
A Kinome-Wide RNAi Screen in Glia Reveals That the RIO Kinases Mediate Cell Proliferation and Survival through TORC2-Akt Signaling in Glioblastoma
Glioblastoma, the most common primary malignant brain tumor, is incurable with current therapies. Genetic and molecular analyses demonstrate that glioblastomas frequently display mutations that activate receptor tyrosine kinase (RTK) and Pi-3 kinase (PI3K) signaling pathways. In Drosophila melanogaster, activation of RTK and PI3K pathways in glial progenitor cells creates malignant neoplastic glial tumors that display many features of human glioblastoma. In both human and Drosophila, activation of the RTK and PI3K pathways stimulates Akt signaling along with other as-yet-unknown changes that drive oncogenesis. We used this Drosophila glioblastoma model to perform a kinome-wide genetic screen for new genes required for RTK - and PI3K-dependent neoplastic transformation. Human orthologs of novel kinases uncovered by these screens were functionally assessed in mammalian glioblastoma models and human tumors. Our results revealed that the atypical kinases RIOK1 and RIOK2 are overexpressed in glioblastoma cells in an Akt-dependent manner. Moreover, we found that overexpressed RIOK2 formed a complex with RIOK1, mTor, and mTor-complex-2 components, and that overexpressed RIOK2 upregulated Akt signaling and promoted tumorigenesis in murine astrocytes. Conversely, reduced expression of RIOK1 or RIOK2 disrupted Akt signaling and caused cell cycle exit, apoptosis, and chemosensitivity in glioblastoma cells by inducing p53 activity through the RpL11-dependent ribosomal stress checkpoint. These results imply that, in glioblastoma cells, constitutive Akt signaling drives RIO kinase overexpression, which creates a feedforward loop that promotes and maintains oncogenic Akt activity through stimulation of mTor signaling. Further study of the RIO kinases as well as other kinases identified in our Drosophila screen may reveal new insights into defects underlying glioblastoma and related cancers and may reveal new therapeutic opportunities for these cancers.
Published in the journal:
. PLoS Genet 9(2): e32767. doi:10.1371/journal.pgen.1003253
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003253
Summary
Glioblastoma, the most common primary malignant brain tumor, is incurable with current therapies. Genetic and molecular analyses demonstrate that glioblastomas frequently display mutations that activate receptor tyrosine kinase (RTK) and Pi-3 kinase (PI3K) signaling pathways. In Drosophila melanogaster, activation of RTK and PI3K pathways in glial progenitor cells creates malignant neoplastic glial tumors that display many features of human glioblastoma. In both human and Drosophila, activation of the RTK and PI3K pathways stimulates Akt signaling along with other as-yet-unknown changes that drive oncogenesis. We used this Drosophila glioblastoma model to perform a kinome-wide genetic screen for new genes required for RTK - and PI3K-dependent neoplastic transformation. Human orthologs of novel kinases uncovered by these screens were functionally assessed in mammalian glioblastoma models and human tumors. Our results revealed that the atypical kinases RIOK1 and RIOK2 are overexpressed in glioblastoma cells in an Akt-dependent manner. Moreover, we found that overexpressed RIOK2 formed a complex with RIOK1, mTor, and mTor-complex-2 components, and that overexpressed RIOK2 upregulated Akt signaling and promoted tumorigenesis in murine astrocytes. Conversely, reduced expression of RIOK1 or RIOK2 disrupted Akt signaling and caused cell cycle exit, apoptosis, and chemosensitivity in glioblastoma cells by inducing p53 activity through the RpL11-dependent ribosomal stress checkpoint. These results imply that, in glioblastoma cells, constitutive Akt signaling drives RIO kinase overexpression, which creates a feedforward loop that promotes and maintains oncogenic Akt activity through stimulation of mTor signaling. Further study of the RIO kinases as well as other kinases identified in our Drosophila screen may reveal new insights into defects underlying glioblastoma and related cancers and may reveal new therapeutic opportunities for these cancers.
Introduction
Glioblastoma (GBM), the most common primary malignant brain tumor, infiltrates the brain, grows rapidly, and is refractory to current therapies. Signature genetic lesions in GBM include amplification, mutation, and/or overexpression of receptor tyrosine kinases (RTKs), such as EGFR and PDGFRα, as well as activating mutations in components of the PI-3 kinase (PI3K) pathway (reviewed in [1]). More than 40% of GBMs show EGFR gene amplification, and these amplification events are often accompanied by mutations in EGFR [1]. The most prevalent mutant form of EGFR is ΔEGFR (EGFRvIII, de2-7EGFR, EGFR*), an intragenic truncation mutant that displays constitutive kinase activity [2]. ΔEGFR and other constitutively active mutant forms of EGFR found in GBMs potently drive tumor cell survival, migration, and proliferation [2], [3]. The most frequent mutation in the PI3K pathway in GBM is loss of the PTEN lipid phosphatase, which results in unopposed signaling through PI3K and robust stimulation of Akt, especially in the context of EGFR activation [1]. In mouse models, co-activation of these pathways in glia, glial progenitor cells, and/or neuro-glial stem cells induces GBM [4], [5], [6], [7]. However, the full range of signaling events acting downstream of or in combination with EGFR and PI3K to drive oncogenesis remain to be determined. While several normal effectors of RTK and PI3K signaling, such as Ras, Akt, and mTor, are used by EGFR and PI3K in GBM and are required for gliomagenesis [1], constitutive activation of RTK and PI3K pathways may evoke changes distinct from those induced by normal developmental signaling. Notably, treatments with pharmacologic inhibitors of EGFR or mTor are cytostatic at best in a subset of patients, indicating that other, unidentified factors or compensatory signals affect the survival and growth of tumor cells [8].
To uncover new factors required for EGFR - and PI3K - mediated gliomagenesis, we developed a GBM model in Drosophila melanogaster [9]. Drosophila offers several advantages for modeling cancers like GBM. Flies have orthologs for 75% of human disease genes [10], including nearly all known gliomagenic genes; signaling pathways are highly conserved; versatile genetic tools are available for cell-type specific gene manipulation [11], ; and Drosophila neural cell types are homologous to their mammalian counterparts [13], [14]. While a Drosophila model cannot address all aspects of human GBM, our model recapitulates important pathologic features. Specifically, constitutive activation of EGFR-Ras and PI3K signaling in Drosophila glial progenitor cells gives rise to proliferative, invasive neoplastic glia that create transplantable malignant tumors [9]. These tumors are induced through activation of a synergistic genetic network composed of downstream pathways commonly mutated and/or activated in human GBMs, such as Akt and mTor signaling [9]. However, activating these known downstream pathways alone or in combination is not sufficient to induce glial neoplasia in Drosophila, indicating that additional, as yet unidentified, genetic pathways are involved in transformation. Thus, we undertook genetic screens using our Drosophila GBM model to discover new genes underlying EGFR and PI3K mediated neoplastic transformation, and tested whether human orthologs of the genes identified in Drosophila represent new human genes involved in GBM.
Our analyses in both Drosophila and human systems uncovered that the RIOK1 and RIOK2 kinases drive the survival and proliferation of GBM cells. RIOK1 and RIOK2 are members of the RIO (right open reading frame) family of atypical protein kinases, named for yeast (S. cerevisiae) Rio1p and Rio2p, respectively [15]. The RIOK1 and RIOK2 proteins are highly conserved, and are present in all phylogenetic kingdoms, from yeast to mammals among eukaryotes. Kinases in this family are characterized by the presence of the RIO kinase domain, a kinase fold structurally homologous to eukaryotic serine-threonine protein kinase domains, but that lacks classic activation and substrate binding loops (reviewed in [15]). While these kinases undergo autophosphorylation and phosphorylate nonspecific substrates in vitro, the actual in vivo substrates of RIO kinases are unknown [15]. In both yeast and human cells, RIOK1 and RIOK2, which are not functionally redundant, are required for processing of the 18S rRNA and cytoplasmic maturation of the 40S ribosomal subunit, although neither kinase is an integral component of the ribosome [16], [17], [18], [19]. Recent studies demonstrate that, in yeast, Rio2p also transiently associates with immature ribosomes to block translation initiation, although how RIOK2 is regulated in this context is unclear [20]. To date, several studies have provided suggestive evidence that the RIO kinases could be involved in RTK and PI3K signaling: RIOK2 becomes rapidly phosphorylated in response to EGFR stimulation; Rio2p binds to Tor2p, an ortholog of the mTor kinase, and RIOK1 is required for the proliferation and survival of Ras-dependent cancer cells [21], [22], [23]. However, to date, no specific function has been ascribed to RIOK1 or RIOK2 in the context of EGFR or PI3K signaling.
In this manuscript we demonstrate that RIOK1 and RIOK2 become overexpressed in GBM tumor cells relative to normal brain cells; that RIOK1 and RIOK2 overexpression occurs in response to constitutive Akt signaling; that RIOK2 forms a complex with RIOK1, mTor, and other signaling components to drive activation of Akt signaling and tumorigenesis; and that, in GBM cells, RIOK1 or RIOK2 loss causes a reduction in Akt signaling and provokes p53-dependent apoptosis, cell cycle exit, and chemosensitivity through the RpL11-dependent ribosomal stress checkpoint. Our data demonstrate that the RIO kinases play a key role in Akt-mediated transformation of GBM cells.
Results
A kinome-wide screen for modifiers of glial neoplasia
To discover new genes involved in glial pathogenesis, we performed a genetic screen using our Drosophila GBM model. Co-overexpression of constitutively active forms of Drosophila EGFR (dEGFRλ) and the PI3K catalytic subunit p110α (dp110CAAX) stimulates malignant transformation of post-embryonic larval glia, inducing lethal glial neoplasia (Figure 1A–1C) (characterized in detail in [9]). Using this larval Drosophila GBM model, we performed an RNAi-based modifier screen for genes that suppress (inhibit) or enhance (worsen) neoplastic phenotypes caused by constitutive EGFR and PI3K signaling. In this scheme, which is an enhancer-suppressor screen, modifier kinases that block/inhibit fly glial neoplasia when their expression is reduced are referred to as ‘suppressors,’ and modifier kinases that exacerbate neoplasia when their expression is reduced are referred to as ‘enhancers.’ This is in keeping with standard Drosophila nomenclature, in which genes are classified by their loss-of-function phenotypes. As a side-note, in this context, the term suppressor does not refer to mammalian tumor suppressors.

To enrich for new pathway components, we screened nearly all of the kinases encoded in the Drosophila genome (Table S1). Our choice to screen kinases was based on four considerations: (1) kinases regulate a broad array of biological and cellular processes, including those underlying oncogenesis; (2) kinases are highly conserved between Drosophila and humans such that every Drosophila kinase has a clear human ortholog; (3) as a group, they are well characterized, facilitating functional analysis; and (4) drug discovery efforts are focused on development of specific kinase inhibitors.
We tested 553 conditional RNAi constructs targeting 223 of the 243 kinases in the fly genome (Tables S1, S2) [24], [25]. RNAi constructs were expressed specifically within the glial lineage and were tested for their phenotypic effects on proliferation, migration, morphology, and/or viability of dEGFRλ;dp110CAAX neoplastic glia. The specificity of modifier loci was confirmed by testing multiple RNAi constructs, dominant negative constructs, and/or available mutant alleles to determine if they produced analogous phenotypes in the dEGFRλ;dp110CAAX model (Tables S2, S3). Control assays were performed for the effects of modifier RNAi constructs when expressed specifically in other cell types, including normal glia, neuroblasts (Drosophila neural stem cells), and neurons in order to distinguish those RNAi constructs that caused non-specific toxicity from RNAi constructs that caused specific changes in neoplastic glia (Table S4, Text S1). Constructs that caused early organismal lethality in all cellular contexts tested were excluded from analysis (Tables S2, S4).
To test whether modifiers act in oncogenic signaling downstream of specific EGFR mutations found in human GBM, we created flies that overexpress human ΔEGFR. Glial-specific expression of ΔEGFR caused lethal glial neoplasia phenotypes, alone and in combination with dp110CAAX, that were similar to dEGFRλ, and these phenotypes required EGFR kinase activity and core PI3K effectors, such as dAkt (Figure 1A–1F, Figure S1, Table S5). We tested modifier RNAi constructs for the ability to alter glial-specific ΔEGFR and ΔEGFR;dp110CAAX phenotypes (Table S5); the results mirrored their genetic interactions with dEGFRλ;dp110CAAX (Table S2, S5), indicating that modifiers identified in the screen are common to neoplastic phenotypes conferred by both Drosophila and human EGFR.
We identified a total of 45 modifier genes (Figure 1I, Tables S2 and S3). Suppressor RNAi constructs targeting 39 genes reduced neoplasia and induced smaller brain size and lower glial cell numbers relative to dEGFRλ;dp110CAAX controls, whereas enhancer RNAi constructs targeting 6 genes worsened tumorigenesis and neoplasia and induced increased glial cell numbers, and/or aberrant glial morphologies (Figure 1A–1D, 1I, Tables S2 and S3). Modifiers that suppressed glial neoplasia include genes identified in previous studies, including dAkt [9] (Tables S2, S5). A small subset of suppressor constructs caused strong phenotypes in the context of constitutive EGFR-PI3K signaling. Constructs targeting three modifier loci, Raf, Src42A, and Taf1, rescued dEGFRλ;dp110CAAX animals to adult viability, allowing neoplastic glia to differentiate and function normally despite the presence of dEGFRλ and dp110CAAX (Table S2). Constructs targeting two of the strongest modifiers, CG11660 and CG11859 (the Drosophila orthologs of the RIOK1 and RIOK2 kinases, respectively), caused severe reduction in brain size and glial cell number when combined with dEGFRλ;dp110CAAX, ΔEGFR;dp110CAAX, and/or ΔEGFR, as compared to wild-type control animals (Figure 1A–1H, Figure S1). dRIOK2 knockdown gave a stronger effect (Figure 1D, 1G). In contrast, RNAi constructs targeting dRIOK1 and dRIOK2 did not produce a dramatic growth reduction when targeted to normal glia (Figure 1E, Table S4), indicating that dRIOK1 or dRIOK2 knockdown does not simply cause nonspecific cellular toxicity.
Modifier kinases were classified by bioinformatic annotations using Flybase, Gene Ontology, KEGG pathways, the STRING database [26], and comparisons with other Drosophila RNAi screens. These classifications (Figure 1J, Figure S2, Table S6) show that kinases with core functions in the RTK and PI3K pathways were highly represented, validating our model and screening methodology [27], [28], [29]. Notably, very few of our modifiers have emerged from Drosophila RNAi screens for cell viability (Table S7) [30], indicating that most of the modifiers are not generically required for cell survival. The largest group of modifiers have roles in cell proliferation (Figure 1J, Table S6), and many of these yielded reduced cell lineages upon knockdown in neuroblasts (Table S4) [31], consistent with known requirements for RTK and PI3K signaling in neural progenitor cells [9], [32]. Several of the modifiers involved in cell proliferation, such as warts (Table S2), are also components of the hippo pathway, a pathway with a documented role in glial cell proliferation [33]. Broad comparisons with orthologs from species such as yeast (S. cerevisiae), revealed modifiers kinases implicated in protein translation, such as dRIOK1 and dRIOK2 [16], [20], or in cell shape change and migration. Finally, comparison to human kinases shows that the majority of our modifier kinases have orthologs previously implicated in GBM (Figure 1J, Figure S2, Table S7), leaving a set of 16 novel modifiers, including dRIOK1 and dRIOK2.
Overexpression of RIOK kinases in human GBM correlates with Akt activity
Novel modifier kinases identified in our Drosophila screens may represent human kinases directly involved in GBM pathogenesis. Kinases that block fly glial neoplasia when their expression is reduced are of interest because their human orthologs may be promising new targets for therapeutic inhibition. There are 27 human orthologs for the 16 novel Drosophila modifier kinases (Table S8). To determine if any of these human kinases are expressed or mutated in GBM, we analyzed tumor genomic databases, proteomic atlases, and GBM cell lines, which provided suggestive evidence that 12 modifier orthologs are subject to genetic alteration and/or elevated gene or protein expression in GBMs (see Text S1, Tables S9 and S10, Figures S3 and S4). Among these, RIOK1 and RIOK2 showed increased protein expression consistent with involvement in GBM. Given that loss of the dRIOKs strongly and specifically blocks growth and survival of EGFR and PI3K mutant glia, and that recent publications suggest that the RIO kinases may contribute to EGFR and/or mTor signaling [21], [22], the functional roles of RIOK1 and RIOK2 in GBM were of particular interest.
A range of GBM cells and cell lines were examined to determine how RIOK1 and RIOK2 expression correlated with tumor cell genotype and phenotype. Our analyses showed that RIOK1 and RIOK2 were expressed in PTEN-null U87MG GBM cells and were upregulated in U87MG cells engineered to express ΔEGFR at levels detected in tumors [34] (Figure 2A). RIOK1 and RIOK2 were also upregulated in GBM tumors with EGFR overexpression/mutation as well as activated Akt (Figure 2B), although these correlations were not clear in all specimens. Primary neurosphere cultures, which are composed of neural stem cell-like human GBM cells propagated in EGF-supplemented media [35], [36], [37], can maintain mutations/gene expression found in their parent tumors [37]. Neurosphere cultures showed strong RIOK1 and RIOK2 expression (Figure 2C); these included neurosphere lines with ΔEGFR, as well as neurosphere lines displaying PDGFRα overexpression, PTEN loss, and/or other mutant forms of EGFR (Figure 2C). In a panel of standard GBM cell lines, RIOK1 and RIOK2 showed strong expression in cell lines known to harbor PTEN and/or EGFR mutations, and RIOK1 and RIOK2 expression was comparatively lower in a GBM cell line with intact PTEN (Figure S5) [38], [39]. In contrast, RIOK1 and RIOK2 were nearly undetectable in mixed glial cultures freshly derived from adult human cortex (Figure 2D). Thus, RIOK1 and RIOK2 overexpression appeared to be correlated with RTK mutation/overexpression and/or PTEN loss in GBM tumor cells.

To determine if elevated RIOK1 and/or RIOK2 expression in GBM cells depends on EGFR and/or PI3K signaling, neurosphere lines and U87MG-ΔEGFR cells were treated with relevant inhibitors (Figure 2E and 2G, Figure S6). RIOK1 and RIOK2 levels decreased upon either growth factor withdrawal or gefitinib treatment of primary neurosphere cultures (Figure 2E), indicating that their up-regulation can be EGFR-dependent. Consistent with this, Pten−/−; Ink4a/arf−/− mouse astrocytes transformed by ΔEGFR showed increased RIOK1 and RIOK2 levels, which were reduced by gefitinib treatment (Figure 2F). RIOK1 and RIOK2 protein levels also decreased in neurosphere cells and U87MG-ΔEGFR cells treated with inhibitors of the p110 PI3K catalytic subunit, such as BEZ-235, and inhibitors of Akt, such as A443654 (Figure 2G) [40]. siRNA-mediated Akt knockdown or restoration of PTEN function in U87MG-ΔEGFR cells also reduced RIOK protein levels (Figure 2H, Figure S7). Treatments with p110 and Akt inhibitors also demonstrated that p110 and Akt signaling is required for RIOK1 and RIOK2 expression in PDGFRα-overexpressing neurospheres (Figure 2G). Taken together, these data indicate that RIOK1 and RIOK2 overexpression in GBM cells is driven by Akt activity downstream of RTK mutation/overexpression and/or PTEN loss.
However, the role of factors that act downstream of Akt was less clear (mTor inhibitors did not always reduce RIOK levels, see Figure S6), suggesting that RIOK1 or RIOK2 levels may be directly regulated by Akt. Given that mRNA expression levels of RIOK1 and RIOK2 did not show significant upregulation in tumor samples with PTEN and/or EGFR alterations (Table S9), and given that RIOK1 and/or RIOK2 levels decline after short-term treatments with Akt inhibitors (Figure S7), we hypothesized that Akt signaling may regulate RIOK2 and/or RIOK1 levels by modulating protein stability post-translationally. Consistent with this, addition of a proteosome inhibitor, MG132, prevented the reduction in RIOK1 and/or RIOK2 protein levels observed upon A443654 treatment or PTEN add-back (Figure S7). RIOK2 has several mapped serine phosphorylation sites (www.phosphosite.org), including putative Akt target sites (Figure S8). However, mutation of this single site did not abolish detection of RIOK2 by a phospho-Akt-substrate antibody (Figure S9). Thus, Akt-mediated regulation of RIO kinase levels does not hinge on phosphorylation at a single residue in RIOK2, and likely involves a more complex mechanism that requires more investigation.
To confirm that the RIOKs are expressed in GBM, we performed immunohistochemistry (IHC) for RIOK2 on a group of typed tumor specimens (RIOK1 antibodies were unsuitable for IHC). Xenograft specimens of GBM39, which is ΔEGFR-positive [41], showed RIOK2 expression in tumor cells, with cells displaying diffuse cytoplasmic and sub-surface RIOK2 localization (Figure 3A). In contrast, murine stromal cells had little or no RIOK2 immunoreactivity, although the antibody can detect mouse RIOK2. ΔEGFR-positive and EGFR-overexpressing specimens displayed strong, but sometimes heterogeneous, cytoplasmic RIOK2 immunoreactivity, ranging from the giant cell to the small cell populations (Figure 3B–3E, Figures S10 and S11), with the strongest expression in mitotic cells and densely cellular pseudopallisades (Figure 3C and 3D, Figure S10). Heterogeneity in RIOK2 expression may possibly reflect heterogeneity of RTK expression in tumors [42], [43], or may reflect upregulation of RIOK2 in actively cycling cells given the increased immunoreactivity observed in mitotic cells. Cytoplasmic localization of RIOK2 in GBM is consistent with observations of RIO kinase localization in yeast and human cells [16], [17], [18]. In contrast, RIOK2 did not show appreciable immunoreactivity in neural cells in matched normal control brain (n = 14) (Figure 3F, 3H), and did not show immunoreactivity in tumor stroma (Figure 3B), demonstrating that RIOK2 upregulation is tumor-specific. Akt signaling in tumors was assessed by staining for Akt phosphorylated at Serine-473 (Akt-S473-P, example shown in Figure 3I), and EGFR status of tumors was primarily assessed with staining for EGFR phosphorylated on Tyrosine-1068 (EGFR-Y1068-P, example shown in Figure 3J), which indicates EGFR activation. Statistical analysis demonstrated that RIOK2 expression was significantly correlated with EGFR status in tumor specimens (Figure 3L), although some EGFR-negative tumors also showed RIOK2 immunoreactivity (Figure 3G, 3L), while some EGFR-negative tumors did not (Figure 3K). The correlation of RIOK2 expression with EGFR activity is likely secondary to Akt-mediated regulation of RIOK2: RIOK2-expressing specimens positive EGFR-Y1068-P always showed staining for Akt-S473-P (n = 20). Indeed, all specimens that showed RIOK2 immunoreactivity, whether EGFR-positive or EGFR-negative, showed staining for Akt-S473-P (Figure 3L).

RIOK2 overexpression in astrocytes induces invasive glial tumors and TORC2-Akt activation
To determine if elevated levels of RIOK2 drives oncogenic changes in mammalian cells, we tested the effects of RIOK2 overexpression in astrocytes. Pten−/−; Ink4a/arf−/− murine astrocytes, which are immortalized by tumor suppressor mutations common in GBM, express little endogenous RIOK2 and are not gliomagenic in intracranial grafts assays [44]. In two experiments, mice intracranially grafted with Pten−/−; Ink4a/arf−/− astrocytes overexpressing RIOK2 showed symptoms of hydrocephaly and neurological deficits 3 weeks following implantation, unlike mice grafted with Pten−/−; Ink4a/arf−/− control astrocytes. Histological analysis showed that control Pten−/−; Ink4a/arf−/− astrocytes yielded no intracranial tumors in 12 total animals tested, whereas RIOK2overexpresion; Pten−/−; Ink4a/arf−/− astrocytes formed invasive high-grade glial tumors composed of invasive spindle-shaped cells in 7 out of 10 total animals tested (p<.001 by chi-squared test) (Figure 4A, 4B).
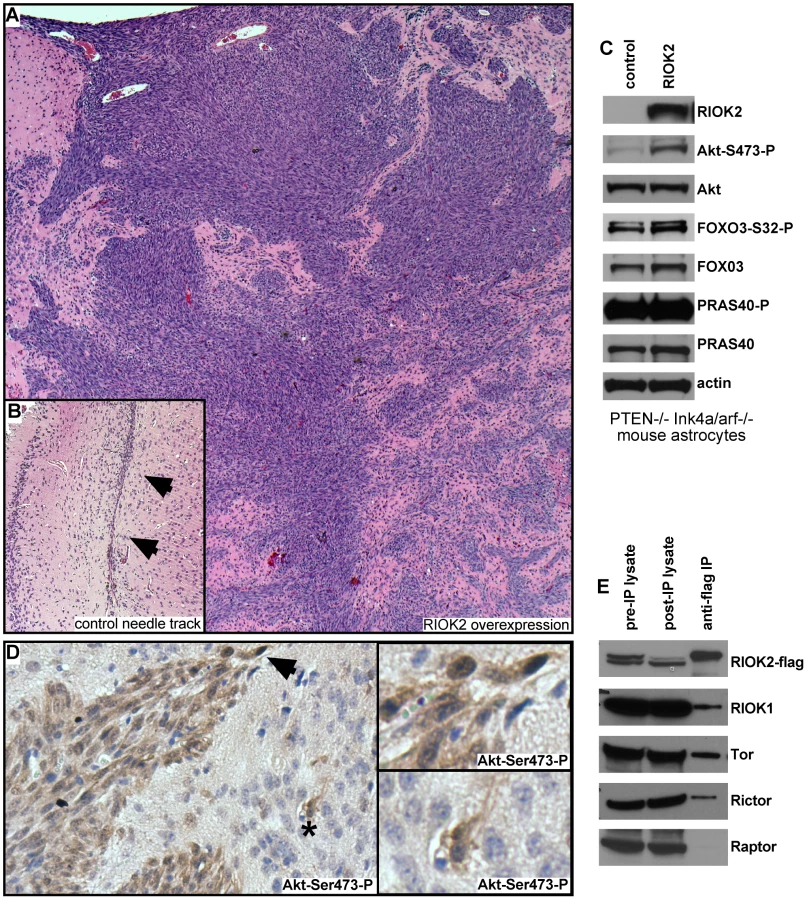
In GBM, ΔEGFR drives strong Akt activation in the context of PTEN loss, and can drive gliomagenic transformation of Pten−/−; Ink4a/arf−/− astrocytes [6], [45]. Thus, we wondered whether RIOK2 overexpression also drives astrocyte transformation by activating Akt. Consistent with this, RIOK2overexpression; Pten−/−; Ink4a/arf−/− astrocytes displayed increased phosphorylation of Akt at Serine-473 (Figure 4C), and tumor tissue from RIOK2overexpression; Pten−/−; Ink4a/arf−/− cells showed specific staining for Akt-Ser473-P (Figure 4D). Phosphorylation of Serine-473 is required for Akt activity towards select substrates such as FOXO3 [46], a direct Akt substrate that governs GBM cell tumorigenicity [47]. FOXO3 also displayed increased phosphorylation RIOK2overexpression; Pten−/−; Ink4a/arf−/− astrocytes (Figure 4C). However, we did not observe increased phosphorylation of all Akt targets, including PRAS40, with RIOK2 overexpression, suggesting that the effect of RIOK2 on phosphorylation of Akt targets was selective to Serine-473 dependent substrates [46].
Akt is phosphorylated on Serine-473 by mTor-complex-2 (TORC2), a multi-protein complex composed of the mTor kinase and several other signaling components, including Rictor [46], a protein that becomes elevated in glioblastomas that also drives gliomagenesis when overexpressed in astrocytic cells [48], [49]. In yeast proteomic analyses, Rio2p has been shown to bind to Rio1p and Tor2, the yeast mTor ortholog that forms the equivalent of TORC2 [22]. From human cells overexpressing RIOK2, RIOK2 co-immunoprecipitated with RIOK1, mTor, and Rictor, a protein which is definitive of the TORC2 complex (Figure 4E) [46]. mTor also associates with another complex, mTor-complex-1 (TORC1), which phosphorylates other mTor substrates, such as EIF-4E, and is composed of signaling components including the Raptor protein [46]. Raptor did not co-immunoprecipitate in the RIOK2-RIOK1-mTor-Rictor complex (Figure 4E), suggesting that RIOK2 specifically associates with TORC2. Taken together, these data suggest that RIOK2 directly binds to TORC2 to stimulate phosphorylation of Akt at Serine-473 and activation of Akt towards select substrates, such as FOXO3, and that this process may directly involve RIOK1 recruitment.
Requirement for RIOK kinases for proliferation and survival in GBM cells
Given that knockdown of their Drosophila cognates yields growth reduction of neoplastic glia, we tested RNAi constructs targeting human orthologs of novel suppressor kinases for their requirement in GBM cell survival and proliferation (Text S1, Figure S12). Among these, RIOK1 or RIOK2 knockdown yielded strong effects, inhibiting U87MG-ΔEGFR and U87MG proliferation (Figure 5A, 5B). ΔEGFR-positive neurosphere cultures, such as GBM301 and GBM39, also showed a pronounced apoptotic response to RIOK2 or RIOK1 knockdown, with RIOK2 loss yielding stronger effects (Figure 5C, 5D). In U87MG cells, which are dependent on Akt signaling for growth [50], RIOK1 or RIOK2 RNAi provoked G2 cell cycle arrest and reduced proliferation (Figure 5A–5B). The phenotypes caused by RIOK1 and RIOK2 knockdown were observed in other GBM cell lines that are PTEN and/or EGFR mutant, such as A172 (Figure 6, Figure S14, data not shown). Of note, RIOK2 knockdown typically triggered a reduction in RIOK1 expression, regardless of the RIOK2 RNAi constructs used, suggesting that RIOK2 regulates RIOK1 protein levels (Figure 5D, Figure S13, see also Figure 6).
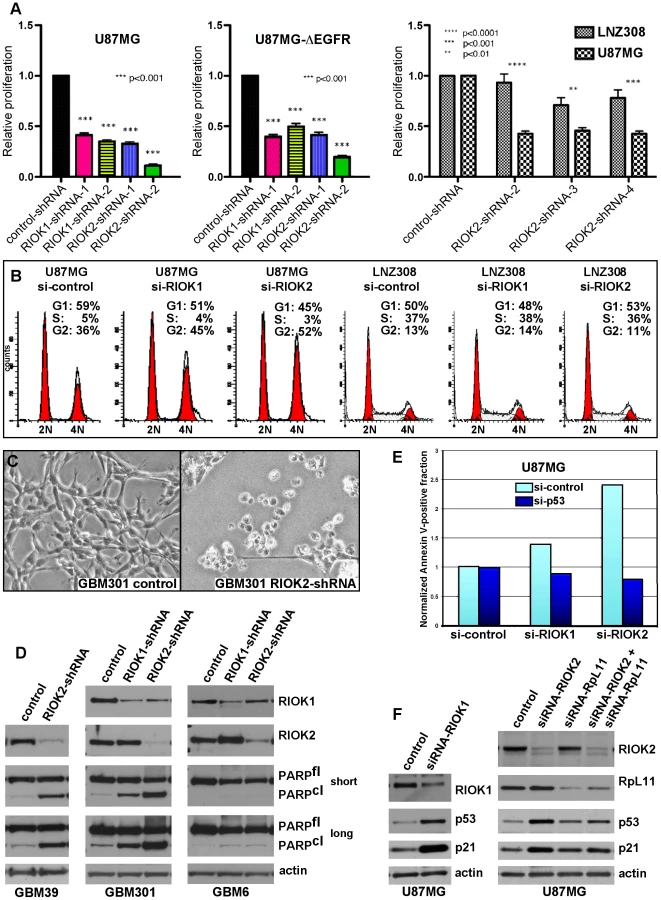

Given that the RIO kinases have been found to stimulate ribosome maturation, we initially suspected that functional reduction of RIOK1 or RIOK2 may cause generic cellular toxicity. However, in testing multiple GBM cell lines that showed strong RIOK expression, we found that a subset of GBM cells were far less affected by knockdown of RIOK1 and RIOK2. GBM6, a ΔEGFR-positive neurosphere line, did not undergo apoptosis upon RIOK1 or RIOK2 knockdown (Figure 5D). Moreover, LNZ308 cells, which are PTEN mutant [38], did not show cell cycle defects or strong reduction of RIOK1 expression with RIOK2 knockdown (Figure 5B). Both GBM6 and LNZ308 are also mutant or null for p53, whereas GBM cells that show cell cycle defects and apoptosis upon RIOK loss, such as U87MG, are wild-type for p53 [38], [41]. We observed a similar lack of apoptosis upon RIOK1/RIOK2 loss in other p53 mutant/null GBM cells, such as U373 (data not shown). This implies that the survival and proliferation defects induced by RIOK1 and RIOK2 loss rely on p53. Consistent with this, concomitant knockdown of p53 with RIOK1 or RIOK2 in U87MG cells blocks the apoptosis observed upon RIOK2 or RIOK1 knockdown alone (Figure 5E).
In human cells, RIOK1 and RIOK2 transiently associate with immature cytoplasmic 40S ribosomal subunits to promote their maturation and stimulate rRNA processing, like their yeast counterparts [18], [19], [22], [51]. Defects in ribosome biogenesis and rRNA processing can activate a p53-dependent ribosomal-stress checkpoint to suppress growth and induce cell cycle arrest and apoptosis, a process that relies on p53 upregulation and transcriptional activation mediated by release of the RpL11 ribosomal protein (reviewed in [52], [53]). In U87MG cells and other GBM cell lines, RIOK1 or RIOK2 knockdown induced up-regulation of p53 and the p21 cdk inhibitor, a p53 transcriptional target (Figure 5F, Figure S13, S14), and coincident knockdown of RpL11 and RIOK1 or RIOK2 blocked induction of p53 and p21 (Figure 5F and Figure S14). Therefore, RIOK1 or RIOK2 loss leads to p53 activation, which requires the p53-RpL11-dependent ribosomal stress checkpoint.
Loss of RIOK function chemosensitizes GBM cells in a p53-dependent manner
p53 is often downregulated in GBM tumors and tumor cells with activated Akt, PTEN loss, and/or EGFR mutation/overexpression [54]. Yet, the majority of tumors with PTEN loss or EGFR mutation/amplification have intact p53 loci (EGFR: 85–92%, PTEN: 92–95%) [55], [56]. Given that RIOK loss upregulates p53 levels, we tested whether knockdown of RIO kinases could potentiate the response of GBM cells to treatments with DNA-damaging agents such as doxorubicin, which cooperates with p53 to provoke apoptosis, [57], [58] and temozolomide, which is a DNA alkylator used to treat GBM. In GBM cells wild-type for p53, such as GBM301 and U87MG, knockdown of RIOK1 or RIOK2 potentiated apoptotic responses to doxorubicin and/or temozolomide (Figure 6A–6B, Figure S15). In contrast, cells mutant for p53, such as LNZ308 cells, did not show apoptosis upon RIOK1 or RIOK2 knockdown and doxorubicin-temozolomide treatments (Figure 6A). Therefore, inhibition of the RIO kinases chemosensitizes EGFR - and/or PTEN mutant GBM cells.
Our results suggest that elevated p53 activity can potentiate elimination of EGFR and/or PTEN mutant GBM cells. One way to increase p53 levels and activity is with nutlin-3, a small molecule which is know to cause cell cycle arrest and sensitivity to DNA-damaging agents in U87MG cells [59]. However, nutlin-3 did not provoke the same cell cycle defects observed with RIO kinase knockdown, despite inducing high levels of p53 and p21 (Figure S16). Thus, other changes induced by RIO kinase loss must contribute to cell cycle arrest and apoptosis.
Loss of RIOK function antagonizes Akt signaling
We tested for signaling alterations that occur upon RIOK1 or RIOK2 knockdown that would explain reduced proliferation and survival of GBM cells. The caspase inhibitor ZVAD was used to dampen apoptosis and thus preserve signaling defects. Compared to controls, RIOK1 or RIOK2 knockdown led to reduced phosphorylation of Akt at Serine-473 and reduced phosphorylation of Akt target proteins such as FOXO3 (Figure 6C, Figure S17). This occurred in both p53 wild-type and p53 mutant GBM cells (Figure 6C, Figure S17). Serine-473 is phosphorylated by Tor complex 2, (TORC2) [46], and in yeast and human cancer cells, TORC2 phosphorylation of Akt is stimulated by mature ribosomes, which can bind to both TORC2 and Akt to mediate their interaction, and TORC2 activity is blocked by defects in ribosome biogenesis [60]. Given that RIOK1 and RIOK2 loss causes defects in 40S ribosome maturation [17], [18], [19], and that we discovered that RIOK1 and RIOK2 bind to TORC2 components, we hypothesized that RIOK1 and RIOK2 knockdown interferes with TORC2 activity. Consistent with this, other readouts of TORC2 activity, such as phosphorylation and levels of NDRG1 [60], were reduced (Figure 6C, Figure S17), demonstrating that TORC2 activity is downregulated by RIOK1 and RIOK2 loss. This is consistent with recent findings demonstrating a requirement for TORC2 signaling in Drosophila glial neoplasia as well as human GBM cells [9], [48], [49]. However, the effects of RIOK loss on Akt signaling were not limited to the TORC2-dependent substrates. Phosphorylation of other Akt substrates, such as PRAS40 and TSC2, can also be reduced upon RIO kinase knockdown (Figure 6C). Thus, RIOK1 and RIOK2 are necessary for Akt signaling in GBM cells. Over-all, our results strongly suggest that functional reduction of RIOK1 and RIOK2 results in loss of Akt activity and p53 activation to drive cell cycle arrest, chemosensitivity, and apoptosis in Akt-dependent GBM cells with intact p53 (Figure 7).

Discussion
From a Drosophila genetic screen, we identified genes encoding 16 novel kinases that affect EGFR - and PI3K - dependent neoplastic glial transformation. We found that a subset of human orthologs for these novel kinases, including RIOK1 and RIOK2, are subject to alterations in GBM. RIOK1 and RIOK2, two related and highly conserved atypical kinases, become upregulated in an Akt-dependent manner in GBM cells. Our results show that RIOK2 forms a complex with RIOK1 and TORC2 signaling components, drives activation of TORC2-dependent Akt signaling, and stimulates glial tumorigenesis. Furthermore, we found that, in GBM cells, RIOK1 or RIOK2 loss causes a reduction in Akt signaling towards TORC2-depdendent targets and provokes p53-dependent apoptosis, cell cycle exit, and chemosensitivity. Thus, our loss-of-function and gain-of-function data imply that RIOK2 creates a feedforward loop that promotes and maintains Akt activity, and disruption of this loop is sufficient to trigger chemosensitivity and apoptosis in Akt-dependent GBM cells with intact p53 (Figure 7). Our results may have broad relevance to other cancers since RIOK2 is strongly expressed in a range of other more common tumor types associated with high Akt activity, such as breast and prostate cancers (Figure S18). Further study of the RIO kinases as well as other kinases identified in our Drosophila screen may reveal new insights into the signaling defects underlying GBM and related cancers.
RIOK1 and RIOK2 upregulation was associated with Akt activity in both GBM tumor specimens and cultured cells, and our results show that Akt signaling regulates RIO kinase protein stability, although the exact mechanism by which Akt regulates RIO kinase levels remains undetermined. RIOK2 has several putative and mapped phosphorylation sites, including at least one putative Akt phosphorylation site (www.phosphosite.org, Figure S8). Other studies show that RIOK2 phosphorylation can be stimulated by EGFR, and can be carried out by Polo-like kinase 1 [21], [61], and perhaps these events contribute to Akt-mediated regulation of RIO kinase levels. Of note, though standard GBM cells lacking PTEN showed high levels of RIO kinase expression, non-transformed astrocytes lacking PTEN did not show high levels of endogenous RIO kinase protein expression relative to astrocytes with intact PTEN. Therefore, other factors present in GBM cells must also contribute to elevated RIO kinase levels.
To date, published studies show that the RIO kinases act as ribosome assembly factors that transiently associate with the 40S subunit to promote ribosome maturation and translation initiation [17], [18], [20]. Given that mature ribosomes are required for TORC2 activation and Akt phosphorylation at Serine-473 [60], disruption of Akt signaling upon RIOK knockdown may be a result of defective ribosome biogenesis caused by RIO kinase loss. However, the RIO kinases may have a much more direct role in promoting and maintaining Akt activity given that RIOK2 binds to RIOK1 and to components of the TORC2 complex, which is consistent with recent studies in yeast showing that Rio2p can bind to Tor2 [22]. Given that Rio2p is released from mature ribosomes in a regulated process [20], it is possible that the reason mature ribosomes promote TORC2 signaling is because they release free cytoplasmic RIOK2 that then stimulates TORC2 assembly or activity. The specific interplay between the RIO kinases and mTor signaling, ribosome biogenesis, protein translation, and Akt signaling will require additional investigation in the context of both normal and abnormal PI3K and RTK signaling, and may involve other as yet undetermined factors.
Although RIOK1 and RIOK2 loss can cause defects in ribosome maturation [17], [18], in GBM cells the effects of RIO loss are not generic and instead are genotype-specific: p53 null mutant GBM cells showed no major cell cycle defects or apoptosis upon loss of these kinases. This specificity is derived from p53 upregulation and activation induced by the RpL11 ribosomal protein in response to RIOK loss. In humans, activation of the RpL11-p53-dependent ribosomal-stress checkpoint is associated with diseases caused by ribosomal protein haploinsufficiency, such as Diamond-Blackfan anemia, which are characterized by stem and progenitor cell failure [52], [53]. Similarly, in Drosophila, haploinsufficiency of genes that encode ribosomal proteins retards developmental cell proliferation [62]. Given that cancer cells share many properties with stem and progenitor cells, induction of the RpL11-p53 ribosomal stress checkpoint may prove useful to deplete cancer cells. Indeed, recent experimental evidence indicates that the RpL11-p53-dependent ribosomal stress checkpoint suppresses tumorigenesis in mouse cancer models [63]. Moreover, several chemotherapeutic drugs induce the ribosomal stress checkpoint [64], [65]. However, many of these drugs have deleterious effects unrelated to ribosomal stress, limiting their use. More specific induction of the ribosomal stress checkpoint, perhaps through RIO kinase inhibition, may prove therapeutically useful for GBM as well as related cancers.
The importance of RIO kinases in cancer cell survival has been validated in independent studies. RIOK2 was recently identified in an RNAi-based screen for kinases that are required for survival of glioma stem-like cells, which confirms our results, although the functionality of RIOK2 in glioma was not explored [66]. In addition, RIOK1 was identified in a cell-based RNAi screen for genes required for Ras-mediated cell survival, although the functionality of RIOK1 was not explored in this study [23]. Of note, almost all other published cell culture-based RNAi screens in GBM cell lines did not pick up RIOK1 or RIOK2 because these screens were not designed to distinguish between kinases that were required for genotypic-specific survival or growth of GBM cell lines, and instead focused on kinases that showed a common requirement in all glioblastoma cell lines tested, be they mutant or wild-type for p53, EGFR, or PTEN [67], [68], [69]. Our results, which are derived from independent multidisciplinary assays, are the first to establish functional connections between the RIO kinases, oncogenesis, Akt signaling, and the RpL11-p53-dependent ribosomal stress checkpoint (Figure 7). We envision that RIOK loss-of-function phenotypes in GBM cells are due to the combined effects of Akt inhibition and p53 induction, which together stimulate apoptosis and cell cycle exit of EGFR - and PTEN - mutant GBM cells, which share a common dependence on Akt signaling (Figure 7). Further studies to address the mechanisms by which the RIO kinases modulate Akt and p53 activity may lead to important new insights into the interactions between both of these pathways in both normal and cancer cells.
Materials and Methods
Fly stocks, genetics, culture conditions, immunohistochemistry, and gefitinib treatment
Flies were cultured at 25°C unless otherwise noted. Genotypes were established by standard genetics. Larval brain phenotypes were assessed and imaged as previously described [9]. Stocks were obtained from VDRC, NIG, and Bloomington stock centers (Table S1). wor-Gal4 lines were from C. Doe. To create UAS-ΔEGFR constructs, a full-length human ΔEGFR cDNA was cloned into pUAS-T, and fly stocks with stable insertions were created.
The screen was based on crosses (see Text S1 for genetic methodology) that generated progeny containing a single RNAi construct exclusively expressed in GFP-labeled glia along with dEGFRλ and dp110CAAX. Transgenes were overexpressed using the glial-specific repo-Gal4 transcriptional driver. Screening was performed using fluorescence microscopy to visualize GFP-labeled glia in living larvae, and phenotypes were confirmed with confocal microscopy. Each positive-scoring RNAi construct was tested at least twice. Positive scoring RNAi constructs were also tested in wild-type glia, neuroblasts, and neurons (Text S1, Table S4)
Mammalian tissue culture techniques and RNAi
Established primary neurosphere cultures (gifts of H. Kornblum) were maintained as previously described in DMEM/F12 medium supplemented with bFGF and EGF [35], [41]. Neurosphere cultures of GBM39 and GBM6 were created from serial xenografts of human GBMs (gifts of C.D. James). Cultured normal human glia were derived from a fresh surgical specimen of normal human cortex (gift of J. Olson) procured under a protocol approved by the Emory University institutional review board. Cultured mouse PTEN−/−; Ink4a/arf−/− astrocytes (gift of R. Bachoo) were maintained in DMEM with 10% serum. The RIOK2 cDNA (Origene) was overexpressed in PTEN−/−; Ink4a/arf−/− astrocytes from the pBabe retroviral vector.
The following drugs were used: Nutlin-3 (Cayman), MG132, Akt inhibitor IV (Calbiochem), temozolomide (Tocris), doxorubicin, rapamycin, PP242 (Santa Cruz), PI-103, ZVAD (Enzo), A443654 (gift of Greg Riggins), gefitinib (LC Laboratories), BEZ-235 (Biovision), LY294002 (Cell Signaling Technology), MK-2206, and GDC-0941 (Selleck). Doses of LY294002, BEZ-235, PI-103, GDC-0941, and MK-2206 used on GBM cells were determined by using dose response assays to find the concentrations at which cells showed substantial reduction (approximately <20% of normal) in Akt-mediated phosphorylation of PRAS40 (as detected by immunoblot).
Lentiviral shRNA pLKO.1 plasmids were obtained from the Broad Institute of MIT. RIOK1 shRNAs: TRCN0000196278 and TRCN0000196981. RIOK2 shRNAs: TRCN0000197250, TRCN0000196672, and TRCN0000196684. pLKO.1-GFP and a nontargeting shRNA against lacZ (in pLKO.1) were used as controls. Lentivirus was produced and used as per standard protocols (Sigma). Knockdown was evident by western blot 96 hrs post-infection. For neurosphere cultures, lentivirus was prepared in DMEM/F12 without serum, and infections were done on cells were plated adherently [36].
For siRNAs, all constructs were transfected at 50–100 µM with RNAimax (Invitrogen). Unless otherwise noted, siRNA-treated cells were harvested at 72 hrs post-transfection. 2 sets of pooled siRNAs were tested each for RIOK1 and RIOK2 (Dharmacon), and two different nontargeting siRNAs against GFP or luciferase were used as controls (Dharmacon). For knockdown of p53, p53 siRNAs were used and compared to matched control nontargeting siRNAs (Cell Signaling Technologies). Target sequences are listed in Text S1. For dual-knockdown experiments, U87MG cells were preferred because, with the necessary higher doses of siRNAs, U87MG-ΔEGFR cells showed nonspecific alterations in ΔEGFR expression that affected RIOK levels.
Immunoblot analysis
Cells were lysed in RIPA buffer and cleared lysates were subjected to standard immunobloting. The following primary antibodies were used: RIOK1 (Novus), RIOK2 (Sigma), p53 (Santa Cruz), p21, EGFR (BD), actin (DSHB), RpL11 (Invitrogen), NRBP2 (Abcam), STK17a/DRAK1 (Anaspec), PDGFRα, VRK1, CDK9, CDK7, STK17B/DRAK2, TLK1, phospho-Akt(S473), phospho-Akt(T308), phospho-PRAS40(T246), phospho-FOXO1(T24)/FOXO3(T32), Akt, phospho-NDRG1(T346), NDRG1, PRAS40, phospho-4E-BP1, 4E-BP1, PARP, cleaved caspase, FOXO3, TSC2, phospho-TSC2, mTor, phospho-mTor(T2448) (Cell Signaling Technologies)
WST1 and FACS assays
For WST-1 assays, cell lines were infected with lentiviral shRNA constructs and placed under selection for 48 hrs. Following selection, cells were plated for WST1 assays for cell proliferation/viability as per manufacturer's instructions (Clontech). For flow cytometry (FACS) analysis of DNA content, cells were dissociated and stained with propidium iodide (PI). For FACS analysis for apoptosis, cells were treated with indicated siRNAs or lentiviral vectors and stained with Annexin V-FITC and PI or 7AAD (Invitrogen, BD Biosciences). Assays were performed on a FACScaliber II flow cytometer and data were collected using FACSdiva software (BD Biosciences). Cell cycle profiles were generated using ModFit LT (Verity Software House). In all cases, at least 5000 cells were analyzed per sample.
RIOK2 overexpression and immunoprecipitation
293T cells were transiently transfected with Myc-DDK-tagged RIOK2 constructs. Cells were lysed in 50 mM HEPES pH 7.5, 150 mM NaCl, 5 mM EDTA, 1 mM DTT buffer with protease and phosphatase inhibitors [22]. RIOK2 was immunoprecipitated with M2-agarose (Sigma) from cleared lysates, and washed immunoprecipitates were subjected to immunobloting.
Biopsy lysates and immunohistochemistry on tumor samples
Human brain tumor biopsies and tissues were obtained from the Brain Tumor Translational Resource under a protocol approved by the University of California, Los Angeles institutional review board. Paraffin embedded human brain tumor specimens and tumor tissue microarrays with matched control tissue were prepared and sectioned using the UCLA Pathology Histology and Tissue Core Facility. Immunohistochemical staining was performed as previously described [70] or as specified by manufacturer guidelines (Sigma). The results were scored by neuropathologists according to standard clinical criteria, and images of RIOK2 immunoreactivity were taken on an Olympus DP72.
Mouse intracranial graft assays
For orthotopic implantation of mouse astrocytes engineered in vitro, low passage cells (no more than 8–10 passages) were used in two separate experiments. 1×105 cells in 5 µl of PBS were injected stereotactically 2 mm lateral to the midline and 1 mm anterior to the bregma into the brains of 5–6 week old athymic nu/nu mice. Mice were monitored and all animals were sacrificed upon evidence of neurological symptoms in experimental groups such that all samples were time-matched. Brains were removed for processing and histological analysis. Sections were scored independently by two neuropathologists for the presence of tumors and injection-associated needle scars. Animals injected with RIOK2-expresing cells that developed tumors outside of the brain (n = 1) were excluded from the final tally. All animal experiments were approved and conducted according to animal welfare guidelines of the IACUC at the University of California, San Diego.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. FurnariFB, FentonT, BachooRM, MukasaA, StommelJM, et al. (2007) Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev 21 : 2683–2710.
2. NishikawaR, JiXD, HarmonRC, LazarCS, GillGN, et al. (1994) A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc Natl Acad Sci U S A 91 : 7727–7731.
3. MukasaA, WykoskyJ, LigonKL, ChinL, CaveneeWK, et al. (2010) Mutant EGFR is required for maintenance of glioma growth in vivo, and its ablation leads to escape from receptor dependence. Proc Natl Acad Sci U S A 107 : 2616–2621.
4. HollandEC, CelestinoJ, DaiC, SchaeferL, SawayaRE, et al. (2000) Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat Genet 25 : 55–57.
5. MarumotoT, TashiroA, Friedmann-MorvinskiD, ScadengM, SodaY, et al. (2009) Development of a novel mouse glioma model using lentiviral vectors. Nat Med 15 : 110–116.
6. BachooRM, MaherEA, LigonKL, SharplessNE, ChanSS, et al. (2002) Epidermal growth factor receptor and Ink4a/Arf: convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell 1 : 269–277.
7. HollandEC, HivelyWP, DePinhoRA, VarmusHE (1998) A constitutively active epidermal growth factor receptor cooperates with disruption of G1 cell-cycle arrest pathways to induce glioma-like lesions in mice. Genes Dev 12 : 3675–3685.
8. CloughesyTF, YoshimotoK, NghiemphuP, BrownK, DangJ, et al. (2008) Antitumor activity of rapamycin in a Phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS Med 5: e8 doi:10.1371/journal.pmed.0050008.
9. ReadRD, CaveneeWK, FurnariFB, ThomasJB (2009) A Drosophila model for EGFR-Ras and PI3K-dependent human glioma. PLoS Genet 5: e1000374 doi:10.1371/journal.pgen.1000374.
10. ReiterLT, BierE (2002) Using Drosophila melanogaster to uncover human disease gene function and potential drug target proteins. Expert Opin Ther Targets 6 : 387–399.
11. St JohnstonD (2002) The art and design of genetic screens: Drosophila melanogaster. Nat Rev Genet 3 : 176–188.
12. DietzlG, ChenD, SchnorrerF, SuKC, BarinovaY, et al. (2007) A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 448 : 151–156.
13. BellenHJ, TongC, TsudaH (2011) 100 years of Drosophila research and its impact on vertebrate neuroscience: a history lesson for the future. Nat Rev Neurosci 11 : 514–522.
14. FreemanMR, DohertyJ (2006) Glial cell biology in Drosophila and vertebrates. Trends Neurosci 29 : 82–90.
15. LaRonde-LeBlancN, WlodawerA (2005) A family portrait of the RIO kinases. J Biol Chem 280 : 37297–37300.
16. VanrobaysE, GelugneJP, GleizesPE, Caizergues-FerrerM (2003) Late cytoplasmic maturation of the small ribosomal subunit requires RIO proteins in Saccharomyces cerevisiae. Mol Cell Biol 23 : 2083–2095.
17. WidmannB, WandreyF, BadertscherL, WylerE, PfannstielJ, et al. (2011) The kinase activity of human Rio1 is required for final steps of cytoplasmic maturation of 40S subunits. Mol Biol Cell
18. ZempI, WildT, O'DonohueMF, WandreyF, WidmannB, et al. (2009) Distinct cytoplasmic maturation steps of 40S ribosomal subunit precursors require hRio2. J Cell Biol 185 : 1167–1180.
19. BaumasK, SoudetJ, Caizergues-FerrerM, FaubladierM, HenryY, et al. (2012) Human RioK3 is a novel component of cytoplasmic pre-40S pre-ribosomal particles. RNA Biol 9 : 162–174.
20. StrunkBS, NovakMN, YoungCL, KarbsteinK (2012) A translation-like cycle is a quality control checkpoint for maturing 40S ribosome subunits. Cell 150 : 111–121.
21. OlsenJV, BlagoevB, GnadF, MacekB, KumarC, et al. (2006) Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127 : 635–648.
22. BreitkreutzA, ChoiH, SharomJR, BoucherL, NeduvaV, et al. (2010) A global protein kinase and phosphatase interaction network in yeast. Science 328 : 1043–1046.
23. LuoJ, EmanueleMJ, LiD, CreightonCJ, SchlabachMR, et al. (2009) A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 137 : 835–848.
24. ManningG, PlowmanGD, HunterT, SudarsanamS (2002) Evolution of protein kinase signaling from yeast to man. Trends Biochem Sci 27 : 514–520.
25. MorrisonDK, MurakamiMS, CleghonV (2000) Protein kinases and phosphatases in the Drosophila genome. J Cell Biol 150: F57–62.
26. SzklarczykD, FranceschiniA, KuhnM, SimonovicM, RothA, et al. (2011) The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res 39: D561–568.
27. BjorklundM, TaipaleM, VarjosaloM, SaharinenJ, LahdenperaJ, et al. (2006) Identification of pathways regulating cell size and cell-cycle progression by RNAi. Nature 439 : 1009–1013.
28. Bettencourt-DiasM, GietR, SinkaR, MazumdarA, LockWG, et al. (2004) Genome-wide survey of protein kinases required for cell cycle progression. Nature 432 : 980–987.
29. FriedmanA, PerrimonN (2006) A functional RNAi screen for regulators of receptor tyrosine kinase and ERK signalling. Nature 444 : 230–234.
30. BoutrosM, KigerAA, ArmknechtS, KerrK, HildM, et al. (2004) Genome-wide RNAi analysis of growth and viability in Drosophila cells. Science 303 : 832–835.
31. NeumullerRA, RichterC, FischerA, NovatchkovaM, NeumullerKG, et al. (2011) Genome-Wide Analysis of Self-Renewal in Drosophila Neural Stem Cells by Transgenic RNAi. Cell Stem Cell 8 : 580–593.
32. Sousa-NunesR, YeeLL, GouldAP (2011) Fat cells reactivate quiescent neuroblasts via TOR and glial insulin relays in Drosophila. Nature 471 : 508–512.
33. ReddyBV, IrvineKD (2011) Regulation of Drosophila glial cell proliferation by Merlin-Hippo signaling. Development 138 : 5201–5212.
34. HuangHS, NaganeM, KlingbeilCK, LinH, NishikawaR, et al. (1997) The enhanced tumorigenic activity of a mutant epidermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phosphorylation and unattenuated signaling. J Biol Chem 272 : 2927–2935.
35. LaksDR, Masterman-SmithM, VisnyeiK, AngenieuxB, OrozcoNM, et al. (2009) Neurosphere formation is an independent predictor of clinical outcome in malignant glioma. Stem Cells 27 : 980–987.
36. PollardSM, YoshikawaK, ClarkeID, DanoviD, StrickerS, et al. (2009) Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell 4 : 568–580.
37. LeeJ, KotliarovaS, KotliarovY, LiA, SuQ, et al. (2006) Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 9 : 391–403.
38. IshiiN, MaierD, MerloA, TadaM, SawamuraY, et al. (1999) Frequent co-alterations of TP53, p16/CDKN2A, p14ARF, PTEN tumor suppressor genes in human glioma cell lines. Brain Pathol 9 : 469–479.
39. CiesielskiMJ, FenstermakerRA (2000) Oncogenic epidermal growth factor receptor mutants with tandem duplication: gene structure and effects on receptor function. Oncogene 19 : 810–820.
40. GalliaGL, TylerBM, HannCL, SiuIM, GirandaVL, et al. (2009) Inhibition of Akt inhibits growth of glioblastoma and glioblastoma stem-like cells. Mol Cancer Ther 8 : 386–393.
41. PanditaA, AldapeKD, ZadehG, GuhaA, JamesCD (2004) Contrasting in vivo and in vitro fates of glioblastoma cell subpopulations with amplified EGFR. Gene Chromosome Canc 39 : 29–36.
42. IndaMD, BonaviaR, MukasaA, NaritaY, SahDW, et al. (2010) Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes Dev 24 : 1731–1745.
43. SzerlipNJ, PedrazaA, ChakravartyD, AzimM, McGuireJ, et al. (2012) Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proc Natl Acad Sci U S A 109 : 3041–3046.
44. ChenAJ, PaikJH, ZhangH, ShuklaSA, MortensenR, et al. (2012) STAR RNA-binding protein Quaking suppresses cancer via stabilization of specific miRNA. Genes Dev 26 : 1459–1472.
45. YingH, ZhengH, ScottK, WiedemeyerR, YanH, et al. (2010) Mig-6 controls EGFR trafficking and suppresses gliomagenesis. Proc Natl Acad Sci U S A 107 : 6912–6917.
46. GuertinDA, StevensDM, ThoreenCC, BurdsAA, KalaanyNY, et al. (2006) Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell 11 : 859–871.
47. SunayamaJ, SatoA, MatsudaK, TachibanaK, WatanabeE, et al. (2011) FoxO3a functions as a key integrator of cellular signals that control glioblastoma stem-like cell differentiation and tumorigenicity. Stem Cells 29 : 1327–1337.
48. TanakaK, BabicI, NathansonD, AkhavanD, GuoD, et al. (2011) Oncogenic EGFR signaling activates an mTORC2-NF-kappaB pathway that promotes chemotherapy resistance. Cancer Discov 1 : 524–538.
49. BashirT, CloningerC, ArtinianN, AndersonL, BernathA, et al. (2012) Conditional astroglial rictor overexpression induces malignant glioma in mice. PLoS ONE 7: e47741 doi:10.1371/journal.pone.0047741.
50. FurnariFB, LinH, HuangHS, CaveneeWK (1997) Growth suppression of glioma cells by PTEN requires a functional phosphatase catalytic domain. Proc Natl Acad Sci U S A 94 : 12479–12484.
51. VanrobaysE, GleizesPE, Bousquet-AntonelliC, Noaillac-DepeyreJ, Caizergues-FerrerM, et al. (2001) Processing of 20S pre-rRNA to 18S ribosomal RNA in yeast requires Rrp10p, an essential non-ribosomal cytoplasmic protein. EMBO J 20 : 4204–4213.
52. FumagalliS, ThomasG (2011) The role of p53 in ribosomopathies. Semin Hematol 48 : 97–105.
53. NarlaA, EbertBL (2010) Ribosomopathies: human disorders of ribosome dysfunction. Blood 115 : 3196–3205.
54. SimmonsML, LambornKR, TakahashiM, ChenP, IsraelMA, et al. (2001) Analysis of complex relationships between age, p53, epidermal growth factor receptor, and survival in glioblastoma patients. Cancer Res 61 : 1122–1128.
55. McLendonR, FriedmanA, BignerD, Van MeirEG, BratDJ, et al. (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455 : 1061–1068.
56. OhgakiH, DessenP, JourdeB, HorstmannS, NishikawaT, et al. (2004) Genetic pathways to glioblastoma: a population-based study. Cancer Res 64 : 6892–6899.
57. LoweSW, RuleyHE, JacksT, HousmanDE (1993) p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell 74 : 957–967.
58. StuppR, MasonWP, van den BentMJ, WellerM, FisherB, et al. (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352 : 987–996.
59. Villalonga-PlanellsR, Coll-MuletL, Martinez-SolerF, CastanoE, AcebesJJ, et al. (2011) Activation of p53 by nutlin-3a induces apoptosis and cellular senescence in human glioblastoma multiforme. PLoS ONE 6: e18588 doi:10.1371/journal.pone.0018588.
60. ZinzallaV, StrackaD, OppligerW, HallMN (2011) Activation of mTORC2 by association with the ribosome. Cell 144 : 757–768.
61. LiuT, DengM, LiJ, TongX, WeiQ, et al. (2011) Phosphorylation of right open reading frame 2 (Rio2) protein kinase by polo-like kinase 1 regulates mitotic progression. J Biol Chem 286 : 36352–36360.
62. MarygoldSJ, RooteJ, ReuterG, LambertssonA, AshburnerM, et al. (2007) The ribosomal protein genes and Minute loci of Drosophila melanogaster. Genome Biol 8: R216.
63. MaciasE, JinA, DeisenrothC, BhatK, MaoH, et al. (2010) An ARF-independent c-MYC-activated tumor suppression pathway mediated by ribosomal protein-Mdm2 Interaction. Cancer Cell 18 : 231–243.
64. BurgerK, MuhlB, HarasimT, RohrmoserM, MalamoussiA, et al. (2010) Chemotherapeutic drugs inhibit ribosome biogenesis at various levels. J Biol Chem 285 : 12416–12425.
65. SunXX, DaiMS, LuH (2007) 5-fluorouracil activation of p53 involves an MDM2-ribosomal protein interaction. J Biol Chem 282 : 8052–8059.
66. GoidtsV, BageritzJ, PuccioL, NakataS, ZapatkaM, et al. (2012) RNAi screening in glioma stem-like cells identifies PFKFB4 as a key molecule important for cancer cell survival. Oncogene 31 : 3235–3243.
67. WurdakH, ZhuS, RomeroA, LorgerM, WatsonJ, et al. (2010) An RNAi screen identifies TRRAP as a regulator of brain tumor-initiating cell differentiation. Cell Stem Cell 6 : 37–47.
68. WiedemeyerWR, DunnIF, QuayleSN, ZhangJ, ChhedaMG, et al. (2010) Pattern of retinoblastoma pathway inactivation dictates response to CDK4/6 inhibition in GBM. Proc Natl Acad Sci U S A 107 : 11501–11506.
69. KimYW, LiuTJ, KoulD, TiaoN, FerozeAH, et al. (2011) Identification of novel synergistic targets for rational drug combinations with PI3 kinase inhibitors using siRNA synthetic lethality screening against GBM. Neuro Oncol 13 : 367–375.
70. MellinghoffIK, WangMY, VivancoI, Haas-KoganDA, ZhuS, et al. (2005) Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med 353 : 2012–2024.
71. RaynaudFI, EcclesSA, PatelS, AlixS, BoxG, et al. (2009) Biological properties of potent inhibitors of class I phosphatidylinositide 3-kinases: from PI-103 through PI-540, PI-620 to the oral agent GDC-0941. Mol Cancer Ther 8 : 1725–1738.
72. HiraiH, SootomeH, NakatsuruY, MiyamaK, TaguchiS, et al. (2010) MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther 9 : 1956–1967.
73. UhlenM, OksvoldP, FagerbergL, LundbergE, JonassonK, et al. (2010) Towards a knowledge-based Human Protein Atlas. Nat Biotechnol 28 : 1248–1250.
74. ChooAY, YoonSO, KimSG, RouxPP, BlenisJ (2008) Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc Natl Acad Sci U S A 105 : 17414–17419.
75. ObenauerJC, CantleyLC, YaffeMB (2003) Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res 31 : 3635–3641.
76. HornbeckPV, KornhauserJM, TkachevS, ZhangB, SkrzypekE, et al. (2011) PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res 40: D261–270.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 2
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Complex Inheritance of Melanoma and Pigmentation of Coat and Skin in Grey Horses
- Coordination of Chromatid Separation and Spindle Elongation by Antagonistic Activities of Mitotic and S-Phase CDKs
- Autophagy Induction Is a Tor- and Tp53-Independent Cell Survival Response in a Zebrafish Model of Disrupted Ribosome Biogenesis
- Assembly of the Auditory Circuitry by a Genetic Network in the Mouse Brainstem
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy