
The Ubiquitin Ligase Subunit Acts in Target Tissue to Restrict Tracheal Terminal Cell Branching and Hypoxic-Induced Gene Expression
The Drosophila melanogaster gene archipelago (ago) encodes the F-box/WD-repeat protein substrate specificity factor for an SCF (Skp/Cullin/F-box)-type polyubiquitin ligase that inhibits tumor-like growth by targeting proteins for degradation by the proteasome. The Ago protein is expressed widely in the fly embryo and larva and promotes degradation of pro-proliferative proteins in mitotically active cells. However the requirement for Ago in post-mitotic developmental processes remains largely unexplored. Here we show that Ago is an antagonist of the physiologic response to low oxygen (hypoxia). Reducing Ago activity in larval muscle cells elicits enhanced branching of nearby tracheal terminal cells in normoxia. This tracheogenic phenotype shows a genetic dependence on sima, which encodes the HIF-1α subunit of the hypoxia-inducible transcription factor dHIF and its target the FGF ligand branchless (bnl), and is enhanced by depletion of the Drosophila Von Hippel Lindau (dVHL) factor, which is a subunit of an oxygen-dependent ubiquitin ligase that degrades Sima/HIF-1α protein in metazoan cells. Genetic reduction of ago results in constitutive expression of some hypoxia-inducible genes in normoxia, increases the sensitivity of others to mild hypoxic stimulus, and enhances the ability of adult flies to recover from hypoxic stupor. As a molecular correlate to these genetic data, we find that Ago physically associates with Sima and restricts Sima levels in vivo. Collectively, these findings identify Ago as a required element of a circuit that suppresses the tracheogenic activity of larval muscle cells by antagonizing the Sima-mediated transcriptional response to hypoxia.
Published in the journal:
. PLoS Genet 9(2): e32767. doi:10.1371/journal.pgen.1003314
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003314
Summary
The Drosophila melanogaster gene archipelago (ago) encodes the F-box/WD-repeat protein substrate specificity factor for an SCF (Skp/Cullin/F-box)-type polyubiquitin ligase that inhibits tumor-like growth by targeting proteins for degradation by the proteasome. The Ago protein is expressed widely in the fly embryo and larva and promotes degradation of pro-proliferative proteins in mitotically active cells. However the requirement for Ago in post-mitotic developmental processes remains largely unexplored. Here we show that Ago is an antagonist of the physiologic response to low oxygen (hypoxia). Reducing Ago activity in larval muscle cells elicits enhanced branching of nearby tracheal terminal cells in normoxia. This tracheogenic phenotype shows a genetic dependence on sima, which encodes the HIF-1α subunit of the hypoxia-inducible transcription factor dHIF and its target the FGF ligand branchless (bnl), and is enhanced by depletion of the Drosophila Von Hippel Lindau (dVHL) factor, which is a subunit of an oxygen-dependent ubiquitin ligase that degrades Sima/HIF-1α protein in metazoan cells. Genetic reduction of ago results in constitutive expression of some hypoxia-inducible genes in normoxia, increases the sensitivity of others to mild hypoxic stimulus, and enhances the ability of adult flies to recover from hypoxic stupor. As a molecular correlate to these genetic data, we find that Ago physically associates with Sima and restricts Sima levels in vivo. Collectively, these findings identify Ago as a required element of a circuit that suppresses the tracheogenic activity of larval muscle cells by antagonizing the Sima-mediated transcriptional response to hypoxia.
Introduction
Metabolically active tissues require an adequate supply of dioxygen (O2) for metabolic production of ATP by aerobic glycolysis and as a necessary substrate in a variety of enzymatic reactions (reviewed in [1]). Consequently, cells in metazoan organisms have evolved a conserved hypoxia-response mechanism that senses low O2 (or hypoxia) and modulates cellular metabolism and signaling in response to this environmental challenge. Activation of this adaptive mechanism results in changes in transcription that allow organisms to adapt to O2 conditions that might otherwise be incompatible with normal development and homeostasis (reviewed in [2]). In most metazoans, these changes include elevated expression of factors involved in oxygen-independent ATP production, increased expression of oxygen-carrying hemoglobin-like molecules and increased branching of O2-carrying tubular organs, the net effect of which is to reduce overall O2 demand and increase O2 delivery.
Molecular mechanisms through which changes in O2 concentration alter metabolism and drive increased tubular branching are conserved through the metazoan tree to include invertebrates like the fruit fly Drosophila melanogaster (reviewed in [3]). A key element of this mechanism is the hypoxia-inducible factor-1 [4]–[7] (HIF-1 or Drosophila HIF [dHIF] in flies), which is a heterodimeric transcription factor composed of an oxygen-regulated HIF–1α subunit and a constitutively expressed HIF–1β subunit. In Drosophila these subunits are respectively encoded by the similar (sima) [8], [9] and tango (tgo) [10]–[12] genes. The HIF-1/dHIF heterodimer is required for cellular adaptation to hypoxic conditions [4]–[7] and is regulated mainly at the level of HIF–1α stability [reviewed in 13]). In normoxic conditions, HIF–1α is hydroxylated at conserved proline residues by the 2-oxoglutarate/Fe(II)-dependent prolyl-4-hydroxylase family member HIF prolyl hydroxylase (HPH) [14], [15]. Prolyl-hydroxylation of HIF–1α facilitates binding with the von Hippel Lindau (VHL) E3-ubiquitin ligase subunit, and subsequent polyubiquitination and proteasome-dependent degradation of HIF-1α [16]–[20]. Drosophila Sima is controlled by a well-conserved version of this pathway involving the HPH homolog fatiga (fga), and the Drosophila VHL homolog, dVHL [14], [21]–[24]. Because the HPH enzymatic activity is dependent upon the availability of oxygen [14], [15], the HPH/VHL pathway effectively functions as a sensor of cellular oxygen levels, allowing HIF–1α/Sima stabilization only in hypoxic conditions and preventing HIF activity in normoxic cells [reviewed in 2]. Mutations in the VHL gene stabilize HIF-1α and are associated with a dominantly inherited hereditary cancer syndrome in humans that predisposes to a variety of malignant and benign tumors of the eye, brain, spinal cord, kidney, pancreas, and adrenal glands [25]. Excess HIF-1α can promote several important aspects of cancer biology, including the metabolic switch to anaerobic glycolysis characteristic of tumor cells [i.e. the Warburg effect; 26], neoangiogenesis, and increased tumor metastasis [reviewed in 13], [27], [28].
The invertebrate response to hypoxia mirrors key features of the mammalian hypoxic response [3], [29], [30]. Hypoxia stabilizes Sima and induces expression of genes that include homologs of mammalian HIF targets, such as lactate dehydrogenase (LDH) [31]. Hypoxic treatment of flies also produces physiological changes reminiscent of the mammalian hypoxic response [32], including altered metabolism and reduced oxygen consumption [33]–[36]. Adult Drosophila respond to hypoxia by entering into state of stupor characterized by low or undetectable neurological activity that allows them to tolerate extended periods of low oxygen [34], and recovery from this state is dependent upon genes necessary for survival in low-oxygen conditions [31]–[33], [35]. Hypoxia also induces a neoangiogenesis-like process in Drosophila involving increased branching of the tracheal system, an open network of interconnected, epithelial tubes that duct gases in and out of the animal [reviewed in 37]. Drosophila larvae reared in chronic hypoxia show increased branching of cells at the tip of each tracheal branch termed ‘terminal tip’ cells, whereas those raised in chronic hyperoxia show a reciprocal decrease in the extent of terminal branch elaboration [22], [38]. This increased larval tracheal branching in low O2 involves the FGF receptor homolog breathless (btl) [39] and the FGF ligand branchless (bnl) [40]: hypoxic exposure results in a sima-dependent increase in expression of btl in tracheal cells and bnl in peripheral oxygen-deficient tissues [22], [38]. Bnl then acts on tracheal terminal tip cells, which express Btl [41], [42], to induce fine tubular extensions that project toward Bnl-expressing cells. These terminal branches serve as the primary site of gas exchange between the tracheal system and internal tissues. When the oxygen demand is met, Bnl and Btl expression decreases, thereby limiting hypoxia-induced tracheal growth. This oxygen responsiveness allows for growth of tracheal terminal branches specifically to localized areas of hypoxia in order to shape the mature tracheal architecture and to increase oxygen-delivery capacity in hypoxic conditions.
In addition to the oxygen-dependent HPH/VHL pathway, mammalian HIF-1 is regulated by VHL-independent mechanisms that are incompletely understood [43], [44]. Recent studies have linked HIF–1α turnover to phosphorylation by the GSK3ß kinase and subsequent binding of the ubiquitin ligase subunit Fbw7 [45], [46], which is a sequence and functional ortholog of the Drosophila Archipelago (Ago) protein. Intriguingly Drosophila Ago binds and stimulates turnover of the Trachealess protein (Trh), which is a Sima/HIF-1α sequence homolog, in embryonic tracheal cells [47]. Genetic data show ago and dVHL also coregulate oxygen-sensitivity in the developing embryonic tracheal arbor [48].
In light of these connections, we have tested the requirement for ago in oxygen-sensitive stages of larval tracheal development and find evidence that ago is an antagonist of dHIF during the larval stage. Genetic manipulations that reduce ago function within post-mitotic larval muscle cells lead to a sima-dependent increase in the branching of nearby terminal cells. This phenotype is not suppressed by a trh allele that suppresses branch defects in ago mutant embryonic tracheal cells [47], but rather correlates with elevated expression of the Sima-induced gene bnl expression in larval muscle cells and genetic dependence on bnl. At an organismal level, reducing ago activity results in constitutive expression of some dHIF target genes in normoxia, increases the sensitivity of others to mild hypoxic stimulus, and allows adult flies to recover more rapidly from hypoxic stupor than normal flies. Significantly, non-cell autonomous effects of ago and dVHL alleles on terminal branching are synergistic, suggesting that the Ago and dVHL proteins co-regulate dHIF. Consistent with this, Ago protein can be found in a complex with Sima in larval extracts and loss of Ago elevates Sima levels in peripheral tissues. Collectively these findings define an important role for Ago as a required antagonist of the Sima-dependent hypoxic response during the larval stage of Drosophila development.
Results
Loss of ago results in increased branching of tracheal terminal cells
Heterozygosity for a null allele of ago sensitizes the Drosophila embryonic tracheal system to mild hypoxia [48]. To determine whether ago is also involved in hypoxia responsiveness in the subsequent larval stage, it was necessary to generate an allele of ago that allowed development beyond the late embryonic lethality associated with ago null alleles [49]. This was achieved by transposase-mediated imprecise excision of EP(3)1135, a P-element located 16 base pairs (bp) upstream of the ago genomic locus (Bloomington Drosophila Stock Center [BDSC]) that behaves genetically as a weak ago hypomorph. Excision of EP(3)1135 produced a 603 bp deletion removing the first exon of the ago-RC transcript (Figure 1A–1B) that was designated agoΔ3–7. The effect of agoΔ3–7 on patterns of ago transcription was determined by quantitative real-time PCR (qRT-PCR). Of the three predicted ago transcripts (ago-RA, ago-RB, and ago-RC) only RA and RC are detected in whole larvae (Figure 1C). Consistent with the location of the deletion in the agoΔ3–7 allele, the ago-RC transcript is specifically absent in agoΔ3–7 mutant larvae while expression of the ago-RA transcript is unaffected. Notably, the ago-RA and RC transcripts display inverse expression patterns: ago-RA is approximately 3-fold more abundant than ago-RC in imaginal discs and larval brain and ventral nerve cord, but ago-RC is 3-fold more abundant than ago-RA in filleted larval body wall preparations (Figure 1D). The agoΔ3–7 allele is thus a tissue - and transcript-specific allele that primarily reduces ago expression in peripheral tissues such as body wall muscle.

Approximately 49% of agoΔ3–7 homozygotes or trans-heterozygotes in combination with the null alleles ago1 and ago3 die as pupae (Table 1) and the remainder die as late 2nd and 3rd instar larvae (data not shown). Those that live to late 3rd instar show tracheal phenotypes (Figure 2 and Table 2). The most prevalent of these phenotypes is an approximate doubling of the number of cytoplasmic branches elaborated from multiple subtypes of terminal cells, including those found along the lateral trunk that serve to oxygenate the ventrolateral body wall muscles: LH cell terminal branching increases from 20.4±0.64 branches (n = 33) in control larvae to 39.5±1.59 branches (n = 34) in agoΔ3–7/1 larvae (p = 2.6×10−16) (Figure 2A–2C and Table 2), and LG cell terminal branching increases from 19.6±0.54 branches (n = 33) in control larvae to 36.8±1.94 branches (n = 31) in agoΔ3–7/1 larvae (p = 9.5×10−13) (Figure 2C). Notably, the magnitude of these increases in terminal cell branch number is similar to that seen in larvae grown in hypoxic conditions [22], [38]. Loss of ago function also causes additional tracheal branch phenotypes in approximately 25% of larvae, including the appearance of terminal branch tangles (Figure 2D) and the development of ‘ringlet’-shaped ganglionic branches (Figure 2F), which resemble phenotypes seen in hypoxic larvae or those in which Sima is activated by genetic disruption of the Fga/dVHL regulatory pathway [22]. Given the transcript - and tissue-specific nature of the agoΔ3–7 allele, these tracheal phenotypes support the hypothesis that ago has a non-autonomous role in in restricting terminal branching.



ago acts non-autonomously to restrict post-embryonic tracheal branching
Although the agoΔ3–7/1 larval phenotypes are reminiscent of hypoxia-induced tracheal growth, they do not exclude the possibility that an earlier developmental requirement for ago (e.g. in the embryo) affects later branching events in the larva. To test the temporal requirement for ago in regulating tracheal terminal branching patterns, a dominant negative ago transgene (UAS-agoΔF) [47], [50] was combined with the hs-Gal4 driver to produce animals in which ago activity could be antagonized at later developmental stages by application of a heat-shock. Whereas control and hs>agoΔF larvae show similar LH cell branch number prior to transgene induction (22.2±0.89 branches [n = 27] vs 21.7±0.69 branches [n = 24]), administration of a transient heat-shock to hs>agoΔF larvae is sufficient to drive an increase in terminal branching throughout the tracheal system (effects on LG and LH cells quantified in Figure 3A and 3B). LH cell branching is increased 24 hrs post heat-shock in hs>agoΔF larvae (40.2±1.48 branches [n = 24]; (p = 3.0×10−14 relative to no heat-shock) but remains unchanged in control larvae (22.6±0.67 branches [n = 24]). Thus animals that complete embryonic and early larval development with wt ago activity can be induced to undergo excess branching by transient expression of an ago dominant-negative allele.
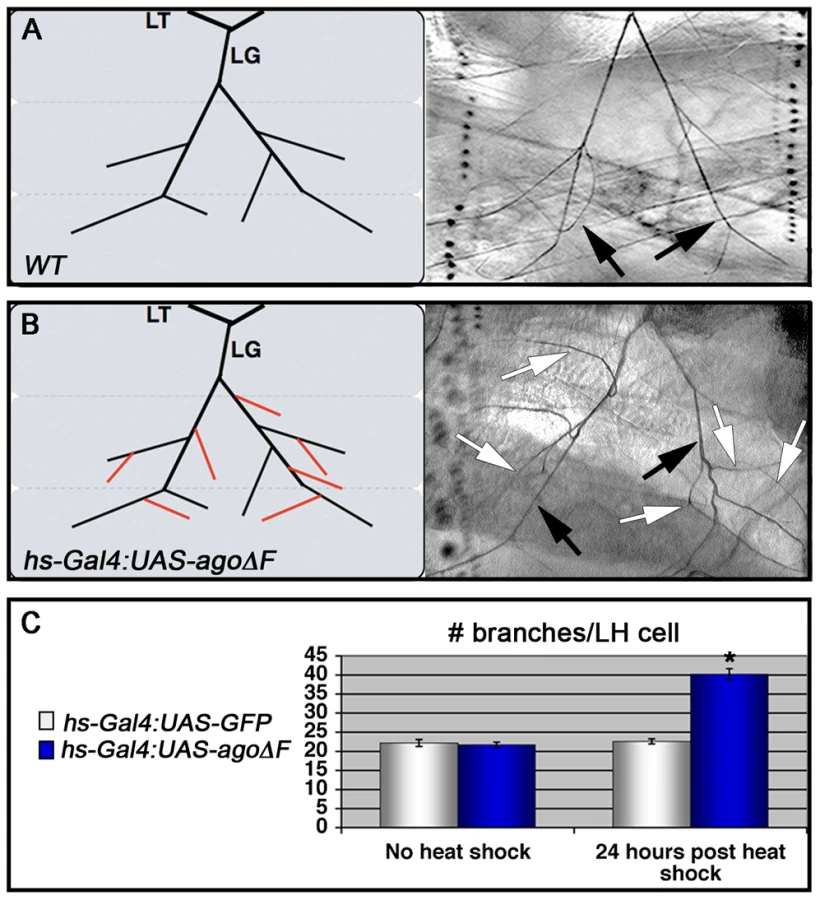
Excess terminal branch phenotypes in hs>agoΔF and agoΔ3–7 animals may reflect a requirement for ago in either tracheal or non-tracheal cell types. To directly test whether ago activity is required in non-tracheal tissue to restrict branching, the agoΔF transgene was driven with the 5053A-Gal4 driver (5053A>agoΔF), which is expressed specifically in ventrolateral body wall muscle 12 (VLM12) and has been used to study non-cell autonomous tracheogenic activity of the Btl/Bnl pathway [38]. The VLM12 muscle expresses endogenous, nuclear Ago protein (Figure 4C–4D) and is normally tracheated by the LF and LH cells (Figure 4A). The 5053A>agoΔF genotype approximately doubles the number of LF and LH tracheal branches that terminate on VLM12 (Figure 4B) relative to either the adjacent muscle (VLM13) or to control larvae expressing a nuclear-localized GFP (nlsGFP) (5.11±0.16 branches [n = 54] in control vs 9.54±0.29 branches [n = 50] in agoΔF, p = 4.67×10−24) (Table 3). This degree of excess branching produced by muscle-specific expression of the agoΔF transgene is similar to that produced by organism-wide depletion of the Ago-RC isoform with the agoΔ3–7 genomic allele. These combined genetic data provide evidence that Ago is required within larval body wall muscle cells to restrict the post-embryonic branching of nearby tracheal terminal cells.


Muscle expression of Ago targets
ago mutations lead to tissue-specific activation of factors normally degraded by the SCF-Ago ubiquitin ligase, including the proliferative proteins CycE and dMyc in larval imaginal discs [49], [50] and the transcription factor Trachealess in tracheal cells [47]. Although the expression patterns of these proteins in body wall muscle are not well defined, we wished to test whether ectopic expression of CycE, dMyc, Trh, or the SCF-Fbw7 target Notch [reviewed in 51] was even capable of conferring a non-cell autonomous tracheogenic activity to VLM12. To this end, each of these factors was individually overexpressed using the 5053A-Gal4 driver (Table 3). Muscle-specific expression of trh or Notch failed to stimulate excess terminal branch growth. The inability of trh to affect tracheal recruitment to VLM12 contrasts with its ability to phenocopy ago mutant phenotypes in the embryonic trachea [47] and further suggests that the ago larval tracheal role is from separable from its embryonic role. Muscle-specific expression of dMyc also had no effect on the degree of terminal cell branching, despite a 28.5% increase in the 2-dimensional size of the VLM12 muscle (Table 4). Notably, increased tracheation of VLM12 driven by agoΔF occurs without an increase in the size of the VLM12 muscle, which is consistent with no role for post-mitotic growth in this phenotype (Table 4). 5053A>cycE does increase terminal branch number, although to a lesser degree than agoΔF. However, CycE protein levels are not obviously affected by expression of agoΔF (Figure S1), suggesting that deregulated CycE is an unlikely cause of the non-autonomous effect of ago alleles on terminal cell branching.

ago restricts dHIF activity and bnl expression in larvae
The similarity of ago mutant terminal branching phenotypes to those induced by hypoxia suggests that ago may antagonize the dHIF pathway. To test the genetic relationship between ago and sima in larval tracheal branching, the sima07607 loss-of-function allele [23] was introduced into the 5053A>agoΔF and agoΔ3–7/1 genetic backgrounds. Heterozygosity for sima (i.e. sima07607/+) dominantly suppressed the agoΔF VLM12 phenotype (Table 3) by decreasing terminal branch number from 9.54±0.29 (n = 50) to 6.34±0.27 branches (n = 53, p = 4.36×10−12), and also suppressed the excess and overlapping terminal branching seen in agoΔ3–7/1 larvae (Table 2 and Figure 5A–5B; white arrow in 5A indicates a ringlet-shaped ganglionic branch) from 39.5±1.59 (n = 34) to 29.0±1.48 branches per LH cell (n = 34, p = 7.45×10−6). In addition, the sima07607 allele dominantly delayed the lethal phase of both agoΔ3–7 homozygotes and agoΔ3–7/1 or agoΔ3–7/3 trans-heterozygotes (Table 1). Reciprocally, ectopic expression of sima in the VLM12 muscle (5053A>sima) increased tracheal recruitment in normoxic conditions (Table 3). Muscle cells are thus distinct from ectodermal cells, which do not recruit branching following overexpression of sima [22].

To more directly assess dHIF activity in ago mutant animals, the transcription of the Drosophila LDH gene (dLDH) was measured in the body wall muscle of agoΔ3–7 and control larvae. LDH is a well-validated HIF target in vertebrates and invertebrates, and HIF-responsive elements from the LDH promoter have been used as the basis of HIF activity reporters in many different systems including Drosophila [e.g. 24]. This analysis showed a 27.3-fold increase in dLDH transcription in ago mutant larval body wall muscle preparations but no equivalent upregulation in steady-state levels of the sima mRNA (Figure 5C). Sima-driven expression of the FGF ligand bnl is a key element of the hypoxic response among non-tracheal cells [22], [38], [52]. The bnlP1 loss-of-function allele dominantly suppressed both the 5053A>agoΔF VLM12 phenotype (Table 3), from 9.54±0.29 (n = 50) to 6.54±0.28 branches (n = 54, p = 5.99×10−11) and the agoΔ3–7/1 excess branching phenotype (Table 2), from 39.5±1.59 (n = 34) to 28.4±1.80 branches per LH cell (n = 29, p = 1.75×10−5). In parallel, qRT-PCR detected an ∼50% upregulation of bnl transcription in body wall muscle of agoΔ3–7 larvae relative to control muscle (Figure 5C). Previous studies using a genomic duplication of the bnl locus have demonstrated that a similar 50% increase in bnl gene-dosage is sufficient to elicit excess tracheal terminal cell branching [38]. Thus reduced ago function in body wall muscle is associated with ectopic expression of the dHIF target dLDH, increased levels of the bnl mRNA, and a genetic dependence on sima and bnl.
ago acts with dVHL to restrict tracheal terminal branching
The data above suggests that ago alleles might exhibit functional interactions with components of the Fga/dVHL pathway, which controls Sima stability and activity in vivo [21]–[23], [53], [54]. A previously characterized dVHL RNAi knockdown transgene (dVHLi) [48]) was used with the 5053A-Gal4 driver to reduce dVHL expression in VLM12. Consistent with the role of dVHL upstream of sima, the 5053A>dVHLi genotype showed an increase in terminal branching relative to a non-specific RNAi control (Figure 6A, and Table 3) (5.28±0.20 branches [n = 40] in Adf1i control vs 7.48±0.21 branches [n = 89] in dVHLi, p = 1.27×10−9, Figure 6). The dVHLi and agoΔF transgenes were then co-expressed with 5053A-Gal4 to determine their ability to enhance VLM12 tracheogenic activity (Figure 6C–6D). The 5053A>agoΔF,VHLi compound genotype shows a synergistic increase in the number of branches that terminate on VLM12 (Table 3), but also leads to a phenotype not seen in either individual genotype: whereas expression of agoΔF or dVHLi individually increase terminal branching of LF and LH onto VLM12, the agoΔF+dVHLi combination also recruits ectopic branches from the LG lateral terminal cell (as seen in the two different focal planes of a single agoΔF+dVHLi-expressing VLM12 muscle; Figure 6C–6D) which normally bypasses VLM12. This ectopic LG recruitment phenotype occurs in approximately 10% of agoΔF+dVHLi VLM12 muscles and is also observed upon 5053A-Gal4 driven expression of bnl [38] or sima (data not shown). Thus dVHL and ago are individually required to restrict the ability of muscle cells to recruit new branch growth, and combined reduction of ago and dVHL activity leads to increased tracheogenic signals emanating from body wall muscle.

To further define the relationship between ago and dVHL in terminal branching, transgenes expressing each factor were tested for rescue of VLM12-branching phenotypes produced by reducing the function of the other (Table 3). Expression of wild type dVHL led to a 66% suppression of the agoΔF branching phenotype (p = 6.55×10−12); reciprocally, over-expression of wild type ago showed a 54% suppression of the dVHLi branching phenotype (p = 2.73×10−4). Thus, each gene can to some degree ameliorate non-autonomous branching phenotypes produced by loss of the other in the VLM12 segment.
Ago controls transcriptional and organismal responses to hypoxia
In view of the genetic and molecular links between ago, sima, dLDH, and dVHL, the organism-wide transcriptional response to hypoxia was examined in ago mutant animals. Drosophila respond to varying degrees of hypoxia by driving transcription of distinct sets of target genes at differing oxygen concentrations, including those involved in metabolic adaptation and survival in low oxygen [31], [32]. A subset of hypoxia-inducible genes was selected for this analysis based on their differential transcription in hypoxic adult Drosophila [31] and predicted links to known mechanisms of the hypoxic response. These included dLDH, which plays a role in the metabolic switch to high flux glycolysis [reviewed in 55], [56], lysyl oxidase (lox), a HIF target in mammalian cells that plays a role in hypoxia-induced changes in cell adhesion [57], [58] and vascular remodeling [59], and dHIG1 (CG11825), the Drosophila homolog of Hypoxia Induced Gene-1 (HIG1), which is induced by HIF and promotes cell survival [60]. qRT-PCR analysis was carried out for each of these genes under conditions of decreasing environmental oxygen (21%, 5%, 0.5%) in whole control larvae or whole agoΔ3–7 larvae (Figure 7A–7B). We find that each of these genes is differentially induced in hypoxia in a manner consistent with findings in adult Drosophila [31] and can be ectopically induced by the agoΔ3–7 allele. dLDH is minimally transcribed in normoxic control larvae, and with progressively higher transcription as the oxygen level falls (1.6 and 2.7-fold increases in 5% and 0.5% O2 respectively, Figure 7A), confirming that dLDH transcription increases with increasing dHIF activity. In agoΔ3–7 homozygous animals, dLDH expression is increased 8.1-fold in whole normoxic larvae (this lower fold induction in the whole larva relative to the ∼27-fold enrichment seen in dissected body wall muscle in Figure 5C is presumably a reflection of the tissue-specific nature of the agoΔ3–7 allele), and is increased approximately 14-fold in agoΔ3–7/1 larvae relative to control larvae at both 5% and 0.5% O2 (Figure 7B, top panel). Thus ago restricts dLDH expression activity across a broad range of oxygen concentrations. The lox gene is normally only up-regulated in whole control larvae by strong hypoxia (4.4-fold induction at 0.5% O2; Figure 7B). The agoΔ3–7 allele leads to a 2.2-fold increase in lox transcription in normoxia, and lox transcription reaches near maximal levels at 5% O2; the 3.4-fold induction seen in ago mutants in 5% O2 is not significantly different from that seen in control larvae at 0.5% O2 (Figure 7B, middle panel). This pattern suggests that the lox promoter is induced by levels of dHIF activity achieved in moderate hypoxic conditions, and that this threshold is more easily reached in ago mutants. The dHIG1 gene displays a more exaggerated version of the lox response pattern: dHIG1 mRNA levels are only induced strongly (19.9-fold) in whole control larvae by 0.5% O2 (Figure 7A); the agoΔ3–7 allele is not sufficient to drive ectopic dHIG1 transcription in normoxic conditions but it is sufficient to sensitize the dHIG1 promoter to reduced O2 levels such that maximal dHIG1 expression is now achieved at a ten-fold higher O2 concentration than normal (Figure 7B, bottom panel). In addition to dLDH, lox, and dHIG1, three other genes also induced by hypoxia, hairy, amy-p and thor genes [31], are also moderately up-regulated in normoxic agoΔ3–7 mutant larvae (Table S1). Reducing ago activity is thus sufficient to alter the threshold required to drive expression of multiple hypoxia-inducible genes.

Adult Drosophila respond to prolonged periods of oxygen deprivation by entering into a state of hypoxic stupor characterized by inactivity and reduced oxygen consumption [34]. Many mutations have been identified that slow this hypoxic recovery [32], [33], [35], but few mutations have been described that enhance it. Under our standard laboratory conditions, control adult flies enter stupor after approximately fifteen to twenty minutes in a 0.5% O2 environment and remain unconscious until re-oxygenation. We assayed control +/+ adults, agoΔ3–7/+ adults, and adults trans-heterozygous for the agoΔ3–7 allele and the ago hypomorphic allele EP(3)1135 (BDSC) for recovery time following acute hypoxia (1 hour at 0.5% O2) (Figure 7C). agoΔ3–7/EP(3)1135 flies display no obvious developmental phenotypes and enter into hypoxic stupor at the same rate as control flies (data not shown); however, they recover significantly faster than either control +/+ or agoΔ3–7/+ adults. Linear regression analysis indicates that the time for 50% recovery is reduced from 4.5±0.75 minutes in control +/+ flies, to 1.4±0.16 minutes in agoΔ3–7/EP(3)1135 flies (p = 0.0015). The agoΔ3–7/EP(3)1135 population also reaches 100% recovery after 10 minutes of re-oxygenation, whereas neither the control +/+ or agoΔ3–7/+ populations achieved 100% recovery by the end of the 15 minute measurement period (data not shown). Thus, the genetic evidence of a role for ago as a regulator of dHIF-regulated branching in the larval tracheal arbor is paralleled at the organismal level by an enhanced transcriptional sensitivity to hypoxia and an increased ability of flies to recover from a transient hypoxic challenge.
Ago associates with Sima and suppresses Sima levels
To test the molecular relationship between Sima and Ago, Sima levels were assessed in two ways: by immunoflourescent staining of VLM12 muscles expressing the UAS-agoΔF transgene and by Western blotting of lysates of agoΔ3–7 larvae (Figure 8). Fluorescence microscopy confirms that a previously described anti-Sima antibody [24] detects high levels of transgenically expressed Sima in the VLM12 nuclei of 5053A>sima muscles, and that endogenous Sima is not readily detectable by this method of analysis in the nuclei of adjacent non-transgenic muscles (Figure 8A). Following expression of the agoΔF dominant-negative transgene (5053A>agoΔF), a fraction of VLM12 nuclei accumulate anti-Sima reactive epitopes (see arrows, Figure 8B). This same anti-Sima antibody detects elevated levels of an ∼110 kD molecular weight band in agoΔ3–7 filleted 3rd instar pelts relative to wt control pelts (Figure 8C). This ∼110 kD band is absent in lysates of sima07607 larvae (Figure 8D, lane 1 vs. 2), and is specifically enriched in precipitates of an anti-Ago polyclonal antibody from lysates of hypoxic larvae (Figure 8D, lane 5). Collectively, these molecular data indicate that Ago can associate with Sima in larval lysates, and that Ago limits Sima levels in developing tissues.

Discussion
The selective stabilization of the Sima/HIF–1α transcription factor in hypoxia plays a key role in the response of metazoan organisms to low oxygen concentrations by its ability to induce a program of hypoxia-specific gene expression [reviewed in 2]. Evidence suggests that in Drosophila, Sima plays a dual role in the post-mitotic growth of tracheal terminal branch cells toward hypoxic peripheral tissues by acting within both the ‘signaling’ hypoxic peripheral cells and in the ‘responsive’ terminal tips cells [reviewed in 37].
Our data implicate the Ago-SCF ubiquitin ligase as a required regulator of Sima during hypoxia sensing in peripheral cells, but do not rule out an additional role for Ago within tracheal terminal tip cells which contributes to their ectopic branching in ago mutant larvae (see below). Phenotypes produced by muscle-specific expression of an ago dominant-negative allele, or by a genomic allele that specifically affects ago expression in peripheral tissues, are phenocopied by overexpression of Sima (this study) or the FGF homolog Bnl [38]. These non-cell autonomous effects of ago alleles on terminal branching are accompanied by a strong induction in peripheral tissues of the dHIF target dLDH, and can be dominantly suppressed by an allele of sima. ago alleles induce expression of a set of dHIF-inducible hypoxia-response genes in normoxia that includes dLDH, and this is paralleled at the organismal level by an enhanced ability of ago mutant flies to the recover from hypoxic stupor. ago alleles are thus among the first genetic alterations shown to enhance the recovery of adult Drosophila from hypoxic exposure. Within larval muscle, ago appears to inhibit sima in parallel to dVHL, which targets the Sima/HIF–1α protein for constitutive degradation in normoxia [reviewed in 3]. Consistent with this, we find evidence that Ago can associate with Sima and limits its levels in vivo. Collectively these data significantly expand the known role of Ago in organism development by demonstrating that it is required in an apparently novel pathway that collaborates with dVHL to inhibit Sima-regulated hypoxic gene expression in peripheral tissues.
Though the work presented here focuses on the ‘tracheo-attractant’ effects of reducing ago expression in body wall muscle, this may be just one manifestation of roles Ago plays in controlling hypoxia-regulated gene expression. Indeed, reducing ago function has a quantitatively stronger effect on terminal branching than a genomic duplication of the bnl locus [38], suggesting either that ago also act within tracheal cells to limit branching [as in 47], [48] or that a larger set of dHIF target genes contribute to the effect. Consistent with this latter hypothesis, normoxic ago mutant larvae display ectopic induction of hypoxia-responsive metabolic genes such as dLDH, lox, hairy, amy-p and thor. Based on this profile, it appears that ago mutant larvae reared in normoxia elevate expression of bnl but also engage a metabolic switch to high-flux glycolysis that is characteristic of hypoxic cells [32], [33], [35], [36], [61]. Future studies will be required to assess the full effect of these transcriptional changes on the behavior of terminal tracheal cells and the tissues into which they project.
In wild type animals, the transcriptional response of cells to hypoxia is graded such that different target genes are induced across a range of environmental O2 concentrations [31]. In ago mutants, this differential induction is largely abolished such that expression of genes such as lox and dHIG1 is virtually indistinguishable at 5% and 0.5% O2. Thus, ago appears to be required both for inhibition of hypoxia-inducible genes in normoxia and for the graded expression of hypoxia-inducible genes under variable levels of oxygen deprivation. We hypothesize that this graded sensitivity is normally a product of the interaction between the Ago and Fga/dVHL regulatory mechanisms. The HPH/VHL pathway has been demonstrated to act in a graded manner, such that it degrades HIF–1α efficiently in normoxia, but is progressively less efficient as the oxygen concentration drops [62]. This leads to a gradient of HIF activity that is presumably required for the differential induction of target genes. We hypothesize that ago acts in parallel to dVHL to dampen Sima/HIF-1 activity across a range O2 concentrations, and that Ago may function as a dHIF regulatory mechanism at very low O2 concentrations in which the HPH/dVHL pathway is hypothesized to be inactive [62]. Thus, the absence of Ago allows a mild hypoxic stimulus (∼e.g. 5% O2) to be translated into levels of dHIF-dependent gene expression that would normally only result from much stronger hypoxic exposure. The data presented here support this prediction, with the end result that the transcriptional response profile of hypoxia-response genes in ago mutant larvae is shifted toward induction by more mild stimuli.
The molecular mechanism(s) underlying the genetic relationship between ago and sima in tracheal branching appears to involve a physical association of their encoded proteins that modulates Sima levels. Given that Ago is a ubiquitin ligase specificity factor, these data are consistent with a model in which Ago supports Sima polyubiquitination and turnover. Recent studies have identified the human Ago ortholog Fbw7 as a HIF–1α interacting factor and have proposed that Fbw7 promotes HIF-1α turnover following GSK3ß phosphorylation in cultured cells [45], [46]. Phenotypic predictions made by this molecular model appear to be confirmed by the ago terminal cell branching phenotypes documented here. Intriguingly, RNAi depletion of GSK3ß/shaggy or a proteasome subunit in VLM12 also elevates the number of tracheal branches that terminate on this muscle (Table S2). However Fbw7 is implicated in the proteolytic destruction of two transcription factors, the Notch intracellular domain (NICD) [reviewed in 51] and sterol-regulatory enhancer binding protein (SREBP) [63], that indirectly modulate HIF-dependent hypoxic gene expression in eukaryotic cells [64], [65]. Ago could thus theoretically influence hypoxic gene expression via these paths as well. Future biochemical studies will be required to clarify the full range of Ago molecular targets that contribute to its role in hypoxic gene expression.
The well-studied anti-proliferative role of ago is conserved in its human ortholog Fbw7, which is mutationally inactivated in a wide spectrum of primary human cancers [reviewed in 51]. Some cancer cells engage a program of gene expression that supports a switch to high-flux glycolysis (a phenomenon termed ‘Warburg effect’ [26]) and are more resistant to transient hypoxia than normal cells [reviewed in 1]. Both of these properties can now, to some degree, also be associated with ago loss in Drosophila. In view of the functional conservation between SCF-Ago and SCF-Fbw7 in degradation of shared oncogenic targets [49], [50] and the proposed role of Ago/Fbw7 in Sima HIF-1α turnover [this study and 45], [46], our data raise the interesting possibility that sensitization to mild hypoxia may be a feature of Fbw7 mutations in vertebrates as well. If so, then tumor suppressive properties of Fbw7 may derive in part from its established anti-proliferative role and in part due to modulation of HIF-regulated angiogenic and metabolic pathways.
Materials and Methods
Stocks, genetics, and statistics
The FRT80B and w1118 strains were used as wild type controls. The ago1 and ago3 alleles have been previously described [49]. The agoΔ3–7 allele was identified as an imprecise excision of the agoEP(3)1135 transposon. Alleles used in this study: bnlP1, sima07607, agoEP(3)1135 (all from the Bloomington Drosophila Stock Center) and 1-eve-1 [66]. The following transgenes were also used: UAS-agoΔF and UAS-ago [47]; UAS-CycE, UAS-dMyc, UAS-N, UAS-nlsGFP (all from the Bloomington Drosophila Stock Center), UAS-trh [67], UAS-sima [24], UAS-dVHL [21], UAS-Adf1RNAi and UAS-sggRNAi (Vienna Drosophila RNAi Center), UAS-dVHLRNAi [48], hs-Gal4, and 5053A-Gal4 (from the Bloomington Drosophila Stock Center). Statistical comparisons were made using Student's t-Test (Microsoft Excel) with the indicated significance levels.
Hypoxia treatments
Hypoxia treatments were performed in a sealed Modular incubator chamber (Billups-Rothenberg Inc., Del Mar, CA) with separate gas intake and exhaust openings. Internal O2 concentration was measured with an electronic O2 sensor (OX-01, RKI Instruments, Inc., Union City, CA). To assay recovery from hypoxia, 5–7 day old adult flies were put into plain glass tubes in groups of 9–15. The flies were then placed into the hypoxia chamber at 0.5% O2 for one hour and then removed to normoxia. Following hypoxic treatment, >99% of the flies (178 of 179) had fallen into hypoxic stupor. Recovery time was defined as the time required for each individual fly to resume walking following re-oxygenation.
Reverse transcription and quantitative real-time PCR (qRT–PCR)
Total RNA was isolated from dissected third instar larval body wall muscles. cDNA was reverse-transcribed using random hexamer primers (Invitrogen) with Superscript II Reverse Transcriptase (Invitrogen). dVHL and ß-tubulin transcripts were then amplified with gene-specific primers. For quantification of mRNA levels, total RNA was isolated from whole third instar larvae or dissected larval tissues and reverse transcribed as described above. Levels of Arp87c, ago-RA, -RB and -RC, dLDH, sima, bnl, lox, hairy, dHIG1, thor and amy-p were then assayed with gene-specific primers using the SYBR green method of quantitative real-time PCR on a Roche LightCycler 480 machine. Transcript abundance was normalized to levels of Arp87c as in [31].
Imaging of third instar larval trachea
To image the larval tracheal system, third instar larvae were dissected in cold PBS and fixed in 4% paraformaldehyde. Air-filled tracheal branches were imaged using bright-field microscopy. and assembled using Photomerge (Adobe Photoshop CS).
Immunohistochemistry, Western blotting, and immunoprecipitation
Third instar larvae were dissected in cold PBS, fixed in 4% paraformaldehyde and incubated with guinea pig anti-CycE (1∶500) or rabbit anti-Sima (1∶1000). Secondary antibodies (anti-guinea pig conjugated to Cy3 or anti-rabbit conjugated Cy5) were used as recommended (Jackson ImmunoResearch). To assess Sima protein levels in third instar larvae, larval pelt extracts were prepared in sample buffer and resolved on 7.5% SDS-PAGE prior to Western blotting with rabbit anti-Sima (1∶1000) [24] and developed with anti-rabbit HRP (1∶1000; Jackson ImmunoResearch). Whole larval extracts were immunoprecipitated with guinea pig anti-Ago polyclonal sera (1∶1000) [47] prior to immunoblotting with anti-Sima antibody.
Supporting Information
{kind=link}
Zdroje
1. DenkoNC (2008) Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer 8 : 705–713.
2. KaelinWGJr, RatcliffePJ (2008) Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell 30 : 393–402.
3. GorrTA, GassmannM, WappnerP (2006) Sensing and responding to hypoxia via HIF in model invertebrates. J Insect Physiol 52 : 349–364.
4. WangGL, JiangBH, RueEA, SemenzaGL (1995) Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A 92 : 5510–5514.
5. WangGL, JiangBH, SemenzaGL (1995) Effect of altered redox states on expression and DNA-binding activity of hypoxia-inducible factor 1. Biochem Biophys Res Commun 212 : 550–556.
6. WangGL, SemenzaGL (1995) Purification and characterization of hypoxia-inducible factor 1. J Biol Chem 270 : 1230–1237.
7. SemenzaGL, WangGL (1992) A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol 12 : 5447–5454.
8. BaconNC, WappnerP, O'RourkeJF, BartlettSM, ShiloB, et al. (1998) Regulation of the Drosophila bHLH-PAS protein Sima by hypoxia: functional evidence for homology with mammalian HIF-1 alpha. Biochem Biophys Res Commun 249 : 811–816.
9. NambuJR, ChenW, HuS, CrewsST (1996) The Drosophila melanogaster similar bHLH-PAS gene encodes a protein related to human hypoxia-inducible factor 1 alpha and Drosophila single-minded. Gene 172 : 249–254.
10. SonnenfeldM, WardM, NystromG, MosherJ, StahlS, et al. (1997) The Drosophila tango gene encodes a bHLH-PAS protein that is orthologous to mammalian Arnt and controls CNS midline and tracheal development. Development 124 : 4571–4582.
11. OhshiroT, SaigoK (1997) Transcriptional regulation of breathless FGF receptor gene by binding of TRACHEALESS/dARNT heterodimers to three central midline elements in Drosophila developing trachea. Development 124 : 3975–3986.
12. MaE, HaddadGG (1999) Isolation and characterization of the hypoxia-inducible factor 1beta in Drosophila melanogaster. Brain Res Mol Brain Res 73 : 11–16.
13. WeidemannA, JohnsonRS (2008) Biology of HIF-1alpha. Cell Death Differ 15 : 621–627.
14. BruickRK, McKnightSL (2001) A conserved family of prolyl-4-hydroxylases that modify HIF. Science 294 : 1337–1340.
15. EpsteinAC, GleadleJM, McNeillLA, HewitsonKS, O'RourkeJ, et al. (2001) C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 107 : 43–54.
16. IvanM, KondoK, YangH, KimW, ValiandoJ, et al. (2001) HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292 : 464–468.
17. JaakkolaP, MoleDR, TianYM, WilsonMI, GielbertJ, et al. (2001) Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292 : 468–472.
18. CockmanME, MassonN, MoleDR, JaakkolaP, ChangGW, et al. (2000) Hypoxia inducible factor-alpha binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein. J Biol Chem 275 : 25733–25741.
19. OhhM, ParkCW, IvanM, HoffmanMA, KimTY, et al. (2000) Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat Cell Biol 2 : 423–427.
20. MaxwellPH, WiesenerMS, ChangGW, CliffordSC, VauxEC, et al. (1999) The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399 : 271–275.
21. ArquierN, VigneP, DuplanE, HsuT, TherondPP, et al. (2006) Analysis of the hypoxia-sensing pathway in Drosophila melanogaster. Biochem J 393 : 471–480.
22. CentaninL, DekantyA, RomeroN, IrisarriM, GorrTA, et al. (2008) Cell autonomy of HIF effects in Drosophila: tracheal cells sense hypoxia and induce terminal branch sprouting. Dev Cell 14 : 547–558.
23. CentaninL, RatcliffePJ, WappnerP (2005) Reversion of lethality and growth defects in Fatiga oxygen-sensor mutant flies by loss of Hypoxia-Inducible Factor-alpha/Sima. EMBO Rep 6 : 1070–1075.
24. Lavista-LlanosS, CentaninL, IrisarriM, RussoDM, GleadleJM, et al. (2002) Control of the hypoxic response in Drosophila melanogaster by the basic helix-loop-helix PAS protein similar. Mol Cell Biol 22 : 6842–6853.
25. KondoK, KaelinWGJr (2001) The von Hippel-Lindau tumor suppressor gene. Exp Cell Res 264 : 117–125.
26. WarburgO (1956) On respiratory impairment in cancer cells. Science 124 : 269–270.
27. RankinEB, GiacciaAJ (2008) The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ 15 : 678–685.
28. ZhouJ, SchmidT, SchnitzerS, BruneB (2006) Tumor hypoxia and cancer progression. Cancer Lett 237 : 10–21.
29. RomeroNM, DekantyA, WappnerP (2007) Cellular and developmental adaptations to hypoxia: a Drosophila perspective. Methods Enzymol 435 : 123–144.
30. HoogewijsD, GeuensE, DewildeS, VierstraeteA, MoensL, et al. (2007) Wide diversity in structure and expression profiles among members of the Caenorhabditis elegans globin protein family. BMC Genomics 8 : 356.
31. LiuG, RoyJ, JohnsonEA (2006) Identification and function of hypoxia-response genes in Drosophila melanogaster. Physiol Genomics 25 : 134–141.
32. ZhouD, XueJ, LaiJC, SchorkNJ, WhiteKP, et al. (2008) Mechanisms underlying hypoxia tolerance in Drosophila melanogaster: hairy as a metabolic switch. PLoS Genet 4: e1000221 doi:10.1371/journal.pgen.1000221
33. HaddadGG, SunY, WymanRJ, XuT (1997) Genetic basis of tolerance to O2 deprivation in Drosophila melanogaster. Proc Natl Acad Sci U S A 94 : 10809–10812.
34. HaddadGG, WymanRJ, MohseninA, SunY, KrishnanSN (1997) Behavioral and Electrophysiologic Responses of Drosophila melanogaster to Prolonged Periods of Anoxia. J Insect Physiol 43 : 203–210.
35. MaE, HaddadGG (1997) Anoxia regulates gene expression in the central nervous system of Drosophila melanogaster. Brain Res Mol Brain Res 46 : 325–328.
36. MaE, XuT, HaddadGG (1999) Gene regulation by O2 deprivation: an anoxia-regulated novel gene in Drosophila melanogaster. Brain Res Mol Brain Res 63 : 217–224.
37. CentaninL, GorrTA, WappnerP (2010) Tracheal remodelling in response to hypoxia. J Insect Physiol 56 : 447–454.
38. JareckiJ, JohnsonE, KrasnowMA (1999) Oxygen regulation of airway branching in Drosophila is mediated by branchless FGF. Cell 99 : 211–220.
39. KlambtC, GlazerL, ShiloBZ (1992) breathless, a Drosophila FGF receptor homolog, is essential for migration of tracheal and specific midline glial cells. Genes Dev 6 : 1668–1678.
40. SutherlandD, SamakovlisC, KrasnowMA (1996) branchless encodes a Drosophila FGF homolog that controls tracheal cell migration and the pattern of branching. Cell 87 : 1091–1101.
41. GuilleminK, GroppeJ, DuckerK, TreismanR, HafenE, et al. (1996) The pruned gene encodes the Drosophila serum response factor and regulates cytoplasmic outgrowth during terminal branching of the tracheal system. Development 122 : 1353–1362.
42. SamakovlisC, ManningG, StenebergP, HacohenN, CanteraR, et al. (1996) Genetic control of epithelial tube fusion during Drosophila tracheal development. Development 122 : 3531–3536.
43. FlugelD, GorlachA, MichielsC, KietzmannT (2007) Glycogen synthase kinase 3 phosphorylates hypoxia-inducible factor 1alpha and mediates its destabilization in a VHL-independent manner. Mol Cell Biol 27 : 3253–3265.
44. ZhangD, LiJ, CostaM, GaoJ, HuangC (2010) JNK1 mediates degradation HIF-1alpha by a VHL-independent mechanism that involves the chaperones Hsp90/Hsp70. Cancer Res 70 : 813–823.
45. FlugelD, GorlachA, KietzmannT (2012) GSK-3beta regulates cell growth, migration, and angiogenesis via Fbw7 and USP28-dependent degradation of HIF-1alpha. Blood 119 : 1292–1301.
46. CassavaughJM, HaleSA, WellmanTL, HoweAK, WongC, et al. (2011) Negative regulation of HIF-1alpha by an FBW7-mediated degradation pathway during hypoxia. J Cell Biochem 112 : 3882–3890.
47. MortimerNT, MobergKH (2007) The Drosophila F-box protein Archipelago controls levels of the Trachealess transcription factor in the embryonic tracheal system. Dev Biol 312 : 560–571.
48. MortimerNT, MobergKH (2009) Regulation of Drosophila embryonic tracheogenesis by dVHL and hypoxia. Dev Biol 329 : 294–305.
49. MobergKH, BellDW, WahrerDC, HaberDA, HariharanIK (2001) Archipelago regulates Cyclin E levels in Drosophila and is mutated in human cancer cell lines. Nature 413 : 311–316.
50. MobergKH, MukherjeeA, VeraksaA, Artavanis-TsakonasS, HariharanIK (2004) The Drosophila F box protein archipelago regulates dMyc protein levels in vivo. Curr Biol 14 : 965–974.
51. WelckerM, ClurmanBE (2008) FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer 8 : 83–93.
52. GhabrialA, LuschnigS, MetzsteinMM, KrasnowMA (2003) Branching morphogenesis of the Drosophila tracheal system. Annu Rev Cell Dev Biol 19 : 623–647.
53. AdryanB, DeckerHJ, PapasTS, HsuT (2000) Tracheal development and the von Hippel-Lindau tumor suppressor homolog in Drosophila. Oncogene 19 : 2803–2811.
54. AsoT, YamazakiK, AigakiT, KitajimaS (2000) Drosophila von Hippel-Lindau tumor suppressor complex possesses E3 ubiquitin ligase activity. Biochem Biophys Res Commun 276 : 355–361.
55. SemenzaGL (2007) Hypoxia-inducible factor 1 (HIF-1) pathway. Sci STKE 2007: cm8.
56. SemenzaGL (2007) Life with oxygen. Science 318 : 62–64.
57. ErlerJT, BennewithKL, NicolauM, DornhoferN, KongC, et al. (2006) Lysyl oxidase is essential for hypoxia-induced metastasis. Nature 440 : 1222–1226.
58. ErlerJT, GiacciaAJ (2006) Lysyl oxidase mediates hypoxic control of metastasis. Cancer Res 66 : 10238–10241.
59. RodriguezC, AlcudiaJF, Martinez-GonzalezJ, RaposoB, NavarroMA, et al. (2008) Lysyl oxidase (LOX) down-regulation by TNFalpha: a new mechanism underlying TNFalpha-induced endothelial dysfunction. Atherosclerosis 196 : 558–564.
60. WangJ, CaoY, ChenY, GardnerP, SteinerDF (2006) Pancreatic beta cells lack a low glucose and O2-inducible mitochondrial protein that augments cell survival. Proc Natl Acad Sci U S A 103 : 10636–10641.
61. AzadP, HaddadGG (2009) Survival in acute and severe low o environment: use of a genetic model system. Ann N Y Acad Sci 1177 : 39–47.
62. JiangBH, SemenzaGL, BauerC, MartiHH (1996) Hypoxia-inducible factor 1 levels vary exponentially over a physiologically relevant range of O2 tension. Am J Physiol 271: C1172–1180.
63. SundqvistA, Bengoechea-AlonsoMT, YeX, LukiyanchukV, JinJ, et al. (2005) Control of lipid metabolism by phosphorylation-dependent degradation of the SREBP family of transcription factors by SCF(Fbw7). Cell Metab 1 : 379–391.
64. HughesAL, ToddBL, EspenshadePJ (2005) SREBP pathway responds to sterols and functions as an oxygen sensor in fission yeast. Cell 120 : 831–842.
65. GustafssonMV, ZhengX, PereiraT, GradinK, JinS, et al. (2005) Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev Cell 9 : 617–628.
66. PerrimonN, NollE, McCallK, BrandA (1991) Generating lineage-specific markers to study Drosophila development. Dev Genet 12 : 238–252.
67. JinJ, AnthopoulosN, WetschB, BinariRC, IsaacDD, et al. (2001) Regulation of Drosophila tracheal system development by protein kinase B. Dev Cell 1 : 817–827.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 2
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Complex Inheritance of Melanoma and Pigmentation of Coat and Skin in Grey Horses
- Coordination of Chromatid Separation and Spindle Elongation by Antagonistic Activities of Mitotic and S-Phase CDKs
- Autophagy Induction Is a Tor- and Tp53-Independent Cell Survival Response in a Zebrafish Model of Disrupted Ribosome Biogenesis
- Assembly of the Auditory Circuitry by a Genetic Network in the Mouse Brainstem
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy