
Single Transmembrane Peptide DinQ Modulates Membrane-Dependent Activities
The functions of several SOS regulated genes in Escherichia coli are still unknown, including dinQ. In this work we characterize dinQ and two small RNAs, agrA and agrB, with antisense complementarity to dinQ. Northern analysis revealed five dinQ transcripts, but only one transcript (+44) is actively translated. The +44 dinQ transcript translates into a toxic single transmembrane peptide localized in the inner membrane. AgrB regulates dinQ RNA by RNA interference to counteract DinQ toxicity. Thus the dinQ-agr locus shows the classical features of a type I TA system and has many similarities to the tisB-istR locus. DinQ overexpression depolarizes the cell membrane and decreases the intracellular ATP concentration, demonstrating that DinQ can modulate membrane-dependent processes. Augmented DinQ strongly inhibits marker transfer by Hfr conjugation, indicating a role in recombination. Furthermore, DinQ affects transformation of nucleoid morphology in response to UV damage. We hypothesize that DinQ is a transmembrane peptide that modulates membrane-dependent activities such as nucleoid compaction and recombination.
Published in the journal:
. PLoS Genet 9(2): e32767. doi:10.1371/journal.pgen.1003260
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003260
Summary
The functions of several SOS regulated genes in Escherichia coli are still unknown, including dinQ. In this work we characterize dinQ and two small RNAs, agrA and agrB, with antisense complementarity to dinQ. Northern analysis revealed five dinQ transcripts, but only one transcript (+44) is actively translated. The +44 dinQ transcript translates into a toxic single transmembrane peptide localized in the inner membrane. AgrB regulates dinQ RNA by RNA interference to counteract DinQ toxicity. Thus the dinQ-agr locus shows the classical features of a type I TA system and has many similarities to the tisB-istR locus. DinQ overexpression depolarizes the cell membrane and decreases the intracellular ATP concentration, demonstrating that DinQ can modulate membrane-dependent processes. Augmented DinQ strongly inhibits marker transfer by Hfr conjugation, indicating a role in recombination. Furthermore, DinQ affects transformation of nucleoid morphology in response to UV damage. We hypothesize that DinQ is a transmembrane peptide that modulates membrane-dependent activities such as nucleoid compaction and recombination.
Introduction
Exposure of E. coli to DNA damaging agents induces the SOS response, which is under control of the RecA and LexA regulatory proteins. The SOS response upregulates gene functions involved in numerous cellular processes such as nucleotide excision repair (NER), UV induced mutagenesis, recombination, inhibition of cell division and replication. The LexA repressor downregulates more than 50 SOS genes by binding to the operator sequence in their promoter regions [1], [2]. SOS inducers (e.g. UV) cause replication blocks and generate RecA/ssDNA nucleoprotein filaments that mediate auto-proteolysis of the LexA repressor.
Both NER and recombination are required to maintain DNA integrity. NER repairs numerous lesions introducing helical distortions, in which UvrA, UvrB and UvrC work in sequential steps to recognize and remove the lesion. The RecBCD complex is the major component for initiation of recombinational repair (RR) of DNA double strand breaks (DSBs) by processing a blunt dsDNA end into a dsDNA molecule possessing a 3′-terminated ssDNA tail. As part of this process RecBCD mediates RecA filamentation required for presynaptic processing of dsDNA ends.
Most of the characterized LexA regulated genes play important roles in the physiology of E. coli, but there are still several genes of unknown function. One of these uncharacterized genes is dinQ, which is located in the 823 bp region between arsR and gor, 78.58 min on the E. coli chromosome. DinQ is predicted to encode an open reading frame (ORF) of 49 aa 139 nt downstream from a LexA operator sequence [1]. Small proteins of less than 50 amino acids are important in cellular processes such as regulation, signalling and antibacterial action [3]–[5]. More than 50 chromosomally encoded small proteins with a validated expression of less than 50 aa have been identified so far in E. coli [6]–[10]. Several of the newly discovered peptides are hydrophobic single transmembrane helices belonging to toxin-antitoxin systems.
In this work we characterize the arsR-gor intergenic region in which an endonucleolytic product of dinQ is translated into a small hydrophobic peptide of 27 aa. DinQ is under LexA control and antisense regulation by a novel small RNA, agrB. DinQ is localized in the inner membrane as a single transmembrane peptide that modulates nucleoid compaction and conjugal recombination.
Results
The arsR-gor intergenic region encodes two small RNAs, agrA and agrB, which are transcribed in the opposite direction to dinQ
The SOS inducible dinQ gene in E. coli was identified in a search for new LexA regulated genes [1]. The dinQ gene was found in the 823 bp arsR-gor intergenic region (Figure 1A), encoding a ∼330 nt transcript with a putative LexA operator sequence (heterology index (HI) = 4.83) in the promoter region and two putative ORFs of 18 and 49 amino acids. No biological function, phenotype or significant homologies to proteins with known function were associated with DinQ [1]. Except for the dinQ gene, no other genes have been reported in the arsR-gor intergenic region. We performed a search for promoter and transcriptional terminator sequences in the arsR-gor intergenic region. As expected this search identified the dinQ LexA operator sequence identified earlier in a screen for LexA regulated genes in E. coli [1]. However, a second operator sequence for LexA (HI = 13.82) in close proximity to the first was identified (Figure 1A and 1B). Further, we identified putative −10 and −35 sequences corresponding to the dinQ promoter which overlaps both operator sequences, and a putative dinQ terminator sequence a few nucleotides downstream of the translational stop codon of the gor gene (Figure 1B). Finally, the sequence search identified two new small noncoding RNAs, termed agrA and agrB (arsR-gor region gene A and B, respectively), containing consensus like −10 and −35 sequences and rho independent terminator sequences (Figure 1A and 1B). AgrA and agrB are transcribed in the opposite direction of dinQ but encode no putative ORFs. Thirty-one nucleotides at the 5′ end of agrA and agrB show antisense complementarity to dinQ (Figure 1B and 1C). Twenty-five out of 31 nucleotides show complementarity to agrA while 30 out of 31 nucleotides show complementarity to agrB. This putative base pairing is indicative of possible RNA interference with the dinQ transcript. It thus appears that the arsR-gor region contain one protein coding gene, dinQ, and two small non-coding RNAs, agrA and agrB, with antisense complementarity to dinQ.

Five dinQ transcripts
To examine a potential role for the small non-coding RNAs agrA and agrB in regulating dinQ, three single mutants (dinQ, agrA and agrB), one double mutant (agrAB) and one triple mutant (dinQ agrAB) were generated (Table S1). To estimate the approximate size of the dinQ, agrA and agrB transcripts, northern blots with total RNA isolated from UV exposed (and unexposed) wild type and mutant strains (dinQ, agrA and agrB) were hybridized with radiolabeled riboprobes against the respective genes (Figure 2A). The dinQ probe generates five specific signals (a–e), in which the main transcript (a) migrates according to the expected size of full-length dinQ, ∼330 nt. DinQ-b, -c, -d and -e migrates as transcripts of about 290 nt, 250 nt, 200 nt and 130 nt, respectively, according to the size marker. All signals are absent in the dinQ mutant demonstrating that all five transcripts are derived from dinQ. The full-length dinQ product is 3 - and 4.6-fold upregulated in the agrA and agrB mutants respectively under normal growth (without UV exposure). Notably, the dinQ-b signal is 4.8 - and 3-fold upregulated in the agrB mutant as compared to the wild type and agrA mutant respectively. The dinQ-c product is not detectable in the agrB mutant. Further, the dinQ transcripts are induced in response to UV in wild type and agrA but not in agrB. These data indicate a regulatory mechanism by RNA interference, in which the agrA and agrB interfere differently with the dinQ transcript. Further, primer extension and 3′ mapping of dinQ RNA revealed transcript starts at 0, +44 and +125 corresponding to the estimated size of dinQ-a, -b and –d, respectively (Figure 2B). In agreement with the northern analysis we find that the +44 primer extension product (dinQ-b) is upregulated in the agrB mutant as compared to wild type and agrA mutant. The primer extension could not identify any products corresponding to dinQ-c or -e. The agrAB probes showed that the agrB transcript migrates slightly slower than the agrA transcript, and none of the transcripts were regulated in response to UV irradiation (Figure 2A, middle panel). The agrA transcript is upregulated 9.5 times in the unexposed agrB mutant as compared to wild type, indicating that the absence of agrB somehow promotes agrA RNA stability or transcription of agrA. Further, primer extension revealed that the sequence of transcription start was identical for the agrA and agrB genes (Figure 2C). 3′ mapping of agrA and agrB showed different transcription stops around the rho terminator (data not shown), indicating that the transcripts are processed/terminated differently at the 3′end. In summary, these data demonstrate that both agrA and agrB downregulate the level of dinQ full-length transcript whereas agrB is particularly important for down regulation of +44 dinQ (dinQ-b).

Deleting agrB results in UV sensitivity
The 31 nt antisense region in agrB gene (Figure 1C) indicate a function in antisense regulation of dinQ via RNA interference. This antisense sequence is partially complementary in agrA (Figure 1C) suggesting that both the agrA and agrB transcripts could base pair with the dinQ transcript. To ensure the function of dinQ when generating mutants, the deletions of agrA and agrB were made without destroying the dinQ promoter. DinQ belongs to the LexA regulon in E. coli [1] which regulates the SOS response. Several mutants of the SOS response, which play a direct role in DNA repair, display UV sensitivity. To examine the role of the dinQ-agrAB locus in the SOS response we tested the UV sensitivity of the various mutants (Figure 3A). The agrB single mutant and agrAB double mutant showed a significant increase in UV sensitivity compared to the isogenic wild type. In contrast, the agrA and dinQ single mutants and the dinQ agrAB triple mutant showed no UV sensitivity. Further we examined UV survival of the agrB mutant carrying a plasmid expressing the agrB gene (Figure S1), demonstrating that the mutant recovered completely. These data indicate a role for agrB in protection against UV exposure, in which the agrB transcript modifies the dinQ transcript. According to the northern analysis (Figure 2A) agrB represses accumulation of the +44 dinQ/dinQ-b transcript. It thus appears that the +44 dinQ product mediates the UV sensitivity of the agrB mutant.

AgrB counteracts the UV sensitivity induced by dinQ
To further investigate the role of the arsR-gor intergenic region in UV protection, we cloned agrA, agrB and dinQ separately or the entire region containing both small RNAs and the dinQ gene into the cloning vector pKK232-8. Wild type cells transformed with pKK232-8-dinQ or pKK232-8 (control plasmid) showed the same viability during normal growth conditions (data not shown). In contrast, it was not possible to transform pKK232-8-dinQ into the agrB mutant (Table S2), indicating that the endogenous level of agrB in wild type cells is sufficient to inhibit dinQ expression from the pKK232-8-dinQ construct during normal growth. However, wild type transformed with pKK232-8-dinQ showed increased sensitivity to UV as compared to wild type transformed with the control plasmid (pKK232-8) (Figure 3B). When the wild type was transformed with constructs expressing agrA or agrB they did not increase sensitivity of the cells to UV (data not shown). Interestingly, wild type cells transformed with the plasmid carrying the entire arsR-gor locus, expressing all three genes, showed no UV sensitivity, demonstrating that agrAB can neutralize the UV sensitizing effect of dinQ. The agrB mutant (Figure 3A) and the wild type cells transformed with pKK232-8-dinQ (Figure 3B) displayed similar sensitivity to UV. These results suggest that the agrB transcript counteracts the UV sensitivity induced by dinQ expression.
Slow growth of the agrB mutant
During construction of the agrB and agrAB mutants we observed that they form small colonies when plated on LB agar. To further investigate this growth phenotype we compared the growth rate in LB medium of the agrB, agrA and dinQ mutants and their isogenic wild type. OD600 was measured during growth and a sample was diluted and plated for viable counts. This experiment showed that only the agrB single mutant and agrAB double mutant grow more slowly than the wild type cells (Figure S2A). Also in glucose-CAA medium agrB mutant cells grew more slowly than wild type cells (Figure S2B and S2C). In another set of experiments we utilized flow cytometry to analyze whether DNA replication was affected in the growth impaired agrB mutant. We found that cells were smaller than normal with a reduced DNA concentration (Figure S2D). The total time for replication from origin to terminus was shorter in the mutant and the number of origins and replication forks per cell were fewer compared to the wild type. There was also a considerable heterogeneity in the observed reduction in cellular DNA concentration. This heterogeneity could be due to cell-to-cell differences in expression of the DinQ peptide.
DinQ is translated from an alternative GTG start codon
The mechanism underlying the UV sensitive phenotype of dinQ in a multicopy situation or under constitutive upregulation in an agrB mutant is not clear. The dinQ gene contains two putative ORFs in which the second ORF contains three putative start codons (Figure 3C). In this work the corresponding peptides are termed DinQ I-IV. None of the putative DinQ peptides show homology to known peptides. To examine if any of the ORFs mediate the UV sensitivity shown by dinQ, each of the putative DinQ peptides (I–IV) were cloned into the expression vector pET28b(+) and expressed under control of IPTG. DinQ I displayed no increased sensitivity in absence or presence of UV, suggesting that the putative peptide translated from ORF I (Figure 3C) does not induce DinQ toxicity (Figure 3D and data not shown). In contrast, we observed that DinQ peptides II, III and IV showed a strong toxic/growth inhibitory effect even in absence of UV, demonstrating that the C-terminal amino acid sequence translated from start codon IV of the second ORF is sufficient to induce DinQ toxicity (Figure 3D). Next, expression of DinQ II was titrated with increasing concentrations of IPTG in absence or presence of UV (Figure 3E), showing that DinQ is highly toxic to the cells at very low doses of IPTG induction and the toxicity was UV independent.
In another set of experiments we used a coupled in vitro transcription/translation E. coli T7 S30 extract to examine translation from pET28b(+) constructs encoding the putative peptides predicted from DinQ I–IV. Expression of DinQ I could not be detected whereas DinQ II–IV were highly expressed (Figure 4A, lanes 2–5). Notably, extracts with the DinQ IV construct produced two peptides of approximately 7.0 and 5.0 kDa, in which the smallest peptide indicates a fifth start codon. A closer inspection of the dinQ sequence uncovered a Shine Dalgarno motif within the DinQ IV sequence in optimal position to initiate translation at a GTG (termed codon V in Figure 3C), which encodes a putative peptide of 27 aa, termed DinQ V. To examine if the DinQ V peptide induces toxicity we cloned the sequence into pET28b(+), transformed the construct into wild type cells and monitored cell survival under the control of IPTG induction in presence or absence of UV exposure. DinQ V expression induced cell killing independently of UV treatment, suggesting that the sequence of peptide V is sufficient to mediate DinQ toxicity (Figure 3C and 3D). In vitro transcription/translation assays with the pET28b(+) DinQ V construct produced a peptide corresponding to the predicted molecular weight of peptide V (Figure 4A, lane 6).

Northern analysis identified five dinQ transcripts (Figure 2A; dinQ a–e), in which the start site were determined for transcript a, b and d. To assess the translational activity of the in vivo dinQ transcripts a, b and d we synthesized the corresponding PCR products carrying the T7 RNA polymerase promoter and added E. coli T7 S30 extract. Only the dinQ-b/+44 transcript was translationally active, generating a peptide with a molecular weight similar to DinQ V, whereas the other transcripts were translationally inert (Figure 4A, lanes 7–10). Thus, it appears in vivo that the biologically active DinQ peptide (peptide V) is translated from the post transcriptionally modified +44 dinQ RNA.
To examine endogenous expression of DinQ in vivo, a 3×FLAG tag was inserted chromosomally in frame with the C-terminal of the dinQ gene in the wild type and agrB mutant. Western analysis revealed a faint band for the FLAG tagged DinQ peptide in the agrB mutant while the peptide was barely detectable in wild type (Figure 4B, lanes 5 and 6). In UV treated cells the DinQ level was about two fold higher in the agrB mutant as compared to wild type (Figure 4B, lanes 3 and 4). It thus appears that the phenotypes of the agrB mutant are not due to polar effects of extensive overexpression of DinQ.
Further, we introduced a chromosomal stop codon in the Lys4 position of dinQ in the agrB mutant and wild type (Figure 3C, base labeled in red). Survival experiments showed that UV resistance was restored to wild type level in the agrB mutant (Figure 4C), indicating that the UV sensitivity of the agrB mutant is caused by translation of a functional DinQ peptide. Next, we introduced three chromosomal point mutations in the dinQ up-stream sequence predicted to be involved in base pairing with agrB (Figure 3C, bases labeled in red). Exposure of these strains to UV in the agrB mutant background showed wild type levels of survival (Figure 4C). All together these results suggest that DinQ is translated into a peptide of 27 aa and the agrB-dinQ RNA interference is important for correct regulation of DinQ translation.
DinQ is localized to the inner membrane
To determine the intracellular localization of DinQ, western analysis was performed on extracts after subcellular fractionation. As antibodies against the native DinQ peptides could not be obtained, we introduced a 3×FLAG epitope at the N-terminal of DinQ (peptide V). Spot assays on LB agar containing IPTG showed that the FLAG tagged peptide induced the same toxicity as the native peptide, demonstrating that the N-terminal FLAG tag had no effect on DinQ toxicity (data not shown). Cells were harvested at several time points after IPTG induction to test the level of expression by western analysis of whole cell extracts. The FLAG-DinQ peptide could not be detected before induction, but showed strong signals 5 to 40 min after induction (data not shown). To examine subcellular fractionation, antibodies against Lep and TolC were used as positive markers for inner and outer membrane fractions, respectively. The western blot showed that the inner and outer membranes are completely separated whereas the cytoplasmic fraction contains some contamination from the inner membrane (Figure 4D, middle panel). DinQ localized to the inner membrane but could not be detected in the outer membrane (Figure 4D, lower panel). The faint signal of the FLAG epitope in the cytoplasmic fraction is possibly due to cross contamination from the inner membrane. These data suggest that DinQ localization is confined to the inner membrane of E. coli.
Computer modelling of DinQ predicts a single transmembrane peptide
Analysis of the DinQ amino acid sequence using the consensus secondary structure prediction tool Jpred3 [11] revealed that DinQ has high propensity to form a single α-helix. All residues except a few on each flanking terminal are predicted with high confidence to belong to the predicted α-helix (Figure 4E). With 20–22 residues in a single α-helix, the DinQ peptide could straightforwardly form a transmembrane helix of 6 full turns spanning more than 30 Å, as shown by modelling of DinQ using a regular α-helical template (Figure 4F). The two positively charged lysine residues (Lys4 and Lys9) are close to the phospholipid head groups, while particularly the charged Glu17, but also Arg20 and Gln24 may form a polar patch that can interact with other membrane embedded proteins (Figure 4F). The predicted single transmembrane peptide supports the localization of DinQ in the inner membrane (Figure 4D).
DinQ is not affecting induction of the SOS response, filamentation, or mutagenesis
Previously, we showed that overexpressing another SOS inducible peptide, TisB, which encodes a small toxic inner membrane peptide, inhibits several SOS functions in wild type E. coli [12]. To determine whether DinQ affected induction of the SOS response we measured the level of recA and lexA mRNA in a mutant which constitutively overexpressed dinQ, agrB mutant or a dinQ deletion mutant (Table S1). Exponentially growing cells were exposed to UV and the amount of recA and lexA mRNA were determined prior to, and 20 min after irradiation by RT-qPCR. The expression levels of recA and lexA were similar in both mutants and wild type indicating that DinQ in contrast to TisB is not affecting regulation of the SOS response (Figure S3).
To investigate a potential role of dinQ in filamentation, we stained cell samples with acridine orange prior to and after UV exposure. All strains showed the same filamentation pattern, in which cells displayed short filaments 1 h after irradiation and long filaments after 2.5 h, indicating that DinQ is not involved in the filamentation process of E. coli (Figure S4). Next, we measured spontaneous and UV induced mutagenesis as the frequency of rifampicin resistant colonies in wild type and the dinQ and agrB single mutants. The results showed no significant differences in mutation frequency in the mutant strains as compared to wild type suggesting that DinQ is not altering spontaneous and SOS induced mutagenesis (data not shown).
DinQ overexpression depolarizes the cell membrane
To examine whether high levels of DinQ induce changes in membrane potential we tested the ability of E. coli cells overexpressing DinQ to take up the dye DiBAC4(3) [bis-(1,3-dibarbituric acid)-trimethine oxanol]. The quantity of dye entering cells is proportional to membrane polarization, the less polarised the membrane the greater the quantity entering the cells and so increased fluorescence intensity due to binding to the membrane and intracellular components [13]. Cells were analyzed 5 and 20 min after IPTG induction of DinQ expression, incubated with DiBAC4(3) for 20 min and analyzed by flow cytometry (Figure 5A). No changes were observed for the plasmid control pET28b(+). However, IPTG induction of DinQ (peptide V) showed a rapid increase in DiBAC4(3) uptake (Figure 5A), suggesting that elevated levels of DinQ depolarize the cell membrane. This data indicates that DinQ overexpression interferes with membrane polarity and could therefore lead to a loss of viability.

DinQ overexpression decreases the intracellular ATP concentration
Subcellular fractionation of E. coli showed that DinQ is localized to the inner membrane (Figure 4D). DinQ is predicted to be a hydrophobic single transmembrane peptide that might compromise inner membrane integrity (Figure 4F). We speculated that if DinQ affected the proton motive force, it would affect ATP production and intracellular ATP concentration. The intracellular ATP concentration was measured in wild type cells and in the agrB mutant, using a quantitative luciferase-based assay. This experiment showed that the concentration of ATP in the agrB mutant was about 50% of the concentration measured in wild type cells (Figure 5B). Further, UV exposure increased the ATP concentration 0.6 fold in both cell types. Thus, it appears that insertion of the DinQ peptide into the inner membrane of E. coli impairs the energy supply in the form of ATP.
DinQ modulates conjugal recombination
The agrB mutant displayed sensitivity to UV suggesting that DinQ could have a role in the repair of UV induced DNA damage. Both nucleotide excision repair (NER) and recombinational repair (RR) counteract the genotoxic effects of UV irradiation. NER is required for the repair of UV induced photoproducts such as thymine dimers and cyclopyrimidine dimers, while RR is required for the repair of strand gaps and double strand breaks. To examine if DinQ is involved in NER we analysed UV sensitivity of the uvrA agrB and uvrA dinQ double mutants as compared to the single mutants. The survival analysis showed an additive effect between agrB and uvrA (Figure 6A), whereas uvrA and dinQ showed no additional effect (Figure S5). These data indicate that elevated levels of DinQ in the agrB mutant sensitize the cell to UV via a pathway which is independent of NER. To examine the role of DinQ in recombination, mutant strains dinQ, agrB, recB, (recB agrB) and (dinQ recB) were exposed to UV irradiation. The double mutant (recB agrB) was slightly more sensitive to UV than the agrB single mutant (Figure 6A), whereas recB and dinQ showed no additional effect (Figure S5).

To further examine the role of DinQ in recombination we performed Hfr conjugation assays with a donor strain containing Tn10, which carries the tetracycline resistance gene integrated in its chromosome and agrB, dinQ and uvrA single mutants as recipient strains. We used the uvrA mutant as control strain since UvrA is not involved in recombination (and carry the kanamycin resistance gene required to detect the recipient). Hfr conjugation of Tn10 was at least 400-fold more efficient in dinQ and uvrA mutants as compared to agrB (Figure 6B), suggesting that elevated levels of DinQ inhibit recombination. To examine if the conjugational process itself is affected in an agrB recipient we performed plasmid conjugation assays with a donor strain carrying an F′-plasmid with tetracycline resistance and the same recipient strains as in the Hfr conjugation experiment. Hfr conjugation differs from F′-plasmid conjugation in that transfer of genes after Hfr requires recombination whereas the F′-plasmid does not recombine in the recipient. In these experiments we find no differences in plasmid conjugation frequencies between the dinQ, uvrA and agrB recipient strains (data not shown). In sum, these results suggest that recombination is inhibited in the agrB mutant during Hfr conjugation, but not in the transfer and uptake of DNA or survival of the agrB recipient. It thus appears that elevated levels of DinQ affect the recombination process.
Extended duration of nucleoid compaction in agrB mutant cells after UV irradiation
In dividing cells, replication forks are stalled by DNA lesions that impair DNA unwinding or block synthesis by the DNA polymerase subunits. In E. coli, UV lesions cause a delay in DNA synthesis for a period of time while stalled forks undergo repair. Fluorescence microscopy of Hoechst stained cells has demonstrated that the DNA often forms a compact structure during this phase, and suggests that the nucleoids undergo a major reorganization after UV exposure [14]. To investigate whether DinQ affects nucleoid organization, we used this technique to examine the shape and size of the nucleoids at different time points after UV exposure. In undamaged cells the nucleoids have characteristic shapes and numbers depending on the growth medium. When grown in glucose-CAA medium most cells have two nucleoids and some (the largest cells) have four (Figure 7A, 0 min). Microscopy of cells 15 min after UV irradiation shows that all the wild type cells had lost the normal nucleoid morphology. In approximately 45% of the cells the nucleoids had been rearranged into a highly compact structure, whereas in the rest of the cells the nucleoids were found to be extended throughout the cells (Figure 7A and 7B). Sixty minutes after UV irradiation all cells were found to contain extended nucleoids. After 90 min approximately 30% of the cells had divided and contained nucleoids with normal morphology. In the agrB mutant the degree of nucleoid compaction was similar to that of wild type cells at 15 min after UV exposure (Figure 7A and 7B). However, in the period from 30 to 90 min 25–30% of the nucleoids of the agrB mutant cells were still locked in a compact state whereas a decreasing number of the wild type cells contained compact nucleoids. The result indicates that the transition from compact to extended nucleoid was inhibited in the agrB mutant.
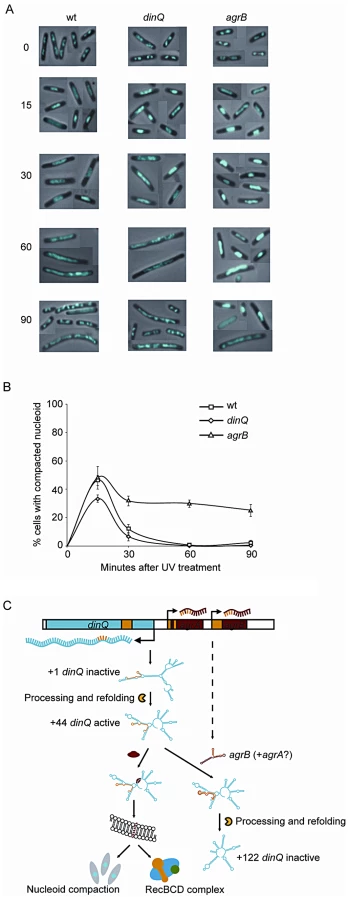
We also investigated the dinQ mutant with respect to nucleoid morphology after UV irradiation. At the 15 min time point approximately 35% of dinQ mutant cells contained a compact nucleoid compared to about 45% of wild type cells (Figure 7B). This indicates that the compaction process might be affected in cells without DinQ. At 30, 60 and 90 min similar numbers of cells with compact and extended nucleoids were found in the dinQ mutant compared to in wild type cells (Figure 7B). The results indicate that cells lacking DinQ have an impaired ability to form a compact nucleoid structure after UV irradiation. Taken together the data reveals that the presence of DinQ is required in order to execute a transformation of nucleoid morphology in response to UV damage, and that overexpression of DinQ leads to a delay in decompaction and extension of the nucleoid during the later stages of the response.
In conclusion, DinQ is under the control of the SOS response and the agrB antisense RNA, and expresses a single transmembrane peptide that has an effect on nucleoid compaction and when overexpressed on conjugal recombination (summarized in Figure 7C).
Discussion
Recently, a search for small proteins in E. coli could not detect any translation of the DinQ ORF [6], [7]. In this paper we characterize the arsR-gor intergenic region of E. coli, which contain the SOS inducible dinQ gene and two constitutively expressed small RNAs, agrA and agrB, with antisense complementarity to the dinQ gene. We show that DinQ is tightly regulated at both the transcriptional and translational level. Five different dinQ transcripts were identified in which only the endonucleolytic +44 transcript (dinQ-b) is translationally active. Further, agrB appears to repress accumulation of dinQ-b by RNA interference. Unexpectedly, DinQ is not translated from any of the three ATG codons within the ORF but from an alternative GTG start codon, encoding a 27 aa peptide which is localized in the inner membrane. The agrB mutant, which expresses elevated levels of dinQ-b, displays increased sensitivity to UV induced DNA damage and an impaired frequency of conjugal recombination. It thus appears that DinQ could be involved in the modulation of homologous recombination. Small single transmembrane peptides such as DinQ may be key regulators of processes at the inner membrane, in which their expression are strictly regulated to avoid toxicity. In summary, the experiments presented in this paper provide insights into the complex regulation of dinQ and suggest a mode of action within the bacterial inner membrane (Summarized in Figure 7C).
Regulation and processing of dinQ RNA
Previous attempts to identify a translation product for dinQ have been unsuccessful, presumably because transcription and translation of dinQ are strictly regulated. First, the promoter region of the dinQ gene contains two LexA operators with different HI, which may suggest differential expression of the transcript early and late in the SOS response. Second, we identified two novel small non coding RNAs agrA and agrB with sequence complementarity to dinQ in the arsR-gor region that regulate dinQ by RNA interference. Notably, only the agrB RNA repressed the translational active +44 dinQ transcript (dinQ-b) whereas both sRNA repressed the primary translationally inactive dinQ transcript. Further, only the agrB antisense RNA counteracts DinQ toxicity. As such the dinQ-agrAB system appears to conform to the definition of a classical type I toxin-antitoxin (TA) system. It thus appears that the dinQ/agrB complex inhibit the endonucleolytic cleavage producing the active mRNA (dinQ-b). Presumably, these antisense sRNAs have been tandemly duplicated in the genome, in which agrA has partly degenerated and appears to be non-functional due to less antisense sequence complementarity with the dinQ sequence as compared to agrB.
The genomic organization and mode of antisense regulation of dinQ in the arsR-gor region resembles regulation of another SOS induced TA system, tisAB [15], [16]. Similar to the dinQ RNA, endonucleolytic processing of the primary tisAB transcript is required to generate an active mRNA producing the toxic TisB peptide. Further, the tisAB locus contains an antisense RNA, IstR-1 that inactivates the translationally active mRNA by RNaseIII dependent cleavage. It appears that agrB may have a similar role in RNase dependent cleavage of the translationally active dinQ mRNA (dinQ-b). In addition, Darfeuille et al revealed that the antisense RNA IstR-1 inhibits translation of the TisB toxin by competing with standby ribosomes binding upstream of the translation initiation region (TIR). It is proposed that binding to the “standby” site is required for initiation of protein synthesis at the highly structured tisB TIR by ribosome sliding to the transiently open TIR [16]. In a similar manner, we speculate that the agrB antisense RNA regulates/inhibits translation from the active +44 transcript by binding a potential “standby” site upstream of the dinQ TIR.
DinQ mode of action
In order to investigate DinQ biochemically, we have attempted to purify DinQ, including fusion peptides. However, all attempts to purify DinQ as well as chemical synthesis of the peptide failed because of the hydrophobic nature of the peptide. Further, a general feature of small hydrophobic peptides including DinQ is the lack of obvious phenotypes associated with their inactivation [17]–[20]. As an alternative strategy we characterized the phenotype of augmented DinQ in an agrB mutant. The DinQ concentration in the agrB mutant after UV treatment is elevated only two fold as compared to the wild type, indicating that the DinQ levels in the agrB mutant is physiologically relevant. Of particular interest was the 400-fold reduction in the recombination frequency in the agrB mutant as compared to wild type cells, suggesting that augmented DinQ inhibits recombination in the agrB mutant. However, genetic data suggests that DinQ may also play a role in UV protective mechanisms independent of recombination.
It appears that the large, ordered hyperstructures involved in homologous recombination are associated with the cell membrane [21]. The hyperstructures are dynamic and their size is dependent on the extent of the initial or ongoing DNA damage. The DinQ peptide is localized in the inner membrane of the cell and it is tempting to speculate about a role for DinQ in regulating DNA repair hyperstructures at the inner membrane. In addition, the prolonged period of nucleoid condensation in the agrB mutant may contribute to the impairment of DNA repair processes. Although such a direct role for DinQ is speculative, several small hydrophobic peptides have been demonstrated to modulate membrane dependent processes. The B1500 protein (65 aa) interacts with the PhoQ sensor [18], the 30 aa protein MgtR (30 aa) interacts with MgtC [19], the KdpF protein (29 aa) is part of the Kdp complex [9] and the SidA protein (29 aa) interacts directly with FtsW and FtsN [22].
The intracellular concentration of ATP was reduced in the agrB mutant compared to the wild type both before and after UV exposure. In wild type cells UV irradiation induces a two fold increase in ATP concentration during the first 20 to 30 min after exposure, and the increase is RecBC dependent [23]. These data suggest that loss of agrB and thereby excess of DinQ limit the cellular energy supply and may also explain some of the observed phenotypes. Our FLAG tag experiments revealed that DinQ increases only two fold in an agrB mutant and this apparently modest increase is sufficient to mediate dramatic effects on conjugal recombination rates, membrane depolarization, ATP levels and nucleoid reorganization. In a wild type cell population the level of DinQ translation is kept strictly under control by the LexA repressor, antisense agrB RNA and dinQ RNA processing, so for the majority of cells DinQ may never reach a level high enough to mediate the effects observed in an agrB mutant. Heterogeneity in the expression of LexA repressed genes has been observed by studying SOS promoter fusions in combination with imaging techniques and a subpopulation of cells clearly have a stronger SOS induction [24]–[26]. It is tempting to speculate that a higher level of DinQ is reached only in a subpopulation of cells where SOS induction is particularly strong or long lasting leading to a permanent or temporary agrB phenotype. Such an effect has been proposed for some toxin/antitoxin pairs in promoting formation of persister cells [27], [28]. To gain a more detailed knowledge about the biological function of DinQ the agrB mutant could be an excellent model for studying the effects of DinQ and similar hydrophobic peptides in bacterial subpopulations.
Materials and Methods
Strains, plasmids, and media
The experiments were carried out in an AB1157 background [29]. Except for chromosomal point mutations and chromosomal 3×FLAG tags all mutants were made in strain BW25113-pKD46 [30] and introduced into AB1157 via T4GT7 transduction [31]. The agrA (BK4042), agrB (BK4043) and dinQ (BK4040) single mutants were made by deleting each of the genes and introducing a kanr cassette. Next, the agrAB double mutant (BK4041) was generated by deleting both genes and introducing a kanr cassette. To construct a triple mutant the entire arsR-gor intergenic region containing dinQ, agrA and agrB was deleted (BK4044) and replaced with the kanr cassette. Table S1 summarizes all strains used and generated in this work. Vector pKK232-8 (10–25 copies pr cell in E. coli) contains a promoter less cat gene allowing selection of DNA fragments containing promoter activity [32]. pBK440 (dinQ-agrAB)/pBK444 (dinQ) is based on the vector pKK232-8 (Pharmacia) with a 2065/415 bp insert respectively from the intergenic region between arsR-gor in MCS, resulting in a plasmid that expresses E. coli dinQ from its own SOS inducible promoter. Cloning primers are listed in Table S3. Expression plasmids pET28b(+)-DinQ I, pET28b(+)-DinQ II, pET28b(+)-DinQ III, pET28b(+)-DinQ IV and pET28b(+)-DinQ V contain the DinQ I to V ORFs inserted in the NcoI-BamHI restriction sites of the pET28b(+) vector (Novagen). Chromosomal point mutations in dinQ to either introduce a premature translational stop codon in DinQ ORFV (K4stop) or introduce three point mutations in the agrB antisense region of dinQ (A108T, C112G, A115G) or to introduce a chromosomal DinQ C-terminal 3×FLAG tag were made by splicing PCR products with overlap extension (SOEing PCR) and recombine the final SOEing PCR product into a MG1655 background as described [30]. All SOEing products contained a flanking kanr cassette close to the arsR gene to facilitate selection of recombinants. To avoid unwanted recombination between the kanr cassette and the point mutations or the 3×FLAG tag during strain construction the SOEing products were transformed into strain BK5444-pKD46 which lacks the chromosomal dinQ-agrAB locus and where insertion/recombination of the SOEing products is possible only in the flanking homologous DNA sequences. The final PCR products were transformed into MG1655 containing pKD46. Cells were cured for pKD46 and insertions verified by PCR and sequencing. Details of strain construction and oligos used are listed in Tables S1 and S3, respectively. GenScript Corp. gene service constructed DinQ II and V with an N-terminal 3×FLAG tag that was inserted in the NcoI-BamHI restriction sites of the pET28b(+) vector (Novagen). Cells were grown in LB - or K-medium [33] with appropriate antibiotics (100 µg/ml ampicillin and 50 µg/ml kanamycin). For the nucleoid compaction studies cells were grown in AB minimal medium [34] supplemented with 1 µg/ml thiamine, 0.2% glucose and 0.5% casamino acids.
In vitro transcription/translation
In vitro transcription/translation on circular pET28b(+) templates or linear PCR products were performed according to Promegas protocols E. coli T7 S30 Extract System for Circular DNA and E. coli S30 Extract System for Linear Templates, respectively, with [14C]-Leucine as radiolabeled amino acid. The translation products were analysed by SDS-PAGE and visualized on Typhoon 9410 (Amersham).
Protein fractionation and membrane localization
Aliquots of exponentially growing ER2566/pET28b(+)-DinQ V were harvested by centrifugation 20 min after IPTG induction (1 mM). Cells were resuspended in 4 ml 50 mM phosphate buffer pH 7.2 and sonicated three times for 15 sec. Further fractionation was performed as described by [20]. Proteins from all fractions were acetone precipitated 1∶1 overnight at -20°C, pellets after centrifugation was resuspended in 4× NuPAGE sample loading buffer (Invitrogen) and loaded onto 10% NuPAGE Novex Bis-Tris gels (Invitrogen).
Flow cytometry
Cells were grown to OD600≈0.4 in LB and induced with IPTG (1 mM). At 0, 5 and 20 min culture samples were diluted 1∶10 in filtered AB minimal medium [34] +10 µg/ml DiBAC4(3) (Sigma-Aldrich). After 20 min incubation in the dark at room temperature, cells were analysed in a Flowcytometry LSRII (Becton Dickinson) equipped with an argon ion laser and a krypton laser (both Spectra Physics). DiBAC4(3) was detected using 488 nm laser. The distribution of DiBAC4(3) fluorescence was plotted on a logarithmic scale. The data obtained was analyzed by winMDI software.
ATP assay
Cell aliquots were harvested before and 20 min after induction with IPTG (1 mM) and washed once in 50 mM Tris-acetate pH 7.75. ATP was extracted from washed cells by 1% trichloroacetic acid (TCA) in 50 mM Tris-acetate pH 7.75 for 10 min. Tris-acetate pH 7.75 was added 1∶10 to obtain optimal pH of 7.75 before mixing with rL/L reagent (ENLITEN ATP assay, Promega) at room temperature. The amount of ATP extracted (RLU value) was measured with 20/20 Luminometer (Turner Designs) and related to the OD600 for each sample.
Frequency of recombination by Hfr conjugation
Aliquots of exponentially growing recipient strains dinQ (BK4040), agrB (BK4043) and uvrA (BK4180) were mixed in equal volumes with donor strains BW7623 (with the tetracycline resistance gene, Tn10, integrated in its chromosome) or ER2738 (carrying a tetracycline resistance conjugative plasmid) and incubated at 37°C for 30 minutes. BW7623 was used to examine chromosomal transfer to the recipient strains (recombination dependent) whereas ER2738 was used for plasmid conjugation. Cells were vortexed thoroughly and spread on selective LB plates. Hfr recombination rate and plasmid conjugation rate was calculated as number of recombinants/conjugated cells pr 106 cells.
Nucleoid compaction
Exponentially growing wt (AB1157), dinQ (BK4040) and agrB (BK4043) cells were UV irradiated with 3 J/m2 while stirring. 1.5 ml samples were taken at 0, 15, 30, 60 and 90 min after irradiation. Washed once and resuspended in 100 µl cold, filtered TE buffer. Then 1 ml of cold, filtered 77% ethanol was added for fixation. Fixed cells were mounted on a poly-L-lysine coated microscope slide and the DNA was stained with Hoechst 33258 (5 µg/ml, Sigma) in mounting medium (40% glycerol in PBS pH 7.5). Visualization of stained cells was performed using a Leica DM6000B phase-contrast/fluorescence microscope equipped with a 63× objective and a BP340-380 excitation filter. Pictures were taken using a Leica DFC350 FX digital camera that was connected to a computerized image analysis system (LAS AF software, version 2.0.0, Leica). The fluorescent image was merged with the phase-contrast image.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Fernandez De HenestrosaAR, OgiT, AoyagiS, ChafinD, HayesJJ, OhmoriH, et al. (2000) Identification of additional genes belonging to the LexA regulon in Escherichia coli. Mol Microbiol 35 : 1560–1572.
2. CourcelleJ, KhodurskyA, PeterB, BrownPO, HanawaltPC (2001) Comparative gene expression profiles following UV exposure in wild-type and SOS-deficient Escherichia coli. Genetics 158 : 41–64.
3. ZuberP (2001) A peptide profile of the Bacillus subtilis genome. Peptides 22 : 1555–1577 doi:DOI:10.1016/S0196-9781(01)00492-2.
4. IbrahimM, NicolasP, BessieresP, BolotinA, MonnetV, et al. (2007) A genome-wide survey of short coding sequences in streptococci. Microbiology 153 : 3631–3644.
5. AlixE, Blanc-PotardABa (2009) Hydrophobic peptides: novel regulators within bacterial membrane. Mol Microbiol 72 : 5–1110.1111/j.1365-2958.2009.06626.x.
6. HemmMR, PaulBJ, SchneiderTD, StorzG, RuddKE (2008) Small membrane proteins found by comparative genomics and ribosome binding site models. Mol Microbiol 70 : 1487–1501.
7. HemmMR, PaulBJ, Miranda-RiosJ, ZhangA, SoltanzadN, et al. (2010) Small Stress Response Proteins in Escherichia coli: Proteins Missed by Classical Proteomic Studies. J Bacteriol 192 : 46–58.
8. BishopRE, LeskiwBK, HodgesRS, KayCM, WeinerJH (1998) The entericidin locus of Escherichia coli and its implications for programmed bacterial cell death. J Mol Biol 280 : 583–596 doi:DOI: 10.1006/jmbi.1998.1894.
9. GasselM, MöllenkampT, PuppeW, AltendorfK (1999) The KdpF Subunit Is Part of the K+-translocating Kdp Complex of Escherichia coli and Is Responsible for Stabilization of the Complex in Vitro. J Biol Chem 274 : 37901–37907.
10. WongRS, McMurryLM, LevySB (2000) ‘Intergenic’ blr gene in Escherichia coli encodes a 41-residue membrane protein affecting intrinsic susceptibility to certain inhibitors of peptidoglycan synthesis. Mol Microbiol 37 : 364–370.
11. ColeC, BarberJD, BartonGJ (2008) The Jpred 3 secondary structure prediction server. Nucleic Acids Res 36: W197–W201.
12. Weel-SneveR, BjorasM, KristiansenKI (2008) Overexpression of the LexA-regulated tisAB RNA in E. coli inhibits SOS functions; implications for regulation of the SOS response. Nucleic Acids Res 36 : 6249–6259.
13. WickensHJ, PinneyRJ, MasonDJ, GantVA (2000) Flow Cytometric Investigation of Filamentation, Membrane Patency, and Membrane Potential in Escherichia coli following Ciprofloxacin Exposure. Antimicrob Agents Chemother 44 : 682–687.
14. OdsbuI, Morigen, SkarstadK (2009) A Reduction in Ribonucleotide Reductase Activity Slows Down the Chromosome Replication Fork but Does Not Change Its Localization. PLoS ONE 4: e7617 doi:10.1371/journal.pone.0007617.
15. VogelJ, ArgamanL, WagnerEG, AltuviaS (2004) The small RNA IstR inhibits synthesis of an SOS-induced toxic peptide. Curr Biol 14 : 2271–2276.
16. DarfeuilleF, UnosonC, VogelJr, WagnerEG (2007) An Antisense RNA Inhibits Translation by Competing with Standby Ribosomes. Mol Cell 26 : 381–392 doi:10.1016/j.molcel.2007.04.003.
17. KawanoM, OshimaT, KasaiH, MoriH (2002) Molecular characterization of long direct repeat (LDR) sequences expressing a stable mRNA encoding for a 35-amino-acid cell-killing peptide and a cis-encoded small antisense RNA in Escherichia coli. Mol Microbiol 45 : 333–349.
18. EguchiY, ItouJ, YamaneM, DemizuR, YamatoF, Okada, et al. (2007) B1500, a small membrane protein, connects the two-component systems EvgS/EvgA and PhoQ/PhoP in Escherichia coli. PNAS 104 : 18712–18717.
19. AlixE, Blanc-PotardAB (2008) Peptide-assisted degradation of the Salmonella MgtC virulence factor. EMBO J 27 : 546–557.
20. UnosonC, WagnerEG (2008) A small SOS-induced toxin is targeted against the inner membrane in Escherichia coli. Mol Microbiol 70 : 258–270.
21. Levin-ZaidmanS, Frenkiel-KrispinD, ShimoniE, SabanayI, WolfSG, MinskyA (2000) Ordered intracellular RecA-DNA assemblies: A potential site of in vivo RecA-mediated activities. Proc Natl Acad Sci U S A 97 : 6791–6796.
22. ModellJW, HopkinsAC, LaubMT (2011) A DNA damage checkpoint in Caulobacter crescentus inhibits cell division through a direct interaction with FtsW. Genes & Development 25 : 1328–1343.
23. BarbeJ, VillaverdeA, CairoJ, GuerreroR (1986) ATP hydrolysis during SOS induction in Escherichia coli. J Bacteriol 167 : 1055–1057.
24. KamensekS, PodlesekZ, GillorO, Zgur-BertokD (2010) Genes regulated by the Escherichia coli SOS repressor LexA exhibit heterogeneous expression. BMC Microbiol 10 : 283 1471-2180-10-283 [pii];10.1186/1471-2180-10-283 [doi].
25. McCoolJD, LongE, PetrosinoJF, SandlerHA, RosenbergSM, et al. (2004) Measurement of SOS expression in individual Escherichia coli K-12 cells using fluorescence microscopy. Mol Microbiol 53 : 1343–1357 10.1111/j.1365-2958.2004.04225.x [doi];MMI4225 [pii].
26. FriedmanN, VardiS, RonenM, AlonU, StavansJ (2005) Precise temporal modulation in the response of the SOS DNA repair network in individual bacteria. PLoS Biol 3: e238 doi:10.1371/journal.pbio.0030238.
27. YamaguchiY, ParkJH, InouyeM (2011) Toxin-antitoxin systems in bacteria and archaea. Annu Rev Genet 45 : 61–79 10.1146/annurev-genet-110410-132412 [doi].
28. DörrT, Vuli-çM, LewisK (2010) Ciprofloxacin Causes Persister Formation by Inducing the TisB toxin in Escherichia coli. PLoS Biol 8: e1000317 doi:10.1371/journal.pbio.1000317.
29. DewittSK, AdelbergEA (1962) The Occurrence of a Genetic Transposition in a Strain of Escherichia Coli. Genetics 47 : 577–585.
30. DatsenkoKA, WannerBL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97 : 6640–6645.
31. WilsonGG, YoungKY, EdlinGJ, KonigsbergW (1979) High-frequency generalised transduction by bacteriophage T4. Nature 280 : 80–82.
32. BrosiusJ (1984) Plasmid vectors for the selection of promoters. Gene 27 : 151–160 doi:DOI: 10.1016/0378-1119(84)90136-7.
33. SeebergE, StrikeP (1976) Excision repair of ultraviolet-irradiated deoxyribonucleic acid in plasmolyzed cells of Escherichia coli. J Bacteriol 125 : 787–795.
34. ClarkDJ, MaaløeO (1967) DNA replication and the division cycle in Escherichia coli. J Mol Biol 23 : 99–112 doi:DOI: 10.1016/S0022-2836(67)80070-6.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 2
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Complex Inheritance of Melanoma and Pigmentation of Coat and Skin in Grey Horses
- Coordination of Chromatid Separation and Spindle Elongation by Antagonistic Activities of Mitotic and S-Phase CDKs
- Autophagy Induction Is a Tor- and Tp53-Independent Cell Survival Response in a Zebrafish Model of Disrupted Ribosome Biogenesis
- Assembly of the Auditory Circuitry by a Genetic Network in the Mouse Brainstem
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy