemia with myeloid/T-lymphoid features. Blood 113 : 2795–2804.
57. SchusterMB, FrankAK, BaggerFO, RapinN, VikesaaJ, et al. (2013) Lack of the p42 form of C/EBPalpha leads to spontaneous immortalization and lineage infidelity of committed myeloid progenitors. Experimental Hematology 41 : 882–e816, 882-893, e816.
58. WeischenfeldtJ, DamgaardI, BryderD, Theilgaard-MonchK, ThorenLA, et al. (2008) NMD is essential for hematopoietic stem and progenitor cells and for eliminating by-products of programmed DNA rearrangements. Genes Dev 22 : 1381–1396.
59. RiegerMA, HoppePS, SmejkalBM, EitelhuberAC, SchroederT (2009) Hematopoietic cytokines can instruct lineage choice. Science 325 : 217–218.
60. LangmeadB, TrapnellC, PopM, SalzbergSL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology 10: R25.
61. ZhangY, LiuT, MeyerCA, EeckhouteJ, JohnsonDS, et al. (2008) Model-based analysis of ChIP-Seq (MACS). Genome biology 9: R137.
62. de HoonMJ, ImotoS, NolanJ, MiyanoS (2004) Open source clustering software. Bioinformatics 20 : 1453–1454.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 1
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- GATA6 Is a Crucial Regulator of Shh in the Limb Bud
- Large Inverted Duplications in the Human Genome Form via a Fold-Back Mechanism
- Differential Effects of Collagen Prolyl 3-Hydroxylation on Skeletal Tissues
- Affects Plant Architecture by Regulating Local Auxin Biosynthesis
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy