
Three Structure-Selective Endonucleases Are Essential in the Absence of BLM Helicase in
DNA repair mechanisms in mitotically proliferating cells avoid generating crossovers, which can contribute to genome instability. Most models for the production of crossovers involve an intermediate with one or more four-stranded Holliday junctions (HJs), which are resolved into duplex molecules through cleavage by specialized endonucleases. In vitro studies have implicated three nuclear enzymes in HJ resolution: MUS81–EME1/Mms4, GEN1/Yen1, and SLX4–SLX1. The Bloom syndrome helicase, BLM, plays key roles in preventing mitotic crossover, either by blocking the formation of HJ intermediates or by removing HJs without cleavage. Saccharomyces cerevisiae mutants that lack Sgs1 (the BLM ortholog) and either Mus81–Mms4 or Slx4–Slx1 are inviable, but mutants that lack Sgs1 and Yen1 are viable. The current view is that Yen1 serves primarily as a backup to Mus81–Mms4. Previous studies with Drosophila melanogaster showed that, as in yeast, loss of both DmBLM and MUS81 or MUS312 (the ortholog of SLX4) is lethal. We have now recovered and analyzed mutations in Drosophila Gen. As in yeast, there is some redundancy between Gen and mus81; however, in contrast to the case in yeast, GEN plays a more predominant role in responding to DNA damage than MUS81–MMS4. Furthermore, loss of DmBLM and GEN leads to lethality early in development. We present a comparison of phenotypes occurring in double mutants that lack DmBLM and either MUS81, GEN, or MUS312, including chromosome instability and deficiencies in cell proliferation. Our studies of synthetic lethality provide insights into the multiple functions of DmBLM and how various endonucleases may function when DmBLM is absent.
Published in the journal:
. PLoS Genet 7(10): e32767. doi:10.1371/journal.pgen.1002315
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002315
Summary
DNA repair mechanisms in mitotically proliferating cells avoid generating crossovers, which can contribute to genome instability. Most models for the production of crossovers involve an intermediate with one or more four-stranded Holliday junctions (HJs), which are resolved into duplex molecules through cleavage by specialized endonucleases. In vitro studies have implicated three nuclear enzymes in HJ resolution: MUS81–EME1/Mms4, GEN1/Yen1, and SLX4–SLX1. The Bloom syndrome helicase, BLM, plays key roles in preventing mitotic crossover, either by blocking the formation of HJ intermediates or by removing HJs without cleavage. Saccharomyces cerevisiae mutants that lack Sgs1 (the BLM ortholog) and either Mus81–Mms4 or Slx4–Slx1 are inviable, but mutants that lack Sgs1 and Yen1 are viable. The current view is that Yen1 serves primarily as a backup to Mus81–Mms4. Previous studies with Drosophila melanogaster showed that, as in yeast, loss of both DmBLM and MUS81 or MUS312 (the ortholog of SLX4) is lethal. We have now recovered and analyzed mutations in Drosophila Gen. As in yeast, there is some redundancy between Gen and mus81; however, in contrast to the case in yeast, GEN plays a more predominant role in responding to DNA damage than MUS81–MMS4. Furthermore, loss of DmBLM and GEN leads to lethality early in development. We present a comparison of phenotypes occurring in double mutants that lack DmBLM and either MUS81, GEN, or MUS312, including chromosome instability and deficiencies in cell proliferation. Our studies of synthetic lethality provide insights into the multiple functions of DmBLM and how various endonucleases may function when DmBLM is absent.
Introduction
Crossover repair of DNA damage is associated with detrimental side effects, including loss of heterozygosity and formation of chromosome rearrangements. This genomic instability is highly deleterious, being linked to loss of cell cycle regulation and cell death; consequently, crossover (CO) formation is strongly suppressed in normal mitotic cells. One source of COs is the recombinational repair of DNA double-strand breaks (DSBs). The most widely cited model for formation of COs during DSB repair involves formation of an intermediate with two four-stranded Holliday junctions (HJs; see Figure S1) [1]. Apparent double-Holliday junction (dHJ) intermediates have been isolated as precursors of meiotic COs in Saccharomyces cerevisiae [2]. Similar structures are also formed during DSB repair in vegetative S. cerevisiae cells, though at a much lower frequency [3].
The BLM helicase has been identified as a key anti-CO factor. Mutations in the human BLM gene lead to Bloom syndrome, which is characterized by reduced size, fertility defects, immunodeficiency, and highly increased risk for a broad spectrum of cancers [4]. On the cellular level, BLM mutation increases COs between sister chromatids and homologous chromosomes, and increases the frequency of deletions and genome rearrangements [5]. This function is widely conserved, as the S. cerevisiae ortholog, Sgs1, also prevents COs during DSB repair [6] and the Drosophila ortholog, DmBLM, prevents both spontaneous and induced mitotic COs [7]. At least two models to explain the anti-CO activity of BLM have been proposed (Figure S1). First, DmBLM has been shown to promote the synthesis-dependent strand annealing (SDSA) pathway for DSB repair (Figure S1) [8]. It has been suggested that BLM's function in this pathway is to dissociate D-loops generated by strand exchange and repair synthesis [9]. Second, BLM has been proposed to catalyze convergent branch migration of the two HJs in the dHJ intermediate and facilitate subsequent decatenation by TOP3α [10]. These hypotheses are supported by in vitro demonstration of D-loop disruption, HJ branch migration, and, together with TOP3α and other proteins, dHJ dissolution activities [11]–[13].
In the absence of BLM, it is thought that COs may be generated through cleavage of a dHJ or similar structure by a HJ resolvase (dHJ resolution). The identity of the hypothesized HJ resolvase remains unknown, but may be one or more of the three structure-selective nuclear endonucleases that have been reported to cleave HJs in vitro: Mus81–Eme1/Mms4, GEN1/Yen1, and SLX4–SLX1. The first of these to be implicated in HJ resolution was Mus81-Eme1/Mms4 [14]. Mus81–Eme1 is required for most meiotic COs in S. pombe and a subset of meiotic COs in several other organisms [14]–[18], and appears to be involved in generating many spontaneous mitotic COs in S. cerevisiae [19]. However, while Mus81–Eme1/Mms4 can cut fully-ligated HJs in vitro, it has more robust activity on other structures, including nicked HJs, 3′ flaps, and structures that mimic replication forks [20]–[24]. Genetic studies implicate this enzyme in replication-associated repair [25]. Mammalian cells mutant for MUS81 or EME1 are hypersensitive to agents that generate DNA damage that blocks replication forks, such as the interstrand crosslinking agent cisplatin [26]; yeast mus81 mutants are hypersensitive to the alkylating agent methyl methanesulfonate (MMS) and UV radiation [27]. Curiously, Drosophila mus81 mutants do not display the same strong hypersensitivities or have defects in generating meiotic COs [28].
The second eukaryotic nuclease found to resolve HJs was initially purified from yeast (Yen1) and human cells (GEN1) by its HJ-resolvase activity. GEN1 and Yen1 both cut HJs in a symmetrical, re-ligatable manner; they also cut 5′ flap and replication fork-like structures, though not as well as they cut HJs [29]. In vivo functions of Yen1/GEN1 are poorly understood. S. cerevisiae yen1 mutants are not hypersensitive to DNA damaging agents and grow normally, but mus81 yen1 double mutants exhibit slow growth [30]. These double mutants also are more hypersensitive to MMS, HN2, camptothecin, and hydroxyurea and have fewer spontaneous mitotic COs than mus81 single mutants [19]. S. pombe lacks a Yen1 ortholog, but expression of human GEN1 rescues the meiotic CO and mutagen sensitivity defects of mus81 mutants [31]. Together, these observations suggest that Yen1/GEN1 primarily plays a backup role to Mus81–Mms4/Eme1.
The most recent enzyme reported to cut HJs in vitro is human BTBD12/SLX4–SLX1 [32]-[34]. Like GEN1, SLX4–SLX1 also cuts 5′ flaps and structures that mimic replication forks. Functions of this enzyme in vivo are also poorly understood. The Drosophila ortholog of SLX4 is MUS312 [35]; mus312 mutants are hypersensitive to agents that induce DNA interstrand crosslinks (ICLs), suggesting that MUS312 is required to repair ICLs [36]. Vertebrate SLX4–SLX1 has also been implicated in ICL repair, based on hypersensitivity to crosslinking agents of cells in which either subunit is knocked down by RNAi [32]–[35]. In support of this conclusion, recent studies have identified mutations in SLX4 in some patients with Fanconi anemia, a disorder associated with an aberrant response to crosslinking agents [37], [38]. Slx1, the catalytic subunit, appears to function solely when dimerized with Slx4, but Slx4 has other nuclease partners [39]. One of these is Rad1–Rad10, an endonuclease that functions in nucleotide excision repair [40]. These dual interactions are conserved in the Drosophila and vertebrate orthologs [32]–[35]. The Drosophila ortholog of Rad1–Rad10, MEI-9–ERCC1, is required for most meiotic COs. Interaction with MUS312 is essential for this function; it has been proposed that MUS312–MEI-9–ERCC1 generates COs by resolving HJs [41], [42], but in vitro analysis of this enzyme has not been published. Mouse SLX4 and the C. elegans ortholog HIM-18 are also involved in generating meiotic COs, though the extent to which different interacting nucleases are involved in these organisms is not yet clear [43], [44].
In fungi, simultaneous loss of both the BLM helicase ortholog, Sgs1, and either Mus81–Eme1/Mms4 or Slx4–Slx1 is lethal [45], [46], [47]. Studies of these synthetic lethal phenotypes have provided additional insights into functions of Sgs1/BLM and these putative HJ resolvases. Likewise, previous studies revealed that mutations in Drosophila mus309, the gene that encodes DmBLM, are synthetically lethal with mutations in mus81 or mus312 [28], [35], [48]. Genetic interactions have also been observed in Bloom syndrome cells in which these nucleases were knocked down singly or in combinations, ranging from modest decreases in sister chromatid exchange to chromosome fragmentation and decreased cell viability [49].
We have now obtained mutations in Drosophila slx1 and Gen. We find that slx1 mus309 mutants are inviable, with phenotypes similar to those of mus312 mus309 mutants. As in yeast, GEN and MUS81 have some overlapping or compensatory functions; however, Gen mutants have more severe hypersensitivities than mus81 mutants, suggesting that GEN plays a more critical role in DNA repair in Drosophila. In support of this conclusion, Gen mus309 mutants are inviable, and die much earlier in development than either mus81; mus309 or mu312 mus309 double mutants. Therefore, three putative HJ resolvases – MUS81–MMS4, GEN, and MUS312–SLX1 are essential in the absence of DmBLM. Each of the double mutants has defects in cell proliferation and features of chromosome instability, though the severities vary from genotype to genotype. The effects of blocking recombination by mutating the gene encoding the ortholog of the strand exchange protein Rad51 (SPN-A in Drosophila) also vary, from nearly complete suppression of defects to selective suppression of a subset of phenotypes. We also analyzed the effects of a mus309 mutation that abolishes a subset of DmBLM functions. Together, our results suggest models for functions of DmBLM in responding to spontaneous replication fork problems, and how MUS81–MMS4, GEN, and MUS312–SLX1 might function in alternative, DmBLM-independent pathways.
Results
mus312 mus309 Synthetic Lethality Is Due to Loss of MUS312-SLX1
We previously reported synthetic lethalities between mutations in mus309, which encodes the Drosophila ortholog of the BLM helicase, and mutations in mus81 or mus312 [28], [35], which encode subunits of putative HJ resolvases [14], [32], [33], [34]. MUS81 is the catalytic subunit of a heterodimeric endonuclease (MUS81–MMS4). MUS312 is a non-catalytic subunit that interacts with at least two nucleases, SLX1 and MEI-9–ERCC1 [35]. It seemed likely that the mus312 mus309 lethality is due to loss of the MUS312–SLX1 nuclease, since S. cerevisiae slx1 sgs1 double mutants are inviable and have phenotypes similar to those of slx4 sgs1 double mutants [47]. To determine the contributions of SLX1 and MEI-9–ERCC1 to mus312 mus309 lethality, we generated double mutants between each of them and mus309. We found that mei-9; mus309 double mutants are viable, consistent with the previous finding that MEI-9–ERCC1 interacts with MUS312 in meiotic recombination, but not in somatic DNA repair [42].
Mutations in Drosophila slx1 (CG18271) have not been reported previously. A complication in generating a mutation in this gene is that the first (non-coding) exon overlaps the first exon of MED31, which is thought to be an essential gene [50]. We therefore generated a synthetic deletion by combining a 30-kb chromosomal deficiency with a transgene that spans the region but on which we disrupted slx1 (Materials and Methods, Figure S2). As an alternative approach, we ordered Targeted Induction of Local Lesions in Genes (TILLING) from the Seattle Drosophila TILLING Project [51]. This effort identified 24 missense mutations in slx1, the most promising being one that changes a conserved phenylalanine residue to isoleucine (F93I; Figure S2). Both slx1F93I and the synthetic deletion are lethal when combined with mus309 mutations. Double mutants die early in the pupal stage, larvae lack imaginal discs, and larval neuroblasts are frequently polyploid. These phenotypes are similar to those of mus312 mus309 double mutants [35, see below]. We conclude that the inviability of mus312 mus309 double mutants is indeed due to simultaneous loss of DmBLM and MUS312–SLX1. In experiments described below, we used mus312 mus309 mutants to further characterize defects caused by loss of DmBLM and MUS312–SLX1.
MUS81–MMS4 and GEN Have Overlapping Functions
Orthologs of MUS81–MMS4 and MUS312–SLX1 have been implicated in HJ resolution. We therefore wanted to determine whether GEN, which is orthologous to the HJ resolvases Yen1 and GEN1, is also essential when DmBLM is absent. Drosophila GEN was initially identified as a novel RAD2/XPG family nuclease [52]. GEN was found to cut flaps and the lagging strand of replication fork-like structures, as well as to have a weak exonuclease activity on nicked substrates [53]. However, no stocks carrying Gen mutations have been reported. To identify such mutations, we screened through a collection of mutagen-sensitive (mus) lines for which the causative mutations had not been mapped [54]. We discovered that the two available mus324 stocks both have mutations in Gen. Although the two alleles, mus324Z4325 and mus324Z5997, were assumed to be independent, they have identical mutations (deletion of ATATAC and insertion of a single G, creating a frameshift at codons 374-5, which is within the conserved nuclease domain), suggesting that they are two isolates of the same mutational event. mus324 mutants are hypersensitive to the crosslinking agent HN2 and to the alkylating agent MMS [54]; we found that these hypersensitivities are uncovered by Df(3L)Exel6103, a deletion that removes Gen and 16 other genes (data not shown). We conclude that mus324 is Gen and hereafter refer to these alleles as GenZ4325 and GenZ5997.
In contrast to the situation in S. cerevisiae, Drosophila Gen mutants are more hypersensitive to MMS and HN2 than mus81 mutants [28,54, S. Bellendir and JS, unpublished data]. As in S. cerevisiae, however, there appears to be overlapping functions for these two nucleases. In the absence of exogenous DNA damage, mus81; Gen double mutants have wild-type survival rates relative to heterozygous siblings, but the eyes of double mutants are reduced in size and exhibit mild roughening (data not shown). This phenotype often results from cell cycle defects and/or increased apoptosis disrupting the highly ordered ommatidia. We quantified apoptosis in larval imaginal discs, which consist of proliferative diploid cells that give rise to adult epidermal structures such as eyes, wings, and legs. Imaginal wing discs from mus81 and Gen single mutants have the same levels of apoptosis as wing discs from wild-type larvae, but double mutants have significantly increased levels (Figure 1). The high level of apoptosis in the double mutant imaginal discs suggests that MUS81 and GEN have shared functions that contribute to cell survival or proliferation even in the absence of exogenously-induced DNA damage.
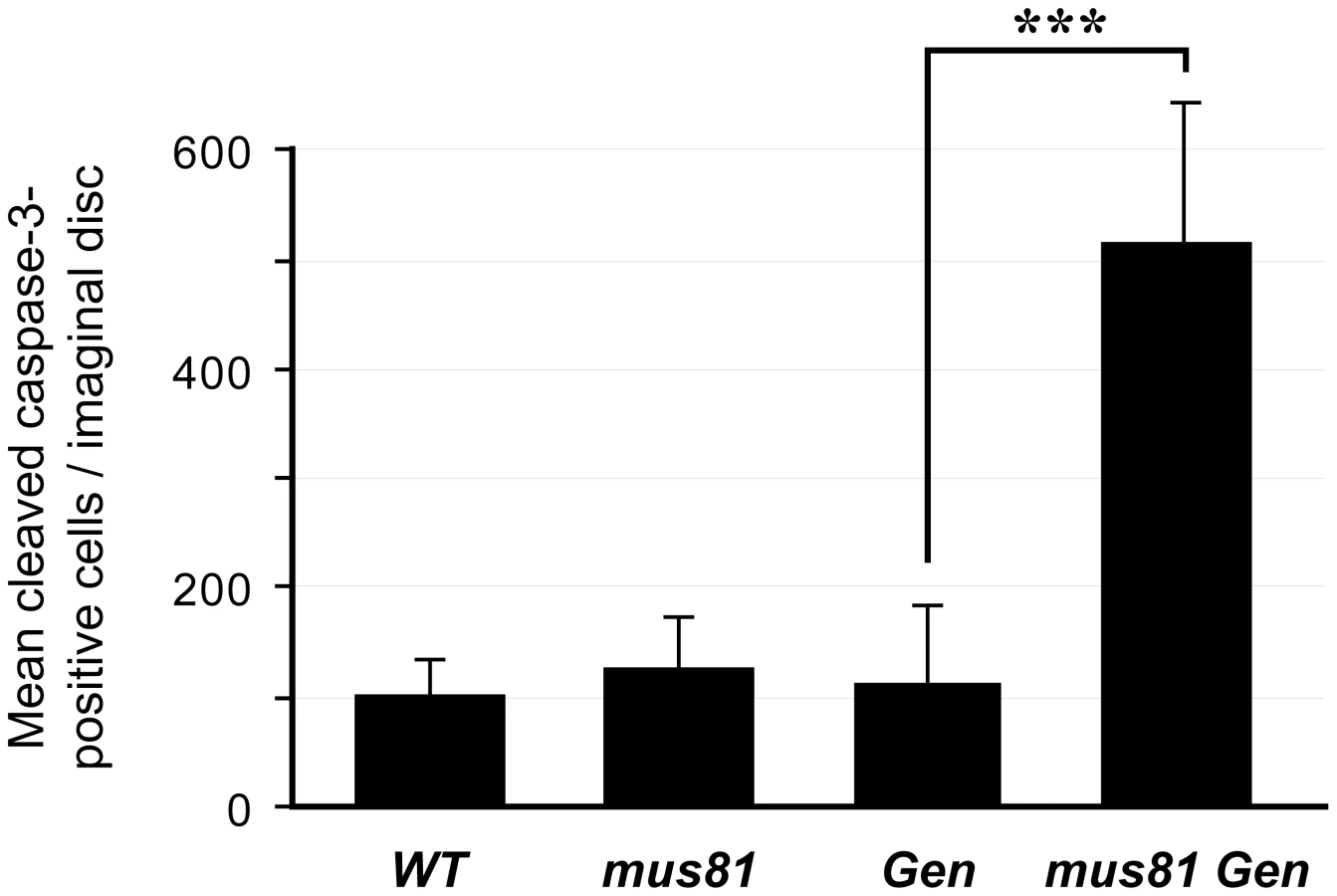
Loss of DmBLM and GEN Is Lethal
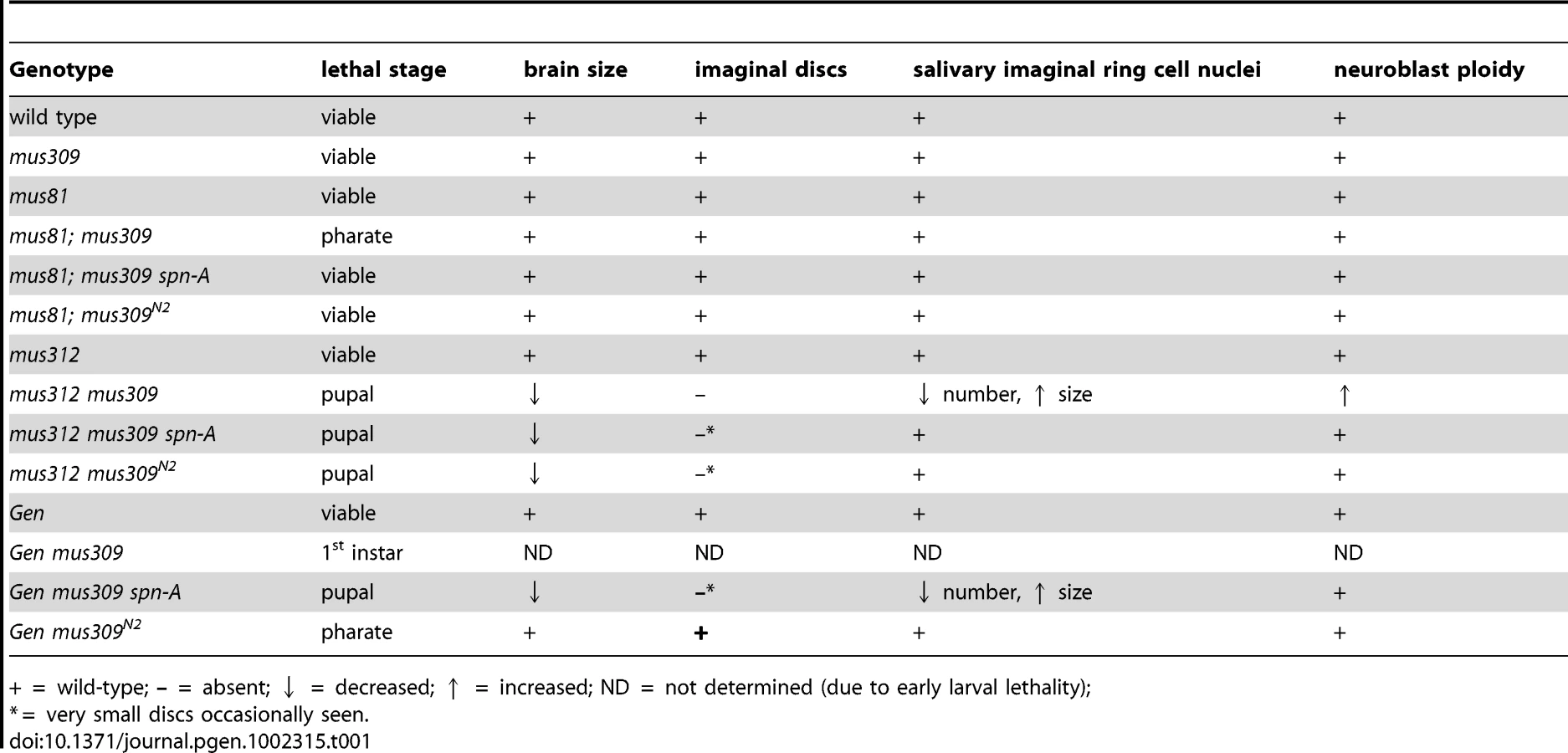
Knowing that two putative HJ resolvases (MUS81–MMS4 and MUS312–SLX1) are required when DmBLM is absent, we wanted to determine whether GEN is also essential when DmBLM is absent. We generated Gen mus309 double mutants and found that they die early in larval development, reaching only the first instar stage (Table 1; Figure 2). This is earlier in development than mus81; mus309 double mutants, which die as pharate adults (adult structures such as wings and eyes are visible within the pupal case, but no adults eclose), and also earlier than mus312 mus309 double mutants, which die at an early pupal stage. As a consequence of the genetic crosses we used (see Materials and Methods), the mus81; mus309 double mutants we analyzed have no maternal contribution of MUS81, but both Gen mus309 and mus312 mus309 double mutant larvae potentially have maternally-deposited wild-type GEN and MUS312 (there is maternal DmBLM in all three cases). The weaker lethal phenotype of mus81; mus309 mutants is therefore consistent with MUS81 contributing less to DmBLM-independent pathways than either GEN or MUS312.

Proliferation Defects in Double Mutants
To gain more insights into the causes of the three synthetic lethalities, we examined several highly proliferative larval tissues. Although most larval growth is due to enlargement of cells undergoing endocycles without mitosis, there is extensive cell proliferation in several tissues, including the neuroblasts of brain, the imaginal discs, and cells in the imaginal ring of the salivary gland. These tissues all appear to be normal in mus81; mus309 double mutants. In contrast, mus312 mus309 mutants have small brains, lack imaginal discs, and have a reduced number of salivary imaginal cells (Figure 3). Nuclei of the remaining salivary imaginal cells appear larger than in wild-type larvae, suggesting increased DNA content. These phenotypes indicate that MUS312–SLX1 has a more critical role in proliferation in the early larva than MUS81–MMS4 (Table 1). Gen mus309 mutants die too early to examine these tissues; it is likely that this early lethality is due to an even more severe defect in cell proliferation.

Chromosome Defects in Double Mutants
We hypothesized that the proliferation defects described above stemmed, at least in part, from unrepaired DNA damage and/or unresolved DNA repair intermediates. To determine whether there were gross chromosomal changes in the double mutants, we arrested larval neuroblasts with colchicine and examined mitotic nuclei for chromosome structural damage and aneuploidy. The frequencies of chromosome aberrations and aneuploidy in mitotic neuroblasts were indistinguishable between wild-type and single mutants for mus81, mus312, mus309, or Gen (Figure 3, Figure 4). In mus81; mus309 double mutants, there was an increased frequency of broken chromosomes (Figure 4A). The mus312 mus309 double mutants showed extreme genome instability: No mitotic nuclei with completely intact chromosomes were detected, and about a third showed polyploidy (Figure 3B, Figure 4). Precise chromosome counts could not be made because of the highly fragmented nature of the chromosomes, but some nuclei appeared to be more than 4N (tetraploid). The early larval death of Gen mus309 mutants precluded us from examining neuroblast chromosomes in this genotype.

Preventing Strand Exchange Suppresses Specific Phenotypes in Double Mutants
In S. cerevisiae, synthetic lethality between sgs1 and mus81 is suppressed by rad51 mutations, but lethality between sgs1 and slx4 is not [45], [55]. The Drosophila ortholog of the strand exchange protein RAD51 is encoded by spn-A [56]. As in yeast, mutation of spn-A suppresses the lethality of mus81; mus309 mutants [28]. A simple interpretation is that strand exchange mediated by SPN-A leads to a toxic intermediate that must be processed by either DmBLM or MUS81; however, a more thorough analysis of multiple genotypes suggested a more complex model [28]. Suppression of lethality of mus81; mus309 by spn-A is not complete, since the triple mutant still has increased apoptosis relative to wild-type larvae and only 70% of the triple mutants survive to adulthood [28]. We found that loss of spn-A also leads to a significant decrease in the number of chromosome breaks in neuroblast cells of mus81; mus309 mutants (Figure 4A). As with viability and apoptosis, suppression is not complete, which suggests that either a subset of the damage that DmBLM and MUS81 are required to process is not generated through a SPN-A-dependent process, or that pathways that operate when SPN-A is unavailable are not sufficient to repair all the spontaneous damage that would normally be processed through strand exchange-mediated pathways.
Mutation of spn-A has a similarly pronounced effect on the phenotype of Gen mus309 mutants. Rather than dying at the first larval instar, the Gen mus309 spn-A mutants survive to the pupal stage (Table 1). Third instar triple mutant larvae have small brains and lack imaginal discs, though very small rudimentary discs are occasionally visible. Salivary imaginal cells are reduced in number and have enlarged nuclei (Table 1). Neuroblasts from Gen mus309 spn-A larvae have numerous chromosome breaks (Table 1, Figure 4A). Thus, preventing strand exchange ameliorates the phenotypes caused by loss of GEN and DmBLM, but the triple mutants still have cell proliferation defects.
In contrast to the profound suppression of defects that arise when DmBLM and either MUS81 or GEN are absent, preventing strand invasion suppressed only some defects caused by loss of both MUS312 and DmBLM. Like mus312 mus309 double mutants, mus312 mus309 spn-A triple mutants lack imaginal discs, have small brains, and die at the pupal stage (Table 1). However, mutation of spn-A does suppresses the chromosome breakage or polyploidy phenotypes (Figure 4), as well as the defect in salivary gland imaginal cells (data not shown). The differences in the effect of spn-A mutations on the various phenotypes suggests that there are multiple circumstances in which either DmBLM or MUS312–SLX1 are essential, but that only a subset of these result from strand exchange.
A mus309 Separation-of-Function Allele Has Less Severe Phenotypes in Double Mutants
Trowbridge et al [28] reported that mus81 mutations are viable with the separation-of-function allele mus309N2. This mutation is an intragenic deletion predicted to remove the first 566 residues of DmBLM but leave the helicase domain intact [7], [28]. Previous studies showed that mus309N2 are similar to null mutants in their inability to repair DNA double-strand gaps by SDSA, their hypersensitivity to ionizing radiation, and their elevated levels of spontaneous mitotic COs [7]. However, maternal-effect embryonic lethality, which is associated with extensive anaphase bridging in early-stage embryos, is substantially reduced in mus309N2 mutants compared to null mutants, though not to wild-type levels [7]. We hypothesized that DmBLM is required during the extremely rapid early embryonic S phases, particularly in the decatenation of converging replication forks, and that DmBLMN2 is capable of carrying out this process, though not with wild-type efficiency [7]. This led to the suggestion that an important function revealed by mus81; mus309 lethality is in processing blocked or regressed replication forks, either by DmBLM-catalyzed migration or by MUS81-dependent cleavage. In this model, mus81; mus309N2 mutants are viable because DmBLMN2 retains the ability to migrate these forks. Thus, the alleviation of maternal-effect lethality in mus309N2 females and the viability of mus81; mus309N2 double mutants suggests that DmBLMN2 retains some replication-fork processing functions. In contrast, the null-equivalent defect in SDSA in mus309N2 mutants suggests that the ability to disrupt D-loops during SDSA is destroyed in DmBLMN2, while the null-equivalent elevation in mitotic COs suggests that DmBLMN2 is also unable to catalyze dHJ dissolution.
To determine the extent to which the activities retained by DmBLMN2 can compensate for the loss of GEN or MUS312–SLX1, we made Gen mus309N2 and mus312 mus309N2 double mutants. Gen mus309N2 mutants are inviable. However, rather than dying as first instar larvae, Gen mus309N2 mutants die later, as pharate adults. These double mutants have apparently normal imaginal disc size, brain size, and number/size of salivary gland imaginal cells (Table 1), but their neuroblasts frequently exhibit chromosome breaks (Figure 4A). The striking differences between Gen mus309 and Gen mus309N2 mutants in their cell proliferation phenotypes and stages of lethality suggest that GEN has an important role in processing replication-associated structures when DmBLM is not available, consistent with the known biochemical activities of GEN [29].
mus312 mus309N2 mutants are also inviable, and are similar to double mutants between mus312 and null alleles of mus309 in that larvae lack imaginal discs and lethality occurs in the pupal stage (Table 1). However, several mutant phenotypes are less severe in mus312 mus309N2 double mutants. Small, severely underdeveloped imaginal discs are sometimes detected in third-instar larvae, and in metaphase neuroblasts there are fewer damaged chromosomes and polyploidy is not seen (Figure 4). These observations suggest that defects in replication contribute to the chromosome breaks, polyploidy, and, perhaps stemming from these aberrations, proliferation defects that are seen in mus312 mus309 double mutants.
Gen mus309N2 mutants have fewer chromosome breaks than mus312 mus309N2 mutants (P = 0.3; P = 0.35 for the differences in frequency of polyploidy), but the latter die earlier. Therefore, chromosome breaks in neuroblasts are not the sole cause of lethality. The early pupal lethality of mus312 mus309N2 mutants is most likely due to the absence of imaginal discs; the reasons for the loss of this tissue are unknown, but are likely due to poor cell proliferation, elevated apoptosis, or both.
Discussion
Our studies of synthetic lethality show that at least three different structure-selective endonuclease are crucial for processing structures that persist or arise when DmBLM is absent. In the absence of induced damage, there are no obvious defects in proliferation in mus81, Gen, or mus312 single mutants, but apoptosis is significantly elevated in mus309 single mutants [28]. These observations suggests that DmBLM has several important functions that operate in the absence of damage induction by exogenous agents, and that the synthetic lethalities we have described are due to loss of secondary, DmBLM-independent pathways. Although our data do not directly implicate specific function, previous studies indicate that BLM functions primarily during S phase, most likely in repair or maintenance of blocked or damaged forks [57]. Based on these considerations, and drawing from previously published models for replication fork repair [61], we suggest functions for DmBLM, MUS81–MMS4, GEN, and MUS312–SLX1 in replication fork management.
Overlapping Functions of GEN and MUS81
In S. cerevisiae, mus81 yen1 double mutants have a slow growth phenotype [58], and we found that Drosophila mus81; Gen double mutants have elevated levels of apoptosis. Thus, in both budding yeast and flies, simultaneous loss of MUS81–MMS4 and Yen1/GEN leads to spontaneous defects in cell proliferation. Although this suggests some functional overlap, the relative contributions of the two enzymes appears to be reversed in these organisms. In yeast, mus81 single mutants are hypersensitive to a number of DNA damaging agents, but yen1 mutants are not [19], [27], [30], [58], whereas in flies, Gen mutants have severe hypersensitivities and mus81 mutants have only weak hypersensitivities [28, 54, S. Bellendir and JS, unpublished data]. It has been proposed that Mus81–Mms4 cuts nicked HJs, but if left uncut (as in mus81 mutants), these are ligated into intact HJs that are cleaved by Yen1 [30], [31], [49].
The in vitro biochemical activities of GEN and MUS81 and the drug sensitivities of single mutants suggest that these enzymes function in replication fork repair. GEN and MUS81–MMS4 cut different sides of replication fork-like substrates in vitro. Functional redundancy could be explained by the ability of either to cut blocked forks (Figure 5A, i); however, in both yeast and Drosophila one enzyme plays a larger role in surviving exogenous DNA damage, suggesting that these enzymes act on structures other than simple stalled forks. An obvious candidate is a regressed fork. Based on in vitro activities, MUS81–MMS4 would be expected to have a preference for forks that are regressed but have not undergone template switching (Figure 5A, ii), whereas GEN would be expected to cut regressed forks in which the leading strand has undergone template switching (Figure 5A, iii). The different biases in enzyme preference might be explained by differing degrees of forks regression in Saccharomyces versus Drosophila.

This model assumes that Drosophila GEN, like Yen1 and human GEN1, is an HJ resolvase. The question of whether GEN is a resolvase has important implications for understanding the partial redundancy between Drosophila GEN and MUS81–MMS4. The rescue of S. pombe mus81 mutant phenotypes by human GEN1 and studies of knockdown of these enzymes in human cells have been interpreted with respect to the HJ-cutting activities [30], [49]; however, a previous study of Drosophila GEN did not detect resolvase activity [53]. That study employed full-length GEN; it is possible that, like human GEN, the unstructured C-terminus must be removed to allow HJ cleavage in vitro [29]. Regardless of whether GEN cuts HJs, it remains possible that the genetic overlap between MUS81 and GEN is due at least in part to cleavage of other substrates that might arise during recombination or replication fork repair.
Roles of GEN and MUS81–MMS4 as Alternatives to DmBLM
Given that GEN and MUS81–MMS4 have some overlapping function(s) and yen1 sgs1 double mutants are viable in S. cerevisiae [30], we were surprised to discover that Gen mus309 double mutants are inviable. In fact, of the three synthetic lethalities we characterized, Gen mus309 double mutants have the most severe developmental phenotype. This suggests that the structures upon which GEN can act are more frequently produced and/or more deleterious when left unprocessed by DmBLM. Conversely, mus81; mus309 mutants die the latest in development and have the least severe defects in proliferation and chromosome stability, suggesting that structures upon which MUS81–MMS4 acts are less frequently produced and/or less deleterious when left unprocessed, or that there are additional repair options available.
Insights into the nature of the structures upon which either DmBLM or one of these endonucleases can act comes from the finding that double mutants with mus309N2 have much milder defects than double mutants with null alleles of mus309 (Table 1, Figure 4). mus309N2 is thought to abolish the DSB repair and dHJ dissolution functions while leaving some replication fork function(s) largely intact [7]. This suggests that synthetic lethality between Gen and mus309 and between mus81 and mus309 are not due to inability to dissolve dHJs or disrupt D-loops, but to inability to process replication fork structures. Additional clues come from the observation that preventing strand exchange partially rescues Gen mus309 and mus81; mus309. In both cases, every phenotype we studied is affected, though rescue is incomplete for each. Incomplete rescue may be because the repair methods that do not rely on strand exchange are themselves problematic, or because some repair intermediates that require either DmBLM or one of these endonucleases are generated by stand exchange and some are not.
A model that is consistent with our findings is illustrated in Figure 5A. It is believed that when a replication fork encounters a block to leading strand synthesis, the fork is regressed so that it is stabilized and so the blockage is accessible for removal (Figure 5A, ii). In some cases, the nascent leading strand may anneal to the nascent lagging strand (Figure 5A, iii). This template switch allows the leading strand to be extended, so that after reversal of the regression the block is bypassed. We hypothesize that DmBLM is required for reversal of regression. Forks that are regressed to various degrees might be cleaved by MUS81–MMS4 (Figure 5A, iv) or by GEN (Figure 5A, v). Regressed forks that are not reversed or cut are toxic and trigger apoptosis. In the absence of both DmBLM and MUS81–MMS4, template switching is still an option, but in the absence of both DmBLM and GEN, there are no further options; hence, Gen mus309 mutants have a more severe defect than mus81; mus309 mutants. DmBLMN2 is capable of reversing regressed forks, and although its activity is less than that of full-length DmBLM, it is sufficient to allow survival of most mus81; mus309N2 individuals to adulthood, and survival of Gen mus309N2 to the pharate adult stage.
Some studies suggest that Rad51 is required to protect blocked forks and perhaps to carry allow regression [59], [60], [61]. If this is true in Drosophila, then mutation of spn-A may suppress defects in double mutants by preventing fork regression, thereby blocking buildup of toxic structures. Blocked forks that are not protected by SPN-A may spontaneously break, giving rise to structures that are similar to those generated by MUS81–MMS4 or GEN cleavage (Figure 5A, dotted line). Several models have been proposed to explain how these broken forks are repaired to allow replication restart [62]. These typically involve strand invasion from the broken end into the intact sister chromatid. In Drosophila, however, we propose that continued replication from adjacent forks or from de novo firing of nearby origins converts the one-ended DSB into a two-ended DSB (Figure 5A, vi, vii). This proposal is consistent with the finding that substantial DNA synthesis persists after induction of S-phase damage in Drosophila [63]. Repair of the two-ended DSB would typically occur through DmBLM-dependent SDSA (see Figure S1). However, if DmBLM is not available to promote SDSA, repair occurs through a pathway that may generate a CO. As a consequence of pairing of homologous chromosomes in proliferating cells in Drosophila, repair will often involve interaction between homologs; this can contribute to the high elevation in mitotic COs in mus309 mutants [7]. In the most popular models, COs arise through resolution of HJ intermediates. It is possible that GEN plays a role in this process and that this also contributes to the early lethality of Gen mus309 double mutants.
Roles of MUS312–SLX1 as an Alternative to DmBLM
The mus312 mus309 synthetic lethality we describe is unique in that it is not alleviated by blocking strand exchange. This has also been reported for S. cerevisiae sgs1 slx4 lethality [47]. Fricke et al. [39] proposed that an important overlapping function between Sgs1 and Slx4–Slx1 is in rDNA replication: Sgs1–Top3 decatenates forks that stall during rDNA replication, but in the absence of Sgs1 these structures are cut by Slx4–Slx1. A similar model has been suggested in S. pombe [64]. McVey et al. [7] hypothesized that DmBLM–TOP3α is required to decatenate converging replication forks during the extremely rapid S phases of early embryonic development. At this stage of development, DNA repair processes seem to be disabled [65], so maternally-deposited DmBLM is essential for early embryonic replication. We hypothesize that DmBLM is still involved in decatenation of problematic fork convergences at later developmental stages, but that DmBLM is no longer essential because a secondary pathway is available: cleavage by MUS312–SLX1 (Figure 5B). Since converging forks are not generated by strand exchange, prevention of strand exchange (through mutation of spn-A) does not rescue lethality. Likewise, mus312 mus309N2 mutants remain inviable because DmBLMN2 is predicted to be unable to interact with TOP3α [7], an interaction that is expected to be essential for decatenation of converging forks.
Interestingly, the chromosome breakage and aneuploidy phenotypes are milder in mus312 mus309N2 and in mus312 mus309 spn-A than in mus312 mus309 null alleles (Table 1, Figure 4). This suggests that there are additional structures, generated by strand exchange but on which DmBLMN2 cannot act, that can be cleaved by MUS312–SLX1. One potential additional function for SLX4–SLX1 is in repairing DNA ICLs [32], [33], [34], [35], [42]. Given the HJ resolvase activity of human SLX4–SLX1, it seems plausible that MUS312–SLX1 cuts a single HJ intermediate or replication fork-like structures formed during ICL repair. It is unclear what defect leads to polyploidy. It has been suggested that prolonged blocks to the completion of DNA replication might prevent cytokinesis, leading to tetraploidy [66]. Consistent with this hypothesis, defects in S-phase-coupled processing of histone mRNAs leads to tetraploidy in Drosophila neuroblasts [67].
Concluding Remarks
We've established that MUS81–MMS4, GEN, and MUS312–SLX1 and are each required in the absence of DmBLM, presumably because these enzymes cleave spontaneously arising DNA structures that are usually acted upon by DmBLM. Although each of these nucleases has been considered primarily as an enzyme that cuts HJs, it is likely that the toxic intermediates that contribute to lethality also include other structures derived from replication forks. We have suggested models to explain the unique functions for each of these nucleases that become essential when DmBLM is absent. Even if these models are correct, it is likely that they describe only a subset of roles for these enzymes. Further studies of cellular phenotypes that occur in mutants lacking various combinations of these enzymes should provide additional insights into the complexities of replication fork repair and the origins and mechanisms of mitotic recombination.
Materials and Methods
Fly Stocks and Culture
Flies were raised on standard cornmeal-based media at 25°C. The following allelic combinations were used: mus312 mutants were heteroallelic for the null alleles mus312D1 (Q226ter) and mus312Z1973 (K143ter); mus309 mutants were heteroalleleic for the null alleles mus309D2 (W922ter) and mus309N1 (Δ 2480bp N-terminus) or mus309D2 and the separation-of-function allele mus309N2 (Δ1537 bp N-terminus); Gen mutants were hemizygous for Gen4325 (mus324Z4325) or Gen5997 (mus324Z5997), over Df(3L)Exel6103 (deletes 19 genes in 64C4-64C8, including Gen); mus81 mutants were homozygous (females) or hemizygous (males) for mus1Nhe1, which has a premature stop codon inserted by targeted recombination [28].
To generate a synthetic deletion of slx1, we first made Df(3R)HKK1 by inducing FLP-mediated recombination between the FRT sequences on P{XP}d03662, inserted at 425,462 (coordinates are from chromosome 3L in release 5.36 of the Drosophila genome) and PBac[37]slx1e01051, inserted at 470,260, in the 3′ untranslated region of slx1 (Figure S2). To complement genes other than slx1 that were deleted in Df(3R)HKK1, we modified the P[acman] clone CH321-44C16 [68], which carries sequences spanning 399,145 to 473,218. We used recombineering to replace 469,261 to 470,077 with a gene conferring bacterial resistance to kanamycin. The deleted region contains almost the entire slx1 coding sequence, but does not overlap with MED31. We were initially unable to get transformants of this large BAC clone, so we also replaced the 39-kb region from 399,284 to 438,520 with the bla gene that confers resistance to ampicillin. The remaining insert spans all annotated genes that are deleted in Df(3R)HKK1,but is deleted for most of slx1. This clone was transformed into a phiC31 attP landing site on 3L (P{CaryP}attP2, in 68A4; injections were done by BestGene, Inc.). We named this integration Dp(3;3)HKK2. Finally, we constructed a recombinant chromosome that has Dp(3;3)HKK2 and Df(3R)HKK1. This chromosome is therefore euploid except for the loss of slx1. Flies homozygous for this chromosome are viable and fertile.
Apoptosis in Imaginal Discs
Discs were harvested in Ringer's buffer from wandering third instar larvae and fixed in 4% formaldehyde in PBST (0.1% Triton-X in PBS) for 45 min. After washing in PBST, the discs were blocked in 5% bovine serum albumin in PBST 1 hr at room temp. They were then stained overnight at 4°C with 1∶500 anti-Cleaved Caspase-3 (Cell Signaling #966S) in PBST. The following day, the discs were stained two hours at room temperature with 1∶1000 the 2° Alexa Fluor 488 goat anti-rabbit (Molecular Probes #A11034). Discs were then washed, fine-dissected, and mounted on a slide with Fluoromount-G (SouthernBiotech #0100-01) and sealed with nail polish. Images were taken with a Nikon Eclipse E800 fluorescent microscope.
Larval Neuroblast Squashes
Third instar larvae brains were dissected and soaked in 0.1 mM colchicine in 0.7% saline for 1.5 hrs, followed by 8 min in 0.5% sodium citrate. Brains were fixed for 20 sec in a 5.5∶5.5∶1 solution of acetic acid: methanol: water. Brains were transferred to a slide and treated with 45% acetic acid for 2 min, then squashed under a siliconized coverslip. The coverslip/slide was placed on dry ice for 10 min, then the coverslip was flicked off and the slide washed in −20°C ethanol then dried for storage at 4°C. The slide was rehydrated for 5 min in 2x SSC (300 mM NaCl, 30 mM sodium citrate, pH 7.0), then stained in 2x SSC plus 1∶10,000 DAPI (1mg/mL) for 5 min, then washed in 2xSSC and air-dried. The slide was mounted with Fluoromount-G (SouthernBiotech #0100-01) and sealed with nail polish. Images were taken with a Nikon Eclipse E800 fluorescent microscope.
Salivary Gland Analysis
Salivary glands were dissected from third instar larvae in Ringer's buffer and fixed for 45 min in 4% formaldehyde in PBST (0.1% Triton-X in PBS). After PBST washes, the glands were stained with 1∶1000 DAPI (1mg/ML) 5 min and washed again. Glands were mounted on slides using Fluoromount-G (SouthernBiotech #0100-01), sealed with nail polish, and imaged on a Nikon Eclipse E800 fluorescent microscope.
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. SzostakJWOrr-WeaverTLRothsteinRJStahlFW 1983 The double-strand-break repair model for recombination. Cell 33 25 35
2. SchwachaAKlecknerN 1995 Identification of double Holliday junctions as intermediates in meiotic recombination. Cell 83 783 791
3. BzymekMThayerNHOhSDKlecknerNHunterN 2010 Double Holliday junctions are intermediates of DNA break repair. Nature 464 937 941
4. GermanJ 1993 Bloom syndrome: a mendelian prototype of somatic mutational disease. Medicine 72 393 406
5. ChagantiRSSchonbergSGermanJ 1974 A manyfold increase in sister chromatid exchanges in Bloom's syndrome lymphocytes. Proc Natl Acad Sci U S A 71 4508 4512
6. IraGMalkovaALiberiGFoianiMHaberJE 2003 Srs2 and Sgs1-Top3 suppress crossovers during double-strand break repair in yeast. Cell 115 401 411
7. McVeyMAndersenSBrozeYSekelskyJ 2007 Multiple functions of Drosophila BLM helicase in maintenance of genome stability. Genetics 176 1979 1992
8. AdamsMDMcVeyMSekelskyJ 2003 Drosophila BLM in double-strand break repair by synthesis-dependent strand annealing. Science 299 265 267
9. McVeyMLaRocqueJRAdamsMDSekelskyJ 2004 Formation of deletions during double-strand break repair in Drosophila DmBlm mutants occurs after strand invasion. Proc Natl Acad Sci U S A 101 15694 15699
10. WuLHicksonID 2003 The Bloom's syndrome helicase suppresses crossing over during homologous recombination. Nature 426 870 874
11. BachratiCZBortsRHHicksonID 2006 Mobile D-loops are a preferred substrate for the Bloom's syndrome helicase. Nucleic Acids Res 34 2269 2279
12. SunHKarowJKHicksonIDMaizelsN 1998 The Bloom's syndrome helicase unwinds G4 DNA. J Biol Chem 273 27587 27592
13. van BrabantAJYeTSanzMGermanIJEllisNA 2000 Binding and melting of D-loops by the Bloom syndrome helicase. Biochemistry 39 14617 14625
14. BoddyMNGaillardPHMcDonaldWHShanahanPYatesJR3rd 2001 Mus81-Eme1 are essential components of a Holliday junction resolvase. Cell 107 537 548
15. BerchowitzLEFrancisKEBeyALCopenhaverGP 2007 The role of AtMUS81 in interference-insensitive crossovers in A. thaliana. PLoS Genet 3 e132 doi:10.1371/journal.pgen.0030132
16. de los SantosTHunterNLeeCLarkinBLoidlJ 2003 The Mus81/Mms4 endonuclease acts independently of double-Holliday junction resolution to promote a distinct subset of crossovers during meiosis in budding yeast. Genetics 164 81 94
17. HigginsJDBucklingEFFranklinFCJonesGH 2008 Expression and functional analysis of AtMUS81 in Arabidopsis meiosis reveals a role in the second pathway of crossing-over. Plant J 54 152 162
18. HollowayJKBoothJEdelmannWMcGowanCHCohenPE 2008 MUS81 generates a subset of MLH1-MLH3-independent crossovers in mammalian meiosis. PLoS Genet 4 e1000186 doi:10.1371/journal.pgen.1000186
19. HoCKMazonGLamAFSymingtonLS 2010 Mus81 and Yen1 promote reciprocal exchange during mitotic recombination to maintain genome integrity in budding yeast. Mol Cell 40 988 1000
20. CicciaAConstantinouAWestSC 2003 Identification and characterization of the human Mus81-Eme1 endonuclease. J Biol Chem 278 25172 25178
21. ConstantinouAChenXBMcGowanCHWestSC 2002 Holliday junction resolution in human cells: two junction endonucleases with distinct substrate specificities. EMBO J 21 5577 5585
22. DoeCLAhnJSDixonJWhitbyMC 2002 Mus81-Eme1 and Rqh1 involvement in processing stalled and collapsed replication forks. J Biol Chem 277
23. GaillardPHNoguchiEShanahanPRussellP 2003 The endogenous Mus81-Eme1 complex resolves Holliday junctions by a nick and counternick mechanism. Mol Cell 12 747 759
24. OsmanFDixonJDoeCLWhitbyMC 2003 Generating crossovers by resolution of nicked Holliday junctions: a role for Mus81-Eme1 in meiosis. Mol Cell 12 761 774
25. OsmanFWhitbyMC 2007 Exploring the roles of Mus81-Eme1/Mms4 at perturbed replication forks. DNA Repair (Amst) 6 1004 1017
26. AbrahamJLemmersBHandeMPMoynahanMEChahwanC 2003 Eme1 is involved in DNA damage processing and maintenance of genomic stability in mammalian cells. EMBO J 22 6137 6147
27. InterthalHHeyerWD 2000 MUS81 encodes a novel helix-hairpin-helix protein involved in the response to UV - and methylation-induced DNA damage in Saccharomyces cerevisiae. Mol Gen Genet 263 812 827
28. TrowbridgeKMcKimKSBrillSSekelskyJ 2007 Synthetic lethality of Drosophila in the absence of the MUS81 endonuclease and the DmBlm helicase is associated with elevated apoptosis. Genetics 176 1993 2001
29. IpSCRassUBlancoMGFlynnHRSkehelJM 2008 Identification of Holliday junction resolvases from humans and yeast. Nature 456 357 361
30. BlancoMGMatosJRassUIpSCWestSC 2010 Functional overlap between the structure-specific nucleases Yen1 and Mus81-Mms4 for DNA-damage repair in S. cerevisiae. DNA Repair (Amst) 9 394 402
31. LorenzAWestSCWhitbyMC 2010 The human Holliday junction resolvase GEN1 rescues the meiotic phenotype of a Schizosaccharomyces pombe mus81 mutant. Nucleic Acids Res 38 1866 1873
32. FekairiSScaglioneSChahwanCTaylorERTissierA 2009 Human SLX4 is a Holliday junction resolvase subunit that binds multiple DNA repair/recombination endonucleases. Cell 138 78 89
33. MuñozIMHainKDeclaisACGardinerMTohGW 2009 Coordination of structure-specific nucleases by human SLX4/BTBD12 is required for DNA repair. Mol Cell 35 116 127
34. SvendsenJMSmogorzewskaASowaMEO'ConnellBCGygiSP 2009 Mammalian BTBD12/SLX4 assembles a Holliday junction resolvase and is required for DNA repair. Cell 138 63 77
35. AndersenSLBergstralhDTKohlKPLaRocqueJRMooreCB 2009 Drosophila MUS312 and the vertebrate ortholog BTBD12 interact with DNA structure-specific endonucleases in DNA repair and recombination. Mol Cell 35 128 135
36. BoydJBGolinoMDShawKESOsgoodCJGreenMM 1981 Third-chromosome mutagen-sensitive mutants of Drosophila melanogaster. Genetics 97 607 623
37. KimYLachFPDesettyRHanenbergHAuerbachAD 2011 Mutations of the SLX4 gene in Fanconi anemia. Nat Genet 43 142 146
38. StoepkerCHainKSchusterBHilhorst-HofsteeYRooimansMA 2011 SLX4, a coordinator of structure-specific endonucleases, is mutated in a new Fanconi anemia subtype. Nat Genet 43 138 141
39. FrickeWMBrillSJ 2003 Slx1-Slx4 is a second structure-specific endonuclease functionally redundant with Sgs1-Top3. Genes Dev 17 1768 1778
40. BardwellAJBardwellLTomkinsonAEFriedbergEC 1994 Specific cleavage of model recombination and repair intermediates by the yeast Rad1-Rad10 DNA endonuclease. Science 265 2082 2085
41. GreenMM 1981 mus(3)312D1, a mutagen sensitive mutant with profound effects on female meiosis in Drosophila melanogaster. Chromosoma 82 259 266
42. YıldızÖMajumderSKramerBCSekelskyJ 2002 Drosophila MUS312 interacts with the nucleotide excision repair endonuclease MEI-9 to generate meiotic crossovers. Mol Cell 10 1503 1509
43. HollowayJKMohanSBalmusGSunXModzelewskiA 2011 Mammalian BTBD12 (SLX4) protects against genomic instability during mammalian spermatogenesis. PLoS Genet 7 e1002094 doi:10.1371/journal.pgen.1002094
44. SaitoTTYoudsJLBoultonSJColaiacovoMP 2009 Caenorhabditis elegans HIM-18/SLX-4 interacts with SLX-1 and XPF-1 and maintains genomic integrity in the germline by processing recombination intermediates. PLoS Genet 5 e1000735 doi:10.1371/journal.pgen.1000735
45. FabreFChanAHeyerWDGangloffS 2002 Alternate pathways involving Sgs1/Top3, Mus81/Mms4, and Srs2 prevent formation of toxic recombination intermediates from single-stranded gaps created by DNA replication. Proc Natl Acad Sci U S A 99 16887 16892
46. KaliramanVMullenJRFrickeWMBastin-ShanowerSABrillSJ 2001 Functional overlap between Sgs1-Top3 and the Mms4-Mus81 endonuclease. Genes Dev 15 2730 2740
47. MullenJRKaliramanVIbrahimSSBrillSJ 2001 Requirement for three novel protein complexes in the absence of the Sgs1 DNA helicase in Saccharomyces cerevisiae. Genetics 157 103 118
48. Johnson-SchlitzDEngelsWR 2006 Template disruptions and failure of double Holliday junction dissolution during double-strand break repair in Drosophila BLM mutants. Proc Natl Acad Sci U S A 103 16840 16845
49. WechslerTNewmanSWestSC 2011 Aberrant chromosome morphology in human cells defective for Holliday junction resolution. Nature 471 642 646
50. BosveldFvan HoekSSibonOC 2008 Establishment of cell fate during early Drosophila embryogenesis requires transcriptional Mediator subunit dMED31. Dev Biol 313 802 813
51. CooperJLTillBJHenikoffS 2008 Fly-TILL: reverse genetics using a living point mutation resource. Fly (Austin) 2 300 302
52. IshikawaGKanaiYTakataKTakeuchiRShimanouchiK 2004 DmGEN, a novel RAD2 family endo-exonuclease from Drosophila melanogaster. Nucleic Acids Res 32 6251 6259
53. KanaiYIshikawaGTakeuchiRRuikeTNakamuraR 2007 DmGEN shows a flap endonuclease activity, cleaving the blocked-flap structure and model replication fork. Febs J 274 3914 3927
54. LaurençonAOrmeCMPetersHKBoultonCLVladarEK 2004 A large-scale screen for mutagen-sensitive loci in Drosophila. Genetics 167 217 231
55. Bastin-ShanowerSAFrickeWMMullenJRBrillSJ 2003 The mechanism of Mus81-Mms4 cleavage site selection distinguishes it from the homologous endonuclease Rad1-Rad10. Mol Cell Biol 23 3487 3496
56. Staeva-VieiraEYooSLehmannR 2003 An essential role of DmRad51/SpnA in DNA repair and meiotic checkpoint control. EMBO J 22 5863 5874
57. CheokCFBachratiCZChanKLRalfCWuL 2005 Roles of the Bloom's syndrome helicase in the maintenance of genome stability. Biochem Soc Trans 33 1456 1459
58. AgmonNYovelMHarariYLiefshitzBKupiecM 2011 The role of Holliday junction resolvases in the repair of spontaneous and induced DNA damage. Nucleic Acids Res
59. BugreevDVRossiMJMazinAV 2011 Cooperation of RAD51 and RAD54 in regression of a model replication fork. Nucleic Acids Res 39 2153 2164
60. SenguptaSLinkeSPPedeuxRYangQFarnsworthJ 2003 BLM helicase-dependent transport of p53 to sites of stalled DNA replication forks modulates homologous recombination. EMBO J 22 1210 1222
61. YoonDWangYStaplefordKWiesmullerLChenJ 2004 P53 inhibits strand exchange and replication fork regression promoted by human Rad51. J Mol Biol 336 639 654
62. BranzeiDFoianiM 2010 Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol 11 208 219
63. JaklevicBRSuTT 2004 Relative contribution of DNA repair, cell cycle checkpoints, and cell death to survival after DNA damage in Drosophila larvae. Curr Biol 14 23 32
64. CoulonSGaillardPHChahwanCMcDonaldWHYatesJR3rd 2004 Slx1-Slx4 are subunits of a structure-specific endonuclease that maintains ribosomal DNA in fission yeast. Mol Biol Cell 15 71 80
65. RaffJWGloverDM 1988 Nuclear and cytoplasmic mitotic cycles continue in Drosophila embryos in which DNA synthesis is inhibited with aphidicolin. J Cell Biol 107 2009 2019
66. GanemNJStorchovaZPellmanD 2007 Tetraploidy, aneuploidy and cancer. Curr Opin Genet Dev 17 157 162
67. SalzlerHRDavidsonJMMontgomeryNDDuronioRJ 2009 Loss of the histone pre-mRNA processing factor stem-loop binding protein in Drosophila causes genomic instability and impaired cellular proliferation. PLoS ONE 4 e8168 doi:10.1371/journal.pone.0008168
68. VenkenKJCarlsonJWSchulzeKLPanHHeY 2009 Versatile P[acman] BAC libraries for transgenesis studies in Drosophila melanogaster. Nat Methods 6 431 434
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 10
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- The Glycobiome Reveals Mechanisms of Pentose and Hexose Co-Utilization in Bacteria
- Global Mapping of Cell Type–Specific Open Chromatin by FAIRE-seq Reveals the Regulatory Role of the NFI Family in Adipocyte Differentiation
- Genetic Determinants of Serum Testosterone Concentrations in Men
- MicroRNA Expression and Regulation in Human, Chimpanzee, and Macaque Brains
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy