
Transcriptome-Wide Mapping of 5-methylcytidine RNA Modifications in Bacteria, Archaea, and Yeast Reveals mC within Archaeal mRNAs
The presence of 5-methylcytidine (m5C) in tRNA and rRNA molecules of a wide variety of organisms was first observed more than 40 years ago. However, detection of this modification was limited to specific, abundant, RNA species, due to the usage of low-throughput methods. To obtain a high resolution, systematic, and comprehensive transcriptome-wide overview of m5C across the three domains of life, we used bisulfite treatment on total RNA from both gram positive (B. subtilis) and gram negative (E. coli) bacteria, an archaeon (S. solfataricus) and a eukaryote (S. cerevisiae), followed by massively parallel sequencing. We were able to recover most previously documented m5C sites on rRNA in the four organisms, and identified several novel sites in yeast and archaeal rRNAs. Our analyses also allowed quantification of methylated m5C positions in 64 tRNAs in yeast and archaea, revealing stoichiometric differences between the methylation patterns of these organisms. Molecules of tRNAs in which m5C was absent were also discovered. Intriguingly, we detected m5C sites within archaeal mRNAs, and identified a consensus motif of AUCGANGU that directs methylation in S. solfataricus. Our results, which were validated using m5C-specific RNA immunoprecipitation, provide the first evidence for mRNA modifications in archaea, suggesting that this mode of post-transcriptional regulation extends beyond the eukaryotic domain.
Published in the journal:
. PLoS Genet 9(6): e32767. doi:10.1371/journal.pgen.1003602
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003602
Summary
The presence of 5-methylcytidine (m5C) in tRNA and rRNA molecules of a wide variety of organisms was first observed more than 40 years ago. However, detection of this modification was limited to specific, abundant, RNA species, due to the usage of low-throughput methods. To obtain a high resolution, systematic, and comprehensive transcriptome-wide overview of m5C across the three domains of life, we used bisulfite treatment on total RNA from both gram positive (B. subtilis) and gram negative (E. coli) bacteria, an archaeon (S. solfataricus) and a eukaryote (S. cerevisiae), followed by massively parallel sequencing. We were able to recover most previously documented m5C sites on rRNA in the four organisms, and identified several novel sites in yeast and archaeal rRNAs. Our analyses also allowed quantification of methylated m5C positions in 64 tRNAs in yeast and archaea, revealing stoichiometric differences between the methylation patterns of these organisms. Molecules of tRNAs in which m5C was absent were also discovered. Intriguingly, we detected m5C sites within archaeal mRNAs, and identified a consensus motif of AUCGANGU that directs methylation in S. solfataricus. Our results, which were validated using m5C-specific RNA immunoprecipitation, provide the first evidence for mRNA modifications in archaea, suggesting that this mode of post-transcriptional regulation extends beyond the eukaryotic domain.
Introduction
5-methylcytidine (m5C) is a modification that occurs both on DNA and RNA. In eukaryotes, this DNA modification has been extensively studied over the past years, and was found to play a crucial role in genomic imprinting, X-chromosome inactivation, and suppression of repetitive elements [1], [2]. Less is known about the distribution and role of m5C sites in RNA. In bacteria, m5C positions were described only in rRNA, whereas in archaea and eukaryotes m5C was mapped to both tRNA and rRNA. In tRNA molecules, m5C sites are typically present at the variable region and the anticodon loop, where they have been shown to stabilize the secondary structure of the tRNA and affect codon identification and tRNA aminoacylation [3]–[5]. For example, m5C at position 40 of the yeast tRNA-Phe enables conformational transition of the entire anticodon loop, inducing Mg2+ binding at a distant site and resulting in structural stabilization [6]. Absence of an m5C modification in the anticodon wobble-base (position 34) of tRNA-Leu in yeast was associated with decreased functionality, observed when nonsense suppressor function was estimated in vivo [7]. Recently, it was shown that various modifications on tRNA, including m5C, are dynamically modulated during cellular responses to several stress conditions, suggesting that these modifications may play a role in cellular response to stress, potentially by mediating translation rates [8], [9]. In rRNA m5C is thought to play a role in translational fidelity [10].
Traditional methods for studying m5C and other RNA modifications include HPLC and mass spectrometry, requiring isolation of the specific RNA molecule being studied to near purity [11], [12]. As opposed to the highly abundant tRNAs and rRNAs, isolation of specific mRNA molecules to purity is experimentally challenging, and hence studies of m5C modifications on mRNA molecules were limited until recently. Indeed, early studies documenting the presence of m5C in mRNA have been controversial; some studies have failed to detect m5C in eukaryotic mRNA [13]–[16], whereas others have identified this modification [17]–[19].
Recently, the presence of m5C in human HeLa mRNA was studied in a global manner, identifying thousands of potential m5C sites within human mRNA [20]. This study utilized RNA bisulfite treatment, which selectively converts C residues, but not m5C, into U residues [21], followed by whole transcriptome sequencing. Such bisulfite treatment is commonly used on DNA sequences when DNA methylation states are studied [22], [23]. However, its usage for RNA m5C methylation interrogation has been limited until recently. As a result, no study has addressed the presence of m5C sites, or any other RNA modification, in prokaryotes in a transcriptome-wide manner, and no modification to bacterial or archaeal mRNA has been described to date.
Here, we set out to generate transcriptome wide maps of m5C RNA modification sites in representative model organisms from all three domains of life. We applied bisulfite treatment on total RNA from these organisms, and generated a computational pipeline that identifies methylated positions after accounting for various artifacts. This approach was highly sensitive, and allowed the identification and quantification of the vast majority of known m5C positions in tRNAs and rRNAs in these organisms, as well as the detection of novel positions in these structural RNAs. Anti-m5C RNA immunoprecipitation confirmed our findings. Intriguingly, we detected m5C sites in several mRNAs in archaea, and identified a sequence motif guiding these RNA methylations. This might be suggestive of an additional layer of regulatory complexity present – potentially having functional consequence – on prokaryotic mRNAs.
Results
To obtain a single-nucleotide resolution overview of m5C sites present in RNA, we extracted total RNA from four model organisms, spanning all three domains of life: the eukaryote Saccharomyces cerevisiae, the archaeon Sulfolobus solfataricus, and the gram positive and negative bacteria, Bacillus subtilis and Escherichia coli, respectively. We treated the RNA with bisulfite using a modified version of the protocol for bisulfite-treatment of DNA,(described in [21]), prepared a pooled cDNA library from the four organisms, and subjected them to massively parallel sequencing (Table 1; Figure 1A). Since all modified sites identified to date have originated from either tRNAs or rRNAs, we used a protocol that enriches for tRNA and rRNA molecules during this initial approach. This allowed us to test the specificity and sensitivity of the assay (Methods). Sequencing reads from this experiment were obtained primarily from tRNA and rRNA, thus validating the approach. Sequencing results gave sufficient coverage (≥5 reads) for all rRNA molecules annotated for the organisms tested, and for 153 of the 187 (82%) annotated tRNA molecules (Table 1).


To identify putative methylated positions, we aligned the reads against genomic DNA that was computationally adjusted so that all C residues were converted to T. Overall, we observed that >98% of all sequenced C residues were converted into T following bisulfite conversion and cDNA sequencing, providing statistical power to identify cytosine residues that reproducibly failed to undergo conversion as putative m5C sites (Table 1). For each genomic cytosine, we calculated the number of reads in which that C did not undergo conversion to T, indicative of methylation (Figure 1). For each such position, we determined its “methylation level”, corresponding to the proportion of transcripts in which that position was methylated, and calculated a p-value based on Fisher's exact test against the null hypothesis that the position was not methylated (Methods). Based on previous experiments using bisulfite sequencing of DNA [24] and RNA [20], where stretches of non-converted Cs were often found in specific reads (presumably reflecting insufficient exposure of a transcript to bisulfite) we filtered out reads in which 3 or more Cs were not converted into Ts, to avoid assignment of m5C modification due to experimental artifacts. Although several modifications other than m5C may result in the absence of C->U conversion following bisulfite treatment, we designated all non-converted sites as putative m5C sites, for the sake of consistency (see Discussion).
We began by examining methylation sites in tRNAs and rRNAs that passed a statistical metric (p<0.01) and in which the methylation levels exceeded 50%. Analysis of the data yielded a total of 10 sites in rRNAs and 85 sites in tRNAs. Below we provide a summary of the identified methylated sites in the different RNA classes in each of the organisms.
Methylation of rRNA molecules
In E. coli we identified rRNA modifications at 3 distinct positions. The identified residues included position 967 in the 16S rRNA subunit, and position 1962 in the 23S rRNA subunit (Figure 2A–B). Both these residues have been previously determined to contain m5C modifications [25]–[27]. In addition, position 1402 in the 16S rRNA of E. coli was identified as having a methylation level of 58%. This position is known to harbor a N4,2-O-dimethylcytidine modification which also is not amenable to bisulfite conversion [21], emphasizing the possibility that a fraction of the sites we detected may reflect modifications other than m5C (see Discussion).

We identified two modified residues at positions 977 and 1411 of the 16S rRNA of B. subtilis (Figure 2C). While neither one of these residues has been previously directly characterized as methylated in B. subtilis, they are positionally orthologous to the previously characterized positions 967 and 1402 in the E. coli 16S rRNA, respectively (above). Not only were the positions conserved, but also the methylation stoichiometry appeared to be conserved between E. coli and B. subtilis, with methylation levels of 98.7% at position 977 compared to 97.5% in E. coli, and 63.7% at position 1411 compared to 58% in E. coli.
In S. solfataricus we identified one m5C site at position 1369 of the 16S rRNA, and another at position 2643 of the 23S rRNA. An additional site with a methylation level of 43% was observed at position 2121 of the 23S rRNA (Figure 2E–F). Position 1369 at the 16S was previously characterized as subjected to an unknown modification [28], whereas no evidence existed for methylation of the 23S sites. Sanger sequencing confirmed that these sites did not undergo bisulfite conversion, providing further support for their methylation (Figure 3). The presence of these modifications is well in line with previous chromatography/mass-spectrometry based analysis, which predicted one m5C site in the 16S subunit of S. solfataricus, and 1–2 additional sites in the 23S subunit [29].

In yeast we identified two modified positions within the 26S rRNA subunit. Position 2278, which was found to have a methylation level of 72%, was previously reported to contain a methylated site [30]. To our knowledge, we are the first to report the presence of a methylation site at position 2870 (methylation level 98%, Figure 2D). Its presence and the nucleotide composition of this detected site (m5C site preceded by a pyrimidine), is consistent with the prediction of an additional m5C site in the 26S unit of yeast rRNA, which was not mapped at the time [30].
Methylation sites within tRNA molecules
Eukaryotic and archaeal tRNA molecules, but not bacterial tRNAs, are known to undergo m5C modifications at specific positions. Consistent with this finding, we identified 46 modified sites in 34 tRNAs in S. solfataricus, and 39 sites in 30 tRNAs in S. cerevisiae. To allow comparative analysis of methylation levels in different tRNAs, we generated a multiple alignment of the archaeal and yeast tRNAs, and color-coded methylated positions along them based on their methylation levels (Figure 4). Notably, in this analysis we examined significant (p<0.01) methylated positions even when methylation levels were below 50%, to allow full comparative inspection of the data.

Consistent with previous findings, the methylated positions in both archaea and yeast were clustered at positions 48–49 along the tRNA molecule (numbers based on E. coli tRNA positions, as in [31]). In archaea most (30/34, 88%) of the tRNAs were methylated at position 48, while about half of these (14/30) were also methylated at position 49. The pattern of m5C modification in yeast was different: usually, either position 48 or position 49 was methylated, with only two tRNAs showing modifications at both positions. Two other tRNAs also contained modifications at position 50 in the variable region.
The relatively high coverage of tens and sometimes hundreds of reads in each tRNA allowed us to quantitatively assess similarities and differences in methylation levels at specific positions (presumably corresponding to the percent of tRNA transcripts modified at those positions). In general, levels of methylation in yeast were significantly higher than those in archaea, with mean values of 96% per position in S. cerevisiae as compared to mean values of 82% observed in S. solfataricus (Mann-Whitney test, p = 3.8e-6). This may imply that the typical single, strongly methylated site in yeast may compensate for the presence of two more weakly methylated sites in S. solfataricus. This interpretation, however, needs to be taken with caution, as other factors may account for the observed differences in methylation levels (see Discussion).
Surprisingly, a small fraction of the tRNAs were found to be devoid of m5C modifications in the variable region, despite the presence of a cytosine in one of the positions 48–50. These include tRNA-AlaAGC,TGC and tRNA-ArgCCT in yeast and tRNA-GluTCC,CTC in S. solfataricus. This selective absence of the modification in a subset of tRNAs might provide a potential anchor towards elucidating the role of this modification on tRNAs. Alternatively, other types of RNA modifications, not detectible through bisulfite-conversion, may replace m5C in these tRNAs.
Several additional methylated positions were observed in our data, including the previously identified methylation in the anticodon (position 34) of yeast tRNA-Leu [32], as well as methylations at position 27 of tRNA-Pro and position 56 of tRNA-Leu. Additional putative m5C sites were observed in S. solfataricus tRNA-TrpCCA and tRNA-LeuTAA, but given their relatively low methylation levels, these sites require further verification. Finally, we failed to detect the known methylation at position 40 of tRNA-Phe [6] due to lack of sequence coverage for this particular tRNA.
Methylation in mRNA molecules
We were next interested in searching for methylated sites within mRNA molecules. Since our initial experiment lacked sufficient sequence coverage to identify such sites within the vast majority of mRNAs due to the high coverage of rRNA and tRNAs, we performed a second bisulfite-seq experiment on the bacterial and the archaeal RNA using size-selected total RNA (>200 bp) to deplete tRNAs, combined with rRNA depletion protocols to enrich for mRNAs (Methods). For S. cerevisiae we made use of a dataset of short sequences previously obtained using bisulfite-treatment of polyA-selected RNA [33].
We used the computational pipeline outlined above to analyze the libraries obtained from the mRNA enrichment protocols. Coverage along mRNA in this experiment was substantially improved, with sufficient coverage (mean coverage ≥5 reads/nucleotide) in 1,394 protein coding genes in B. subtilis, 1,110 genes in S. solfataricus, and 4,489 genes in yeast. In E. coli, due to very high levels of ribosomal RNA reads (>99%), only 287 protein-coding genes had sufficient coverage (Table 2).

Despite significant coverage of most yeast genes, we were only able to detect a single event of m5C on an mRNA in that organism, occurring on a protein of unknown function. No m5C events were found in the expressed bacterial mRNAs that were covered in our experiment, although the lack of coverage for most E. coli genes prevents us from drawing a strong conclusion as to the possibility of methylated sites in mRNAs for that organism.
In S. solfataricus, the mRNAs of two protein coding genes contained a nucleotide methylated to a level of >50%, and 12 additional genes contained nucleotides with methylation levels of 20–50% (Table 3). Many of the genes harboring the m5C modification were enzymes involved in energy and lipid metabolism (notably oxidoreductases and dehydrogenases), which might imply a possible role for this modification in regulating specific metabolic processes. Interestingly, all 14 m5C positions in S. solfataricus mRNAs were located within a strong sequence motif of AUCGANGU (Figure 5C). This same motif is found at the m5C sites we recorded at positions 2121 and 2643 of the S. solfataricus 23S rRNA (Figure 2). These results suggest that the same machinery (probably a methyltransferase) is responsible for m5C modifications both on the 23S rRNA and on the mRNAs. Moreover, our results imply that this machinery recognizes a consensus sequence of AUCGANGU. The identification of a strong consensus motif reinforces the validity of our bisulfite-sequencing approach in identifying real modified sites on RNA bases, and supports the existence of a single predominant methylation machinery on mRNAs in S. solfataricus.

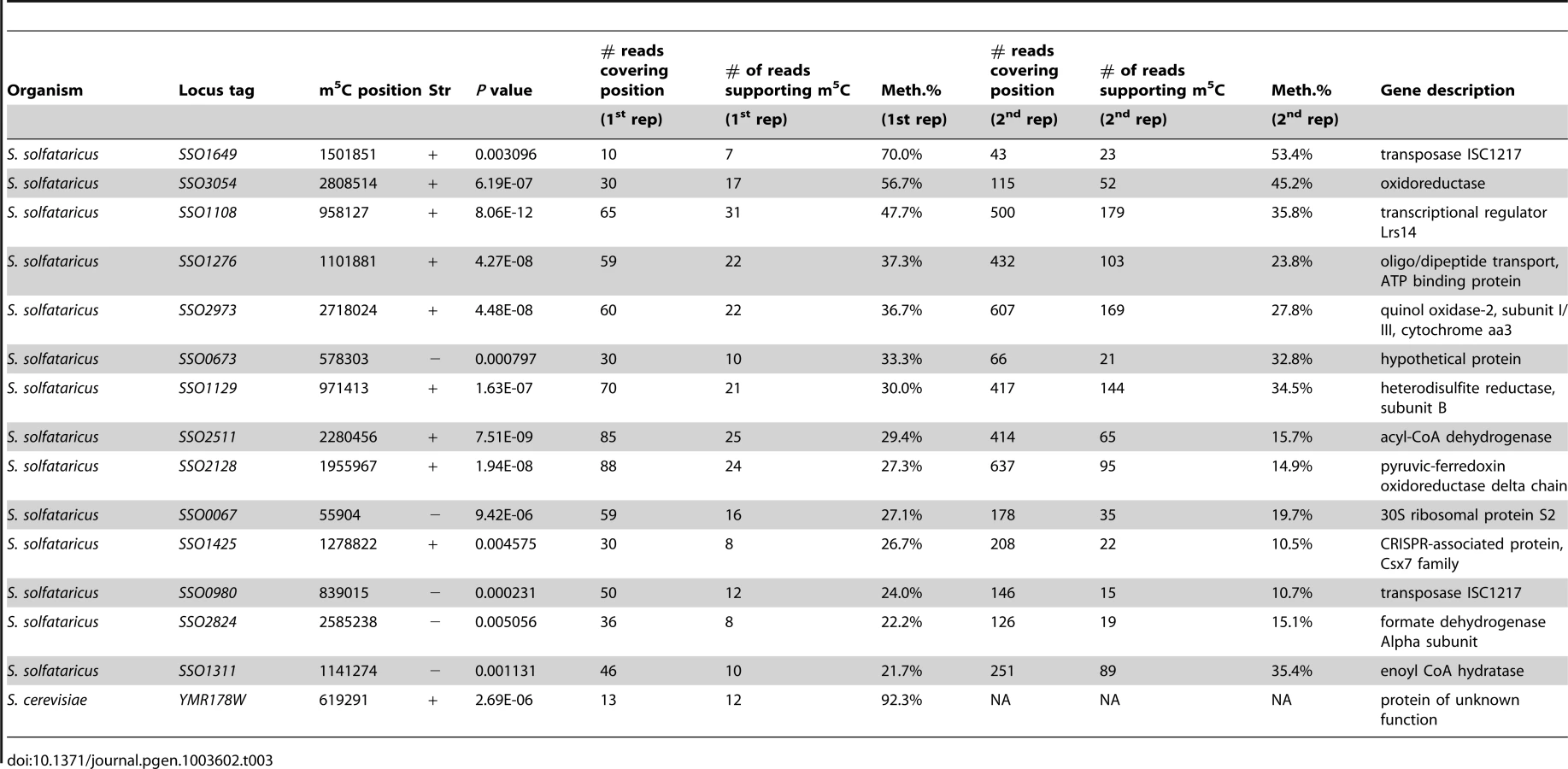
To verify that the m5C modifications we detected reproducibly appear in S. solfataricus mRNAs we sequenced a second bisulfite-treated biological replicate sample of total RNA from S. solfataricus. Indeed, all m5C sites identified in the first experiment were also detected in the second experiment with similar methylation levels but with higher coverage (Table 3). To rule out the possibility that the consensus motif we identified is intrinsically resistant to bisulfite conversion, we mixed the second S. solfataricus RNA sample, prior to bisulfite conversion, with two synthesized 200 nt RNA fragments each harboring a common representation of the consensus motif (either ATCGAGGT or ATCGAAGG; Methods). Despite very deep coverage obtained for the two artificial consensus-bearing RNAs (130,000 and 2.8 million reads, respectively), no evidence for m5C was observed within these RNAs, and the percentage of cytosines that were not converted into uracils within the two synthesized consensus motifs was 0.6%, similar to non-methylated residues (Table 2). These results show that the methylation sites we observed in S. solfataricus mRNAs do not represent a motif-dependent artifact.
To determine if the presence of this consensus sequence is sufficient to drive methylation, we searched for the motif within all S. solfataricus protein coding genes. We identified 75 sites that were covered by at least 5 reads (regardless of p-value score). Of these, 31 (41%) were methylated at levels >10%, and 13 (17%) were methylated at levels higher than 20%. These levels are far higher than expected by chance alone: examining all sites (n = 22,665) covered by at least 5 reads, but significantly differing from the consensus sequence, only 5.9% had methylation levels of >10%, and 0.9% of the sites had methylation levels exceeding 20%. These results suggest that the presence of this motif is to a large extent sufficient for recognition and methylation by the putative methyltransferase. However, the varying levels of methylation suggest that additional factors other than the immediate sequence environment – such as RNA availability or more distant motifs – may also play a role in determining methylation levels.
We next examined the relative position of the modified sites within the genes they reside. Methylated bases tended to be localized towards the beginning of the gene (Fig. S1, P = 0.07). The apparently non-uniform distribution of modified sites may provide an angle for future elucidation of the function of this RNA modification.
Direct immunoprecipitation of methylated RNA
Several modifications other than m5C may result in an inability to convert C->U following bisulfite treatment, including 3-methylcytidine, N4-methylcytidine, N4,2′-O-dimethylcytidine and N4-acetylated variants [20], [21]. To further examine the nature of the novel modifications we observed in S. solfataricus RNAs, we set out to conduct direct immunoprecipitation of modified RNA. For this, we used a monoclonal antibody that specifically binds 5-methylcytosine. This antibody is broadly used to specifically detect m5C modifications in DNA (e.g. [34]), but since it was raised against 5-methylcytosine nucleotide conjugated to ovalbumin without the ribose or deoxyribose sugar, it is blind to the DNA/RNA context of the modification and hence binds the RNA form of m5C as well. As a control we used a second antibody that specifically binds 5-hydroxymethylcytosine (hm5C) but not m5C. The hm5C modification is similar to m5C but carries an additional hydroxyl group on top of the added methyl group. Total RNA of S. solfataricus was sheared into ∼100 nt-long fragments (the “input”) and immunoprecipitated using the anti-m5C or anti-hm5C antibodies. Libraries were prepared from immunoprecipitated as well as non-immunoprecipitated input control RNA fragments, and subjected to massively parallel sequencing on the Illumina platform.
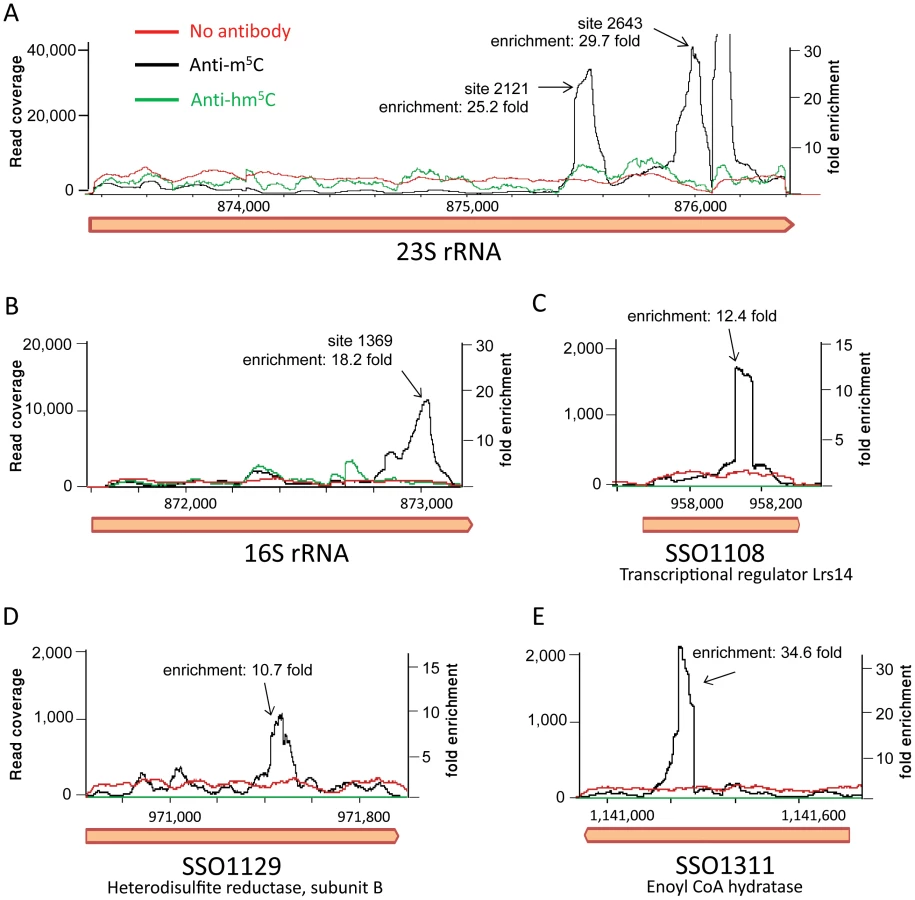
Mapping the resulting reads to the S. solfataricus genome resulted in a highly non-uniform coverage (Fig. 6). Peaks in the coverage were clearly observed in the rRNA at the exact locations identified by the bisulfite sequencing as m5C modified (positions 1369 in the 16S and 2121 and 2643 in the 23S). These peaks represented enrichment of 18–30 fold over the rest of the rRNA molecule, but no enrichment was observed in these positions in the non-immunoprecipitated (input) sample or the sample immunoprecipitated by the hm5C antibody. Most (10/14, 71%) of the sites we identified in the mRNAs of S. solfataricus (Table 3) were also found to be enriched in the immunoprecipitated library (Fig. 6C–E, enrichment of 10–35 fold). These results provide strong independent verification that the sites we identified in the rRNAs and mRNAs of S. solfataricus indeed correspond to m5C modifications.
Discussion
We have employed bisulfite treatment of RNA, combined with high-throughput sequencing, to generate a sensitive map of methyl-modified cytosines in four model organisms across the microbial tree of life. Our map not only recovers the vast majority of known sites within tRNAs and rRNAs in these organisms, but also provides a measurement of methylation efficiency for each position, and reveals novel sites within rRNAs and mRNAs.
Several modifications other than m5C may result in a lack of C→U conversion following bisulfite treatment [20], [21]. Indeed, and in agreement with previously published data [21], position 1402 in E. coli identified in this study is known to harbor an N4,2′-O-dimethylcytidine modification, which is known to be resistant to bisulfite conversion. Therefore, the novel modified positions we report in this study might correspond to modifications other than m5C. To verify that the novel modifications in mRNAs we identified in S. solfataricus indeed represent m5C, we utilized m5C-specific antibodies that are usually used in pulldown experiments of modified DNA molecules that are an epigenetic marker for gene silencing in mammals. Since these antibodies also bind the RNA form of m5C, they formed an ideal tool for independent validation of our bisulfite-based findings. Each of the approaches has its strengths and weaknesses: while the bisulfite conversion provides a single-nucleotide resolution positioning of the modified site, it may also report other types of modifications. The antibody-based approach provides specificity to a single type of modification but with lower resolution of about 100 bp [35]. Therefore, the combination of bisulfite-conversion and RNA-immunoprecipitation provides synergistic results.
In eukaryotes, the chemical modification of mRNAs is emerging as an important factor in the regulation of gene expression. These modifications have been shown to affect mRNA translation, splicing, stability and transport [20], [35], [36]. To our knowledge, our report presents the first evidence for mRNA modification in archaea, opening a window into a possible additional layer of gene regulation in archaea. Further studies are needed in order to elucidate the possible function of these modifications. In particular, profiling of the methylation status across different cellular states, or in response to external stimuli, will allow investigation of the extent to which this modification is dynamically regulated. Knockout studies can provide better understanding of the enzymes involved in mediating this modification across different organisms and will be crucial for understanding its function.
Our identification of a defined sequence motif associated with all mRNA methylated positions in S. solfataricus strongly suggests that these modifications indeed occur in vivo and are not an artifact of the detection method. This motif, along with the recently reported sequence motif associated with m6A modifications on mammalian mRNAs [35], are the only reported cases of clearly defined linear sequence motifs directing multiple RNA methylation events. Indeed, studies on various tRNA and rRNA modifying enzymes had suggested preference for local RNA structures rather than for a specific sequence [25], [37], [38]. In this respect, it is possible that there is a fundamental difference between tRNA and rRNA molecules, in which modifications are often sequentially added according to folding and maturation levels, therefore demanding structural dependency, and mRNAs, in which the linear sequence may be the most important determinant.
The motif we identified in S. solfataricus, AUCGANGU, is also found at the modified positions C2121 and C2643 in the 23S rRNA. Therefore, it is likely that the same methyltransferase responsible for modification of these rRNA residues also methylates the positions we detected in mRNAs. It is possible that the modifications we find on mRNAs might reflect a “spillover” of this enzymes' activity onto non-specific mRNA substrates. However, no methylated site on S. solfataricus mRNAs resembles the modified sequence of the 16S rRNA in that organism, suggesting a specific activity for the 23S methylase, but not for the 16S methylase, on mRNA positions. Interestingly, recent work by Squires and coworkers similarly found that m5C modifications on human mRNAs are mediated by the RNA methyltransferase NSUN2, previously known to act only on human tRNAs [39].
Our approach not only detects methylated positions in RNAs, but also provides a quantitative measure of the fraction of transcripts harboring this methylation (to which we refer as “methylation level”). The close similarities in rRNA methylation levels of orthologous positions between the gram positive bacterium B. subtilis and the gram negative E. coli reinforces the validity of this quantitative measure. Nevertheless, this measure should be taken with caution, as factors such as miniscule DNA contamination (which is non-methylated), for example, could contribute noise to this measurement. In addition, bulky modifications (which are prevalent in tRNAs) may hinder reverse transcriptase processivity, and hence, completely modified tRNA molecules may be under-represented in the sequenced data. Therefore, the stoichiometric measurements we recorded might be biased. Methodologies that are not based on reverse transcription should be applied to decisively determine methylation stoichiometry.
An additional limitation of our approach is that it will fail to detect positions methylated at low levels, given the conservative cutoff we set of filtering out rarely methylated positions. Indeed, apart from the positions we described with high methylation levels, there were thousands of additional positions with significant p values, but with much lower methylation levels. About 95% of these sites had methylation levels lower than 10%, including many putative positions in rRNAs and tRNAs. As most of these positions have not been previously reported, despite the fact that tRNAs and rRNAs have been extensively characterized, it is highly probable that the majority of these sites represent artifacts from various sources. This notwithstanding, this group contains a single site in the E. coli 16S rRNA (position 1407), which is, indeed, known to be modified. This suggests that there may be additional real methylated nucleotides within this group. However, our approach is currently unable to differentiate between rarely methylated positions and experimental artifacts.
The emergence of ultra-high throughput sequence interrogation technologies is revolutionizing research on RNA modifications. Application of such technologies, including RNA-seq and RIP-seq, to study such modifications in eukaryotes has recently revealed that mRNAs carry a plethora of conserved modifications, such as A-to-I [40], m6A [35] and m5C [20]. Nevertheless, the field of mRNA modifications is still in its infancy, and even in eukaryotes, where several modifications have already been extensively characterized, their functional consequences are still poorly understood. Our report that mRNAs of archaea also carry such modifications raises the intriguing possibility that mRNA modifications in prokaryotes form an additional layer of gene regulation that has not yet been addressed. Whether the m5C modifications are functionally relevant, and whether other RNA modifications also exist on prokaryotic mRNAs, remains to be determined.
Methods
RNA samples preparation
Escherichia coli (MG1655) cells were grown in LB medium to log phase at 37°C. Bacillus subtilis str. 168 cells were grown in LB medium to mid log phase at 37°C. S. solfataricus P2 (DSMZ 1617) cells were grown in defined modified Brock's mineral medium with final pH 3.5, and S. cerevisiae (BY4741) cells were grown in YPD medium to log phase at 30°C. All cells pellets were suspended in RNAlater (Ambion, AM7022) for 30 min at room temperature. Pre-treatments for RNA isolation included Lysozyme digestion for E. coli cells, glass beads vortex for B. subtilis and Lyticase digestion for S. cerevisiae. Total RNA was then extracted using Tri-Reagent (Molecular Research Center Inc.) according to manufacturer's instructions. RNA samples were treated with Turbo DNA-free kit (Ambion) and rRNA was removed from the E.coli and B.subtilis samples using MICROBExpress mRNA enrichment kit (Ambion).
Bisulfite treatment
Bisulfite conversion was performed based on the protocol by Schaefer et al. [21]. Briefly, one microgram of each RNA sample was incubated in DNA protect buffer and bisulfite mix, EpiTect Bisulfite Kit (Qiagen), through 6 cycles of denaturation step at 70°C, followed by a deamination reaction step of 1 hour at 60°C. The RNA was purified from the bisulfite reaction mix using Micro Bio-Spin 6 columns (Bio-Rad) and treated with 0.5M Tris–HCl, pH 9 at 37°C for 1 hour. RNA was then washed on YM-10 microcon (Millipore) 5 times with 0.5 ml ultra-pure water. RNA samples were analyzed using the Bioanalyzer (Agilent) to assess degradation status.
cDNA synthesis and Sanger sequencing
For all 4 organisms, untreated RNA and bisulfite-treated RNA were used as templates for cDNA synthesis using SuperScript II RT (Invitrogen) and random hexamers according to manufacturer's protocol. cDNA was amplified by PCR (ABgene) using regular primers (for untreated RNAs) and primers specific to cytosine-deaminated sequences (for bisulfite treated RNAs). In order to improve sequencing results, generic M13 sequences (containing all 4 bases) were combined with the 5′ end of all the deaminated primer sequences. PCR products were separated on agarose gel and extracted using gel extraction kit (Qiagen). Sanger sequencing of the PCR product was conducted from the M13 primer sequence.
Library construction and Illumina sequencing
Equal amounts (50 ng) of each of the four organism's bisulfite-treated total RNA were mixed together. The mixed bisulfite-treated RNA sample was used as a template for cDNA library preparation according to the mRNA-seq Illumina protocol, omitting the polyA-based mRNA purification step. In brief, RNA was first fragmented by divalent cations at 94°C for 5 min. Double stranded cDNA was generated using SuperScriptII and random primers. cDNA was then end-repaired, adenylated and end-ligated to adapters. Following gel separation, a ∼200 bp fragment was gel-purified. The cDNA library was further amplified and sequenced using 40 single-read cycles on a Genome Analyser II (Illumina).
Alignment of reads
40-nt long reads were aligned using Novoalign (Novocraft Technologies Sdn Bhd, http://www.novocraft.com) to bisulfite-converted genomes of Bacillus subtilis (NC_000964), Escherichia coli (NC_000913), Sulfolobus solfataricus (NC_002754) and Saccharomyces cerevisiae (NC_001133-NC001148) downloaded from the NCBI website. Reads that did not align to the reference sequence at their original length, were iteratively trimmed by two base-pairs from the end of the read and then realigned, as long as their length exceeded 35 nt. To allow identification of methylated positions in tRNAs and rRNAs occurring in multiple copies in the genome, reads mapping to multiple regions in the genome were randomly assigned to one such region. Parameters used for the indexing step were “–b”, and for alignment were “-t 60 -h 120 -b 4 -l 35 -s 2 -F STDFQ -r Random -u 6”. For the transcriptome-based study of rRNAs and tRNAs, reads were aligned against a database consisting of unique copies of all fully processed tRNAs obtained from the tRNAdb [41], and against a single representative of each of the 5S, 16S and 23S rRNA genes in each of the four organisms. Identical alignment parameters were set as above. The rational for this approach was based on two considerations: First, to prevent dilution of reads to multiple, identical copies of the same tRNAs and rRNAs, and second, to prevent loss of reads due to differences between the transcriptome and the genome, such as in cases of tRNA introns that are excised during maturation of the tRNA molecule.
Identification of candidate methylated positions
For each cytosine in the genome in both orientations, the number of reads in which the cytosine underwent conversion to uridine (suggesting that it was not methylated), and those in which it was not converted (suggesting methylation) were counted. To eliminate artifacts of various sources, the following filters were applied: (1) identical reads were considered a single read, to eliminate PCR amplification artifacts, (2) reads with ≥3 unconverted cytosines were eliminated, as they might reflect transcripts that for did not obtain sufficient exposure to bisulfite, (3) only positions with a sequencing quality >20 were counted, to eliminate low-quality positions. In the transcriptome based analysis we did not apply the first filter, since for the highly expressed rRNA and tRNA molecules identical reads generally do not reflect PCR artifacts, but merely reflect the high coverage of these genes. Each position was then assigned a methylation level, equivalent to the proportion of reads in which it was not converted, and a p-value determining the significance of the number of converted and non-converted reads was assigned using Fisher's exact test against the null hypothesis that all reads were converted. Identified methylation sites that were within 10 nt of an additional site were discarded. Since many genes are present in the genome in multiple copies, following identification of all significantly methylated positions (p<0.01), all positions sharing an identical 31-nt sequence surrounding the identified position were collapsed together into a single sequence. Curation of the data confirmed that this efficiently collapsed together sequences from identical genes. The joint methylation level for each collapsed position was recalculated based on the cumulative number of converted and non-converted reads in its ‘parents’.
Alignment of tRNAs
For the alignment of tRNAs from tRNAdb [41], a multiple sequence alignment of tRNAs was generated using mafft v6.850b, with the parameters “–maxiterate 1000 –localpair”.
Analysis of the consensus motif in S. solfataricus
The motif AUCG-A/U-G/UG-U/G was searched in all protein coding genes in which the potentially methylated ‘C’ in the center of this motif was covered by at least 3 reads, and methylation ratios were recorded. As a negative control the analysis was repeated taking only motifs with at least three mismatches compared to the consensus (specifically, demanding a “C” at position 0, and that position −2 was not an “A”, −1 not a “T”, and +1 not a “G”). For the analysis presented in Fig. S1, we assembled a dataset of 38 sites harboring a consensus and with a methylation P value<0.05. Notably, to gain more statistical power for the downstream analysis, here we did not set a threshold on the minimal methylation level. For each site, its relative position within the gene was calculated, from a scale of 0 to 1 (representing the 5′ and 3′ ends of a gene, respectively). We compared the distributions of these methylation sites to 22,665 controls lacking a consensus site, and obtained marginally significant results (t-test, P = 0.07).
RNA spikes
Two ∼200 bp sequences from the E.coli genome, containing the consensus sequence found in Sulfolobus solfataricus, were amplified via PCR from the genome of E. coli MG1655 (primers: p1_fwd TAATACGACTCACTATAGGGTCATGCACGGTGTCGTTATT; p1_rev AAAGGTTTCCATGTCGAACG; p2_fwd TAATACGACTCACTATAGGGGCTGTGGTGATCAGTGTGCT; p2_rev TGGCGTTGATAAAACTGACG). These amplified sequences were used as template for an in-vitro transcription reaction using MaxiScript kit (Ambion, AM1344) to produce RNA transcripts. These RNA transcripts were spiked into Sulfolobus solfataricus total RNA prior to the bisulfite conversion treatment. Reads obtained from this library were aligned separately against the S. solfataricus genomes, and against a synthetic genome comprising the two E. coli templates. Non-converted cytosines were quantified using the pipeline and filters described above.
RNA immunoprecipitation
Sulfolobus solfataricus total RNA was chemically fragmented (Ambion, AM8740) to an average size of ∼100 bp and was subjected to immunoprecipitation with an anti m5C monoclonal antibody (Diagenode, MAb-081-010) or anti-hm5C polyclonal antibody (Diagenode, pAb-HMC-050) based on the protocol described in ref [35], using two rounds of IP. The precipitated RNA was used for dsDNA Illumina library construction. Illumina MiSeq sequencing of 30 bp paired-end was used to characterize rRNA modifications. Then, Illumina HiSeq run was conducted on the same library with single-end 50 bp reads to gain enough coverage to analyze the mRNA positions.
Supporting Information
Zdroje
1. BasuR, ZhangLF (2011) X chromosome inactivation: a silence that needs to be broken. Genesis 49 : 821–834.
2. RigalM, MathieuO (2011) A “mille-feuille” of silencing: epigenetic control of transposable elements. Biochim Biophys Acta 1809 : 452–458.
3. HelmM (2006) Post-transcriptional nucleotide modification and alternative folding of RNA. Nucleic Acids Res 34 : 721–733.
4. MotorinY, HelmM (2010) tRNA stabilization by modified nucleotides. Biochemistry 49 : 4934–4944.
5. SquiresJE, PreissT (2010) Function and detection of 5-methylcytosine in eukaryotic RNA. Epigenomics 2 : 709–715.
6. ChenY, Sierzputowska-GraczH, GuentherR, EverettK, AgrisPF (1993) 5-Methylcytidine is required for cooperative binding of Mg2+ and a conformational transition at the anticodon stem-loop of yeast phenylalanine tRNA. Biochemistry 32 : 10249–10253.
7. StrobelMC, AbelsonJ (1986) Effect of intron mutations on processing and function of Saccharomyces cerevisiae SUP53 tRNA in vitro and in vivo. Mol Cell Biol 6 : 2663–2673.
8. ChanCT, DyavaiahM, DeMottMS, TaghizadehK, DedonPC, et al. (2010) A quantitative systems approach reveals dynamic control of tRNA modifications during cellular stress. PLoS Genet 6: e1001247.
9. SchaeferM, PollexT, HannaK, TuortoF, MeusburgerM, et al. (2010) RNA methylation by Dnmt2 protects transfer RNAs against stress-induced cleavage. Genes Dev 24 : 1590–1595.
10. ChowCS, LamichhaneTN, MahtoSK (2007) Expanding the nucleotide repertoire of the ribosome with post-transcriptional modifications. ACS Chem Biol 2 : 610–619.
11. DouthwaiteS, KirpekarF (2007) Identifying modifications in RNA by MALDI mass spectrometry. Methods Enzymol 425 : 1–20.
12. EmmerechtsG, BarbeS, HerdewijnP, AnneJ, RozenskiJ (2007) Post-transcriptional modification mapping in the Clostridium acetobutylicum 16S rRNA by mass spectrometry and reverse transcriptase assays. Nucleic Acids Res 35 : 3494–3503.
13. DesrosiersR, FridericiK, RottmanF (1974) Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci U S A 71 : 3971–3975.
14. DesrosiersRC, FridericiKH, RottmanFM (1975) Characterization of Novikoff hepatoma mRNA methylation and heterogeneity in the methylated 5′ terminus. Biochemistry 14 : 4367–4374.
15. WeiCM, GershowitzA, MossB (1976) 5′-Terminal and internal methylated nucleotide sequences in HeLa cell mRNA. Biochemistry 15 : 397–401.
16. PerryRP, KelleyDE, FridericiK, RottmanF (1975) The methylated constituents of L cell messenger RNA: evidence for an unusual cluster at the 5′ terminus. Cell 4 : 387–394.
17. DubinDT, StollarV (1975) Methylation of Sindbis virus “26S” messenger RNA. Biochem Biophys Res Commun 66 : 1373–1379.
18. DubinDT, TaylorRH (1975) The methylation state of poly A-containing messenger RNA from cultured hamster cells. Nucleic Acids Res 2 : 1653–1668.
19. DubinDT, StollarV, HsuchenCC, TimkoK, GuildGM (1977) Sindbis virus messenger RNA: the 5′-termini and methylated residues of 26 and 42 S RNA. Virology 77 : 457–470.
20. SquiresJE, PatelHR, NouschM, SibbrittT, HumphreysDT, et al. (2012) Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res 40 : 5023–5033.
21. SchaeferM, PollexT, HannaK, LykoF (2009) RNA cytosine methylation analysis by bisulfite sequencing. Nucleic Acids Res 37: e12.
22. ZhangY, RohdeC, TierlingS, StamerjohannsH, ReinhardtR, et al. (2009) DNA methylation analysis by bisulfite conversion, cloning, and sequencing of individual clones. Methods Mol Biol 507 : 177–187.
23. KruegerF, KreckB, FrankeA, AndrewsSR (2012) DNA methylome analysis using short bisulfite sequencing data. Nat Methods 9 : 145–151.
24. WarneckePM, StirzakerC, SongJ, GrunauC, MelkiJR, et al. (2002) Identification and resolution of artifacts in bisulfite sequencing. Methods 27 : 101–107.
25. AndersenNM, DouthwaiteS (2006) YebU is a m5C methyltransferase specific for 16 S rRNA nucleotide 1407. J Mol Biol 359 : 777–786.
26. SmithJE, CoopermanBS, MitchellP (1992) Methylation sites in Escherichia coli ribosomal RNA: localization and identification of four new sites of methylation in 23S rRNA. Biochemistry 31 : 10825–10834.
27. TscherneJS, NurseK, PopienickP, MichelH, SochackiM, et al. (1999) Purification, cloning, and characterization of the 16S RNA m5C967 methyltransferase from Escherichia coli. Biochemistry 38 : 1884–1892.
28. CzerwoniecA, Dunin-HorkawiczS, PurtaE, KaminskaKH, KasprzakJM, et al. (2009) MODOMICS: a database of RNA modification pathways. 2008 update. Nucleic Acids Res 37: D118–121.
29. NoonKR, BruengerE, McCloskeyJA (1998) Posttranscriptional modifications in 16S and 23S rRNAs of the archaeal hyperthermophile Sulfolobus solfataricus. J Bacteriol 180 : 2883–2888.
30. VeldmanGM, KlootwijkJ, de RegtVC, PlantaRJ, BranlantC, et al. (1981) The primary and secondary structure of yeast 26S rRNA. Nucleic Acids Res 9 : 6935–6952.
31. MotorinY, LykoF, HelmM (2010) 5-methylcytosine in RNA: detection, enzymatic formation and biological functions. Nucleic Acids Res 38 : 1415–1430.
32. MotorinY, GrosjeanH (1999) Multisite-specific tRNA:m5C-methyltransferase (Trm4) in yeast Saccharomyces cerevisiae: identification of the gene and substrate specificity of the enzyme. Rna 5 : 1105–1118.
33. LevinJZ, YassourM, AdiconisX, NusbaumC, ThompsonDA, et al. (2010) Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nat Methods 7 : 709–715.
34. YamashitaS, HosoyaK, GyobuK, TakeshimaH, UshijimaT (2009) Development of a novel output value for quantitative assessment in methylated DNA immunoprecipitation-CpG island microarray analysis. DNA Res 16 : 275–286.
35. DominissiniD, Moshitch-MoshkovitzS, SchwartzS, Salmon-DivonM, UngarL, et al. (2012) Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485 : 201–206.
36. SieCP, KuchkaM (2011) RNA editing adds flavor to complexity. Biochemistry (Mosc) 76 : 869–881.
37. HoriH, YamazakiN, MatsumotoT, WatanabeY, UedaT, et al. (1998) Substrate recognition of tRNA (Guanosine-2′-)-methyltransferase from Thermus thermophilus HB27. J Biol Chem 273 : 25721–25727.
38. MorinA, AuxilienS, SengerB, TewariR, GrosjeanH (1998) Structural requirements for enzymatic formation of threonylcarbamoyladenosine (t6A) in tRNA: an in vivo study with Xenopus laevis oocytes. Rna 4 : 24–37.
39. BrzezichaB, SchmidtM, MakalowskaI, JarmolowskiA, PienkowskaJ, et al. (2006) Identification of human tRNA:m5C methyltransferase catalysing intron-dependent m5C formation in the first position of the anticodon of the pre-tRNA Leu (CAA). Nucleic Acids Res 34 : 6034–6043.
40. LiJB, LevanonEY, YoonJK, AachJ, XieB, et al. (2009) Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science 324 : 1210–1213.
41. JuhlingF, MorlM, HartmannRK, SprinzlM, StadlerPF, et al. (2009) tRNAdb 2009: compilation of tRNA sequences and tRNA genes. Nucleic Acids Res 37: D159–162.
42. CrooksGE, HonG, ChandoniaJM, BrennerSE (2004) WebLogo: a sequence logo generator. Genome Res 14 : 1188–1190.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 6
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- BMS1 Is Mutated in Aplasia Cutis Congenita
- Sex-stratified Genome-wide Association Studies Including 270,000 Individuals Show Sexual Dimorphism in Genetic Loci for Anthropometric Traits
- Distinctive Expansion of Potential Virulence Genes in the Genome of the Oomycete Fish Pathogen
- Distinct Neuroblastoma-associated Alterations of Impair Sympathetic Neuronal Differentiation in Zebrafish Models
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy