
Neuronal Target Identification Requires AHA-1-Mediated Fine-Tuning of Wnt Signaling in
Electrical synaptic transmission through gap junctions is a vital mode of intercellular communication in the nervous system. The mechanism by which reciprocal target cells find each other during the formation of gap junctions, however, is poorly understood. Here we show that gap junctions are formed between BDU interneurons and PLM mechanoreceptors in C. elegans and the connectivity of BDU with PLM is influenced by Wnt signaling. We further identified two PAS-bHLH family transcription factors, AHA-1 and AHR-1, which function cell-autonomously within BDU and PLM to facilitate the target identification process. aha-1 and ahr-1 act genetically upstream of cam-1. CAM-1, a membrane-bound receptor tyrosine kinase, is present on both BDU and PLM cells and likely serves as a Wnt antagonist. By binding to a cis-regulatory element in the cam-1 promoter, AHA-1 enhances cam-1 transcription. Our study reveals a Wnt-dependent fine-tuning mechanism that is crucial for mutual target cell identification during the formation of gap junction connections.
Published in the journal:
. PLoS Genet 9(6): e32767. doi:10.1371/journal.pgen.1003618
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003618
Summary
Electrical synaptic transmission through gap junctions is a vital mode of intercellular communication in the nervous system. The mechanism by which reciprocal target cells find each other during the formation of gap junctions, however, is poorly understood. Here we show that gap junctions are formed between BDU interneurons and PLM mechanoreceptors in C. elegans and the connectivity of BDU with PLM is influenced by Wnt signaling. We further identified two PAS-bHLH family transcription factors, AHA-1 and AHR-1, which function cell-autonomously within BDU and PLM to facilitate the target identification process. aha-1 and ahr-1 act genetically upstream of cam-1. CAM-1, a membrane-bound receptor tyrosine kinase, is present on both BDU and PLM cells and likely serves as a Wnt antagonist. By binding to a cis-regulatory element in the cam-1 promoter, AHA-1 enhances cam-1 transcription. Our study reveals a Wnt-dependent fine-tuning mechanism that is crucial for mutual target cell identification during the formation of gap junction connections.
Introduction
According to the neuron doctrine, the neural network is a linkage of discrete nerve cells, which are connected to one another indirectly through chemical synapses or directly through electrical synapses, also known as gap junctions. To wire the neuronal network, many neurons extend neurites (axons and dendrites) over great distances, bypassing numerous potential but inappropriate targets to reach the correct area. Over the past two decades, a number of important long-range and short-range environmental cues that regulate neurite guidance have been discovered, including Netrins, Semaphorins, Slits and Ephrins [1]. The list of guidance cues was further expanded by the finding that morphogens such as Wnt and Shh can also guide neurites in specific directions [2], [3]
After neurites from both synaptic partners reach the targeting field, a more refined target identification process must occur to establish the final connection. An array of cell adhesion molecules, including cadherins and immunoglobulin super-family proteins, are responsible for the direct selective cell-cell attraction between specific synaptic partner cells [4], [5]. Transcription factors also control synaptic connectivity. For example, the C. elegans homeodomain protein UNC-4, together with transcription factors HB9 and Groucho, is involved in the formation of neuronal gap junctions between the interneuron AVA and the motor neuron VA [6]–[9]. A recent study further revealed that UNC-4 antagonizes Wnt signaling to regulate this synaptic choice [10]. In addition, molecules involved in axon guidance play a significant role in the refinement of target identification. During visual system development, for instance, axons of retinal ganglion cells initially project into the optic tectum at incorrect positions along the medial-lateral tectal axis, but later correct this error by altering their trajectory or extending collateral branches at right angles [11]. Ephrins, a class of membrane-bound short-range signaling proteins, are involved in this fine-tuning process [12]. Netrin is a secreted chemotropic factor that can act as an attractive or repellent long-range cue during axon guidance. A recent study showed that the connectivity between AIY and RIA interneurons in C. elegans requires the coordination of Netrin-mediated short-range signals in both neurons [13]. In the target region, the same cues may steer neurites from both the pre-synaptic neuron and its target cell. In the formation of neuronal gap junctions, neurites of both reciprocal target cells are in close proximity and are thus very likely under the control of the same environmental signals. Therefore, to achieve effective connectivity, the spatial, temporal, and cell-type-specific responsiveness within both neurites must be tightly regulated and precisely coordinated. However, the detailed mechanism underlying this coordination is largely unknown.
Wnt signaling has recently been linked to axon guidance and synapse patterning [14], [15]. By binding to the extracellular cysteine-rich domain (CRD) of the Frizzled receptor (Frz) and the co-receptor LRP, Wnt activates Dishevelled (Dsh) and triggers downstream events. Receptor tyrosine kinase-like orphan receptor (Ror) proteins also bind Wnt and participate in multiple Wnt-mediated biological processes [16], [17]. In C. elegans, CAM-1, the homolog of mammalian Ror2, can function as a Wnt or Frizzled antagonist in cell migration and vulval development, or as a Wnt co-receptor regulating nerve ring organization and axon outgrowth [18]–[21]. In mammals the non-canonical Wnt5a-Ror2-Dsh pathway inhibits the canonical Wnt/β-catenin pathway [22]. The Wnt pathway is also modulated by various activators, including R-spondin and Norrin, and inhibitors, such as DKK, sFRP, WIF, SOST, and Tiki1 [23]. Tight transcriptional regulation of these modulators is important for proper Wnt signaling. For example, the GATA-type transcription factor Trps1 activates expression of the Wnt inhibitors WIF and DKK4 and is essential for vibrissa follicle morphogenesis in mice [24]. Another GATA transcription factor, GATA6, negatively regulates the level of DKK1 in pancreatic cancer [25]. The PAS-bHLH family protein HIF-1 is required for hypoxia-induced transcription of cam-1 in C. elegans [26]. However, the detailed molecular mechanisms underlying transcription-mediated regulation of Wnt activity during neural connection remain to be further elucidated.
Here, we show that C. elegans BDU and PLM neurons connect to each other through gap junctions and that Wnt signaling is important for BDU-PLM connectivity. We found that the PAS-bHLH transcription factor AHA-1 and its partner AHR-1 function cell-autonomously within BDU and PLM to regulate expression of the Wnt antagonist CAM-1. Together, our results reveal that transcription-mediated fine-tuning of Wnt signaling is responsible for target identification by BDU and PLM neurons.
Results
Development of the BDU-PLM neuronal connection
The BDU neurons are a pair of interneurons with cell bodies situated laterally in the anterior body of C. elegans. From its cell body, each BDU neuron projects an anterior process and a posterior process (Figure 1A). Electron microscopy (EM) reconstruction studies have revealed that BDU mainly receives chemical synaptic input from the mechanosensory neurons ALM and AVM through its anterior process around the nerve ring region [27]. However, these EM studies appear to fail to track the BDU posterior process after it turns away from the lateral nerve [27]. Here, using various GFP reporters (Altun and Hall, 2013 in WormAtlas http://www.wormatlas.org/ and this study), we show that the BDU posterior process extends all the way to the mid-body position and then turns towards the ventral-lateral nerve, where PLM runs. The functional properties of the BDU posterior process are unclear. PLMs are a pair of sensory neurons that transduce touch stimuli in the posterior part of the worm body to guide forward movement [28]. Each PLM neuron has its cell body located in the tail region and sends out an anterior process to the mid-body region of the animal. Before reaching the vulva region, this anterior neurite bifurcates. One branch goes ventrally and forms en passant chemical synapses with neurons in the ventral nerve cord (Figure 1A “b” region). The other extends continuously forward and terminates anterior of the vulva at a relatively fixed position (Figure 1A “a” region). The neural connection and the function of this anterior lateral branch are not known. When we examined nervous system organization using a cellular green fluorescent protein (GFP) marker (Punc-86::MYR::GFP) that highlights both BDU and PLM neurons, we found that the BDU posterior process touches the anterior lateral branch of the PLM neuron. To visualize the BDU and PLM junction region unambiguously, we simultaneously introduced into worms two fluorescent markers, Punc-53::GFP and Pmec-7::mCherry, that label BDU and PLM respectively (Figure 1B and 1C). As shown in Figure 1D, the terminus of the BDU posterior process overlaps with the tip of the PLM neuron.

We next followed the development of BDU and PLM neurites. Using the Punc-86::MYR::GFP marker, we found that in 73% (n = 45) of newly hatched L1 larvae, the BDU and PLM neurites were already touching each other. We therefore dissected eggs from gravid adults and followed the development of BDU and PLM in living embryos. BDU and PLM neurons are born at about the same time during the embryonic 1.5 to 2 fold stage, around 450 min after the first cell division (Figure 1E). Two hours after birth, the BDU neuron has completed its posterior migration and leaves an anterior neurite behind (∼570 min after the first cell division). No posterior BDU process, however, can be seen by the end of this two-hour period. In contrast, the PLM cell starts sending out the anterior neurite once it is born, and by ∼570 min this neurite has already grown to a significant length (Figure 1F). After its posterior migration is finished, the BDU neuron begins to project the posterior neurite (Figure 1G). By the end of embryonic elongation or during the early L1 stage, the posterior BDU neurite is in touch with the PLM anterior process (Figure 1H).
We then tested whether PLM influences BDU neurite outgrowth and vice versa. ced-3 encodes a caspase that is crucial for apoptosis and causes cell death when its expression level is up-regulated [29], [30]. When ced-3 was over-expressed in PLM using the Pmec-7 promoter, PLM neurons were specifically eradicated, but BDU still projected a single long posterior neurite to the target region (Figure 1I and 1J, n = 29–38). Similarly, in the absence of BDU, PLM still extended a single anterior process just like in wild-type animals (Figure 1K and 1L, n = 25–39). For both BDU and PLM, we found no obvious alteration in cell body position, neurite branching, or the orientation of neurite outgrowth when the target neuron was removed (Figure 1I and 1K). These results indicate that BDU and PLM do not play a significant role in directing long-range growth of the target cell to its destination. Interestingly, we found that when PLM was removed, the BDU process was longer at various developmental stages compared to untreated animals (Figure 1J), suggesting that BDU may have a more active role than PLM in establishing the BDU-PLM connection after the neurites reach the target region.
Gap junctions form between the BDU and PLM neurons
A nerve cell is functionally connected to its target cell through chemical synapses and/or electrical synapses. To determine the biochemical nature of the BDU-PLM connection, we first tested whether BDU connects with PLM through chemical synapses. One characteristic feature of chemical synapses is the enrichment of synaptic vesicles at the pre-synaptic site. RAB-3 is a synaptic vesicle-associated protein that is widely present in all kinds of neurons [31]. While RAB-3 signal was clearly evident along the ventral nerve cord where PLM forms chemical synapses (Figure 1A “b” region and Figure 2C), no accumulation of RAB-3 puncta was found at the posterior termini of BDU neurons or at the anterior tips of PLM neurons (Figure 2A, 2B and 2C). Thus, BDU and PLM neurons may not connect to each other through chemical synapses.

We then asked whether BDU connects to PLM by forming gap junctions. Gap junctions are complex multi-unit plasma membrane structures that are composed of innexins in worms. The C. elegans genome contains 25 innexin genes, among which unc-9 is one of the few that are widely expressed in the nervous system [32]. We generated a functional UNC-9::GFP fusion protein and expressed it in BDU (Punc-53::UNC-9::GFP) or PLM (Pmec-7::UNC-9::GFP). UNC-9::GFP signal was present in a punctate structure at the terminus of the BDU posterior process and at the tip of the PLM anterior neurite (Figure 2D and 2E). We next labeled PLM neurite and BDU gap junctions simultaneously, and found that the gap junction GFP puncta in BDU co-localized with the tip of the anterior PLM process (Figure 2F). Furthermore, we found that the UNC-9::GFP cluster is absent when BDU or PLM is removed, implying that neuron-neuron contact may be required for assembling the gap junction cluster (Figure 2G and 2H).
Next, we examined during which developmental stage the BDU-PLM gap junction connection is formed. Live imaging showed that the UNC-9::GFP cluster was not detected before the BDU and PLM neurites contacted each other (Figure 3A). Soon after or at the same time that BDU and PLM made contact, the UNC-9::GFP cluster appeared in the neuron-neuron contact region and persisted into adulthood (Figure 3A). In Enabled/VASP mutant unc-34 animals, which are defective in neurite outgrowth, BDU and PLM failed to contact each other and no UNC-9::GFP cluster could be identified (Figure 3B). The fact that direct membrane contact is required for UNC-9::GFP clustering indicates that BDU and PLM connect through gap junctions. In addition, we used an UNC-9 antibody to detect endogenous gap junctions [33] and found that it highlights punctate structures at the BDU-PLM junction, but not in other regions along the BDU or PLM processes (Figure 3C and 3D).
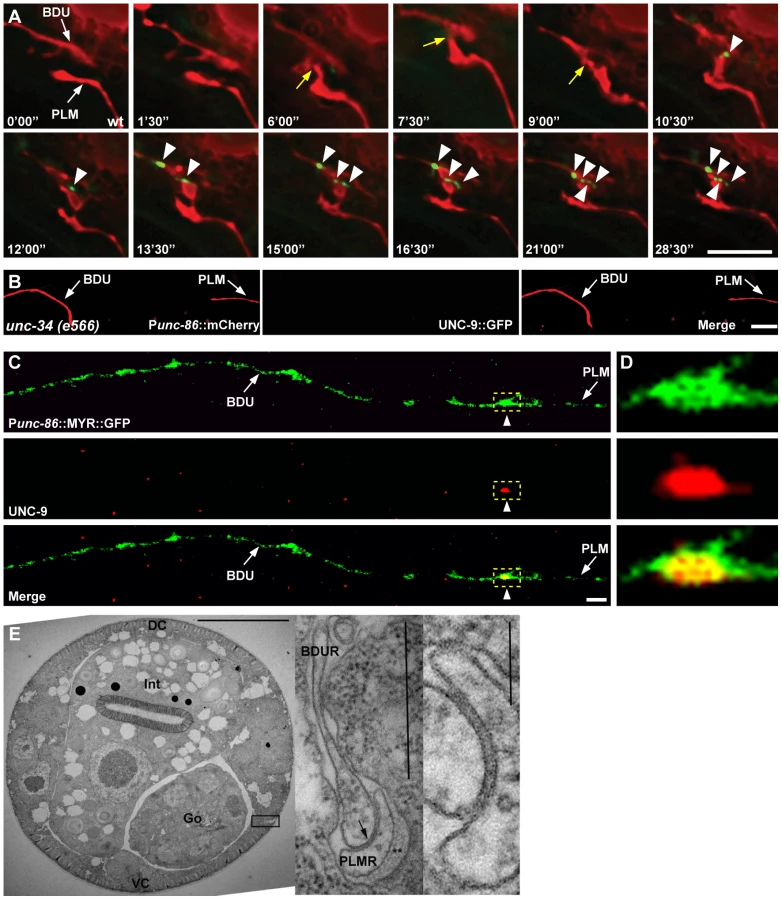
Finally, we performed electron microscopy (EM) analysis. The anterior process of PLM could be readily identified in any thin section due to the presence of large diameter microtubules within the process, and the secretion of an electron dense “mantle” protein into the space between this process and the surrounding hypodermis. Except for the gap junction found at the PLM anterior tip (as shown in Figure 3E), this portion of the PLM neurite showed no other synaptic interactions, and never encountered another neuron process except for a few passing circumferential commissures that run periodically between the ventral and dorsal nerve cords, and which generally pass along a different route, virtually orthogonal to the orientation of BDU's posterior process in Figure 1A. Together with the UNC-9 antibody staining pattern and UNC-9::GFP localization, this EM data further support the notion that BDU and PLM neurons indeed form gap junctions.
Wnt signaling is involved in BDU-PLM target identification
Next, we took both forward and reverse genetic approaches to dissect the genetic regulation of BDU-PLM contact formation. It has been previously reported that in C. elegans the gap junction components UNC-7 and UNC-9 are required in the AVB interneuron and the B class motor neuron, respectively, for gap junction clusters to form between these neurons [34]. However, it is unclear whether the expression of UNC-7 and UNC-9 affects the contact between these neurons. We therefore tested whether gap junction components are required for BDU-PLM target identification. Among the 25 C. elegans innexins, UNC-7 has been shown to localize in BDU cells [34], while INX-7 and UNC-9 are expressed in both BDU and PLM cells [32]. When we examined unc-7, unc-9, inx-7, unc-9 unc-7, and unc-9;inx-7 mutants, we found that BDU and PLM still made contact (Table S1). In addition, UNC-9::GFP clustered at the interface between BDU and PLM in unc-7 and inx-7 animals (Figure S1A). Since it is difficult to rule out the possibility that other innexins may be present in BDU and/or PLM cells or that multiple innexins may function redundantly, it remains to be determined whether gap junction components can direct BDU-PLM target identification.
Another possibility is that cell adhesion molecules may facilitate BDU-PLM recognition. We examined a panel of mutations in genes encoding cell adhesion molecules, but none displayed obvious defects in BDU-PLM target identification (Table S1).
We then examined a panel of molecules that have been implicated in axon guidance, including components of the Netrin, Slit, Ephrin, and Wnt pathways. In unc-6/Netrin, unc-40/DCC, slt-1/Slit, sax-3/Robo, and vab-1/Ephrin receptor mutant animals, the BDU and PLM neurons were still able to contact each other (Figure S1B). In contrast, several mutants in the Wnt pathway, including lin-44/Wnt, lin-17/Wnt receptor, cam-1/Wnt co-receptor and dsh-1/Dishevelled, displayed defective BDU-PLM contact (Figure 4A, 4B and Figure S1C). In addition, mutations in mig-14 and vps-35, which act upstream to regulate the secretion of Wnts [35], [36], also cause failure of BDU-PLM contact (Figure 4A). Previous studies showed that PLM polarity is influenced by LIN-44/Wnt signaling [36], [37]. Indeed, in the absence of LIN-44/Wnt or its receptor LIN-17/Frizzled, the PLM process is much shorter (Figure 4C). In these mutants, the abnormal BDU-PLM contact may result from cell polarity defects and thus is likely a secondary effect. dsh-1 appears not to affect the polarity of PLM or the length of the anterior PLM neurite, which is indistinguishable from wild type (Figure 4C). However, the BDU neurite length is greatly reduced in dsh-1 mutants (Figure S1D), suggesting that the Wnt signal is required for BDU outgrowth.

We also examined the role of CAM-1, the C. elegans homolog of the CRD domain-containing receptor tyrosine kinase Ror2 [38]. We previously showed that through its intracellular domain, CAM-1 binds to DSH-1 and serves as a Wnt co-receptor in axon outgrowth [21]. CAM-1 has also been implicated in attenuating Wnt signaling during cell migration and vulval development [18], [19]. In cam-1 mutants, both BDU and PLM display normal neuronal polarity and neurite length (Figure S1C). However, in 38% (n = 96) of cam-1(gm122) null mutants, BDU failed to contact PLM (Figure 4A and 4B). cam-1 has been reported to partially affect BDU cell migration [38], but the non-connection phenotype is not entirely correlated with the anterior over-migration defect and 17% of animals (n = 136) with normal BDU cell body position displayed defective BDU-PLM connection. In addition, cwn-1 and cwn-2 mutants show BDU migration defects, but the BDU-PLM contacts are normal (Figure 4A) [39]. Together, these results suggested that cam-1 may affect target identification by BDU and PLM neurons.
Five Wnts, positioned at various locations along the body axis in C. elegans, likely cooperate with each other to regulate tissue development [40]. The maternal lethality of mom-2 precludes loss-of-function analysis. Single mutants of the four other Wnt ligands have either no phenotype or polarity defects (Figure 4A). To address whether Wnt signaling can influence the BDU-PLM contact, we perturbed the distribution of Wnt by over-expression. Previous studies showed that CWN-2 is the only Wnt that is highly expressed in the anterior region of the worm [20], so we used the cwn-2 promoter to ectopically increase the expression level of the other four Wnts (CWN-1, EGL-20, LIN-44, and MOM-2) individually in the anterior region. We found that ectopic expression of EGL-20, LIN-44, CWN-2, and MOM-2 did not lead to any BDU-PLM connection defect (Figure S1E). In contrast, when CWN-1, a Wnt normally expressed in the middle and posterior of the worm body, was expressed in the anterior part using the cwn-2 promoter, both BDU and PLM extended neurites long enough to reach each other, but these neurites failed to contact (Figure 4D and 4E). In addition, over-expression of the downstream effector DSH-1 in both BDU and PLM caused BDU-PLM contact failure, while cell polarity and neurite outgrowth of BDU and PLM were normal (Figure 4D and Figure S1F). We further restricted dsh-1 over-expression to either BDU or PLM and found that this also caused BDU-PLM connection failure (Figure 4D). These observations suggested that the responsiveness of both BDU and PLM to extracellular Wnt signaling must be tightly regulated to ensure precise neuronal connectivity.
The BDU-PLM contact requires aha-1 and ahr-1
To reveal the intrinsic molecular machinery responsible for BDU-PLM connection, we performed a genetic screen for mutants which display BDU-PLM contact defects. From the screen, we isolated a mutant named xd4. xd4 animals generally show wild-type morphology and locomotion, but the BDU-PLM contact is lost (Figure 5A and 5B). Through genetic mapping and transgenic rescue experiments, we found that xd4 is an allele of the aha-1 gene. We identified a G to A nucleotide transition at the second splice donor site in xd4 genomic DNA that results in decreased transcription of the aha-1 gene (Figure 5C and Figure S2A). In xd4 mutants the lengths of BDU and PLM neurites are indistinguishable from wild-type animals (Figure 5D and 5E), suggesting that the xd4 mutation may specifically affect the target identification process. The aha-1(ok1396) allele contains a large deletion (Figure 5C) and causes an early larval arrest phenotype. ok1396 larvae show a similar percentage of defective BDU-PLM contacts to xd4 mutants (Figure 5F). Thus, xd4 may represent a strong loss-of-function allele of aha-1.

aha-1 encodes the sole C. elegans homolog of ARNT (AHR nuclear translocator), which contains a bHLH (basic helix-loop-helix) domain at the N-terminus and a PAS (Per/ARNT/Sim) domain in the middle. ARNT usually forms heterodimers with other PAS domain proteins to regulate gene expression [41]. It has been previously reported that AHA-1 and its partners function together to control neuronal development including neural fate determination [42], [43]. There are five PAS-bHLH family members (AHA-1, AHR-1, HIF-1, CKY-1, and HLH-34) in C. elegans [44]. To address which PAS-bHLH protein is the partner of AHA-1 in regulating BDU-PLM contact, we first evaluated the expression profiles of the corresponding genes. Using a promoter-driven GFP assay, we showed that cky-1 and hlh-34 were restricted to specific tissues or cells, but not BDU or PLM (Figure S2B and S2C). hif-1 has been shown to be expressed in every somatic cell [44], while ahr-1 has been reported to be present in both BDU and PLM neurons [43]. We then examined the loss-of-function phenotypes of ahr-1, hif-1, cky-1, and hlh-34 mutants or RNAi-treated animals. We found that ahr-1 mutants display a phenotype similar to aha-1 mutants, while loss-of-function of hif-1(ia4), cky-1(RNAi), or hlh-34(RNAi) did not lead to any detectable phenotype (Figure 5D–F and Table S1). Furthermore, aha-1 ahr-1 double mutants show a similar phenotype to aha-1 or ahr-1 single mutants (Figure 5F), suggesting that ahr-1 may function together with aha-1 in formation of the BDU-PLM contact.
Since aha-1 and ahr-1 can affect neural cell fate [42], we asked whether the BDU-PLM contact defect in aha-1 and ahr-1 mutants is due to cell fate changes. unc-86 has been shown to be expressed in BDU and PLM, so we used Punc-86::MYR::GFP as a BDU-PLM cell-specific marker in this study [45]. We found that in aha-1 mutants, unc-86 is still present in BDU and PLM cells (Figure 5A and 5B). The mec-7 and mec-18 genes, which are selectively turned on in mechanosensory neurons including PLM, are still expressed in PLM neurons in aha-1 mutant animals (Figure S2D and S2E). We also used the promoters of two BDU markers, unc-53 and nlp-1, to drive GFP expression in aha-1 mutants and found that fluorescent signals from both reporters can be detected in BDU (Figure S2F and S2G). Thus, we found no evidence of cell fate changes. Together, these data suggest that aha-1 and ahr-1 may ensure that BDU and PLM processes contact one another for the creation of a gap junction connection.
AHA-1 functions in both BDU and PLM to regulate neuronal connection
Next, we sought to identify the site at which AHA-1 and AHR-1 act during formation of the BDU-PLM connection. A GFP transgene driven by the aha-1 promoter highlights a wide range of tissues and cells, including neurons, muscle cells, intestine, and epidermis (Figure S2H). We identified the GFP signal in BDU and PLM neurons based on their typical morphology and cell body position (Figure S2I). Consistent with a previous report [44], functional translational AHA-1::GFP is localized in nuclei (Figure S2J). To determine in which tissue or cell aha-1 function is required, we expressed the aha-1 cDNA under the control of various cell-specific promoters in aha-1 mutants. When the aha-1 transgene was expressed in BDU and PLM together, the BDU-PLM connection defect was fully rescued in almost all animals (Figure 5G), suggesting that aha-1 functions cell-autonomously to regulate neuronal connection.
We further asked whether aha-1 acts in BDU or PLM or both. An aha-1 cDNA driven by the unc-53 promoter, which confers expression in BDU, partially rescued the aha-1 mutant phenotype (Figure 5G). Furthermore, when aha-1 was expressed in PLM, the connection between BDU-PLM was also partially recovered (Figure 5G), suggesting that aha-1 may function in both BDU and PLM cells to facilitate the formation of BDU-PLM connection.
AHA-1 directly regulates cam-1 gene expression
cam-1 mutants display a similar phenotype to aha-1 and ahr-1 mutants. Therefore, we tested the genetic relationship between cam-1 and aha-1 or ahr-1. We created double mutants with the cam-1 null allele gm112 and the aha-1(xd4) allele. In cam-1;aha-1 double mutants, BDU-PLM contact defects are moderately increased compared to aha-1(xd4) or cam-1(gm112) single mutants alone (Figure 6A). We further generated ahr-1; cam-1 double mutants and found that the BDU-PLM contact defect in these animals is similar to that in ahr-1 null worms (Figure 6A). These results suggest that cam-1 may act in the same pathway as aha-1 and ahr-1.

Since the AHR/ARNT heterodimer controls gene expression, our results raise the possibility that AHA-1 and AHR-1 may regulate cam-1 transcription in BDU and PLM. To test this, we compared the cam-1 expression level in wild type and aha-1 mutants using GFP reporter and quantitative RT-PCR approaches. In wild-type animals, a GFP reporter driven by a 5.4 kb cam-1b promoter sequence is highly expressed in BDU cells (Figure 6B). However, in aha-1(xd4) animals, the Pcam-1b::GFP intensity is decreased (Figure 6C). We also attempted to examine the Pcam-1b::GFP intensity in PLM, but the cluster of neurons surrounding the PLM region prevented us from carrying out this analysis. We further examined the endogenous cam-1 mRNA level using quantitative RT-PCR and found that the cam-1 mRNA level is significantly decreased in aha-1 mutants compared to wild-type animals (Figure 6D). The cam-1 mRNA level is also reduced in ahr-1 mutants (Figure 6E), suggesting that cam-1 expression is affected by both aha-1 and ahr-1.
Does AHR-1/AHA-1 directly regulate cam-1 gene expression? The AHR/ARNT heterodimer binds a specific DNA sequence, the Xenobiotic Response Element (XRE, 5′-CACGC-3′), to regulate the transcription of its target genes [41]. However, we searched the 5.4 kb promoter region of cam-1 and did not identify any obvious XRE sequence. In mammals, AHR/ARNT heterodimers can be recruited specifically to an enhancer element 5′-CATGN6CT/ATG-3′ by unknown factors, thus promoting transcription [46]. Interestingly, a putative AHR/ARNT enhancer sequence 5′-CATGTTTATCCTTG-3′ is found at position -2720 to -2707 relative to the cam-1 start codon. To test whether AHA-1 indeed regulates cam-1 expression by binding to this enhancer region, we performed chromatin-immunoprecipitation followed by quantitative PCR (ChIP-Q-PCR) in whole worm extracts. AHA-1::GFP immunoprecipitated by GFP antibody shows significantly higher binding affinity to the putative AHR/ARNT enhancer region than to a coding region sequence in the sixth exon of cam-1 corresponding to bases 7161-7134 (Figure 6F). We made a GFP construct driven by the cam-1Δ promoter in which this enhancer region was deleted. The GFP signal intensity of this reporter was no longer influenced by mutation of aha-1 (Figure 6G, n = 48 for wild type and n = 53 for aha-1), further supporting the notion that AHA-1 may regulate cam-1 expression by binding to the enhancer region.
Our genetic analyses indicated that cam-1 likely negatively regulates Wnt signaling during BDU-PLM connection. Therefore, loss-of-function of aha-1 and ahr-1 may create a scenario in which the Wnt signal is enhanced. Consistent with this idea, we found that over-expression of dsh-1 indeed further enhanced the BDU-PLM connection defects in aha-1 and ahr-1 mutants from 40% to 58% and 47% to 82% respectively (Figure 6H). In contrast, increasing the cam-1 level suppressed the aha-1 and ahr-1 mutant phenotypes. As shown in Figure 6H, when cam-1 was over-expressed using its endogenous promoter, the percentage of animals with BDU-PLM connection defects decreased from 40% (n = 126) to 18% (n = 169) in aha-1 mutants and from 47% (n = 109) to 28% (n = 119) in ahr-1 mutants. To determine in which cell the cam-1 suppression effect is achieved, we restricted cam-1 expression to both BDU and PLM cells and found that the aha-1 phenotype was alleviated (Figure 6H). Furthermore, we over-expressed cam-1 within either BDU or PLM and found that this also suppressed the aha-1 mutant phenotype, indicating that cam-1 functions cell-autonomously (Figure 6H). Together, these data strongly suggest that aha-1 and ahr-1 regulate the expression of cam-1 to facilitate the Wnt-mediated fine-tuning of target identification in formation of the BDU-PLM connection.
Discussion
Here we have shown that the C. elegans BDU and PLM neurons are connected by a gap junction, and that a Wnt fine-tuning mechanism is crucial for bringing the mutual target cells into contact during development.
Previous studies using paired recording techniques revealed that in the neocortex, electrical coupling was found exclusively between GABAergic cells of the same class [47], [48]. In C. elegans, the posterior process of BDU and the anterior process of PLM contact multiple tissues and cells during development [27], but the gap junction only forms when these two processes meet each other. This high specificity points to a tight regulation of gap junction formation in vivo. However, the process by which an individual cell is guided to its appropriate partner with which to form an electrical synapse remains poorly understood.
Since oriented neurite growth precedes any physical contact between BDU and PLM neurons, it is possible that one or both of the participating neurons may provide signals to guide its corresponding target cell. Interestingly, when we eliminated BDU or PLM, the remaining cell was still able to grow relatively normally towards the target area. These phenomena can be interpreted as follows: extracellular environmental cues guide BDU and PLM neurites independently to the correct target area and the participating neurons themselves may then promote neuronal connectivity by locally refining the target identification process.
Intriguingly, although many well-known guidance cues such as Netrin, Slit, and Ephrin are not required for BDU-PLM connectivity, disturbing Wnt resulted in BDU-PLM contact failure. Single loss-of-function mutations of mom-2, cwn-1, cwn-2, or egl-20 caused either maternal lethality or no defect in BDU-PLM connection, while the lin-44 mutation severely affected the polarity of PLM, precluding a definite answer to the question of whether Wnt plays an attractive or repulsive role in the BDU-PLM contact process. However, the reduced BDU neurite growth phenotype in dsh-1 mutants suggests that Wnt signaling may be important to guide BDU neurites to the correct target region. In addition, mis-expression of CWN-1, overexpression of DSH-1, or loss-of-function of cam-1, all specifically disrupted the BDU-PLM contact process, indicating that proper Wnt levels are important for the precise targeting between BDU and PLM cells.
How is the Wnt signal integrated within BDU and PLM? We showed that the PAS-bHLH transcription factors AHA-1 and AHR-1 function autonomously within both BDU and PLM to direct neuronal connectivity. Over-expression of cam-1 bypasses the requirement for AHA-1 or AHR-1. We further revealed that AHA-1 promotes cam-1 transcription by associating with a cam-1 enhancer element. These results highlight a local Wnt detection process mediated by the transcription factors AHA-1 and AHR-1. Previous studies have suggested a link between Wnt and AHR. In prostate cancer cells, AHR was identified as a target gene of the Wnt/β-catenin pathway [49]. On the other hand, in liver progenitor cells and MCF-7 cells, AHR activation leads to down-regulation of Wnt/β-catenin signaling [50], [51]. Similarly, in zebrafish, blockage of caudal fin regeneration by the AHR/ARNT pathway activator 2,3,7,8-tetrachlorodibenzo-ρ-dioxin is suppressed by down-regulation of LRP6 [52]. These observations are consistent with our notion that Wnt signaling is regulated by AHR/ARNT.
This fine-tuning of Wnt signal detection through AHA-1/AHR-1-mediated CAM-1 expression regulation may provide a way to direct the process placement, thus to ensure the cell-cell contact between mutual target cells. Transcriptional regulation of Wnt inhibitors has been demonstrated in multiple situations. For instance, the LIM-transcription factor Lhx5 and the homeobox protein Barx1 activate transcription of the secreted Wnt antagonists sFRP1 and sFRP2 in forebrain and gut development respectively [53], [54]. Modifying the expression level of secreted Wnt inhibitors affects many surrounding cells, probably cell-non-autonomously, while regulation of the membrane-bound Wnt antagonist CAM-1 may represent a locally confined, cell type-specific and cell-autonomous mechanism, which is particularly valuable for bringing processes in contact with each other.
What is the biological significance of the BDU-PLM gap junction connection? The anterior neurite of BDU mainly receives chemical synaptic input from the mechanosensory neurons ALM and AVM, which reside in the head region to mediate backward movement in response to touch [27]. The PLM neuron transduces touch stimuli in the posterior part of the worm body to guide forward movement [28]. Four pairs of command interneurons act as the common thread in the neuronal circuit linking mechanosensation to locomotion: AVA and AVD are needed for backward movement, while AVB and PVC are required for forward movement [55]. Here we showed that BDU connects with PLM through gap junctions, while previous studies indicate that BDU also innervates PVC and AVA through chemical synapses [27]. Therefore, BDU likely coordinates touch-responsive backward and forward locomotion through both mechanoreceptor neurons and interneurons (Figure S3).
For more than 50 years, neuronal gap junctions have been known to provide a simple, direct mechanism for information signaling between neurons [56]. Over the past decade, there has been a proliferation of investigations into the structure and function of gap junctions in the nervous system. Single neuron injection with a low molecular weight tracer revealed that gap-junction-mediated coupling is cell type-specific [57]. In the future, we would like to exploit the discovery of the BDU-PLM gap junction to dissect the mechanism of neuron-specific gap junction assembly in vivo. With the powerful genetics of C. elegans, more comprehensive developmental and functional analyses will shed light not only on the common rules governing how individual neurons identify and reach their mutual targets to form gap junctions, but also on the general principles of nervous system organization.
Materials and Methods
C. elegans genetics
C. elegans strains were maintained on NGM plates under standard conditions as described [58]. Mutants and transgenic fluorescence reporters used in these studies are listed here and Table S2: LGI, unc-40 (e271), aha-1(xd4), aha-1(ok1396), ahr-1(ju145), ahr-1(ia3), lin-44(n1792), lin-17(n671), mig-1(e1787), xdIs27(Punc-53::UNC-9::GFP, Podr-1::GFP); LGII, cwn-1(ok546), cam-1(gm122), dsh-1(ok1445), mig-14(ga62), vab-1(e2), vps-35(hu68); LGIV, inx-7(ok2319), kyIs262(Punc-86::MYR::GFP, Podr-1::dsRed), egl-20(n585), cwn-2(ok895), wyIs22(Punc-86::RAB-3::GFP, Podr-1::dsRed); LGV, cfz-2(ok1201), hif-1(ia4), jsIs37[Pmec-7::SNB-1::GFP, lin-15(+)]; LGX, unc-6(ev400), sax-3(ky123), slt-1(eh15), unc-7(e5), unc-9(fc16), unc-9(e101), uIs25[Pmec-18::GFP, dpy-20(+)].
The xd4 mutation of aha-1 was isolated from kyIs262 animals treated with EMS. Briefly, the BDU-PLM neurite contact phenotype of F2 progeny was examined under a fluorescence microscope, and mutant animals were recovered to produce progeny. A total of 2,500 mutagenized haploid genomes were screened. xd4 was mapped to chromosome I. The entire coding region of aha-1 and all exon-intron boundaries were sequenced by amplifying PCR products of the aha-1 gene from mutant animals. RNA interference of cky-1 and hlh-34 was performed in the RNAi-sensitive strain rrf-3(pk1426) mutant as described [59]. For RNA interference, dsRNA was injected into young adult rrf-3(pk1426);kyIs262 animals at the maximum possible concentration. Progeny laid after the first six hours were scored when they grew to the young adult stage.
Molecular biology and transgenes
A 4.3 kb aha-1 genomic DNA fragment containing the promoter and coding region was amplified from N2 genomic DNA to perform the rescue experiment. cDNA clones of aha-1, unc-9, dsh-1, and cam-1 were obtained from Dr. Yuji Kohara (National Institute of Genetics, Mishima, Japan). For expression in both BDU and PLM cells, the unc-86 promoter was used. For PLM-specific expression, a 0.85 kb mec-7 promoter fragment was cloned into the SalI sites of pPD95.75. For BDU-specific expression, a 2.8 kb unc-53 promoter sequence was cloned into the BamHI sites of pPD95.75. The cDNAs of aha-1, dsh-1, and cam-1 were inserted into unc-86, mec-7 or unc-53 promoter-containing vectors to create cell-specific expression constructs. A translational AHA-1::GFP fusion was constructed by cloning a PCR fragment containing the entire 4.3 kb promoter and coding region into the GFP expression vector pPD95.75. The aha-1 expression construct was prepared by cloning a PCR fragment containing the entire 1.5 kb promoter into the BamHI site of pPD95.75. The unc-9 cDNA was cloned in-frame into the BamHI site of pPD95.75 containing the unc-53 promoter to generate Punc-53::UNC-9::GFP. Transgenic animals were generated following standard procedures. In general, plasmid DNAs of interest were used at 1–50 ng/µl, and the co-injection markers Podr-1::dsRed, pRF4 or Podr-1::GFP at 50 ng/µl. xdIs27 is an integrated transgenic line of Punc-53::UNC-9::GFP and was out-crossed three times before being used for subsequent analysis.
Immunohistochemistry
Immunohistochemistry was performed with whole-mount worms according to previous report [60]. For immunofluorescence detection of endogenous UNC-9, worms were sequentially stained with anti-UNC-9 and anti-GFP antibody. The UNC-9-specific antibody was kindly provided by Dr. Zhaowen Wang. Mouse monoclonal antibody against GFP (Santa Cruz) was used to detect the Punc-86::MYR::GFP. Antibodies against UNC-9 or GFP were used at 1∶100 dilution. FITC conjugated goat anti-mouse and Cy3 conjugated goat anti - rabbit (Earthox) secondary antibodies were used at 1∶100 dilution. Samples were viewed with an IX81 Olympus inverted confocal microscope.
Electron microscopy
An archival series of serial thin sections of an L4 stage wild-type C. elegans was closely viewed over a range of about 50 microns (1000 sections) to follow the anterior extent of the PLM dendrites to their most anterior limits. This series of thin sections comes from the archives of the Sydney Brenner lab at MRC/LMB, Cambridge England., which are now kept for curation in the Hall lab. We infer that the second neuron process in Figure 3E comes from BDU, although no attempt was made to trace it across thin sections. Within those serial sections, PLM dendrites formed no other synapses.
Image collection and phenotypic quantification
Animals were mounted on 2% agar pads in M9 buffer containing 1% 1-phenoxy-2-propanol and examined by fluorescence microscopy. Fluorescence photographs were taken using a Zeiss Axioimager A1 with AxioCam digital camera and Axiovision rel. 4.6 software (Carl Zeiss) or an IX81 Olympus inverted confocal microscope. Contact was considered defective if the BDU neurite failed to touch the PLM neurite. To measure neurites, eggs were allowed to hatch for 10 min, and newly hatched L1 animals were examined immediately or after incubating on food for different lengths of time. The lengths of neurites and of whole animals were traced from photographs and measured with NIH Image J software. The length of each neurite was traced from the center of the cell body to the tip of neurite. The length of each animal was measured from the anus to the tip of the nose. Images of BDU-PLM contact formation during embryonic stages and time-lapse live images were collected on a Delta-vision Core imaging system (Applied Precision) with an UPLSApo 100×/1.40 NA oil-immersion objective and a Photometrics CoolSnap HQ camera. Deconvolution and analysis of images were performed with Softworx (Applied Precision). The quantification of Pcam-1::GFP and PΔcam-1::GFP intensity was done double-blind with the same extra-chromosomal lines. All the statistical tests were done with two-tailed Student's t-tests. Unless otherwise indicated, the n-value in this study is around 50.
Quantitative RT-PCR
Quantitative RT-PCR was performed using a Stratagene Mx3000P QPCR System. For each reaction, three independent experiments were carried out in triplicate. 2×TransStart Green qPCR Supermix was used according to the manufacturer's instructions. PCR consisted of 40 cycles of 95°C for 30 s, 56°C for 30 s and 72°C for 30 s. A final cycle (95°C, 1 min; 56°C, 30 s; 95°C, 30 s) generated a dissociation curve to confirm a single product. A threshold cycle (ΔCt) value was obtained by subtracting act-3 Ct values from cam-1 Ct values. kyIs262 was used as a reference. ΔΔCt values were derived by subtracting kyIs262 ΔCt from aha-1 ΔCt and ahr-1 ΔCt. The relative expression of cam-1 was then calculated by 2−ΔΔCt. Primers for cam-1 cDNA amplification were 5′ATCAATAGTGCCGCCAATTC3′ and 5′GTGGAGGTCCGAGATGTTGT3′. Primers for amplification of the internal control act-3 were 5′TCCAAAGGCTAACCGTGAA3′ and 5′GGAAGCGTAGAGGGAGAGG3′.
ChIP-qPCR assay
In vivo ChIP assays were carried out according to a previous report [61]. Briefly, mixed stages of Paha-1::AHA-1::GFP transgenic worms were collected and crosslinked with 2% formaldehyde. After sonication, worm lysates were immunoprecipitated using anti-GFP antibody (Abcam, ab290). The ChIP-DNA and input DNA samples (genomic DNA from the same prep) were subjected to qPCR analysis. A 10 µl PCR reaction with each primer set was run in an Agilent Mx 3000P instrument using TransStart Green qPCR SuperMix (TransGen AQ101) according to the manufacturer's instructions. The PCR program was set as follows: Step 1 : 95°C for 10 min; Step 2 : 95°C for 30 sec; Step 3 : 55°C for 30 sec, Step 4 : 72°C for 30 sec. Steps 2–4 were repeated 40 times.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. DicksonBJ (2002) Molecular mechanisms of axon guidance. Science 298 : 1959–1964.
2. LyuksyutovaAI, LuCC, MilanesioN, KingLA, GuoN, et al. (2003) Anterior-posterior guidance of commissural axons by Wnt-frizzled signaling. Science 302 : 1984–1988.
3. CharronF, SteinE, JeongJ, McMahonAP, Tessier-LavigneM (2003) The morphogen sonic hedgehog is an axonal chemoattractant that collaborates with netrin-1 in midline axon guidance. Cell 113 : 11–23.
4. FannonAM, ColmanDR (1996) A model for central synaptic junctional complex formation based on the differential adhesive specificities of the cadherins. Neuron 17 : 423–434.
5. ShapiroL, LoveJ, ColmanDR (2007) Adhesion molecules in the nervous system: structural insights into function and diversity. Annu Rev Neurosci 30 : 451–474.
6. WhiteJG, SouthgateE, ThomsonJN (1992) Mutations in the Caenorhabditis elegans unc-4 gene alter the synaptic input to ventral cord motor neurons. Nature 355 : 838–841.
7. MillerDM, ShenMM, ShamuCE, BurglinTR, RuvkunG, et al. (1992) C. elegans unc-4 gene encodes a homeodomain protein that determines the pattern of synaptic input to specific motor neurons. Nature 355 : 841–845.
8. WinnierAR, MeirJY, RossJM, TavernarakisN, DriscollM, et al. (1999) UNC-4/UNC-37-dependent repression of motor neuron-specific genes controls synaptic choice in Caenorhabditis elegans. Genes Dev 13 : 2774–2786.
9. Von StetinaSE, FoxRM, WatkinsKL, StarichTA, ShawJE, et al. (2007) UNC-4 represses CEH-12/HB9 to specify synaptic inputs to VA motor neurons in C. elegans. Genes Dev 21 : 332–346.
10. SchneiderJ, SkeltonRL, Von StetinaSE, MiddelkoopTC, van OudenaardenA, et al. (2012) UNC-4 antagonizes Wnt signaling to regulate synaptic choice in the C. elegans motor circuit. Development 139 : 2234–2245.
11. NakamuraH, O'LearyDD (1989) Inaccuracies in initial growth and arborization of chick retinotectal axons followed by course corrections and axon remodeling to develop topographic order. J Neurosci 9 : 3776–3795.
12. ChengHJ, NakamotoM, BergemannAD, FlanaganJG (1995) Complementary gradients in expression and binding of ELF-1 and Mek4 in development of the topographic retinotectal projection map. Cell 82 : 371–381.
13. Colon-RamosDA, MargetaMA, ShenK (2007) Glia promote local synaptogenesis through UNC-6 (netrin) signaling in C. elegans. Science 318 : 103–106.
14. ZouY (2004) Wnt signaling in axon guidance. Trends Neurosci 27 : 528–532.
15. KlassenMP, ShenK (2007) Wnt signaling positions neuromuscular connectivity by inhibiting synapse formation in C. elegans. Cell 130 : 704–716.
16. HikasaH, ShibataM, HirataniI, TairaM (2002) The Xenopus receptor tyrosine kinase Xror2 modulates morphogenetic movements of the axial mesoderm and neuroectoderm via Wnt signaling. Development 129 : 5227–5239.
17. MikelsAJ, NusseR (2006) Purified Wnt5a protein activates or inhibits beta-catenin-TCF signaling depending on receptor context. PLoS Biol 4: e115.
18. GreenJL, InoueT, SternbergPW (2007) The C. elegans ROR receptor tyrosine kinase, CAM-1, non-autonomously inhibits the Wnt pathway. Development 134 : 4053–4062.
19. ForresterWC, KimC, GarrigaG (2004) The Caenorhabditis elegans Ror RTK CAM-1 inhibits EGL-20/Wnt signaling in cell migration. Genetics 168 : 1951–1962.
20. KennerdellJR, FetterRD, BargmannCI (2009) Wnt-Ror signaling to SIA and SIB neurons directs anterior axon guidance and nerve ring placement in C. elegans. Development 136 : 3801–3810.
21. SongS, ZhangB, SunH, LiX, XiangY, et al. (2010) A Wnt-Frz/Ror-Dsh pathway regulates neurite outgrowth in Caenorhabditis elegans. PLoS Genet 6: e1001056.
22. HoHY, SusmanMW, BikoffJB, RyuYK, JonasAM, et al. (2012) Wnt5a-Ror-Dishevelled signaling constitutes a core developmental pathway that controls tissue morphogenesis. Proc Natl Acad Sci U S A 109 : 4044–4051.
23. CruciatCM, NiehrsC (2013) Secreted and transmembrane Wnt inhibitors and activators. Cold Spring Harb Perspect Med 3: a015081.
24. FantauzzoKA, ChristianoAM (2012) Trps1 activates a network of secreted Wnt inhibitors and transcription factors crucial to vibrissa follicle morphogenesis. Development 139 : 203–214.
25. ZhongY, WangZ, FuB, PanF, YachidaS, et al. (2011) GATA6 activates Wnt signaling in pancreatic cancer by negatively regulating the Wnt antagonist Dickkopf-1. PLoS One 6: e22129.
26. ShenC, NettletonD, JiangM, KimSK, Powell-CoffmanJA (2005) Roles of the HIF-1 hypoxia-inducible factor during hypoxia response in Caenorhabditis elegans. J Biol Chem 280 : 20580–20588.
27. WhiteJG, SouthgateE, ThomsonJN, BrennerS (1986) The structure of the nervous system of the nematode Caenorhabditis elegans. Philos Trans R Soc Lond B Biol Sci 314 : 1–340.
28. ChalfieM, SulstonJ (1981) Developmental genetics of the mechanosensory neurons of Caenorhabditis elegans. Dev Biol 82 : 358–370.
29. YuanJY, HorvitzHR (1990) The Caenorhabditis elegans genes ced-3 and ced-4 act cell autonomously to cause programmed cell death. Dev Biol 138 : 33–41.
30. YuanJ, ShahamS, LedouxS, EllisHM, HorvitzHR (1993) The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1 beta-converting enzyme. Cell 75 : 641–652.
31. MahoneyTR, LiuQ, ItohT, LuoS, HadwigerG, et al. (2006) Regulation of synaptic transmission by RAB-3 and RAB-27 in Caenorhabditis elegans. Mol Biol Cell 17 : 2617–2625.
32. AltunZF, ChenB, WangZW, HallDH (2009) High resolution map of Caenorhabditis elegans gap junction proteins. Dev Dyn 238 : 1936–1950.
33. ChenB, LiuQ, GeQ, XieJ, WangZW (2007) UNC-1 regulates gap junctions important to locomotion in C. elegans. Curr Biol 17 : 1334–1339.
34. StarichTA, XuJ, SkerrettIM, NicholsonBJ, ShawJE (2009) Interactions between innexins UNC-7 and UNC-9 mediate electrical synapse specificity in the Caenorhabditis elegans locomotory nervous system. Neural Dev 4 : 16.
35. CoudreuseDY, RoelG, BetistMC, DestreeO, KorswagenHC (2006) Wnt gradient formation requires retromer function in Wnt-producing cells. Science 312 : 921–924.
36. PrasadBC, ClarkSG (2006) Wnt signaling establishes anteroposterior neuronal polarity and requires retromer in C. elegans. Development 133 : 1757–1766.
37. HilliardMA, BargmannCI (2006) Wnt signals and frizzled activity orient anterior-posterior axon outgrowth in C. elegans. Dev Cell 10 : 379–390.
38. ForresterWC, DellM, PerensE, GarrigaG (1999) A C. elegans Ror receptor tyrosine kinase regulates cell motility and asymmetric cell division. Nature 400 : 881–885.
39. ZinovyevaAY, ForresterWC (2005) The C. elegans Frizzled CFZ-2 is required for cell migration and interacts with multiple Wnt signaling pathways. Dev Biol 285 : 447–461.
40. EisenmannDM (2005) Wnt signaling. WormBook 1–17.
41. HahnME (2002) Aryl hydrocarbon receptors: diversity and evolution. Chem Biol Interact 141 : 131–160.
42. HuangX, Powell-CoffmanJA, JinY (2004) The AHR-1 aryl hydrocarbon receptor and its co-factor the AHA-1 aryl hydrocarbon receptor nuclear translocator specify GABAergic neuron cell fate in C. elegans. Development 131 : 819–828.
43. QinH, Powell-CoffmanJA (2004) The Caenorhabditis elegans aryl hydrocarbon receptor, AHR-1, regulates neuronal development. Dev Biol 270 : 64–75.
44. JiangH, GuoR, Powell-CoffmanJA (2001) The Caenorhabditis elegans hif-1 gene encodes a bHLH-PAS protein that is required for adaptation to hypoxia. Proc Natl Acad Sci U S A 98 : 7916–7921.
45. FinneyM, RuvkunG (1990) The unc-86 gene product couples cell lineage and cell identity in C. elegans. Cell 63 : 895–905.
46. SogawaK, Numayama-TsurutaK, TakahashiT, MatsushitaN, MiuraC, et al. (2004) A novel induction mechanism of the rat CYP1A2 gene mediated by Ah receptor-Arnt heterodimer. Biochem Biophys Res Commun 318 : 746–755.
47. GalarretaM, HestrinS (1999) A network of fast-spiking cells in the neocortex connected by electrical synapses. Nature 402 : 72–75.
48. GibsonJR, BeierleinM, ConnorsBW (1999) Two networks of electrically coupled inhibitory neurons in neocortex. Nature 402 : 75–79.
49. ChesireDR, DunnTA, EwingCM, LuoJ, IsaacsWB (2004) Identification of aryl hydrocarbon receptor as a putative Wnt/beta-catenin pathway target gene in prostate cancer cells. Cancer Res 64 : 2523–2533.
50. ProchazkovaJ, KabatkovaM, BryjaV, UmannovaL, BernatikO, et al. (2011) The interplay of the aryl hydrocarbon receptor and beta-catenin alters both AhR-dependent transcription and Wnt/beta-catenin signaling in liver progenitors. Toxicol Sci 122 : 349–360.
51. ZhaoS, KannoY, NakayamaM, MakimuraM, OharaS, et al. (2012) Activation of the aryl hydrocarbon receptor represses mammosphere formation in MCF-7 cells. Cancer Lett 317 : 192–198.
52. MathewLK, SenguptaSS, LaduJ, AndreasenEA, TanguayRL (2008) Crosstalk between AHR and Wnt signaling through R-Spondin1 impairs tissue regeneration in zebrafish. FASEB J 22 : 3087–3096.
53. PengG, WesterfieldM (2006) Lhx5 promotes forebrain development and activates transcription of secreted Wnt antagonists. Development 133 : 3191–3200.
54. KimBM, BuchnerG, MiletichI, SharpePT, ShivdasaniRA (2005) The stomach mesenchymal transcription factor Barx1 specifies gastric epithelial identity through inhibition of transient Wnt signaling. Dev Cell 8 : 611–622.
55. ChalfieM, SulstonJE, WhiteJG, SouthgateE, ThomsonJN, et al. (1985) The neural circuit for touch sensitivity in Caenorhabditis elegans. J Neurosci 5 : 956–964.
56. FurshpanEJ, PotterDD (1957) Mechanism of nerve-impulse transmission at a crayfish synapse. Nature 180 : 342–343.
57. PeinadoA, YusteR, KatzLC (1993) Extensive dye coupling between rat neocortical neurons during the period of circuit formation. Neuron 10 : 103–114.
58. BrennerS (1974) The genetics of Caenorhabditis elegans. Genetics 77 : 71–94.
59. SimmerF, TijstermanM, ParrishS, KoushikaSP, NonetML, et al. (2002) Loss of the putative RNA-directed RNA polymerase RRF-3 makes C. elegans hypersensitive to RNAi. Curr Biol 12 : 1317–1319.
60. NonetML, GrundahlK, MeyerBJ, RandJB (1993) Synaptic function is impaired but not eliminated in C. elegans mutants lacking synaptotagmin. Cell 73 : 1291–1305.
61. ZhongM, NiuW, LuZJ, SarovM, MurrayJI, et al. (2010) Genome-wide identification of binding sites defines distinct functions for Caenorhabditis elegans PHA-4/FOXA in development and environmental response. PLoS Genet 6: e1000848.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 6
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- BMS1 Is Mutated in Aplasia Cutis Congenita
- Sex-stratified Genome-wide Association Studies Including 270,000 Individuals Show Sexual Dimorphism in Genetic Loci for Anthropometric Traits
- Distinctive Expansion of Potential Virulence Genes in the Genome of the Oomycete Fish Pathogen
- Distinct Neuroblastoma-associated Alterations of Impair Sympathetic Neuronal Differentiation in Zebrafish Models
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy