
Evidence for Two Different Regulatory Mechanisms Linking Replication and Segregation of Chromosome II
Understanding the mechanisms that coordinate replication initiation with subsequent segregation of chromosomes is an important biological problem. Here we report two replication-control mechanisms mediated by a chromosome segregation protein, ParB2, encoded by chromosome II of the model multichromosome bacterium, Vibrio cholerae. We find by the ChIP-chip assay that ParB2, a centromere binding protein, spreads beyond the centromere and covers a replication inhibitory site (a 39-mer). Unexpectedly, without nucleation at the centromere, ParB2 could also bind directly to a related 39-mer. The 39-mers are the strongest inhibitors of chromosome II replication and they mediate inhibition by binding the replication initiator protein. ParB2 thus appears to promote replication by out-competing initiator binding to the 39-mers using two mechanisms: spreading into one and direct binding to the other. We suggest that both these are novel mechanisms to coordinate replication initiation with segregation of chromosomes.
Published in the journal:
. PLoS Genet 9(6): e32767. doi:10.1371/journal.pgen.1003579
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003579
Summary
Understanding the mechanisms that coordinate replication initiation with subsequent segregation of chromosomes is an important biological problem. Here we report two replication-control mechanisms mediated by a chromosome segregation protein, ParB2, encoded by chromosome II of the model multichromosome bacterium, Vibrio cholerae. We find by the ChIP-chip assay that ParB2, a centromere binding protein, spreads beyond the centromere and covers a replication inhibitory site (a 39-mer). Unexpectedly, without nucleation at the centromere, ParB2 could also bind directly to a related 39-mer. The 39-mers are the strongest inhibitors of chromosome II replication and they mediate inhibition by binding the replication initiator protein. ParB2 thus appears to promote replication by out-competing initiator binding to the 39-mers using two mechanisms: spreading into one and direct binding to the other. We suggest that both these are novel mechanisms to coordinate replication initiation with segregation of chromosomes.
Introduction
Studies in bacteria as well as in eukaryotes have shown that processes that maintain chromosomes, such as replication, recombination and repair, although able to occur independently of each other, often influence each other. Chromosome segregation is a major maintenance process but our knowledge of it in bacteria is relatively recent. This is because in well-studied bacteria such as Escherichia coli, genes dedicated to the segregation process have not been evident. Such genes were discovered in bacterial plasmids, called parA and parB. Subsequently, their homologs were found in a majority of sequenced bacterial chromosomes [1], [2]. Wherever tested, the chromosomal parAB genes were capable of conferring segregational stability on unstable plasmids bearing parS (“centromere” analogous) sites [3], [4], [5], [6], and made at least some contribution to chromosome segregation [7], [8]. In spite of limited study, it is becoming clear that chromosomal segregation systems can influence and be influenced by other chromosome maintenance processes.
In bacteria, replication and transcription have been proposed to provide motive force in chromosome segregation [9], [10], [11]. Coupled transcription-translation of membrane proteins is also thought to play an important role in chromosome segregation [12], [13]. One of the segregation proteins, ParB, can also spread and silence transcription of genes in its path [14], [15], [16]. The influence of segregation proteins in replication was suggested when ParB was found to load a condensin protein in the vicinity of the replication origin in Bacillus subtilis and in Streptococcus pneumoniae [17], [18], [19]. A more direct role was evident when ParA was found to influence the activity of the initiator DnaA in B. subtilis chromosome replication [20], [21] and in replication of Vibrio cholerae chromosome I (chrI) [22]. Recently, ParB encoded by V. cholerae chromosome II (chrII) was also found to influence chrII replication [23]. Here we report two distinct mechanisms for this ParB-mediated effect.
V. cholerae chrII replication is primarily controlled by its specific initiator protein, RctB [24], [25]. RctB binds to two kinds of site in the replication origin of chrII. One kind, the 11 - or 12-mers, plays both essential and regulatory roles [26]. The other kind, two 39-mers and a 29-mer (a truncated 39-mer), plays only an inhibitory role in replication [26], [27]. One of the 39-mers is situated at a locus called rctA, at one end of the origin, and the other is more centrally located in the origin (Figure 1, top). The 29-mer is located in front of the rctB gene and is involved primarily in autorepression of the gene [27]. The rctA locus contains, in addition to a 39-mer, one of the ParB2 binding sites, parS2-B [28]. It has been reported recently that parS2-B alleviates some of the replication inhibitory activity of rctA in a ParB2-dependent fashion, but the mechanism is unknown [23]. Here we show that ParB2 spreads from parS2-B into the rctA 39-mer, and suggest that the spreading likely interferes with RctB binding to the 39-mer and thereby restrains the inhibitory activity of rctA. Unexpectedly, we also found ParB2 promotes replication by directly binding to the central 39-mer, without requiring spreading from parS2-B. We provide evidence that ParB2 competes with RctB for binding to the central 39-mer specifically and could thereby restrain its activity. In addition to revealing new ways by which a Par protein might influence replication, our results are significant in demonstrating that a segregation protein can bind specifically outside of centromeric sites.

Results
ParB2 can spread into the replication origin of chrII
The origin region of chrII comprises three functional units: A region required for controlling initiation (incII), a region minimally required for initiation (oriII) and a gene required for synthesizing the initiator protein (RctB) (Figure 1, top). The region covering the first two units, which consists mostly of sites for initiator binding, will be referred to as the origin. The rctA locus of incII exerts a strong inhibitory effect on chrII replication because of the 39-mer it contains [26]. The locus also has a site, parS2-B, for binding to the segregation protein ParB2, and the inhibitory effect of rctA is reduced in the presence of ParB2 [23]. Knowing that ParB proteins can spread out from their binding sites to neighboring sequences [14], we asked whether ParB2 spreading from parS2-B over the 39-mer might be a mechanism to control its inhibitory function. The extent of ParB2 spreading was tested by ChIP-chip analysis using antibody against ParB2 (Figure 1, bottom). Spreading was evident on either side of parS2-B (grey profile). In contrast, when the antibody was against RctB, the immunoprecipitated DNA in the origin region was restricted to where RctB has specific binding sites (black profile). These results suggest that ParB2 has the potential to modulate replication initiation activity by interfering with RctB binding.
ParB2 can silence transcription in the origin of chrII
Spreading of proteins across genes can silence them [14]. For example, spreading into plasmid replication genes suitably close to a parS site can be lethal when selective pressure for plasmid retention is applied. By such an experimental test, we found that ParB2 can spread from parS2-B (Figure S1A). Spreading is also suggested by the formation of ParB2-GFP fluorescent foci in parS2-B carrying plasmids (Figure S1B). ParB2 could also silence two promoters, PrctA and PrctB, in the origin of chrII (Figure 1, top). PrctA is proximal to parS2-B whereas PrctB is located about 1 kb away at the other end of the origin. The activity of the promoters was assayed by fusing them to a promoter-less lacZ gene present in a multicopy plasmid in E. coli (Figure 2). The promoter fragments fused to lacZ carried either the entire origin including parS2-B (1A and 1B), or the origin lacking parS2-B (2A and 2B) or no additional DNA (3A and 3B). ParB2 was supplied constitutively in trans at about an order of magnitude higher than the physiological level (monitored by Western blotting; Figure S2), using Ptrc promoter without an intact lac repressor binding-site (lacO1), which makes the promoter unresponsive to IPTG. The presence of ParB2 reduced the activities of PrctA and PrctB significantly, only when the parS2-B site was present (Figure 2, 1A and 1B). These results suggest that ParB2 can spread over the entire origin in the presence of parS2-B, and does not have a significant effect on either promoter in the absence of parS2-B.

Since RctB has numerous binding sites in the origin, it appeared possible that RctB binding to them could counteract ParB2 spreading and reduce silencing of the promoters. This possibility was addressed by supplying RctB from an arabinose-inducible promoter, PBAD [24], [26], [27], [29]. The induction of RctB alone (at about two-fold the physiological level) repressed PrctA marginally and PrctB about two fold (Figure S3, lanes 1 vs. 5). Silencing by ParB2 exceeded 90% for both the promoters (Figure S3, lanes 1 vs. 4). When RctB and ParB2 were supplied together, the repression of both the promoters was reduced about two fold compared to the level achieved with ParB2 alone (Figure S3, lanes 4 vs. 6; also inset). These results indicate that RctB can counteract the ParB2-mediated silencing.
In the results presented above (Figure 2 and Figure S3), the level of ParB2 was about 14-fold the level normally present in V. cholerae (Figure S2). When the concentration was reduced to about 10-fold, ParB2 could silence only the parS2-B proximal PrctA, but not the distal PrctB promoter (Figure S3, lanes 1 vs. 3). This reduced level of ParB2 was used in all subsequent experiments.
ParB2 binds to the central 39-mer in vivo
In order to determine how far ParB2 can spread beyond PrctA, progressively increasing lengths of incII were fused to a foreign reporter promoter, PrepA, itself fused to lacZ [30] (Figure 3). In these experiments, in addition to a plasmid supplying ParB2, another plasmid was used to supply RctB. The two proteins were expressed from inducible promoters, Plac and PBAD, respectively (Figure 3, cartoon at the top right corner).

As expected, neither ParB2 nor RctB influenced the activity of PrepA itself (Figure 3, top panel). In contrast, when rctA was present, ParB2 reduced the activity of PrepA by two-fold (second panel). We believe this is due to ParB2 spreading from parS2-B into PrepA, whose −35 box was only 167 bp away. RctB alone was ineffective, most likely because it does not spread, and its specific binding site, the rctA 39-mer, is well separated (by 85 bp) from the −35 box of PrepA. Supplying RctB was marginally effective in relieving the silencing by ParB2. The next extension of the incII fragment included the 3x11-mers (third panel). Neither ParB2 nor RctB could silence the reporter promoter in this case. This result suggests that the spreading may not extend too far beyond PrctA. A further extension of the incII fragment by only 74 bp that included the central 39-mer, restored ParB2-mediated repression of the reporter promoter (fourth panel). This result was surprising since parS2 sites were not found within incII [28]. The effect of ParB2 was significantly reduced when RctB was supplied, which is to be expected since RctB binds strongly to the central 39-mer [26]. This result suggests that ParB2 and RctB can compete for binding to the central 39-mer. The largest fragment (bottom panel) did not show a significant ParB2 effect on the reporter promoter, suggesting that ParB2 may not spread significantly from the central 39-mer. Together, the results suggest that under the conditions tested, ParB2 affects the origin primarily through interactions near rctA and the central 39-mer. The interaction near the central 39-mer suggests that ParB2 might bind there directly.
ParB2 binds to the central 39-mer in vitro
The possibility of site-specific binding of ParB2 within the origin but outside of parS2-B was tested by EMSA. Several fragments covering the origin were used. Fragments 1 and 2, carrying the parS2-B site (positive controls), showed maximal ParB2 binding (Figure 4). The next significant binding was with the fragment containing the central 39-mer (fragment 5). This fragment contained natural flanking sequences of only 3 bp and 32 bp beyond the central 39-mer. The sequences (3+39+32) are exactly those that were added to the incII fragment of the third panel to generate the silencing-proficient fragment of the fourth panel (Figure 3). We found that the flanking sequences do not contribute to the central 39-mer binding (Figure S4, fragment #1). This result supports the inference from in vivo studies that ParB2 can directly bind to the central 39-mer without requiring parS2-B. Binding to the rctA 39-mer (fragment 3) was considerably weaker, possibly because the two 39-mers have several mismatches between them (Figure 1 of [26]; discussion related to Figure 5 below). The level of binding seen with the rctA 39-mer was comparable to the levels seen with fragments 4, 6, 8 and 9, and the level was marginally above that of the negative control that lacks any chrII sequences, suggesting that ParB2 has significant non-specific DNA binding activity.
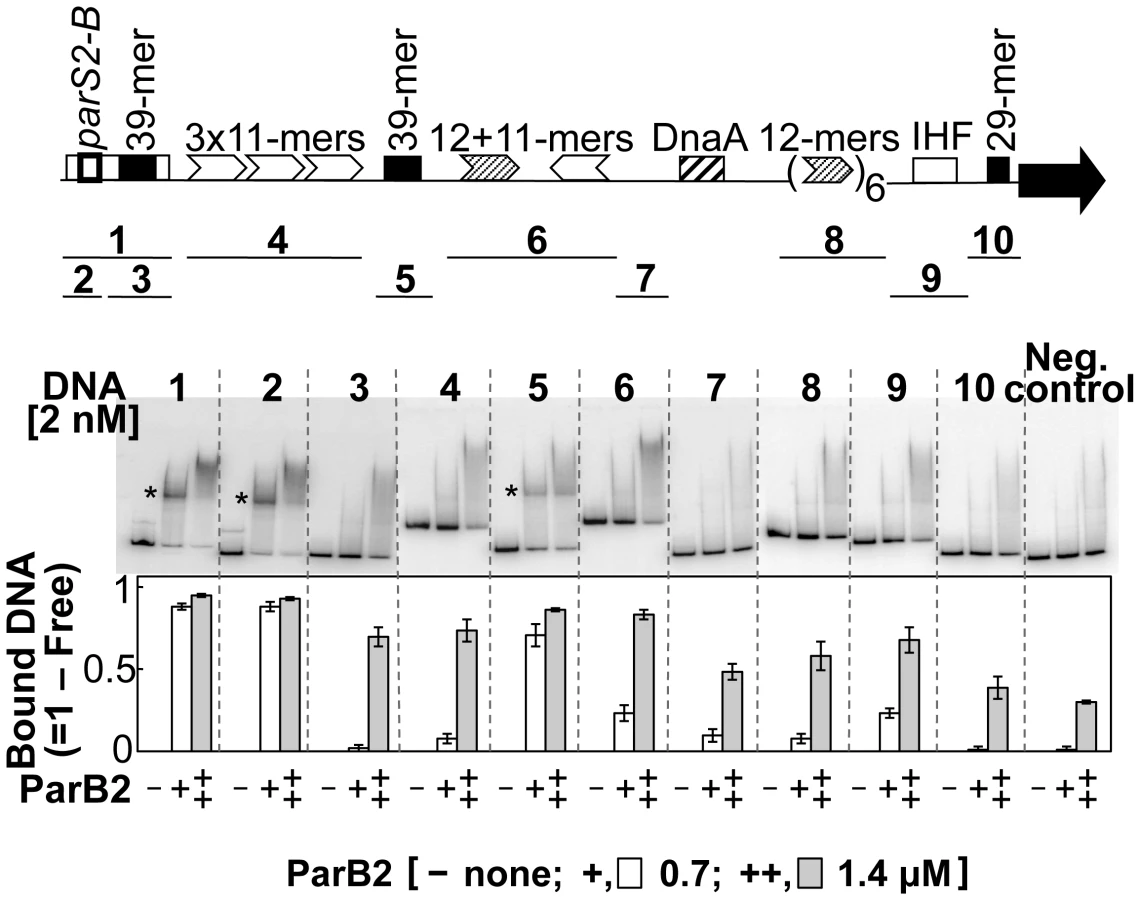

The sequence requirement for specific binding of ParB2 to the central 39-mer was tested by variously mutating the sequence. The 39-mer has two conserved 9 bp direct repeats (called A and B boxes) flanking a 19 bp AT-rich spacer (Figure S4). The presence of both of the repeats and their proper phasing are important for RctB binding [26]. The AT richness of the spacer is also important but not the sequence per se. The parS2 sites are AT-rich inverted repeats, only 15 bp long. Notably, the 39-mer spacer also contains an inverted repeat, which has some similarity to the consensus parS2 site. However, the 39-mer spacer by itself was not sufficient for ParB2 binding; the presence of one of the direct repeats was necessary (Figure S4, fragments 4–6). Either of the direct repeats alone was also not sufficient (fragments 2–3). The inverted repeat feature could also be destroyed without compromising the binding efficiency (fragment 12). When the same fragments were tested for RctB binding, only the ones with the intact 39-mer and 10 bp deletion or addition (fragments 9–11) showed significant binding, as was also found earlier (data not shown; [27]). It appears that while both ParB2 and RctB bind to the 39-mer, the presence of one of the direct repeats is not obligatory for ParB2 binding.
Specific binding of ParB2 to the 39-mer was also verified by DNase I footprinting (Figure S5). Protection by ParB2 was conspicuous at the junction of the first direct repeat (A-box) and the AT-rich spacer. At this junction an intact parS-2B half site, 5′-TGTAAA, is present. This sequence is fully conserved in all 10 parS2 sites that were competent in ParB2 binding [28]. In Figure S4, this half-site sequence was intact in all the binding positive fragments and mutant in all the fragments that failed to show specific binding. The half-site is also mutated to 5′-TTAAAC in the ParB2 binding-negative 39-mer in rctA (Figure 4, fragment #3). The half site thus appears to be necessary for 39-mer binding of ParB2. In further support of this inference, when we restored the original bases to some of the binding-negative 39-mer mutants to regenerate the half-site, binding proficiency was regained (Figure 5, fragments #3–6). Although necessary, the half site was not sufficient for binding ParB2 (fragment #7). We conclude that extension of the half site either to the left or right is necessary. This is not surprising since the affinity drops by orders of magnitude when one half of a dyad symmetric site is mutated [31], [32]. The minimal size of the extensions needed to regain binding activity of ParB2 remains to be determined.
ParB2 and RctB compete for binding to the central 39-mer in vitro
ParB2 and RctB can bind to rctA simultaneously [23] (Figure 6, top panel). This is not surprising since there are 34 bp of spacer sequence between the binding sites of the two proteins, and that ParB2 does not spread in vitro. The sites also remain functional when isolated from each other [33]. On the other hand, at the central 39-mer, the binding sites for the two proteins appeared to be largely overlapping, suggesting that they could not bind simultaneously. This was indeed the result (Figure 6, bottom panel). Even at the higher protein concentrations (++), no new discrete species representative of dual binding was detected. The results indicate that ParB2 and RctB compete for binding to 39-mer, unlike the simultaneous binding that can occur on rctA.

ParB2 can control oriII copy number without requiring parS2-B
We previously showed that the central 39-mer is the most potent replication inhibitory site in incII and it functions through RctB binding [26]. If ParB2 competes with RctB for binding to the central 39-mer, this competition appeared likely to influence oriII activity without requiring the parS2-B site. This prediction was tested by determining the copy number of oriII-driven plasmids (Figure 7). The copy number of oriII plasmids depends on the extent of the incII sequences present [26]. Although the 39-mers are always inhibitory to replication, the 11 - and 12-mers can either promote or inhibit replication depending upon whether the 39-mers are present or not. In the present experiments also, the oriII plasmid copy number first decreased and then increased with increasing deletion of incII (Figure 7, − ParB2 column). When ParB2 was additionally present, the copy number increased significantly in the first two 39-mer-carrying plasmids, the increase being maximal for the plasmid with the lowest copy number (pTVC25). In this plasmid, we suggest that the 39-mer was unencumbered by the 3x11-mers, and was maximally available for binding to ParB2. Together, these results indicate that ParB2 has the potential to facilitate chrII replication by restraining the inhibitory activity of the incII sequences, and can do so whether parS2-B is present or not.

Roadblock of ParB2 spreading compromises the replication stimulatory activity
If ParB2 spreading is one of the mechanisms by which the protein stimulates chrII replication, it might be possible to restrain this activity by placing a roadblock in the path of spreading. To this end, we inserted an array of five P1 RepA binding sites (iterons) between parS2-B and the 39-mer in rctA (Figure 8). The effectiveness of the roadblock in preventing the spreading of P1 ParB protein was demonstrated earlier [34]. Comparison of the top two rows of the Table in Figure 8 shows that in the absence of RepA (that is in the absence of a roadblock), ParB2 was equally efficient in promoting cell growth that depended on the functioning of oriII plasmids. In other words, the P1 iterons in pBJH218 did not compromise ParB2 spreading in the absence of the roadblock. The same two plasmid-carrying cells behaved differently in the presence of RepA (the last two rows). Upon induction of ParB2 production by IPTG, cell growth improved more in the case of pTVC20 than in the case of pBJH218. In other words, ParB2 effect was compromised under the condition the roadblock was expected to be effective. These results are consistent with ParB2 spreading as a mechanism for stimulating chrII replication initiation. Note that some increase of growth rate was seen even when ParB spreading was inhibited by a roadblock (generation time decreased 7% for cells in row #4). This result is not surprising because ParB2 can bind to the central 39-mer without requiring spreading from parS2-B. Overall, the ParB2 effects were modest, which is to be expected because of the existence of multiple controls on chrII replication.

The central 39-mer is not a centromere
In chromosome and plasmid segregation, ParB proteins serve to couple centromeres to ParA proteins (NTPases) and modulate the NTPase activity that is believed to provide the movement required for segregation [8]. The binding ParB2 to a 39-mer raises the possibility of an inherent centromeric function of the site. This was tested by cloning the central 39-mer into a miniF plasmid, which is unstable due to deletion of its own segregation genes [35]. The stability of the miniF plasmid improved with the inclusion of the parS2-B site but not with the 39-mer, when ParA and ParB proteins were supplied in trans (Figure S6). This result suggests that ParB2 binding to parS2-B and the central 39-mer is different in an important respect.
Discussion
Our knowledge of interactions between the processes of replication and segregation of chromosomes in bacteria is rather recent and limited. An interaction between the universal bacterial replication initiator, DnaA, and a segregation protein, ParA, was recently discovered in B. subtilis [20], [21], [22], [36] and later in V. cholerae where it was specific for chromosome I [22]. In both cases, replication initiation was modulated by ParA. A partner segregation protein, ParB, was found to affect replication of chromosome II (chrII) in V. cholerae. This parB, called ParB2, somehow promoted replication by binding to one of its cognate centromeric sites (parS2-B) [23]. Here we show that ParB2 spreads out of this centromeric site into the replication origin of chrII, and suggest that this spreading is a mechanism by which ParB2 promotes replication. We also report an additional mechanism by which ParB2 can promote chrII replication: direct binding to a replication inhibitory site in the origin. To our knowledge, the present results provide the first examples of a replication activation mechanism that is mediated by spreading of the activator from a distant site, and by the specific binding of a segregation protein outside of the centromere. These studies have revealed at least three ways by which segregation proteins can influence replication (Figure 9).

ParB2 interferes with RctB binding
The spreading of ParB2 from a centromeric site into the origin of chrII was evident from in vivo cross-linking experiments (Figures 1,S7), from silencing of promoters within the origin (Figure 2) and from the reduction of reporter promoter activity when natural initiator (RctB) binding sites were present between the centromeric site and the promoter (Figure 3, third panel; Figure S3, insets). This latter result indicates that RctB binding could create a natural roadblock to ParB2 spreading. The fact that the span of silencing lengthened with increased ParB2 concentration (Figure S3) also supports the idea that the underlying mechanism involves spreading along the DNA. Finally, the results of placing an artificial roadblock were also consistent with the spreading mechanism (Figure 8). When a powerful replication inhibitory site (the rctA 39-mer) was present within the span of spreading, growth of cells dependent upon the functioning of the chrII origin improved. It was also reported earlier that ParB2 could increase replication of chrII origin carrying plasmids when they included the adjacent rctA region [23]. This increase was shown to be dependent on the presence of parS2-B. Together with the finding that ParB2 does not directly bind to the rctA 39-mer (Figure 4), and cannot spread from its binding site in the central 39-mer as discussed below (Figures 3 (last panel), S1A, S1B), the simplest explanation of these results is that by spreading from parS2-B, ParB2 compromises the inhibitory activity of the rctA 39-mer by interfering with RctB binding.
ParB2 was also found to reduce the activity of another potent replication inhibitor (the central 39-mer) without requiring the centromeric site and spreading (Figure 3, S1). The latter effect appears to be due to direct binding of ParB2 to the central 39-mer. This mode of ParB2 interaction with the 39-mer most likely also causes interference with RctB binding to this site (Figure 6). Since the 39-mers are the two sites most inhibitory to chrII replication and their activities are mediated through RctB binding, the reduction in binding suffices to explain how ParB2 could promote replication of chrII (Figures 7,8). Interference with binding of regulatory proteins to DNA by the spreading of a competing protein along DNA has also been invoked to explain transcriptional silencing, inhibition of DNA methylation and of DNA gyrase binding, and resistance to DNase I cleavage [37], [38], [39], [40].
ParB2 does not interact with RctB directly
Although the only model we have entertained so far to explain the ParB2 effect is interference with specific binding of RctB, we have also tested whether ParB2 and RctB could interact directly. This possibility was suggested by the finding that ParA influences replication by protein-protein interaction rather than DNA-protein interaction [20], [21], [22], [36]. However, ParB2 did not show any detectable interaction with RctB, as was also reported earlier (Figure S8) [23].
Influence of ParA2 on binding and spreading of ParB2
ParA participates in a number of processes involving ParB [8]. Here, we asked whether binding of ParB2 to the central 39-mer is also influenced by ParA2. The binding was assayed indirectly by fusing a foreign promoter close enough to the 39-mer that ParB2 binding to the site could interfere with the promoter activity. The promoter activity did not change significantly upon supply of ParA2 (Figure S9; data with PrepA). This suggests ParB2 binding to the 39-mer is not influenced by ParA2. We did find a minor influence of ParA2 on PrctA silencing by ParB2 spreading, the basis of which was not explored. In the case of P1 plasmid and B. subtilis chromosome, no ParA effect on ParB spreading was evident [14], [41].
E. coli as a host to study chromosome II
Whereas deletion of parS2-B in V. cholerae was easily tolerated (Figure S7; [23]), deletion of the parB2 gene was essentially lethal [42]. In the absence of ParB2, chrII loss is evident at every cell division that causes a severe growth defect. We were therefore unable to test conveniently the role of ParB2 on replication of chrII in the native host. On the other hand, there was no obvious effect of ParB2 on the growth of E. coli, in which we did most of our experiments. The validity of extrapolating the observations in E. coli to the native host appears warranted by the observation that ParB2 effects were seen in the context of the entire origin (Figure 8; [23]), and by the finding that some of the inferences from the E. coli results were valid when tested in vitro (Figure 6). In the past, wherever chrII replication control was studied in both E. coli and V. cholerae, the results agreed [25], [26], [28], [29]. Nonetheless, the ParB2 concentration required for spreading to proceed just over rctA was an order of magnitude higher than that is normally present in the native host. The reason for this discrepancy is not understood but a possibility is that ParB2 when supplied from a trans source is much less effective. A discrepancy in the amount of protein required from a cis vs. trans source has been noted in the case V. cholerae ParA1 [22] and ParA of Pseudomonas aeruginosa [43]. The production of one of the Par proteins without its partner could also have altered the protein activity and stability. The importance of maintaining the stoichiometry of Par proteins has also been indicated in studies of B. subtilis [20], [44]. Another possibility is that higher protein concentration may be required to bind to a single parS site, as we have used here, than when there exists neighboring sites, as in the native host, that might allow cooperative binding.
Physiological relevance of ParB2 spreading
We show that in wild type V. cholerae cells ParB2 can bind and spread over the entire origin (Figure 1). We detected considerable ParB2 spreading, even with the deletion of the origin proximal centromeric site (parS2-B) (Figure S7). Most likely, this spreading originates from the neighboring parS2-A site 5.7 kb away (Figure 1). The spreading could add an additional layer of control over PrctA by silencing the promoter, which is independently repressed by RctB (Figure S3) [24], [45]. The PrctA activity in turn controls RctB binding to the rctA 39-mer [29]. The multiple feedback loops that operate to control the initiation of replication from the origin of chrII appear securely interlocked with the specific segregation system of this chromosome. The presence of multiple layers of control could compensate for a deficiency in any one of the regulators, and help in homeostasis of origin copy number.
The finding that ParB2 could spread over the entire origin might suggest that it could be a mechanism to promote chromosome segregation. It might increase the effective size of the kinetochore, which might facilitate its interaction with ParA, the essential partner of ParB in chromosome segregation. However, this role has yet to be established [16], [34], [46].
Evolutionary considerations
ParB proteins of plasmids are known to be plasmid-specific and to bind to their cognate sites [47]. This helps to avoid segregation-mediated incompatibility if different plasmids happen to be present in the same host. By the same token, in multichromosome bacteria, the segregation systems should be chromosome-specific. Such is clearly the case in Burkholderia cenocepacia [5] and in V. cholerae [4], [48]. The same ParB protein has been found to bind to variant parS sites but the sites are believed to be descendants of a common ancestor [49]. In this context, it is noteworthy that although the central 39-mer is largely non-homologous to parS2-B, the region of the 39-mer crucial for ParB2 binding shares six bp of perfect identity with parS2-B (Figures 5,S4), suggesting the possibility of an evolutionary link between the sites here also.
Chromosome segregation begins soon after replication initiation, thereby compressing the total time for the completion of these two processes. Their close coordination also allows segregation to proceed in a more orderly fashion than if the substrate for segregation were a pair of completed and entangled sister chromosomes. Here, we have described interactions that might assist in coordinating replication initiation and segregation. In V. cholerae, following replication initiation, the majority of the RctB binding sites (11 - and 12-mers) stay hemi-methylated and are unable to bind the initiator [50]. This stage of the cell cycle should favor spread of ParB2 into the origin (Figure S7), which is likely to favor origin segregation and at the same time discourage premature reinitiation. Spreading of ParB2 towards the origin is apparently prevented later in the cell cycle when the origin is remethylated, allowing RctB binding to 11 - and 12-mers that eventually leads to initiation (Figure 3, 3rd panel). At these latter stages, when spreading is blocked, direct binding of ParB2 to the central 39-mer should favor initiation. Thus, depending upon the stage of the cell cycle, ParB2 appears to play opposite roles in controlling chrII replication, but in such a way as to promote the orderly sequence of chromosome replication followed by segregation. In the case of plasmids, which can complete replication in a tiny fraction of the cell division cycle, such coordination is neither necessary nor evident. We suggest that the acquisition of interactions such as we describe are a feature of the putative adaptation of an acquired plasmid to permanent residency as a second chromosome.
Materials and Methods
Strains and plasmids
V. cholerae and E. coli strains, and plasmids used in this study are listed in Table S1. ChrII fragments were amplified from N16961 (CVC209) DNA by PCR using Phusion High-Fidelity polymerase (NEB, Beverly, MA). The sequences of primers used for PCR are shown in Table S2. For cloning sequences up to 100 bp, complementary oligonucleotides (IDT, Skokie, IL) were used after annealing the two [29]. The exact chrII coordinates of each cloned fragment are given in Table S1.
β-galactosidase assay
This was done in L broth cultures of E. coli strain BR8706 at OD600 between 0.4–0.5, as described [24]. To account for any effect that ParB2 might have on the replication of the lacZ-reporter plasmid, β-galactosidase activities were normalized for the plasmid copy number in all cases. The copy number variation was small; one standard deviation was within 20% of the mean. The copy numbers were measured (see below) from aliquots of the same cultures that were used for β-galactosidase measurements. Some of the cultures were simultaneously monitored for ParB2 amounts by Western blotting (Figure S2). Note that +/ − ParB2 refer to cells carrying pTVC501 (that carries parB2 under IPTG control) with and without IPTG induction, respectively. In Figure 2 and Figure S3 (lanes 4,6), + refers to cells carrying pTVC236, which supplies ParB2 from a constitutive promoter and − refers to cells carrying the empty vector, pACYC184.
Plasmid copy number measurement
The copy number of lacZ-carrying plasmids (Figures 2,3, S3, S9) were measured exactly as described [32]. Briefly, different experimental cultures were grown to log phase and mixed with separately grown cells carrying pNEB193 before plasmid isolation. The latter plasmid helped to account for plasmid loss, if any, during plasmid isolation steps. The copy number of oriII plasmids (Figure 7) was determined similarly except that cells instead of growing in liquid cultures were obtained by washing out colonies from transformation plates directly, to avoid mutant accumulation, as described [26]. The origin fragments were first cloned in a plasmid vector driven by the γ-origin of plasmid R6K, and the clones were maintained in cells that supplied the cognate initiator (π) protein. The clones were electroporated into E. coli (BR8706) carrying pTVC499 that supplied RctB (but no π protein) and pTVC501 that supplied parB2.
EMSA
The DNA probes were made from plasmids by PCR using oligonucleotides TVC286 (5′-TCCGATTACGGCACCAAATCGA-3′) and TVC287 (5′ - AACGTGGATAAACTTCCTGTAAT-3′), which allowed amplification of extra 100 bp of vector sequences from each flank of the region of interest. The PCR products were labeled using 30 units T4 Polynucleotide Kinase (NEB) and 50 µCi of [γ32-ATP] (Perkin-Elmer) and purified by passing through G-50 columns (Roche diagnostics). Binding was done in the presence of 300 ng poly dI-dC. Other details are as described [25], except that the binding reactions were run in 0.5×TBE, which improved ParB2 binding. In Figure 5, the probe was non-radioactive and was visualized with SYBR Gold nucleic acid gel stain (Molecular Probes) at 0.5 mg/ml for 30 min. As non-specific competitor, supercoiled pUC19 DNA was used instead of poly dI-dC, as the former stayed at the top of gels and did not interfere with visualization of probe bands. The images were recorded using Fuji LAS-3000 imaging system.
ChIP-chip assay
ChIP assay was performed as described [50]. Briefly, cells of V. cholerae CVC209 were cultivated in L broth at 37°C to exponential phase and cross-linked with 1% formaldehyde. After cell lysis and sonication, RctB-DNA or ParB2-DNA complexes were immunoprecipitated using RctB or ParB2 antibody, respectively. The precipitated DNA was amplified, labeled and hybridized to a custom Agilent 8 X 60K V. cholerae oligonucleotide microarray representing the whole genome according to the manufacturer's protocol and as described [51]. The custom tiling array contained 60 bp probes specific for both the Crick and Watson strands. The consecutive probes were separated by 140 bp in each strand and by 10 bp between the Watson and Crick strands. Data was extracted using an Agilent scanner and Agilent Feature Extraction program. Individual ChIP (Cy5) and input (Cy3) signals were first normalized with respect to total Cy5 and Cy3 signals, respectively. Fold change was calculated by dividing the normalized Cy5 signals with normalized Cy3 signals. The values are mean from three independent experiments.
Roadblock assay
The effect of roadblock to ParB2 spreading was tested by cloning an array of five consensus P1 plasmid iterons in front of chrII origin of pTVC20, resulting in pBJH218 (Figure 8). Consensus iterons were used to avoid the PrepA promoter present within the array of natural iterons of P1ori. To clone the iterons, pTVC20 was modified by creating a NdeI restriction enzyme site between parS2-B and the 39-mer within rctA, using QuikChange II XL site-directed mutagenesis kit (Agilent Technologies) and oligonucleotides BJH472 and BJH473. To the resulting plasmid (pBJH217), the iteron array, amplified from pALA753 [52] using oligonucleotides BJH475 and BJH476, was cloned at the NdeI site, resulting in pBJH218. The plasmid pair, pTVC20 and pBJH218, was used in the same genetic background as used in Figure 7, and additionally, in an otherwise isogenic host that supplied P1RepA protein from a constitutive promoter (bla-p2 of pBR322). The P1repA gene and the adjoining bla-p2 promoter was cloned in a λD69 vector and the resulting phage (λDKC311 [53]) was used to lysogenize BR8706.
The copy numbers of pTVC20 and pBJH218 were nearly identical, about four-fold lower than pTVC22 used in Figure 7, as was found earlier for pTVC20 [29]. The copy number was estimated to be about one per cell when there should be four oriC. The low copy number of pTVC20 and pBJH218, and the lack of an active partitioning system, made the plasmids unstable (130/130 cells lost the plasmids after seven generations of growth without selection). The effect of ParB2 was therefore checked only under selection. Young colonies (≤1 mm) from selection plates were used to inoculate LB medium with appropriate drugs (ampicillin at 50 µg/ml, chloramphenicol at 25 µg/ml and spectinomycin at 40 µg/ml) and inducers (arabinose at 0.02% and, when desired, IPTG at 100 µM), and the cultures were grown to early log phase (OD600∼0.1) and stored in 0.01 M MgSO4 in ice. For calculating generation times, the cultures were diluted to OD600 = 0.002 and grown to early log phase. Generation times were calculated from OD600 values in the range 0.02–0.2. Saturation of growth was avoided to prevent accumulation of faster growing revertants. Relative colony sizes were also determined from the MgSO4 suspensions on plates with and without IPTG but otherwise identical in volume and contents.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. GerdesK, Moller-JensenJ, Bugge JensenR (2000) Plasmid and chromosome partitioning: surprises from phylogeny. Mol Microbiol 37 : 455–466.
2. LivnyJ, YamaichiY, WaldorMK (2007) Distribution of centromere-like parS sites in bacteria: insights from comparative genomics. J Bacteriol 189 : 8693–8703.
3. YamaichiY, NikiH (2000) Active segregation by the Bacillus subtilis partitioning system in Escherichia coli. Proc Natl Acad Sci U S A 97 : 14656–14661.
4. Saint-DicD, FrushourBP, KehrlJH, KahngLS (2006) A parA Homolog Selectively Influences Positioning of the Large Chromosome Origin in Vibrio cholerae. J Bacteriol 188 : 5626–5631.
5. DubarryN, PastaF, LaneD (2006) ParABS systems of the four replicons of Burkholderia cenocepacia: new chromosome centromeres confer partition specificity. J Bacteriol 188 : 1489–1496.
6. Godfrin-EstevenonAM, PastaF, LaneD (2002) The parAB gene products of Pseudomonas putida exhibit partition activity in both P. putida and Escherichia coli. Mol Microbiol 43 : 39–49.
7. GerdesK, HowardM, SzardeningsF (2010) Pushing and pulling in prokaryotic DNA segregation. Cell 141 : 927–942.
8. MierzejewskaJ, Jagura-BurdzyG (2012) Prokaryotic ParA-ParB-parS system links bacterial chromosome segregation with the cell cycle. Plasmid 67 : 1–14.
9. LemonKP, GrossmanAD (1998) Localization of bacterial DNA polymerase: evidence for a factory model of replication. Science 282 : 1516–1519.
10. DworkinJ, LosickR (2002) Does RNA polymerase help drive chromosome segregation in bacteria? Proc Natl Acad Sci U S A 99 : 14089–14094.
11. LemonKP, GrossmanAD (2000) Movement of replicating DNA through a stationary replisome. Mol Cell 6 : 1321–1330.
12. WoldringhCL (2002) The role of co-transcriptional translation and protein translocation (transertion) in bacterial chromosome segregation. Mol Microbiol 45 : 17–29.
13. LibbyEA, RoggianiM, GoulianM (2012) Membrane protein expression triggers chromosomal locus repositioning in bacteria. Proc Natl Acad Sci U S A 109 : 7445–7450.
14. RodionovO, LobockaM, YarmolinskyM (1999) Silencing of genes flanking the P1 plasmid centromere. Science 283 : 546–549.
15. BartosikAA, LasockiK, MierzejewskaJ, ThomasCM, Jagura-BurdzyG (2004) ParB of Pseudomonas aeruginosa: interactions with its partner ParA and its target parS and specific effects on bacterial growth. J Bacteriol 186 : 6983–6998.
16. KusiakM, GapczynskaA, PlochockaD, ThomasCM, Jagura-BurdzyG (2011) Binding and spreading of ParB on DNA determine its biological function in Pseudomonas aeruginosa. J Bacteriol 193 : 3342–3355.
17. SullivanNL, MarquisKA, RudnerDZ (2009) Recruitment of SMC by ParB-parS organizes the origin region and promotes efficient chromosome segregation. Cell 137 : 697–707.
18. GruberS, ErringtonJ (2009) Recruitment of condensin to replication origin regions by ParB/SpoOJ promotes chromosome segregation in B. subtilis. Cell 137 : 685–696.
19. MinnenA, AttaiechL, ThonM, GruberS, VeeningJW (2011) SMC is recruited to oriC by ParB and promotes chromosome segregation in Streptococcus pneumoniae. Mol Microbiol 81 : 676–688.
20. MurrayH, ErringtonJ (2008) Dynamic control of the DNA replication initiation protein DnaA by Soj/ParA. Cell 135 : 74–84.
21. ScholefieldG, ErringtonJ, MurrayH (2012) Soj/ParA stalls DNA replication by inhibiting helix formation of the initiator protein DnaA. EMBO J 31 : 1542–1555.
22. KadoyaR, BaekJH, SarkerA, ChattorajDK (2011) Participation of Chromosome Segregation Protein ParAI of Vibrio cholerae in Chromosome Replication. J Bacteriol 193 : 1504–1514.
23. YamaichiY, GerdingMA, DavisBM, WaldorMK (2011) Regulatory cross-talk links Vibrio cholerae chromosome II replication and segregation. PLoS Genet 7: e1002189.
24. PalD, Venkova-CanovaT, SrivastavaP, ChattorajDK (2005) Multipartite regulation of rctB, the replication initiator gene of Vibrio cholerae chromosome II. J Bacteriol 187 : 7167–7175.
25. DuigouS, KnudsenKG, SkovgaardO, EganES, Lφbner-OlesenA, et al. (2006) Independent Control of Replication Initiation of the Two Vibrio cholerae Chromosomes by DnaA and RctB. J Bacteriol 188 : 6419–6424.
26. Venkova-CanovaT, ChattorajDK (2011) Transition from a plasmid to a chromosomal mode of replication entails additional regulators. Proc Natl Acad Sci U S A 108 : 6199–6204.
27. Venkova-CanovaT, SahaA, ChattorajDK (2012) A 29-mer site regulates transcription of the initiator gene as well as function of the replication origin of Vibrio cholerae chromosome II. Plasmid 67 : 102–110.
28. YamaichiY, FogelMA, McLeodSM, HuiMP, WaldorMK (2007) Distinct centromere-like parS sites on the two chromosomes of Vibrio spp. J Bacteriol 189 : 5314–5324.
29. Venkova-CanovaT, SrivastavaP, ChattorajDK (2006) Transcriptional inactivation of a regulatory site for replication of Vibrio cholerae chromosome II. Proc Natl Acad Sci U S A 103 : 12051–12056.
30. ChattorajDK, SnyderKM, AbelesAL (1985) P1 plasmid replication: multiple functions of RepA protein at the origin. Proc Natl Acad Sci U S A 82 : 2588–2592.
31. KimB, LittleJW (1992) Dimerization of a specific DNA-binding protein on the DNA. Science 255 : 203–206.
32. DasN, ChattorajDK (2004) Origin pairing (‘handcuffing’) and unpairing in the control of P1 plasmid replication. Mol Microbiol 54 : 836–849.
33. KadoyaR, ChattorajDK (2012) Insensitivity of Chromosome I and the Cell Cycle to Blockage of Replication and Segregation of Vibrio cholerae Chromosome II. MBio 3 : 00067–00012.
34. RodionovO, YarmolinskyM (2004) Plasmid partitioning and the spreading of P1 partition protein ParB. Mol Microbiol 52 : 1215–1223.
35. LemonnierM, BouetJY, LibanteV, LaneD (2000) Disruption of the F plasmid partition complex in vivo by partition protein SopA. Mol Microbiol 38 : 493–505.
36. ScholefieldG, WhitingR, ErringtonJ, MurrayH (2011) Spo0J regulates the oligomeric state of Soj to trigger its switch from an activator to an inhibitor of DNA replication initiation. Mol Microbiol 79 : 1089–1100.
37. YarmolinskyM (2000) Transcriptional silencing in bacteria. Curr Opin Microbiol 3 : 138–143.
38. LynchAS, WangJC (1995) SopB protein-mediated silencing of genes linked to the sopC locus of Escherichia coli F plasmid. Proc Natl Acad Sci U S A 92 : 1896–1900.
39. LimCJ, WhangYR, KenneyLJ, YanJ (2012) Gene silencing H-NS paralogue StpA forms a rigid protein filament along DNA that blocks DNA accessibility. Nucleic Acids Res 40 : 3316–3328.
40. ShinM, LagdaAC, LeeJW, BhatA, RheeJH, et al. (2012) Gene silencing by H-NS from distal DNA site. Mol Microbiol 86 : 707–719.
41. BreierAM, GrossmanAD (2007) Whole-genome analysis of the chromosome partitioning and sporulation protein Spo0J (ParB) reveals spreading and origin-distal sites on the Bacillus subtilis chromosome. Mol Microbiol 64 : 703–718.
42. YamaichiY, FogelMA, WaldorMK (2007) par genes and the pathology of chromosome loss in Vibrio cholerae. Proc Natl Acad Sci U S A 104 : 630–635.
43. LasockiK, BartosikAA, MierzejewskaJ, ThomasCM, Jagura-BurdzyG (2007) Deletion of the parA (soj) homologue in Pseudomonas aeruginosa causes ParB instability and affects growth rate, chromosome segregation, and motility. J Bacteriol 189 : 5762–5772.
44. OguraY, OgasawaraN, HarryEJ, MoriyaS (2003) Increasing the ratio of Soj to Spo0J promotes replication initiation in Bacillus subtilis. J Bacteriol 185 : 6316–6324.
45. EganES, DuigouS, WaldorMK (2006) Autorepression of RctB, an initiator of Vibrio cholerae chromosome II replication. J Bacteriol 188 : 789–793.
46. CastaingJP, BouetJY, LaneD (2008) F plasmid partition depends on interaction of SopA with non-specific DNA. Mol Microbiol 70 : 1000–1011.
47. DabrazhynetskayaA, BrendlerT, JiX, AustinS (2009) Switching protein-DNA recognition specificity by single-amino-acid substitutions in the P1 par family of plasmid partition elements. J Bacteriol 191 : 1126–1131.
48. FogelMA, WaldorMK (2005) Distinct segregation dynamics of the two Vibrio cholerae chromosomes. Mol Microbiol 55 : 125–136.
49. PassotFM, CalderonV, FichantG, LaneD, PastaF (2012) Centromere binding and evolution of chromosomal partition systems in the Burkholderiales. J Bacteriol 194 : 3426–3436.
50. DemarreG, ChattorajDK (2010) DNA adenine methylation is required to replicate both Vibrio cholerae chromosomes once per cell cycle. PLoS Genet 6: e1000939.
51. CamHP, SugiyamaT, ChenES, ChenX, FitzGeraldPC, et al. (2005) Comprehensive analysis of heterochromatin - and RNAi-mediated epigenetic control of the fission yeast genome. Nat Genet 37 : 809–819.
52. BrendlerTG, AbelesAL, ReavesLD, AustinSJ (1997) The iteron bases and spacers of the P1 replication origin contain information that specifies the formation of a complex structure involved in initiation. Mol Microbiol 23 : 559–567.
53. ParkK, ChattorajDK (2001) DnaA boxes in the P1 plasmid origin: the effect of their position on the directionality of replication and plasmid copy number. J Mol Biol 310 : 69–81.
54. ErdmannN, PetroffT, FunnellBE (1999) Intracellular localization of P1 ParB protein depends on ParA and parS. Proc Natl Acad Sci U S A 96 : 14905–14910.
55. TillyK, ChecrounC, RosaPA (2012) Requirements for Borrelia burgdorferi plasmid maintenance. Plasmid 68 : 1–12.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 6
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- BMS1 Is Mutated in Aplasia Cutis Congenita
- Sex-stratified Genome-wide Association Studies Including 270,000 Individuals Show Sexual Dimorphism in Genetic Loci for Anthropometric Traits
- Distinctive Expansion of Potential Virulence Genes in the Genome of the Oomycete Fish Pathogen
- Distinct Neuroblastoma-associated Alterations of Impair Sympathetic Neuronal Differentiation in Zebrafish Models
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy