
Inflammation-Mediated Genetic and Epigenetic Alterations Drive Cancer Development in the Neighboring Epithelium upon Stromal Abrogation of TGF-β Signaling
Deletion of tumor suppressor genes in stromal fibroblasts induces epithelial cancer development, suggesting an important role of stroma in epithelial homoeostasis. However, the underlying mechanisms remain to be elucidated. Here we report that deletion of the gene encoding TGFβ receptor 2 (Tgfbr2) in the stromal fibroblasts (Tgfbr2fspKO) induces inflammation and significant DNA damage in the neighboring epithelia of the forestomach. This results in loss or down-regulation of cyclin-dependent kinase inhibitors p15, p16, and p21, which contribute to the development of invasive squamous cell carcinoma (SCC). Anti-inflammation treatment restored p21 expression, delayed tumorigenesis, and increased survival of Tgfbr2fspKO mice. Our data demonstrate for the first time that inflammation is a critical player in the epigenetic silencing of p21 in tumor progression. Examination of human esophageal SCC showed a down-regulation of TGFβ receptor 2 (TβRII) in the stromal fibroblasts, as well as increased inflammation, DNA damage, and loss or decreased p15/p16 expression. Our study suggests anti-inflammation may be a new therapeutic option in treating human SCCs with down-regulation of TβRII in the stroma.
Published in the journal:
. PLoS Genet 9(2): e32767. doi:10.1371/journal.pgen.1003251
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003251
Summary
Deletion of tumor suppressor genes in stromal fibroblasts induces epithelial cancer development, suggesting an important role of stroma in epithelial homoeostasis. However, the underlying mechanisms remain to be elucidated. Here we report that deletion of the gene encoding TGFβ receptor 2 (Tgfbr2) in the stromal fibroblasts (Tgfbr2fspKO) induces inflammation and significant DNA damage in the neighboring epithelia of the forestomach. This results in loss or down-regulation of cyclin-dependent kinase inhibitors p15, p16, and p21, which contribute to the development of invasive squamous cell carcinoma (SCC). Anti-inflammation treatment restored p21 expression, delayed tumorigenesis, and increased survival of Tgfbr2fspKO mice. Our data demonstrate for the first time that inflammation is a critical player in the epigenetic silencing of p21 in tumor progression. Examination of human esophageal SCC showed a down-regulation of TGFβ receptor 2 (TβRII) in the stromal fibroblasts, as well as increased inflammation, DNA damage, and loss or decreased p15/p16 expression. Our study suggests anti-inflammation may be a new therapeutic option in treating human SCCs with down-regulation of TβRII in the stroma.
Introduction
Mounting evidence supports a cross talk between epithelial and stromal cells in cancer progression mediated by paracrine signals and extracellular matrix components [1], [2]. For example, in a prostate cancer model, epithelial tumor progression induces loss of p53 function in stromal fibroblasts [3]. Conversely, in a breast tumor model, deletion of Pten in stromal fibroblasts promoted tumor progression associated with massive extracellular matrix (ECM) remodeling, immune cell infiltration, increased angiogenesis, and increased recruitment of the Ets2 transcription factor to targeted gene regulation [4]. Likewise, conditional expression of a mutant allele of APC gene in murine uterine stromal cells resulted in endometrial gland hyperplasia progressing to endometrial carcinoma in situ and invasive endometrial adenocarcinoma [5]. Furthermore, deletion of Notch1 in epidermal keratinocytes has significant impact on the stromal microenvironment, promoting skin carcinogenesis [6]. This crosstalk has been witnessed in human sporadic breast cancers: somatic TP53 mutations in stroma but not epithelia were associated with regional nodal metastases [7]. These studies suggest that epithelial and stromal cell signaling influence one another and may co-evolve during the course of tumor progression [2].
The autocrine and paracrine actions of transforming growth factor-β (TGF-β) have been well documented in stromal and tumor cell interaction [8], [9]. Deletion of Tgfbr2 in a variety of epithelial cells results in a more aggressive tumor phenotype in mammary, pancreatic, colon, intestinal, head and neck, anal and genital tumors (reviewed by Yang [9]). The mechanism underlying this observation involves increased infiltration of immune cells in the tumor microenvironment [8]–[11]. Interestingly, conditional knockout of the Tgfbr2 gene in a subset of stromal fibroblasts (FSP1+ cells) contributes to the transformation of epithelia and results in invasive squamous cell carcinoma (SCC) in mouse forestomach [1]. The deletion of Smad4, a downstream mediator of TGF-β signaling in T cells, results in spontaneous epithelial cancers throughout the gastrointestinal tract in mice [12]. Unexpectedly, epithelial-specific deletion of Smad4 did not result in the same tumor phenotype [12]. The impact of stromal TGF-β on epithelial cancers was also demonstrated in a tissue recombination model wherein loss of TβRII function in 50% of immortalized human prostate fibroblasts resulted in malignant transformation of the nontumorigenic human prostate epithelial cells [13]. These studies suggest that stromal loss of TGF-β signaling induces epithelial transformation. One of the mechanisms delineated in these studies involves hepatocyte growth factor (HGF) overproduction by Tgfbr2fspKO stroma and activation of c-MET signaling on adjacent epithelia through paracrine signaling, resulting in epithelial hyperproliferation [1], [14]. However, it is unclear whether changes in stromal cells induce genetic and epigenetic alterations in the epithelial compartment, and if so, what are the underlying molecular mechanisms?
Here we report that stromal deletion of Tgfbr2 induced inflammation resulted in DNA damage, loss of p15 and p16, promoter methylation of p21, and increased epithelial proliferation, i.e. the development of SCC. We showed for the first time that down-regulation of TGF-β signaling in the stroma has significant impact on the genetic and epigenetic components of the adjacent epithelial compartment through inflammation mediated mechanisms. Therefore, therapeutic targeting of inflammation may be a useful strategy in treating human SCCs with down-regulation of TβRII in the stroma.
Results
Deletion of Tgfbr2 in FSP1+ Stromal Cells Induces Loss of p15 and p16 in the Neighboring Epithelial Compartment
Stromal cells and their signaling pathways have significant impact on epithelial tumor progression [4], [12], [13]. Specific deletion of Tgfbr2 in FSP1+ fibroblasts (Tgfbr2fspKO) induced development of SCC in forestomach with 100% penetrance [1] (Figure 1A, left panel). These mice die by 7 weeks with a median survival of 38 days (Log rank p<0.001) (Figure 1A, right panel). Examination of Tgfbr2fspKO forestomach between embryonic day 16 (E16) and 5 weeks of age suggested that hyperplasia began during week 3 and was followed by dysplasia, carcinoma in situ, and invasive SCC (Figure S1A). Here we investigated the molecular mechanisms that are responsible for the development of SCC due to loss of Tgfbr2 in the stromal compartment. We first confirmed the specific deletion of Tgfbr2 in stromal fibroblasts using TβRII immunofluorescence (Figure S1B) and β-galactosidase IHC in FSP1-Cre/Rosa26 reporter mouse tissue (Figure S1C). The absence of p-smad2 nuclear localization in stroma was used as an indicator for the absence of TGF-β signaling (Figure S1D) [1].

Tgfbr2fspKO SCC tumors showed substantial infiltration of CD45+ leukocytes between weeks 3 and 5 compared to Tgfbr2flox/flox littermates (Figure 1B), indicating an inflammatory reaction due to loss of Tgfbr2 in stromal fibroblasts. Inflammation is a critical player in carcinogenesis and is known to cause DNA damage as well as histone modification in cancer [15], [16]. We thus examined DNA damage in forestomach sections of Tgfbr2fspKO and Tgfbr2flox/flox mice using immunofluorescence staining of 8-oxo-2′-deoxyguanosine (8-oxo-dG), a major product of DNA oxidation indicative of DNA damage. Interestingly, DNA damage was initially detected in mice at 3 weeks of age and became progressively worse by 5 weeks (Figure 1C) concomitant with infiltration of CD45+ leukocytes. The expression of γ-H2AX (Figure 1D), a histone molecule associated with DNA double strand breaks, was also increased in Tgfbr2fspKO mice. The 8-oxo-dG and γ-H2AX were not seen in the forestomach of Tgfbr2flox/flox control mice (Figure 1D, left and middle panels). Our data suggest that loss of Tgfbr2 in FSP1+ stromal cells induced inflammation and DNA damage.
DNA damage often results in chromosomal aneuploidy [17] and alteration of epigenetic marks including acetylation, methylation, and ubiquitylation [18]. We evaluated genetic alterations using array-CGH and genomic DNA PCR. We first analyzed epithelial cells isolated from forestomach tumors of Tgfbr2fspKO mice. We found a loss of band C4 of chromosome 4 (Figure S2A), which contains CDK inhibitors Cdkn2b/p15INK4B (p15), Cdkn2a/p16Ink4A (p16), and Cdkn2a/p19Arf. Loss of p15 and p16 tumor suppressor genes is a frequent event in human and mouse cancers [19], [20]. We confirmed loss of p15 and p16 using array-CGH and genomic PCR of epithelial cells from tumor tissue sections of 5 week old Tgfbr2fspKO mice using laser capture microdissection technology (Figure S2B and S2C, Figure 1E). Cdkn2a/p19Arf is an alternative reading frame in the same locus that harbors p16, it is presumably deleted along with p16. These genetic alterations were not detected in the stromal compartment of the forestomach sections from Tgfbr2fspKO mice (Figure S2C and Figure 1E). Western analysis of forestomach tumor tissues from Tgfbr2fspKO mice showed loss of or decreased expression of p15 and p16 proteins (Figure 1F), which was detected from 4 week of age (Figure 1G), one week after the inflammation onset (Figure 1B). p15 and p16 are critical mediators in cell cycle control and are important in suppressing tumor development [19]. Our data suggest that deletion of Tgfbr2 in FSP1+ stromal cells induced a loss of p15 and p16 in epithelial cells.
Alteration of Cell Cycle Mediators and Increased Proliferation in the Forestomach Epithelia of Tgfbr2fspKO Mice
Our data suggest a dysregulation of the G1 cell cycle checkpoint in epithelial cells due to loss of Tgfbr2 in FSP1+ stromal cells. We next examined several critical molecules in cell cycle control. The expression of Cyclin D1 was increased in the forestomach of 5 week old Tgfbr2fspKO compared to Tgfbr2flox/flox mice (Figure 2A). Likewise, expression of phospho-p53 (p-p53) was increased (Figure 2A), likely in response to DNA damage (Figure 1C). No mutation was found in p53 (data not shown). Surprisingly, expression of Cdkn1a/p21 (p21), the downstream mediator of p53, was reduced in the forestomach of Tgfbr2fspKO mice at 3 weeks of age but with more profound reduction at 4 weeks (Figure 2A and 2B). Suspecting epigenetic regulation of p21 expression in tumor cells, we performed pyrosequencing and found an increased methylation of CpG in the p21 promoter in forestomach tumor samples of Tgfbr2fspKO (77%) compared to that of Tgfbr2flox/flox mice (29%) (Figure 2C, left panel). We then treated forestomach tumor epithelial cells (1096, isolated from SCC of FSP-Cre/RII knock out mouse, and kept in low passages) with the DNA methyltransferase inhibitor 5-aza 2′ deoxycytidine (Decitabine) at 5 uM concentration for 48 hours. We observed increased expression of p21 at both the mRNA and protein level, and decreased cell proliferation compared to untreated cells (Figure 2C, right panel, and Figure S3A). Our data suggest that methylation of the p21 promoter likely prevented p53-mediated p21 transcription, resulting in decreased expression of p21 in Tgfbr2fspKO mice. Together, these data support a loss of cell cycle mediators in epithelial cells due to a loss of Tgfbr2 in stromal compartment in Tgfbr2fspKO mice.

We next evaluated cell proliferation in the forestomach of Tgfbr2fspKO and control mice as downregulation of p15, p16 and p21 may lead to increased proliferation. Forestomach tissue from 3, 4, and 5-week old mice, was collected and stained with FSP1 (stroma) and cytokeratin-14 (K14) (epithelium). Using immunofluorescence, we observed hyperplasia at 4 weeks and dysplasia/carcinoma in situ at 5 weeks in the epithelial compartment (Figure 2D), and increased FSP1+ cells in Tgfbr2fspKO mice (Figure 2E). The latter was likely due to deletion of Tgfbr2 and loss of growth inhibition. Co-staining of Ki-67, a marker of cellular proliferation, with K14 or FSP1 showed the extent of proliferation was more pronounced in the epithelial compartment compared to the stromal compartment (Figure 2F). Interestingly, the epithelial compartment had very few apoptotic cells compared to the stromal compartment (Figure S3B). Together, these data suggest that absence of TGF-β signaling in stromal fibroblasts likely induced uncontrolled proliferation and decreased apoptosis in the epithelial compartment.
Inflammatory Mediators and the Tumor Microenvironment of SCC in Tgfbr2fspKO Mice
Inflammation has been shown to play an important role in carcinogenesis. To characterize the role of inflammation in Tgfbr2fspKO mice, we collected forestomach tissue from 4 week old Tgfbr2fspKO and Tgfbr2flox/flox control mice for Western analysis. We found an increased expression of inducible nitrogen oxide synthatase (NOS2), cyclooxygenase 2 (COX2), and nuclear factor (NF) κB subunit (p65) in forestomach epithelail layer samples of 4 week old Tgfbr2fspKO mice compared to control mice (Figure 3A, left and right panels). The increased expression of NOS2, COX2, and p65 was found in both the stromal and epithelial compartments (Figure 3B). IFN-γ and TNF-α were significantly increased in the tumor tissues (Figure 3C), suggesting a type I inflammatory response associated with the development of SCC frequently observed in gastrointestinal cancers [21].

Inflammation promotes migration and infiltration of leukocytes, certain types of which are known to have significant impact on tumor microenvironment and tumor progression. We further characterized the subsets of infiltrating immune cells described earlier (see figure 1). We found a significant increase in CD45+ cells in the forestomach of 5-week-old Tgfbr2fspKO compared to control Tgfbr2flox/flox mice (29.36% vs 8.34%) (Figure 3D). The increased immune cells included Gr-1+ CD11b+ cells (20.52% vs 9.59%), also known as myeloid derived suppressor cells (MDSCs). These MDSCs are mostly monocytic subset Ly6C+CD11b+ (13.81% vs 1.10%) but not Ly6G+CD11b+ cells (Figure 3E). MDSCs have significant impact on tumor microenvironment [8], [21], [22] and are well known for their role in cancer associated immune suppression [23]. In addition, there was a significant presence of TH17 cells that stained for CD4+IL17A+ by immunofluorescence staining in 5-week-old Tgfbr2fspKO compared to control Tgfbr2flox/flox mice (Figure 3F). These findings reveal considerable alteration in the cellular and molecular properties of the tumor microenvironment in Tgfbr2fspKO compared to Tgfbr2flox/flox mice, suggesting that multiple inflammatory mediators interacting with an altered microenvironment are implicated in the progression of SCC following deletion of TGF-β signaling in stromal fibroblasts.
Anti-Inflammation Treatment Significantly Delayed SCC Development and Prolonged Survival of Tgfbr2fspKO Mice
To investigate the role of inflammation in SCC development, Tgfbr2fspKO mice were treated with the COX2 inhibitor (Celecoxib). Celecoxib treatment significantly improved body size and weight, decreased tumor burden, and increased lifespan of Tgfbr2fspKO mice from 28 to 49 days (Figure 4A, left and right panels and Figure S4A and S4B). The median survival of Celecoxib treated Tgfbr2fspKO mice was 53 days compared to 38 days in untreated mice (Log rank p<0.001) (Figure 4A, left and right panels and Figure S4A and S4B). This was accompanied by reduced infiltration of CD45+ leukocytes and decreased hyperplasia (Figure 4B). Surprisingly, treatment with L-NAME, an inhibitor of NOS2, did not significantly affect body weight and tumor burden or lifespan despite decreased serum levels of nitric oxide to normal baseline (Figure S4A and S4B, Figure S5A and S5B).

Alterations in microbial communities, particularly in gastrointestinal (GI) tract, are associated with inflammation and cancer development [24]. To investigate the role of microbiome in the progression of SCC in the Tgfbr2fspKO model, we re-derived the mouse line using super-ovulation and artificial insemination to obtain pups free of Helicobacter (Table S1). The uninfected Tgfbr2fspKO mice displayed significantly improved body size and weight with a median survival time of 47 days compared to 38 days in mice housed under standard conditions (Log rank p<0.001) (Figure 4A, left and right panels and Figure 4B). The tumors in the uninfected mice were characterized by decreased CD45 infiltration, decreased production of COX2 and P-p65 (Figure 4B and Figure S5C); delayed hyperplasia and dysplasia compared to the control mice (Figure 4B). These data suggest an involvement of microflora in the inflammation and SCC development in Tgfbr2fspKO mice. Indeed, alterations in the microflora are associated with inflammation and intestinal metaplasia of the distal esophagus [25].
In addition to a decrease in p53 expression (Figure 4C), Celecoxib treatment decreased COX-2, NOS2 and p65 expression to levels similar to control samples (Figure 4C and Figure S5C). L-NAME treatment decreased the expression of COX-2, NOS2 and p65. However, it did not affect p53 and γ-H2AX production (Figure 4C). Celecoxib treatment and Helicobacter free environment significantly reduced IFN-γ and TNF-α levels (Figure 4D), and decreased 8-oxo-dG production in the forestomach of Tgfbr2fspKO mice (Figure 4E). These data suggest that COX-2 and Helicobacter infection are important mediators in inflammation and SCC progression.
Celecoxib Treatment Significantly Restored the Expression p21 in Tgfbr2fspKO Mice
We showed earlier that the downregulation of p21 was likely mediated by methylation of p21 promoter. We next investigated whether anti-inflammatory treatment would decrease this methylation and increase p21 expression. Celecoxib treated Tgfbr2fspKO mice were evaluated for p21 promoter methylation by pyrosequencing of the epithelial layer from the forestomach tissue. Interestingly, methylation was significantly decreased (p<0.05) at CpG 16 (Figure 5A). Consistent with this finding, the p21 mRNA expression was observed in the laser captured epithelia from Celecoxib treated Tgfbr2fspKO mice (Figure 5B). The p21 mRNA in the stroma was not different from that of the Tgfbr2flox/flox control mice, and was not changed by Celecoxib treatment (Figure 5B). p21 protein expression was also increased upon Celecoxib treatment (Figure 5C). In contrast, p15 expression was not observed (Figure 5D). Together with genomic PCR showing a loss of p15 (Figure 1E), our data indicate that p15 might be genetically deleted. Surprisingly, Celecoxib treatment also resulted in increased expression of p16 protein (data not shown), suggesting a possible methylation of p16 promoter as a result of inflammation, similar to that of p21. Together, our data support that inflammation induced DNA damage, genetic and epigenetic alterations of cell cycle mediators play a critical role in SCC progression in Tgfbr2fspKO mice.

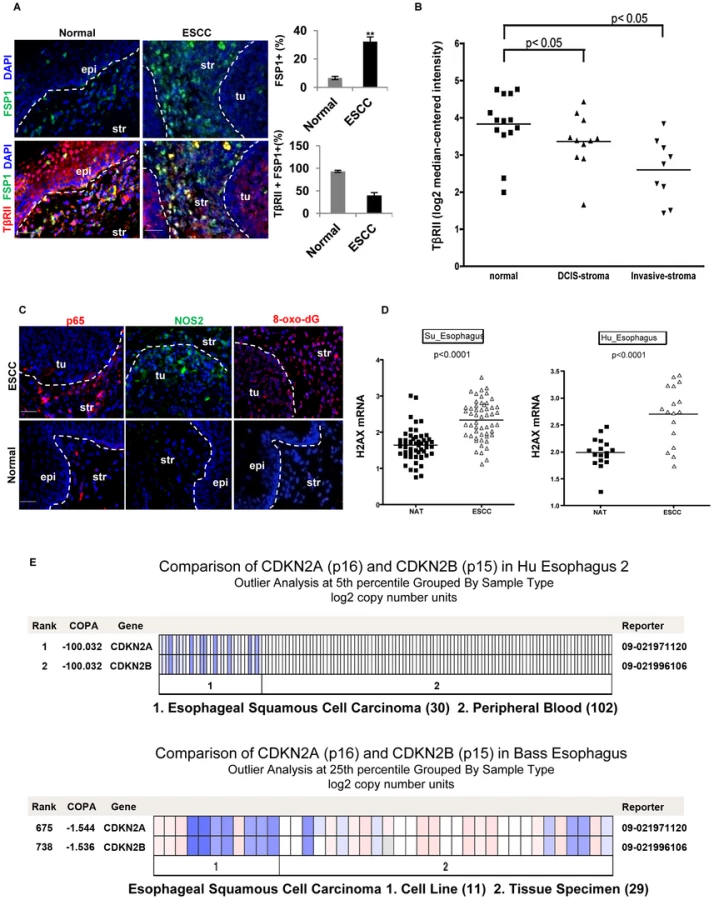
Human Esophageal Squamous Cell Carcinoma (ESCC) Exhibited a Decreased Expression of TβRII in FSP1+ Stromal Cells, Increased Inflammation, and Elevated Production of 8-oxo-dG
The SCC in the forestomach of the Tgfbr2fspKO mice shows similarity to that of human ESCC by way of similar histology and functional behavior. Additionally, downregulation of TGF-β receptors has previously been reported at the invasive front and stroma in human ESCC [26] and prostate cancer [13], [27]. Due to these histological and molecular similarities, we measured the TβRII expression level in FSP1+ stromal cells of eight human ESCC specimens. Adjacent normal tissues from these patients served as a control (Figure S6A). We observed an increased number of FSP1+ cells in the stromal compartment of the ESCC tumors compared to the adjacent normal esophagus (32.2% vs 6.6%, p<0.001) (Figure 6A, upper panels). This data was consistent with the expansion of FSP1+ cells in Tgfbr2fspKO mice (Figure 2E). In these FSP1+ cells, TβRII expression was decreased in tumor esophagus compared to adjacent normal esophagus (39.8% vs 93.2%, p<0.001) (Figure 6A, lower panels). Down-regulation of TβRII was also observed in tumor-associated stroma compared to the adjacent normal in a dataset of breast carcinoma (Ma 4 Breast) (Figure 6B). The expression of p65 and NOS2 was elevated, 8-oxo-dG was increased in both stromal and epithelial compartments (Figure 6C), consistent with findings in the animal model. In order to investigate biomarkers of DNA damage and genetic aberrations in human ESCC, we interrogated the Oncomine database (www.oncomine.com). Expression of H2AX mRNA was significantly upregulated in ESCC (p<0.0001) (Figure 6D). Additionally, p15 and p16 were co-deleted in human ESCC (Figure 6E) and [28]–[30]. These data suggest an association of reduced expression of TβRII in stromal cells with increased inflammation, DNA damage, and genetic alterations in human ESCC, which is consistent with our observations in Tgfbr2fspKO mice.
Discussion
Significant cross-interactions between stroma and tumor cells have been reported in recent studies [1]–[4]. Alteration of tumor suppressor genes in stromal fibroblasts induces epithelial cancer development [4], [5], [7], suggesting an important role of stroma in epithelial homoeostasis. Deletion of TGF-β signaling in the stroma modulates oncogenic potential of neighboring epithelia and induces the development of SCC in the forestomach in a mouse model (Tgfbr2fspKO mice) 1. However, the underlying mechanisms are unclear. In this report, we demonstrate that deletion of Tgfbr2 in the stromal fibroblast resulted in severe inflammation and DNA damage, which induced lost or decreased expression of p15, p16, as well as p21 in the epithelial cell compartment and the subsequent SCC development in forestomach (Figure 7). Our studies provide mechanistic insight to how loss of tumor suppressor in the stromal cells impact epithelial tumorigenesis and progression.

FSP1-cre Mediated Specific Deletion of Tgfbr2
We used FSP1-cre transgenic mice to mediate Tgfbr2 deletion specifically in the stromal fibroblasts. However, no alteration in epithelial TβRII was noticed. We used several approaches including TβRII immunofluorescence, p-smad2 nuclear localization in stroma, and an FSP1-Cre/Rosa26 reporter mouse to verify specificity. FSP1/S100A4 was identified as a specific marker for fibroblasts [31]. The FSP1-cre mediated gene deletion has been widely used in a number of mouse models for fibroblast specific gene deletion [1], [4], [32]. However, several studies reported that a specific subset of inflammatory macrophages co-express FSP1 and F4/80+ under a number of pathological conditions [33], [34]. We did not observe an overlap between FSP1+ cells with F4/80 positive cells (Figure S6B). This is supported by an extensive study regarding comparison overlap between FSP1+ fibroblasts with the macrophage markers, in which they authors demonstrated that F4/80 antibodies can be used to distinguish macrophages from FSP1+ fibroblasts [35]. We thus believe that the FSP1-cre mediated deletion is highly specific in a subset of stromal fibroblasts, with a small possibility in other host cells, and certainly not in tumor cells.
Loss of TGF-β Signaling Induced Inflammation and DNA Damage
TGF-β signaling regulates inflammation, as deletion of Tgfbr2 increased the transcription factor NF-κB [36]. In TGF-β1 deficient mice, inflammation causes precancerous lesions to progress to colon cancer [37]. Inflammation is known to induce DNA damage, genetic alterations and histone posttranslational modifications in mice and human cancers [15], [16], [20], [38]. We found a significant production of 8-Oxo-dG and γ-H2AX, indicating severe DNA damage from 3 weeks of age in the Tgfbr2fspKO mice. This is likely due to production of reactive oxygen and nitrogen species such as nitrogen oxide (NO). DNA damage often results in chromosomal segregation errors and structural alterations including mutations, deletions, amplifications, and balanced/unbalanced chromosomal translocations [17], [20], [39]–[41]. Indeed, we found a loss of p15 and p16 located in mouse chromosome 4 band C4 locus, which is orthologous to chromosome band 9p21 in humans. The loss of these chromosome bands is frequently observed in murine and human cancers [20], [42], [43]. Inflammatory mediated genetic alterations were previously observed in the head and neck tumor mice model, in which SMAD4−/ − deletion in head and neck epithelia resulted in genetic aberrations and deletion of chromosome 4q in SMAD−/ − mice [20]. Conditional deletion of p120 in the esophagus, oral cavity, and forestomach increased the production of proinflammatory cytokine TNF-α [21]. TNF-α, and IFN-γ, are known to induce epithelial dysfunctions and SCC of intestine [44], [45]. Our data and these published reports may provide molecular insight for a previous study that showed inactivation of Smad4 and PTEN in K5+ epithelia induced forestomach SCC development and downregulation of CDK inhibitors [46].
Inflammation, Epigenetic Silencing of p21, and Lost Cell Cycle Control
Deletion of Tgfbr2 induced inflammation was also responsible for the decreased p21 expression in the Tgfbr2fspKO mice. Very interestingly, increased p53 expression in response to DNA damage did not result in elevated expression of p21. Our data demonstrated that the inflammation induced methylation of p21 promoter region. This methylation inhibited the expression of p21, the critical mediator of p53 function. The inhibition of p21 was reversed by treatment with the COX2 inhibitor Celecoxib and 5-aza 2′ deoxycytidine treatment. We believe that the loss of p15 and p16, combined with decreased expression of p21 is critical in dysregulation of cell cycle control. This explains a massive proliferation of epithelia in the forestomach of Tgfbr2fspKO mice. Our data add novel mechanistic insight to the SCC development in addition to the finding of HGF as critical mediator [1] (Figure S7). Our data are supported by studies in which decreased levels or total loss of TGF-β signaling via defects of TGF-β receptors or Smads resulted in inflammation and uncontrolled proliferation of epithelial cells while promoting tumor development [26], [36], [47].
Human ESCC
Our results in Tgfbr2fspKO mice are clearly corroborated by human ESCC. Our studies showed significantly decreased expression of TβRII in FSP1+ stromal cells in human ESCC (Figure 6A). Interestingly, no alteration of TβRII was found in tumor epithelial cells compared to that of adjacent normal tissue [48], [49]. Similar to our mouse model data, elevated expression of inflammatory mediators such as COX2 and CCL2 as well as production of DNA damaging mediators 8-Oxo-dG and γ-H2AX were associated with the development of human ESCC (Figure 6C and 6D) [28], [48], [50]. Methylation of p21 gene promoter was also observed in 56% ESCC [28]. In addition, genetic deletion, loss of heterozygosity, and promoter methylation of p15 and p16 genes was associated with the development of human ESCC [30], [51], [52]. Co-deletion of p15 and p16 has also been found in human ESCC (Figure 6E) and [29], [49], [51]. Notably, approximately, 60% of human SCC including skin, head and neck, esophagus, bronchi, and uterine cervix are associated with the alterations in TGF-β signaling pathway molecules [53]. Indeed, stromal cell signaling has an impact on epithelial carcinogenesis and prediction of clinical outcome of cancer [54]. Furthermore, down-regulation of TβRII in tumor-associated stroma is correlated with poor prognosis in the clinic [55]. The squamous mucosal lining of mouse forestomach is similar to that of human esophagus at both histopathological and molecular levels [56]. Therefore, targeting inflammation may be a strategy to counteract the stromal-epithelial cross-talk in ESCC development.
Methods
Mice
Cre-Tgfbr2flox/flox female and Cre+Tgfbr2flox/wt male mice were kindly provided by Dr. Harold Moses, Vanderbilt Cancer center, Nashville, TN. Mice were bred to yield Tgfbr2fspKO mice. Cre+Tgfbr2flox/wt male mice were crossed with Rosa26 reporter female mice to validate the Cre specificity. All mice were housed at the National Cancer Institute (NCI) animal facility Animal studies were performed under NCI - IACUC approved protocol.
Immunofluorescence and Immunohistochemistry
Forestomach samples were collected from Tgfbr2fspKO and Tgfbr2flox/flox mice, which then fixed, sectioned and stained using H&E. For immunofluorescence studies, the sections were incubated overnight at 4°C with primary antibodies directed against TβRII (1∶50, Santa Cruz), S100A4 (1∶100, Abcam), K14 (1∶100, Covance), F4/80 (1∶100, BD Transduction Laboratories), NOS2 (1∶100, BD Transduction Laboratories), Cox2 (1∶100, Cell Signaling Technology), p65 (1∶100, Cell Signaling Technology), γH2AX (1∶100, Trevigen), CD4 and IL17A (1∶100, Biolegend). Fluorescence-tagged secondary antibodies were used for visualization (anti-rabbit, 1∶1000, Invitrogen or; anti-mouse, Vector lab, respectively). Slides were examined using fluorescence microscopy (Olympus). For immunohistochemistry, slides were incubated with primary antibodies against CD45 (1∶100, BD Pharmigen), Psmad2 (1∶100, Cell Signaling Technology), and β-galactosidase (1∶100, Abcam). Signals were visualized using Vectastain (Vector Lab) and examined under a light microscope (Carl Zeiss). Quantitative data was measured by counting total number of cells expressing the marker out of all the cells in one field by Image J software. Three different fields were evaluated; percentage was calculated from total number of cells counted and averaged for three independent fields.
Apoptosis Assay
Apoptosis was evaluated by TUNEL (Terminal deoxynucleotidyl transferase dUTP nick end labeling) method using “In Situ cell death detection kit, Fluorescein” (Roche Applied Science, Indianapolis, IN, USA). Briefly, tissue sections were pretreated with xylene and ethanol (100%, 95%, 90%, 80%, 70%), washed with 1× PBS, then treated with proteinase K (20 ug/ml in 10 mM Tris-HCL (pH 7.4) and 50 ul tunel mixture for 60 min. Data was acquired using fluorescence microscopy (Olympus). Quantitative data was obtained using Image J software by counting the number of cells expressing the marker out of total cells for each field. Three different fields were evaluated; the data was presented as percentage of stained cells in total cells and averaged for three independent fields.
Flow Cytometry of Tumor Microenvironment
Single cell suspensions were made using fresh forestomach tissue from Tgfbr2fspKO and Tgfbr2flox/flox mice through incubation with Liberase TL (200 U/mL) (Roche Applied Science, Indianapolis, IN) at 37°C for 30 minutes. Forestomach tissue was then crushed and filtered through a 70 µm cell strainer. Cells were labeled with fluorescence-conjugated antibodies against CD45-PE, CD11b-FITC, Gr-1-APC, Ly6G-APC, Ly6C-PE, or 7-AAD (BD Pharmingen). Isotype-matched IgG was used as a control (BD Pharmingen). The flow data was acquired on BD FACS Calibur flow cytometer (BD Biosciences, San Jose, CA) and analyzed using FlowJO.
Western Blotting
The forestomachs were dissected and treated with 0.05% trypsin overnight at 4°C. The forestomach tumor tissues from Tgfbr2fspKO or equivalent normal tissues from Tgfbr2flox/flox mice were separated by peeling them from the stromal and muscle layers. Protein was extracted, and then separated by gel electrophoresis. Membranes were incubated with primary antibodies against NOS2 (1∶100, BD Transduction Laboratories), γH2AX (1∶1000, Trevigen), HGF (1∶1000, Santa Cruz Biotechnology), Cox2, P-p65, P-p53, Cyclin D1, p21, p15, and p16 (all at 1∶1000, Cell Signaling Technology) or β-actin (1∶5000, Sigma), and horseradish peroxidase-conjugated secondary antibody (1∶5000, Biorad). The blots were developed using a SuperSignal West Pico Chemiluminescent substrate kit (Pierce). Images were scanned in a G: Box (Syngene).
Pyrosequencing
DNA was isolated from epithelial layers of forestomach as described in “Western Blotting.” QIAGEN Genomic-tip 20/G (Qiagen, CA, USA) and modified with Epitech Bisulfite kit (Qiagen, CA, USA) were used in pyrosequencing. PCR templates for pyrosequencing analysis were amplified from 10 ng gDNA using Hotstart Taq Mastermix (Qiagen, CA, USA) and 5 pmol of each primer in a total reaction volume of 25 µl. In all, 1 µl of each PCR reaction was analysed on an Agilent 2100 Bioanalyzer (Santa Clara, CA) using a DNA 1000 kit. Pyrosequencing was carried out on 0.15–0.5 pmol of each PCR product using the PyroMark MD System (Qiagen, CA, USA) following the manufacturer's instructions with sequencing primers and assay parameters specific to each methylation site. Resulting pyrograms were analysed using the PyroMark MD 1.0 software in ‘AQ mode’. For each assay, duplicate pyrosequencing analysis was performed, and the average of these was taken to represent the identified percentage methylation of the methylated allele.
Laser Captured Micro-Dissection
Laser capture microdissection of Tgfbr2flox/flox, Tgfbr2fspKO and Celecoxib treated Tgfbr2fspKO mouse tissue was performed using an Arcturus XT (Life Technologies, CA, USA). Frozen tissue sections on PEN membrane frame slides (Applied Biosystems) were H&E stained followed by dehydration using the standard protocol to improve visualization of the cells at the microscope. The epithelia and stroma were identified by morphology, captured using a low-power infrared laser pulse, and transferred onto a cap (Capsure™ Macro LCM Caps, Life Technologies). The DNA was extracted using a QIAamp DNA micro kit (Qiagen, CA, USA).
5-aza 2′ Deoxycytidine Treatments
Primary epithelial cells (1096, isolated from SCC of FSP-Cre/RII knock out mouse, and kept in low passages) were cultured in 6 well plate with seeding density 0.3×106 per well for overnight in DME/F12 medium containing 10% FBS and 1× antibiotics (GIBCO, Life Technologies, CA, USA). DNA methyltransferase inhibitor, 5-aza 2′ deoxycytidine (Decitabine), was added to the culture at 5 uM concentration for 48 hours. Cells were harvested using 0.25% trypsin-EDTA (GIBCO, Life Technologies, CA, USA) and subjected to total RNA extraction by RNeasy mini kit (Qiagen, CA, USA) and protein extraction by standard method.
RNA Isolation and Quantitative RT–PCR (qPCR)
Total RNA was isolated from laser captured micro-dissected tissue samples by Arcturus Picopure RNA isolation Kit (Applied Biosystems). Reverse transcription was performed using oligo dT primers and Superscript II (Invitrogen). The primers for qPCR were mCDKN1A (p21) F: 5′-ACAGGAGCAAAGTGTGCCGTTGT-3′; mCDKN1A (p21) R: 5′ - GCTCAGACACCA GAGTGCAAGACA 3′; mGAPDH F: 5′-ATGACCACAGTCCATGCCATCACT-3′; mGAPDH R: 5′-TGTTGAAGTCGCAGGAGACAACCT-3′. PCR reactions were performed using fast real-time 7500 PCR system (Applied Biosystems). All samples were tested in triplicate. The comparative CT method was used for quantification of gene expression. Gapdh was used as an endogenous reference. Statistical analysis was performed using SDS v2.1 software (Applied Biosystems) according to the manufacturer's instructions.
Bio-Plex Protein Assays
Protein extraction was obtained from forestomach samples of Tgfbr2fspKO and Tgfbr2flox/flox mice, and analyzed for IFN-γ and TNF-α expression, as per manufacturer's instruction. Data was acquired and analyzed using Bio-Plex Manager version 4.0 software (Bio-Rad).
Array-Based Comparative Genomic Hybridization (CGH)
Genomic DNA was isolated from primary tumor cells (1096) from Tgfbr2fspKO mice and primary normal epithelial cells isolated from forestomach epithelial cell layer with a QIAamp DNA Mini Kit according to manufacturer protocol (Qiagen, Valencia, CA). Array-CGH was performed using test DNA from laser captured epithelia and stroma, 1096 primary tumor cell culture, and reference DNA. DNA was labeled with Cy3 or Cy5 fluorescent dyes (Pharmacia, Piscataway, NJ) according to the BioPrime array CGH genomic labeling protocol (Invitrogen, Carlsbad, CA) and cleaned using Microcon YM-30 filters (Millipore, Billerica, MA). Hybridization was carried out using Mouse Genome CGH Microarray 4×44 K from Agilent Technologies (Santa Clara, CA) according to CGH Procedures for Genomic DNA Analysis (Agilent Technologies). Slides were hybridized for 20 hours, washed, and scanned with an Agilent microarray scanner. Data was analyzed using Feature Extraction® and CGH Analytics® software packages (Agilent Technologies). The array-based CGH data is available, GEO accession number: GSE42773.
Genomic PCR
Genomic DNA from laser captured epithelia and stroma described above was used for genomic PCR using the 2× Taq master mix (Gene Script, NJ, USA), 50 ng genomic DNA and Exon 1 specific primers of mouse p15 and p16 genes. The primer sequences were p15: forward - 5′ - GTT GGG CGG CAG CAG TGA C-3′ and reverse - 5′-CCT CCC GAA GCG GTT CAG-3′, p16: forward - 5′-ACT GGT CAC ACG ACT GGG CGA TTG -3′ and reverse - 5′-AAT CGG GGT ACG ACC GAA AGA G-3′. Actin: forward-5′-TCA TCA GGT AGT CAG TGA GGT CGC-3′ and reverse-5′-CAC CAC ACC TTC TAC AAT GAG CTG-3′. The PCR conditions included initial denaturation at 95°C for 5 min, denaturation at 95°C for 1 min, annealing at 60°C for 1 min and extension at 72°C for 1 min for 40 cycles and final extension at 72°C for 7 min. Agarose gel electrophoresis (2%) was used to detect the PCR products and data was recorded using G: Box (syngene).
Mouse Line Re-Derivation
C57BL/6NCr wild type female mice were super-ovulated and crossed with the Fsp-Cre male mice. Four or eight cell embryos were transferred to pathogen free females. After birth, pups were examined for infection and genotypes using ELISA and PCR. Male mice with Cre+ Tgfbr2flox/wt and female mice with Cre - Tgfbr2flox/wt were identified and crossed to obtain pathogen free Tgfbr2fspKO mice. The mice re-derivation was performed in the mice re-derivation core facility located at NCI-Frederick, MD. Mice were transferred and housed in Helicobacter free facility at NIH Bethesda, MD.
Anti-Inflammation Treatment
Tgfbr2flox/flox and Tgfbr2fspKO mice were treated with diet pellets containing Celecoxib at 1500 ppm with (D04090202) or without (AIN 76A D1000) active compound, or Nω-Nitro-L-arginine methyl ester hydrochloride (L-NAME, Sigma) at a dose of 50 mg/kg/day. The pups received the Celecoxib starting the second week after birth, together with the nursing mother mouse in the same cage, due to early inflammation onset (Figure 1B). The treatment continued after weaning. The synergistic effect of L-NAME and Celecoxib was also examined. In addition, Mice housed in the Helicobacter free conditions were treated with Celecoxib to evaluate the cooperative effect in survival and phenotype. For gel diet treatment, after weaning on day 21, Tgfbr2fspKO and Tgfbr2flox/flox pups were fed with gel diet (AIN76A, Clear H2O). All mice were monitored daily and sacrificed with signs of poor health including small size, hunched body, slow movements, and weakness in comparison to healthy littermates.
Analysis of Human Esophageal Squamous Cell Carcinoma (ESCC) and Breast Carcinoma
Human ESCC (n = 8) and adjacent normal FFPE tissue slides (n = 8) were previously described [57] (PMID: 19789312), and were stained to detect TβRII expression in FSP1+ stromal cells. p65, NOS2, and 8-Oxo-dG adducts were also examined for inflammation and DNA damage. The immunofluorescence staining procedures are as described above. Oncomine database (www.oncomine.com) was utilized to evaluate γ-H2AX expression, p15, and p16 loss in human ESCC, specifically mRNA expression datasets, Hu Esophagus (34 samples) and Su Esophagus 2 (106 samples), and DNA copy number datasets, Hu Esophagus 2 (blood vs LCM tumor sample) and Bass Esophagus (cell lines and tissue specimens). Dot plots of H2AX mRNA expression are presented as log2 median-centered intensity. The dataset (PMID: 19789312) from Oncomine was analyzed using GraphPad Prism 5.0 and two-tailed, paired t-test. Heat map of copy number loss of p15 and p16 in ESCC was obtained from the same dataset that measure DNA copy number on a SNP microarray platform. The expression of TβRII in tumor-associated stroma vs adjacent normal was also analyzed using Oncomine mRNA dataset Ma 4 Breast Carcinoma.
Statistical Analysis
Data was analyzed using the Student t-test, and was expressed as mean ± SE. Differences were considered statistically significant at p<0.05. Mouse survival data was examined using SPSS 16 software, and is presented as Kaplan Meier curve. A log rank test was used to calculate statistical differences in survival and median survival of the different groups.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. BhowmickNA, ChytilA, PliethD, GorskaAE, DumontN, et al. (2004) TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science 303 : 848–851.
2. WallaceJA, LiF, LeoneG, OstrowskiMC (2011) Pten in the breast tumor microenvironment: modeling tumor-stroma coevolution. Cancer Res 71 : 1203–1207.
3. HillR, SongY, CardiffRD, Van DykeT (2005) Selective evolution of stromal mesenchyme with p53 loss in response to epithelial tumorigenesis. Cell 123 : 1001–1011.
4. TrimboliAJ, Cantemir-StoneCZ, LiF, WallaceJA, MerchantA, et al. (2009) Pten in stromal fibroblasts suppresses mammary epithelial tumours. Nature 461 : 1084–1091.
5. TanwarPS, ZhangL, RobertsDJ, TeixeiraJM (2011) Stromal deletion of the APC tumor suppressor in mice triggers development of endometrial cancer. Cancer Res 71 : 1584–1596.
6. DemehriS, TurkozA, KopanR (2009) Epidermal Notch1 loss promotes skin tumorigenesis by impacting the stromal microenvironment. Cancer Cell 16 : 55–66.
7. PatocsA, ZhangL, XuY, WeberF, CaldesT, et al. (2007) Breast-cancer stromal cells with TP53 mutations and nodal metastases. N Engl J Med 357 : 2543–2551.
8. BierieB, StoverDG, AbelTW, ChytilA, GorskaAE, et al. (2008) Transforming growth factor-beta regulates mammary carcinoma cell survival and interaction with the adjacent microenvironment. Cancer Res 68 : 1809–1819.
9. YangL (2010) TGFbeta and cancer metastasis: an inflammation link. Cancer Metastasis Rev 29 : 263–271.
10. BierieB, ChungCH, ParkerJS, StoverDG, ChengN, et al. (2009) Abrogation of TGF-beta signaling enhances chemokine production and correlates with prognosis in human breast cancer. J Clin Invest 119 : 1571–1582.
11. IjichiH, ChytilA, GorskaAE, AakreME, BierieB, et al. (2011) Inhibiting Cxcr2 disrupts tumor-stromal interactions and improves survival in a mouse model of pancreatic ductal adenocarcinoma. J Clin Invest 121 : 4106–4117.
12. KimBG, LiC, QiaoW, MamuraM, KasprzakB, et al. (2006) Smad4 signalling in T cells is required for suppression of gastrointestinal cancer. Nature 441 : 1015–1019.
13. FrancoOE, JiangM, StrandDW, PeacockJ, FernandezS, et al. (2011) Altered TGF-beta signaling in a subpopulation of human stromal cells promotes prostatic carcinogenesis. Cancer Res 71 : 1272–1281.
14. ChengN, ChytilA, ShyrY, JolyA, MosesHL (2008) Transforming growth factor-beta signaling-deficient fibroblasts enhance hepatocyte growth factor signaling in mammary carcinoma cells to promote scattering and invasion. Mol Cancer Res 6 : 1521–1533.
15. BlancoD, VicentS, FragaMF, Fernandez-GarciaI, FreireJ, et al. (2007) Molecular analysis of a multistep lung cancer model induced by chronic inflammation reveals epigenetic regulation of p16 and activation of the DNA damage response pathway. Neoplasia 9 : 840–852.
16. Mendez-AcunaL, Di TomasoMV, PalittiF, Martinez-LopezW (2010) Histone post-translational modifications in DNA damage response. Cytogenet Genome Res 128 : 28–36.
17. CrastaK, GanemNJ, DagherR, LantermannAB, IvanovaEV, et al. (2012) DNA breaks and chromosome pulverization from errors in mitosis. Nature 482 : 53–58.
18. LukasJ, LukasC, BartekJ (2011) More than just a focus: The chromatin response to DNA damage and its role in genome integrity maintenance. Nat Cell Biol 13 : 1161–1169.
19. KrimpenfortP, IjpenbergA, SongJY, van der ValkM, NawijnM, et al. (2007) p15Ink4b is a critical tumour suppressor in the absence of p16Ink4a. Nature 448 : 943–946.
20. BornsteinS, WhiteR, MalkoskiS, OkaM, HanG, et al. (2009) Smad4 loss in mice causes spontaneous head and neck cancer with increased genomic instability and inflammation. J Clin Invest 119 : 3408–3419.
21. StairsDB, BayneLJ, RhoadesB, VegaME, WaldronTJ, et al. (2011) Deletion of p120-catenin results in a tumor microenvironment with inflammation and cancer that establishes it as a tumor suppressor gene. Cancer Cell 19 : 470–483.
22. YangL, DebuskLM, FukudaK, FingletonB, Green-JarvisB, et al. (2004) Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell 6 : 409–421.
23. GabrilovichDI, NagarajS (2009) Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 9 : 162–174.
24. PlottelCS, BlaserMJ (2011) Microbiome and malignancy. Cell Host Microbe 10 : 324–335.
25. YangL, FrancoisF, PeiZ (2012) Molecular pathways: pathogenesis and clinical implications of microbiome alteration in esophagitis and Barrett esophagus. Clin Cancer Res 18 : 2138–2144.
26. FukaiY, FukuchiM, MasudaN, OsawaH, KatoH, et al. (2003) Reduced expression of transforming growth factor-beta receptors is an unfavorable prognostic factor in human esophageal squamous cell carcinoma. Int J Cancer 104 : 161–166.
27. LiX, PlacencioV, IturreguiJM, UwamariyaC, Sharif-AfsharAR, et al. (2008) Prostate tumor progression is mediated by a paracrine TGF-beta/Wnt3a signaling axis. Oncogene 27 : 7118–7130.
28. LingY, HuangG, FanL, WeiL, ZhuJ, et al. (2011) CpG island methylator phenotype of cell-cycle regulators associated with TNM stage and poor prognosis in patients with oesophageal squamous cell carcinoma. J Clin Pathol 64 : 246–251.
29. BassAJ, WatanabeH, MermelCH, YuS, PernerS, et al. (2009) SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat Genet 41 : 1238–1242.
30. XingEP, NieY, SongY, YangGY, CaiYC, et al. (1999) Mechanisms of inactivation of p14ARF, p15INK4b, and p16INK4a genes in human esophageal squamous cell carcinoma. Clin Cancer Res 5 : 2704–2713.
31. StrutzF, OkadaH, LoCW, DanoffT, CaroneRL, et al. (1995) Identification and characterization of a fibroblast marker: FSP1. J Cell Biol 130 : 393–405.
32. ScholtenD, OsterreicherCH, ScholtenA, IwaisakoK, GuG, et al. (2010) Genetic labeling does not detect epithelial-to-mesenchymal transition of cholangiocytes in liver fibrosis in mice. Gastroenterology 139 : 987–998.
33. OsterreicherCH, Penz-OsterreicherM, GrivennikovSI, GumaM, KoltsovaEK, et al. (2011) Fibroblast-specific protein 1 identifies an inflammatory subpopulation of macrophages in the liver. Proc Natl Acad Sci U S A 108 : 308–313.
34. BoomershineCS, ChamberlainA, KendallP, Afshar-SharifAR, HuangH, et al. (2009) Autoimmune pancreatitis results from loss of TGFbeta signalling in S100A4-positive dendritic cells. Gut 58 : 1267–1274.
35. InoueT, PliethD, VenkovCD, XuC, NeilsonEG (2005) Antibodies against macrophages that overlap in specificity with fibroblasts. Kidney Int 67 : 2488–2493.
36. MonteleoneG, MannJ, MonteleoneI, VavassoriP, BremnerR, et al. (2004) A failure of transforming growth factor-beta1 negative regulation maintains sustained NF-kappaB activation in gut inflammation. J Biol Chem 279 : 3925–3932.
37. EngleSJ, OrmsbyI, PawlowskiS, BoivinGP, CroftJ, et al. (2002) Elimination of colon cancer in germ-free transforming growth factor beta 1-deficient mice. Cancer Res 62 : 6362–6366.
38. PangB, ZhouX, YuH, DongM, TaghizadehK, et al. (2007) Lipid peroxidation dominates the chemistry of DNA adduct formation in a mouse model of inflammation. Carcinogenesis 28 : 1807–1813.
39. Padilla-NashHM, HathcockK, McNeilNE, MackD, HoeppnerD, et al. (2012) Spontaneous transformation of murine epithelial cells requires the early acquisition of specific chromosomal aneuploidies and genomic imbalances. Genes Chromosomes Cancer 51 : 353–374.
40. MeiraLB, BugniJM, GreenSL, LeeCW, PangB, et al. (2008) DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J Clin Invest 118 : 2516–2525.
41. JanssenA, van der BurgM, SzuhaiK, KopsGJ, MedemaRH (2011) Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science 333 : 1895–1898.
42. HerzogCR, WisemanRW, YouM (1994) Deletion mapping of a putative tumor suppressor gene on chromosome 4 in mouse lung tumors. Cancer Res 54 : 4007–4010.
43. MontagnaC, AndrechekER, Padilla-NashH, MullerWJ, RiedT (2002) Centrosome abnormalities, recurring deletions of chromosome 4, and genomic amplification of HER2/neu define mouse mammary gland adenocarcinomas induced by mutant HER2/neu. Oncogene 21 : 890–898.
44. LiuY, HanZP, ZhangSS, JingYY, BuXX, et al. (2011) Effects of inflammatory factors on mesenchymal stem cells and their role in the promotion of tumor angiogenesis in colon cancer. J Biol Chem 286 : 25007–25015.
45. Resta-LenertS, BarrettKE (2006) Probiotics and commensals reverse TNF-alpha - and IFN-gamma-induced dysfunction in human intestinal epithelial cells. Gastroenterology 130 : 731–746.
46. TengY, SunAN, PanXC, YangG, YangLL, et al. (2006) Synergistic function of Smad4 and PTEN in suppressing forestomach squamous cell carcinoma in the mouse. Cancer Res 66 : 6972–6981.
47. AchyutBR, YangL (2011) Transforming growth factor-beta in the gastrointestinal and hepatic tumor microenvironment. Gastroenterology 141 : 1167–1178.
48. SuH, HuN, YangHH, WangC, TakikitaM, et al. (2011) Global gene expression profiling and validation in esophageal squamous cell carcinoma and its association with clinical phenotypes. Clin Cancer Res 17 : 2955–2966.
49. HuN, CliffordRJ, YangHH, WangC, GoldsteinAM, et al. (2010) Genome wide analysis of DNA copy number neutral loss of heterozygosity (CNNLOH) and its relation to gene expression in esophageal squamous cell carcinoma. BMC Genomics 11 : 576.
50. HashimotoN, InayamaM, FujishimaM, ShiozakiH (2007) Clinicopathologic significance of expression of cyclooxygenase-2 in human esophageal squamous cell carcinoma. Hepatogastroenterology 54 : 758–760.
51. XingEP, NieY, WangLD, YangGY, YangCS (1999) Aberrant methylation of p16INK4a and deletion of p15INK4b are frequent events in human esophageal cancer in Linxian, China. Carcinogenesis 20 : 77–84.
52. HuN, WangC, SuH, LiWJ, Emmert-BuckMR, et al. (2004) High frequency of CDKN2A alterations in esophageal squamous cell carcinoma from a high-risk Chinese population. Genes Chromosomes Cancer 39 : 205–216.
53. GlickAaVWC (2011) Signaling Pathways in Squamous Cancer Springer.
54. FinakG, BertosN, PepinF, SadekovaS, SouleimanovaM, et al. (2008) Stromal gene expression predicts clinical outcome in breast cancer. Nat Med 14 : 518–527.
55. BacmanD, MerkelS, CronerR, PapadopoulosT, BruecklW, et al. (2007) TGF-beta receptor 2 downregulation in tumour-associated stroma worsens prognosis and high-grade tumours show more tumour-associated macrophages and lower TGF-beta1 expression in colon carcinoma: a retrospective study. BMC Cancer 7 : 156.
56. GartnerK, PfaffJ (1979) The forestomach in rats and mice, a food store without bacterial protein digestion. Zentralbl Veterinarmed A 26 : 530–541.
57. MatheEA, NguyenGH, BowmanED, ZhaoY, BudhuA, et al. (2009) MicroRNA expression in squamous cell carcinoma and adenocarcinoma of the esophagus: associations with survival. Clin Cancer Res 15 : 6192–6200.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 2
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Complex Inheritance of Melanoma and Pigmentation of Coat and Skin in Grey Horses
- Coordination of Chromatid Separation and Spindle Elongation by Antagonistic Activities of Mitotic and S-Phase CDKs
- Autophagy Induction Is a Tor- and Tp53-Independent Cell Survival Response in a Zebrafish Model of Disrupted Ribosome Biogenesis
- Assembly of the Auditory Circuitry by a Genetic Network in the Mouse Brainstem
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy