
Genetic Diversity in Cytokines Associated with Immune Variation and Resistance to Multiple Pathogens in a Natural Rodent Population
Pathogens are believed to drive genetic diversity at host loci involved in immunity to infectious disease. To date, studies exploring the genetic basis of pathogen resistance in the wild have focussed almost exclusively on genes of the Major Histocompatibility Complex (MHC); the role of genetic variation elsewhere in the genome as a basis for variation in pathogen resistance has rarely been explored in natural populations. Cytokines are signalling molecules with a role in many immunological and physiological processes. Here we use a natural population of field voles (Microtus agrestis) to examine how genetic diversity at a suite of cytokine and other immune loci impacts the immune response phenotype and resistance to several endemic pathogen species. By using linear models to first control for a range of non-genetic factors, we demonstrate strong effects of genetic variation at cytokine loci both on host immunological parameters and on resistance to multiple pathogens. These effects were primarily localized to three cytokine genes (Interleukin 1 beta (Il1b), Il2, and Il12b), rather than to other cytokines tested, or to membrane-bound, non-cytokine immune loci. The observed genetic effects were as great as for other intrinsic factors such as sex and body weight. Our results demonstrate that genetic diversity at cytokine loci is a novel and important source of individual variation in immune function and pathogen resistance in natural populations. The products of these loci are therefore likely to affect interactions between pathogens and help determine survival and reproductive success in natural populations. Our study also highlights the utility of wild rodents as a model of ecological immunology, to better understand the causes and consequences of variation in immune function in natural populations including humans.
Published in the journal:
. PLoS Genet 7(10): e32767. doi:10.1371/journal.pgen.1002343
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002343
Summary
Pathogens are believed to drive genetic diversity at host loci involved in immunity to infectious disease. To date, studies exploring the genetic basis of pathogen resistance in the wild have focussed almost exclusively on genes of the Major Histocompatibility Complex (MHC); the role of genetic variation elsewhere in the genome as a basis for variation in pathogen resistance has rarely been explored in natural populations. Cytokines are signalling molecules with a role in many immunological and physiological processes. Here we use a natural population of field voles (Microtus agrestis) to examine how genetic diversity at a suite of cytokine and other immune loci impacts the immune response phenotype and resistance to several endemic pathogen species. By using linear models to first control for a range of non-genetic factors, we demonstrate strong effects of genetic variation at cytokine loci both on host immunological parameters and on resistance to multiple pathogens. These effects were primarily localized to three cytokine genes (Interleukin 1 beta (Il1b), Il2, and Il12b), rather than to other cytokines tested, or to membrane-bound, non-cytokine immune loci. The observed genetic effects were as great as for other intrinsic factors such as sex and body weight. Our results demonstrate that genetic diversity at cytokine loci is a novel and important source of individual variation in immune function and pathogen resistance in natural populations. The products of these loci are therefore likely to affect interactions between pathogens and help determine survival and reproductive success in natural populations. Our study also highlights the utility of wild rodents as a model of ecological immunology, to better understand the causes and consequences of variation in immune function in natural populations including humans.
Introduction
Individuals vary in their resistance to infectious disease. Much of this variation is genetic and, in natural populations, considerable attention has focussed on the potential for pathogens to act as a selective force on genetic diversity [1], [2]. However, studies into the role of host genetics in infectious disease resistance in natural populations have thus far concentrated almost exclusively on genes of the Major Histocompatibility Complex (MHC) which, although important, account for only a fraction of the genetic variation in pathogen resistance [3], [4]. By contrast, a large body of literature now identifies numerous loci associated with resistance to specific pathogens in humans and domestic livestock [5]–[8]. This has obvious applied importance in establishing the genetic basis of variation in pathogen susceptibility, identifying individuals most vulnerable to infection and in determining novel molecular mechanisms of resistance. However, humans and domesticated animals inhabit environments that are divorced from the ecological context in which their immune system has evolved, typically lacking, for example, the nutritional and environmental stresses characteristic of natural populations [9]. In addition, studies of humans or domesticated animals have typically focussed on resistance to a single pathogen, whereas individuals in natural populations will be subject to concurrent or sequential infection by multiple pathogen species [10]. Thus, the selective forces and mechanisms that drive the genetic diversity involved in pathogen resistance are best studied in the context of the natural environment. More widely, differences between individuals in their ability to resist infection may have important consequences for their fitness, either in terms of survival or the ability to attract a mate and to reproduce. As such, discovering genes associated with pathogen resistance in natural populations has the potential to enable the identification of at-risk individuals or groups, to uncover novel mechanisms associated with infectious disease susceptibility or pathogenesis, and to help uncover the genetic basis of sexual selection in natural populations [4], [11].
In this paper we concentrate primarily on genes encoding cytokines. Cytokines are secreted signalling molecules that facilitate communication between cells of the immune system during both innate and acquired immune responses [12]. They are critical in the polarization, amplification and modulation of immune responses and help to determine which effector mechanisms are employed, and to what extent, in response to a given immune challenge [13]. Numerous studies in humans and domesticated animals have associated genetic variants at cytokine loci with susceptibility to specific pathogens or with host immunopathology [14]–[19]. These associations arise because different cytokine alleles vary in their immune activity or responsiveness, with consequent downstream effects on the development of immune effector functions [17], [20]. In natural populations too, genetic variation at cytokine loci could therefore underlie important differences between individuals in their immune responses or resistance to pathogens. For example, the balance between cytokines promoting Th1 - and Th2-type immune responses (which are broadly effective against microparasites and macroparasites, respectively) and, in turn, the modulation of these responses by regulatory T cells, is a major factor in the outcome of the host response to infection [21], [22]. Any genetic variants that influence this balance and the resulting immune profile of an individual are likely therefore to be a major factor in determining host susceptibility [23], [24]. As different arms of the immune response are counter-regulated, and different types of pathogen require different effector responses [25], cytokine variation may be particularly relevant in natural populations infected by multiple pathogen species. Thus, one cytokine genotype may upregulate a set of immune responses effective against one class of pathogen, but this may lead to downregulation of responses that are effective against others.
To our knowledge, only two previous studies have examined how genetic variation at cytokine loci impacts on pathogen resistance in wild populations [26], [27]. Both were on ungulates (free-living Soay sheep and African buffalo) and each associated microsatellite polymorphism within a single cytokine gene (Interferon gamma) with resistance to gastrointestinal nematodes. Here we test, in a natural population of field voles (Microtus agrestis), whether coding-region polymorphisms within a wide range of cytokine and other putative resistance genes are associated with, first, differential expression of immunological parameters associated with distinct components of the immune response and, second, resistance to infection for multiple pathogen species. We use a population of wild, individually monitored field voles as a study system, where we have previously demonstrated strong interactions among the multiple pathogens infecting this host [10] and for which we have developed assays to quantify activity of distinct immunological components [28]. We find repeated associations between genetic variants at three cytokine loci with both differential immunological activity and resistance to multiple pathogens, the effects of which are of comparable magnitude to other intrinsic factors more usually considered in studies of infectious disease resistance, such as sex and body weight.
Results
Gene fragments from 12 field vole immune genes were sequenced, yielding 26 single nucleotide polymorphisms (SNPs) from a total of 6,629 bp good quality coding sequence data (Table 1). Eighteen SNPs from eight of these genes were successfully genotyped in a total of 665 field voles. Several preliminary analyses were performed to check for any errors in the genotyping data, including testing for departures from Hardy-Weinberg equilibrium, the use of positive and negative genotyping controls and checking for unusual patterns of missing data. No unusual patterns were observed and all replicate genotype samples (n = 395) were successfully typed in duplicate. In addition, no genotypes were called from any of the negative controls. Finally, all animals that were genotyped at a sham Il10 SNP were correctly called as A∶A homozygotes.

Significant linkage disequilibrium (LD) was observed between most markers located within the same gene (Table S1). No significant LD was seen between the two genotyped Il12b SNPs, or between several pairwise comparisons involving the Tlr2 1383 G/A and Tlr2 1706 G/A SNPs with other Tlr2 markers. However, it should be noted that, first, minor allele frequencies are low at these loci and may not therefore give reliable estimates of LD, and second, significant LD was observed between other SNPs located within the Tlr2 gene (Table S1). No LD was detected between SNPs located in different genes.
Immunological variation
We tested whether genetic diversity within cytokine and other immune genes was associated with variation in immunological parameters, in animals killed as part of a cross-sectional study (n = 307). The immunological parameters were chosen to represent different functional arms of the immune system, and were measured as normalized mRNA expression levels, relative to a reference, pooled cDNA sample, for a suite of cytokine and transcription factor genes. Further details can be found in Materials and Methods and in [28]. Given the extent of LD observed within genes, and to reduce the number of statistical tests performed, we primarily examined the effect of haplotypes followed as necessary by post hoc single SNP analyses (see Materials and Methods). We first controlled for non-genetic intrinsic and extrinsic effects on immunological parameters. A summary of non-genetic factors associated with immunological parameters can be found in Table S2 and a more detailed analysis in [28]. The additional effect of genotype on each immunological parameter was then fitted under (i) an additive model, where the value of an immunological parameter is linearly related to the number of copies of a haplotype present; and (ii) a heterozygosity model, where heterozygotes have either increased or decreased value of an immunological parameter relative to homozygotes (i.e. molecular heterosis). Such heterosis effects have been reported acting within cytokine and other complex regulatory gene networks, possibly arising from increased stability of expression afforded by heterozygotes in a variable environment [29]–[31]. Significant associations were found between immunological parameters and genetic variation at the cytokine genes Il1b, Il2 and Il12b. Associations were limited to these genes; no immune associations were observed with genetic variation at Tgfb1 or Tnf, or with the non-cytokine membrane-bound genes (Slc11a1, Tlr2 and Tlr4).
Polymorphism within the Il1b locus was strongly associated with variation in its own mRNA expression levels where, under an additive model, individuals with the GAT haplotype were associated with the highest expression (Table 2). This was the only gene where variation within the locus was associated with differential expression of that same gene. It is unlikely that the polymorphisms typed within Il1b in this study could themselves affect transcription, as they are located in the exonic regions of the gene. Furthermore, the individual SNP that exhibited the greatest effect (Il1b 324 C/T; Table S3) is synonymous and therefore does not result in an amino acid change in the translated protein. It is then perhaps more likely that the causative mutation is an un-typed polymorphism linked to the genotyped SNPs, particularly as LD is strong within this gene (Table S1). In humans, IL1B mRNA expression has been shown to be a heritable trait and polymorphisms within the promoter region (in particular the −31 T/C SNP) are thought to be functional [17]. It is possible that the field vole Il1b gene contains a polymorphism of similar effect. Identifying the precise causative mutation and the molecular mechanism of altered Il1b expression is however beyond the scope of this study. The Il1b locus was also associated with 96 hour unstimulated Gata3 expression, with heterozygotes less likely to demonstrate a measurable Gata3 response (Table 2), but no associations with single SNPs were observed. Marginally non-significant associations were observed between Il1b and expression of Ifng (p = 0.070) and Tgfb1 (p = 0.056) under additive models, where for both genes the Il1b GGT haplotype was associated with decreased mRNA transcription. IL-1β is an important, pleiotropic mediator of the innate response and a potent pro-inflammatory cytokine. It is feasible, therefore, that polymorphism within this gene should have a discernible effect on several immunological parameters with which it interacts.

Variation within the Il2 gene was associated with Il10 expression under a heterozygosity model, with Il2 heterozygotes exhibiting lower levels of Il10 expression than homozygotes (Table 2). Il2 heterozygotes also tended to have lower Il1b expression, but this trend was marginally non-significant (p = 0.070). Unlike Il1b, variation within the Il2 gene was not associated with its own transcription levels. The observed interaction between Il2 variation and expression of Il10 might then relate to functional consequences of heterozygosity in Il2 other than altered transcription levels. The two cytokines are very likely to indirectly influence each other's expression and biological activity at the protein level; IL-10 is an anti-inflammatory cytokine which inhibits the production of many other cytokines, including IL-2 [32], [33]. There is then scope for a genetic influence on the relationship between these cytokines, but further experimental work would be needed to elucidate the precise mechanism of such an interaction in the field vole.
There were strong associations between the Il12b locus and expression of both Il1b and Il2 (Table 2). The most highly significant associations were observed under an additive model for Il1b expression, with the commonest Il12b GC haplotype linked to the lowest levels of Il1b expression, and the rarest GT haplotype associated with the greatest increase in Il1b mRNA levels. Further, under a heterozygosity model for Il2 expression, Il12b heterozygotes exhibited significantly decreased Il2 transcription. Here, however, it should be noted that because CC and GT Il12b haplotypes are rare (0.03 and 0.07, respectively, Table S10), these haplotypes virtually always occur as heterozygotes with the major GC haplotype. As a result, the heterozygote and additive models for Il12b genotype cannot be reliably distinguished and exhibit similar levels of statistical support in models of both Il1b and Il2 expression. Linkage disequilibrium between SNPs within Il12b was low, and the observed genetic effects appear to result from associations with different SNPs. The rare C variant at the Il12b 278 G/C SNP was associated with markedly decreased expression of Il2, while the minor T allele at the Il12b 704 C/T locus exhibited significantly lower levels of Il1b expression (Table S3). Both these SNPs are nonsynonymous and may therefore be the most likely of those examined to represent causal mutations. Indeed, the Il12b 704 C/T SNP is predicted to have a major impact on the activity of the translated protein, based on multiple alignments and biochemical and physical characteristics of the amino acid replacements [34] (Turner et al., in review). Effect sizes of the observed genetic associations with immunological parameters were equal to or greater than those related to important non-genetic factors (Table 3).

Pathogen resistance
We hypothesized that genetic variation at cytokines affecting immunological parameters would also affect pathogen resistance. We therefore tested for associations between genotypic variation and pathogen resistance in, first, the cross-sectional study in which immunological parameters were measured (n = 307), and second, a longitudinal study from the same study area (n = 358). Again, we first controlled for confounding non-genetic factors that may influence pathogen resistance (summarized in Tables S4, S5, S6, S7). We then tested for genetic associations as described for immunological parameters, above. We found that genetic variation at the cytokines Il1b, Il2 and Il12b, which showed associations with immunological parameters, were also associated with extensive variation in resistance to a range of pathogens (Table 4). Although some other genes (Tnf, Slc11a1 and Tlr2) also exhibited associations with certain pathogen species (Figure 1), the majority of associations were observed for the cytokines Il1b, Il2 and Il12b and, in each case, against multiple pathogen species. As these were also the only genes associated with variation in immunological parameters, we will therefore concentrate primarily on these.

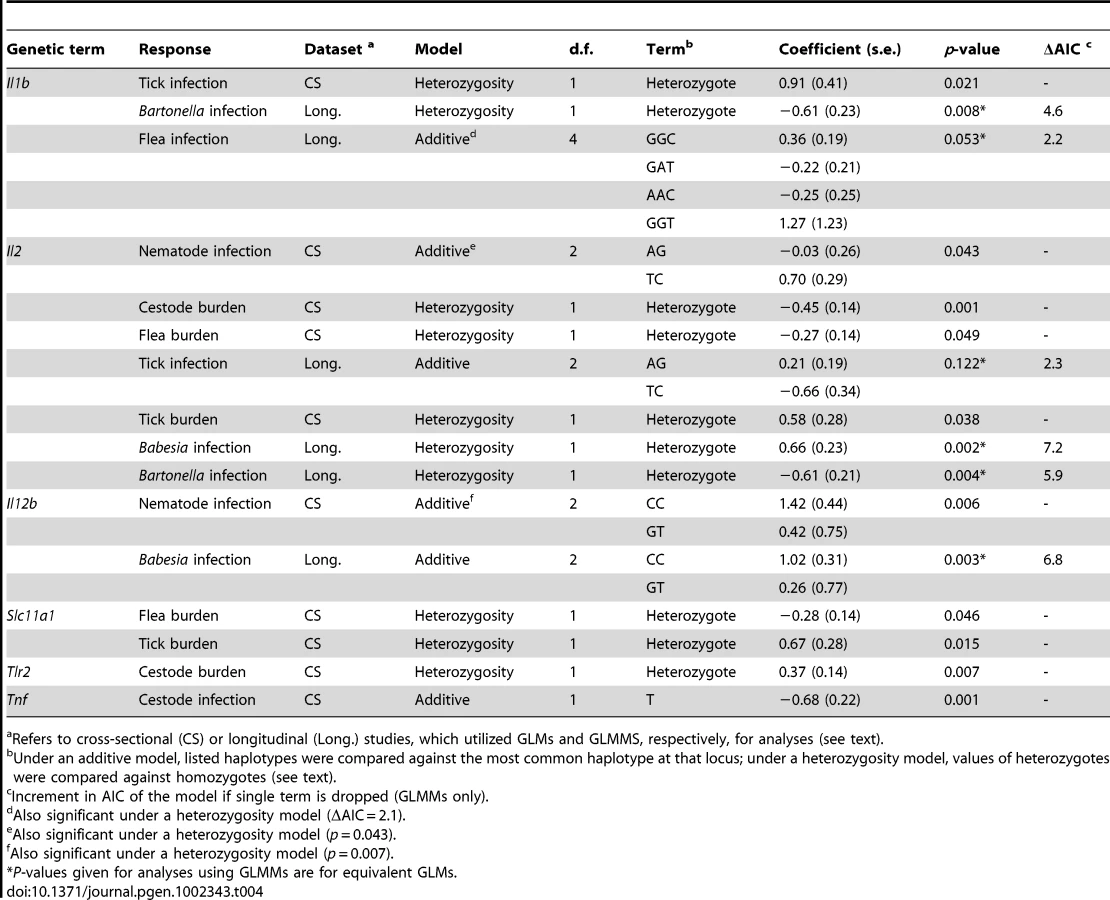
Il1b haplotype was associated with variation in the probability of infestation by ticks and fleas and infection with Bartonella (Table 4). Il1b-heterozygous individuals were more likely to be infested with ticks and less likely to be infected with Bartonella or infested with fleas. Furthermore, under an additive model, voles that carried the GGC or GGT haplotypes were more likely to be infested with fleas. Post hoc single SNP analyses revealed that the association with the probability of flea infestation was largely attributable to the nonsynonymous Il1b 253 A/G SNP, with individuals carrying the G allele at this locus more likely to be infested (Table S8).
Polymorphism within the Il2 gene was associated with resistance to a wide range of pathogens (Table 4). Significant associations under additive models were observed between Il2 and infection by nematodes and tick infestation; the TC haplotype was associated with an increased chance of infection by nematodes but, in the longitudinal study, a decreased probability of tick infestation. Voles heterozygous at the Il2 locus were more likely to harbour a Babesia microti infection but less likely to be infected with Bartonella. Furthermore, in the cross sectional study, heterozygosity at the Il2 locus was associated with lower numbers of cestodes and fleas, but an increased tick burden (Table 4).
Variation within the vole Il12b gene was associated with resistance to nematodes and to Babesia microti, each under an additive model (Table 4). In both cases, individuals carrying the Il12b CC haplotype had an increased probability of infection. Single SNP analyses revealed that these associations were due solely to the Il12b 273 G/C SNP, where the C allele increased probability of infection; no effect of the Il12b 704 T/C SNP was observed (Table S8). In all cases, the effect sizes found for cytokine variants were of comparable magnitude to those found for other intrinsic, but non-genetic, factors such as sex and body weight (Table 5). Only season, an extrinsic factor, consistently showed stronger effects on probability of pathogen infection. Although no previous studies have examined associations between Il1b, Il2, or Il12b and infectious disease in a wild species, their roles in the shaping of immune responses means it is not difficult to imagine that polymorphism within these genes may affect pathogen resistance, particularly given the associations with immunological parameters described above.

Discussion
Twin aims of this study were, first, to expand research on wildlife immunogenetics away from concentrating solely on the MHC, and second, to transfer the broader research conducted on laboratory species into the more ecologically valid arena of natural populations. We have previously demonstrated strong interactions between the multiple pathogens infecting the Kielder voles [10] and associations between seasonal and non-genetic host factors and individual variation in immunological parameters [28]. In the present study, we have explicitly controlled for such confounding non-genetic factors and have demonstrated that genetic variation at cytokine genes provides a source for phenotypic variation in both immunological parameters and in pathogen resistance in the natural environment. We have concentrated primarily on cytokines as these genes play critical roles in many physiological and immunological processes, yet, despite being well studied in humans and model organisms, have thus far been overlooked in studies of disease resistance in natural populations.
The analyses of this paper necessarily required a relatively large number of statistical tests of association, yet explicitly correcting for this (by Bonferroni methods) would likely be too conservative and could lead to the rejection of truly significant genetic associations. However, non-independence among tests – inherent in any study of a cytokine network – prevents the assignment of accurate false discovery rates. Instead, we emphasize in this discussion only those genes that exhibited repeated associations with both immunological parameters and pathogen resistance, since while single associations are inevitably prone to Type I Errors, these are unlikely where associations are repeated and consistent.
Polymorphisms within three cytokine genes, Il1b, Il2 and Il12b, were associated with variation in important components of the immune response in our natural populations of field voles (Figure 1). Effects were localized to these genes; no associations with immunological parameters were observed either for other cytokines or for any of the membrane-bound genes. Furthermore, variation within these three genes were also associated with resistance to multiple pathogen species which, taken together, is consistent with effects of genetic variation in pathogen resistance being mediated by variation in systemic immune responsiveness between cytokine genotypes. IL-1β, IL-2 and IL-12 are all broadly immunostimulatory cytokines which, through acting on the same cells, interact indirectly with one another during both the innate and adaptive immune response [16], [32]. In this study, these three genes exhibited associations with each other (Table 2; Figure 1), which is suggestive of a possible shared physiological pathway in which interactions may occur within the field vole. The IL-12 p40 subunit encoded by Il12b also forms part of the related cytokine IL-23, which is another mediator of the inflammatory response and is involved in the production of Th17 cells [35], [36]. There are then a number of possible pathways in which Il1b, Il2 and Il12b may interact in vivo, giving rise to the observed associations. Elucidating the precise mechanisms and pathways underlying such immunological interactions in wild rodents represents an exciting avenue for future work. The fact that associations are localized to these three cytokines also argues against these associations having arisen spuriously due to demographic factors, such as population subdivision or inbreeding, since any such demographic factors would act equally at all loci [37], not just Il1b, Il2 and Il12b. Furthermore, any systematic differences between sites were tested and controlled for in our statistical models. Finally, in a separate study, we show that there is no evidence for subdivision or inbreeding in the populations studied, based on the markers presented here [34] (Turner et al., in review).
The magnitude of the effects observed for genetic variation at Il1b, Il2 and Il12b on immunological parameters and pathogen resistance were of comparable size to those observed for other important intrinsic factors, such as sex and weight, which are more usually considered in studies of infectious disease susceptibility. Cytokine polymorphism therefore represents an important source of variation between individuals in immune function and pathogen resistance. This variation may, in part, be maintained by exposure to multiple pathogen species, such that a genotype conferring resistance to one pathogen species may confer increased susceptibility to a second (e.g. Il2 TC haplotypes are associated with lower probability of tick infestation but an increased risk of nematode infection, Table 4). These associations may represent an example of antagonistic pleiotropy, which has been proposed as a mechanism by which genetic diversity may be maintained within a population [38], [39]. In support of this, we have shown elsewhere that high sequence and allelic diversity observed within field vole Il1b and Il2 genes appears to have been maintained through natural selection [34] (Turner et al., in review). Alongside their direct immunological roles, cytokines also have a wide-range of non-immune physiological effects including influences on thermoregulation, appetite and fatigue [40]. It is therefore likely that an alteration in the action of cytokines will have a more significant effect on host pathology than simply its impact on infectious disease susceptibility [41]. Natural selection for resistance to a pathogen may lead to an increase in frequency of alleles that are detrimental in the absence of infection [42]. These antagonistic effects on fitness may be particularly likely to occur for immune gene polymorphisms, as many immune responses to infection, such as inflammation, can actually be harmful to host tissues. It is known in humans that the magnitude of cytokine-mediated inflammatory responses has a genetic basis and, while an effective immune response is essential for the clearance of pathogenic organisms, too strong a response can lead to significant immunopathology [43]. It is notable in this respect that Il2 polymorphism was associated both with resistance to a wide range of pathogens and to Il10 expression (Table 2 and Table 4), and that IL-10 is an anti-inflammatory cytokine associated with preventing immunopathology [44]. The complex nature of the often antagonistic genetic associations observed for Il1b, Il2 and Il12b may therefore represent a balance between normal physiological functioning and an ability to deal effectively with an unpredictable and dynamic range of co-infecting pathogens.
Our results suggest that wildlife immunogenetics should broaden its scope beyond MHC genes [4] and that cytokine variation in natural populations may have a number of equally important consequences. First, cytokine variation may help to provide populations with resilience against infectious disease [41] and so is likely to be an important source of genetic variation to conserve in endangered species. Second, our results suggest that cytokines may represent a source of ‘good genes’ as predicted by sexual selection and the immunocompetence handicap hypothesis; genes that protect males against pathogens prevalent in the environment may allow them to invest more in sexually selected traits, thereby indicating their fitness to females [45], [46].
Finally, we note that by studying wild rodent populations we have the opportunity to extend laboratory immunology to the field [47]. Wild rodents exhibit increased and much more variable immune responses than their laboratory relatives [48]. To better understand variation in susceptibility to infection – including genetic components of this variation – we must therefore examine immune responses within the ecological context in which they have evolved. In addition, because of their relatedness to traditional laboratory species, the transfer of laboratory-based immunology and genetics to wild rodents should be relatively straightforward. We also highlight the importance of measuring multiple immune parameters in natural populations to move beyond the simplified view of ‘immunocompetence’ as a single trait [12], [49]. Thus, while we do not underestimate the importance of simple measures of immunity (such as phytohemagglutinin-induced swelling) which have thus far predominated in ecological studies, a one-off measure of a single aspect of immunity is unlikely to give an accurate representation of an individual's ‘immunocompetence’ [50]. The ability to monitor the various arms of the immune response (innate, adaptive, regulatory etc.), and account for individual variation, will help us to better define the immune status of hosts and may therefore increase our understanding of how host immune profile influences life-history traits, co-infection dynamics, susceptibility to pathogens and disease transmission [12], [25], [28]. As cytokine function tends to be conserved across taxa, incorporating measurements of these important molecules into studies of ecological immunology will offer insights into many different host-parasite interactions [49]. Wild rodents may therefore provide a new model in ecological immunology, with great potential to begin to bridge the gap in our understanding between the mechanistic insights of immunity gained through studies on laboratory rodents and the variation in infectious disease susceptibility and immune responsiveness observed in humans and other natural populations.
Materials and Methods
Ethics statement
All animal procedures were performed under UK Home Office licence (40/3235) and with approval from the University of Liverpool Animal Welfare Committee.
Study site and animals
Naturally-occurring field voles were sampled, first, by a cross-sectional study involving destructive sampling of field voles followed by both immunological and pathogen assays (n = 307) [28] and, second, by a longitudinal capture-recapture study accompanied by pathogen assays (n = 358). Briefly, two grassy clear-cut sites within Kielder Forest, UK, designated SQC (55.2549, −2.6116) and BLB (55.2457, −2.6108), were sampled monthly. For the cross-sectional study (February to November 2008 and February and March 2009), curvilinear transects of 100 Ugglan special live capture traps (Grahnab, Sweden), arranged at 5–10 m intervals, were placed around the margins of each habitat in order to sample a very large area of the habitat and provide data representative of the whole clear-cut population. For the longitudinal study (March to October 2008), a rectangular 0.375 ha, 150-trap (10×15) live-trapping grid was placed centrally in each habitat, and individual voles were tagged by injecting a subcutaneous Passive Induced Transponder (PIT) tag (AVID plc., Uckfield, UK) under the skin at the back of the neck, and subsequently identified via their unique nine-digit code with hand-held scanners (AVID). In November 2008 and March 2009, larger samples of animals were captured and killed from both transect and central grid habitats at both sites, including animals that had been previously marked with transponders and processed as part of the capture-recapture study. On capture, each animal was weighed, sexed and examined for ectoparasites (see ‘Pathogen assays’, below). Reproductive maturity was also ascertained; males were classed as sexually mature if they possessed an adult coat and showed external signs of descended testes, while females were designated as mature if they had an adult coat along with a perforate vagina and/or an open pubic symphysis and evidence of lactation. Animals caught as part of the longitudinal study were sampled non-invasively by acquiring a small amount of blood from the tip of the tail, which was placed immediately in RNAlater (Qiagen, Crawley, UK) and stored at −80°C as soon as possible.
SNP discovery and genotyping
We chose an initial panel of 16 candidate genes, 13 cytokines and 3 non-cytokines (Table 1). The identification of single nucleotide polymorphisms (SNPs) within these genes is described fully elsewhere [34] (Turner et al., in review). Briefly, degenerate primers were designed from alignments of rat and mouse coding sequence and used to amplify cDNA from approximately 12 individual field vole spleens. We were able to amplify and sequence 12 of our original set of 16 candidate genes, designing genomic assays for eight of these. These included the following five cytokine genes; Interleukin 1, beta (Il1b), Il2, Il12b, Transforming growth factor, beta 1 (Tgfb1) and Tumour necrosis factor (Tnf). SNPs from three non-cytokine genes were also analysed; Solute carrier family 11a, member 1 (Slc11a1), Toll-like receptor 2 (Tlr2) and Tlr4. These genes have previously been implicated in pathogen resistance but, unlike cytokines, are all membrane bound and so lack systemic activity. No SNPs were identified in Ifng, Il5 or Il10, and we could not successfully genotype the single Il18 SNP; these genes were therefore excluded from this study. 9 lists the primers and PCR conditions used to amplify the gene products above. We were unable to amplify the field vole cytokine genes Il1a, Il4, Il12a and Il13 despite several attempts to redesign primers and optimize PCR conditions.
Genomic DNA was extracted from the livers of voles caught and killed as part of the cross-sectional study (n = 307), using DNeasy Blood and Tissue Kit (Qiagen). For the non-invasive longitudinal samples (n = 358), gDNA was extracted from tail blood using DNAzol BD Reagent (Invitrogen, Paisley, UK), specifically using the protocol of Mackey et al. [51], which is modified for extracting DNA from small volumes of blood; the resulting gDNA pellet was resuspended in 12.5 µl water and 1 µL of this solution was used as a template for whole genome amplification, using the illustra GenomiPhi V2 DNA amplification kit (GE Healthcare, Buckinghamshire, UK). Genotyping was performed by KBioscience (Hoddesdon, UK; http://www.kbioscience.co.uk) utilizing the KASPar SNP genotyping system, their own novel form of competitive allele-specific PCR, and incorporated duplicate samples, negative controls (water and blank whole genome amplification reactions) and a positive control (a monomorphic site in Il10 to check for miscalling rates) to validate reproducibility.
Preliminary analyses
Several preliminary analyses were performed as checks on the SNP genotyping data, including examination of controls and for unusual patterns of missing data. Deviations from Hardy-Weinberg equilibrium were tested for in GENEPOP 4.0 [52]–[54], using the Markov chain method to evaluate exact tests. The extent of linkage disequilibrium (LD) between SNPs was analysed using LinkDos [55], which computes genotypic or composite linkage disequilibrium estimates from diploid data [56]–[58]. Because of the large number of LD tests performed, a sequential Bonferroni correction [59], [60] was applied to the resulting p-values. Pairwise LD measurements were calculated for every pair of SNPs, to test the hypotheses that SNPs within the same gene would demonstrate high LD – and thus have greater power to pick up a phenotypic association if a causal mutation was up or downstream from the genotyped polymorphism – and that SNPs located in different genes would not demonstrate LD and genetic loci were therefore independent.
Haplotypes were inferred from genotypic data using algorithms provided by PHASE v2.1 [61], [62] within DnaSP v5 [63], using the default settings of 100 main iterations, a thinning interval of 1 and 100 burn-in iterations. To check reliability of the haplotype reconstruction, the algorithm was run five times for each data set and checked for consistency across the runs; no modifications of the default settings were necessary. PHASE imputes haplotypes even when genotypic data for an individual is missing; however, if more than one SNP per gene was missing for a particular individual the resulting inferred haplotype was deemed unreliable and excluded from further analyses. A summary of haplotype frequencies for each gene can be found in Table S10.
Immunological assays
Relative mRNA expression of several cytokines and immunity-related transcription factors was measured using quantitative real-time PCR (Q-PCR), from cultured leucocytes isolated from spleens of field voles killed as part of the cross sectional study (n = 307) [28]. Messenger RNA accumulations were measured from both unstimulated cells, reflecting constitutive levels of mRNA expression, and from cells cultured with various immunostimulatory molecules, under the assumption that observed ex vivo responses reflect the potential for that response to occur in vivo [28]. We note that protein-based assays, such as ELISAs, were not available for this species.
A summary of the immunological assays can be found in Table S11. mRNA accumulations of the following genes were measured at 24 hours: Il1b, Interferon regulatory factor 5 (Irf5; a transcription factor which regulates the production of type I interferons and inflammatory cytokines [64]), Tgfb1 and Il10, for both constitutive expression and for splenocyte cultures stimulated with either the TLR2 agonist HKLM (heat-killed Listeria monocytogenes) or the synthetic TLR7 agonist, imiquimod (InvivoGen, San Diego, USA). The production of Irf5 mRNA in the TLR2-stimulated culture and Il1b in TLR7-stimulated cells did not demonstrate a positive response to the stimuli after analysis of 100 individuals and so these measurements were ceased [28].
mRNA expression was measured at 96 hours for Ifng, Il2, T-box21 (Tbx21/Tbet; a transcription factor which is specific to the Th1 cell subset [65]), Gata binding protein 3 (Gata3; a transcription factor associated with the Th2 cell subset [66]), Forkhead box p3 (Foxp3; another transcription factor, associated with regulatory T cells [67]), Tgfb1 and Il10. Expression levels were measured in both unstimulated splenocyte cultures and cultures stimulated with the mitogen phytohaemagglutinin (PHA), which preferentially induces proliferation of activated CD4+ helper T cells [68]. For all assays, target genes were first normalized to the endogenous control gene Ywhaz (tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, zeta polypeptide) and then measured and analysed as normalized expression relative to a reference, pooled cDNA sample (the ΔΔ Ct method [69]). This method allows one to compare relative, normalized gene expression levels between animals [28].
Pathogen assays
On capture, all animals were thoroughly examined for ticks and fleas. Counts of three species of flea (Peromyscopsylla spectabilis, Ctenophthalmus nobilis vulgaris, Megabothris walkeri) were grouped as a single ‘flea’ variable in order to simplify further analyses, under the assumption that they have a similar effect on the host. Field voles caught as part of the cross-sectional study were killed by an overdose of chloroform followed by exsanguination. Each animal was then dissected and examined for infection by nematodes and cestodes. Total abundances were measured and analysed for several species; however, in order to simplify analyses and improve sample size, data were reduced and recorded as presence or absence of cestodes or nematodes. In addition, overall burdens of all cestodes were analysed for each infected vole. Animals caught as part of the longitudinal study were sampled non-destructively via tail bleeds and could not therefore be examined for helminths, with the exception of those animals captured and killed in November 2008 and March 2009.
Field voles from both studies were tested for infection by two blood-borne pathogens using specific PCR protocols described previously [70], [71]: Babesia microti, a tick-transmitted protozoan parasite, and Bartonella spp., which are flea-borne, gram-negative bacteria. Although up to five species of Bartonella circulate concurrently in rodent communities in the UK [72]–[74], all species are thought to have similar life-cycles and are predicted to interact with the host immune system in a similar way [75]; therefore, individual voles were recorded as either positive or negative for the presence of Bartonella spp.
Statistical analysis
Statistical modelling was performed using R version 2.10 [76]. The primary aim of the analyses was to examine the relationship between genetic variation and both immune function and pathogen resistance, whilst controlling for confounding non-genetic factors. The approach, for each response variable, was first to construct a minimal model containing only non-genetic terms. To this, each genetic term was added in turn to assess their significance in explaining the remaining variance for a given response variable. We chose to primarily examine genetic associations using haplotypes rather than single SNPs, for several biological and statistical reasons. First, the functional and biologically relevant product of a gene, the protein, consists of chains of amino acids whose sequences correspond to haplotypes inherited from each parent [77]. Second, variation in populations is structured into haplotype blocks which are likely to be transmitted as a single unit [77], [78]. Third, utilizing haplotypes comprised of multiple SNPs reduces the number of tests performed in comparison to testing each individual SNP. Finally, it has been recognized that haplotype-based studies are more powerful for detecting phenotypic associations resulting from untyped causal mutations in LD with genotyped SNPs/haplotypes [78], [79]. However, in addition to haplotype-based analyses, post hoc single SNP analyses were also carried out where appropriate in an attempt to identify any potential causal mutations.
Host immunological parameters were measured via the quantification of expression levels of a number of immune genes, with multiple, positively correlated measurements taken of mRNA expression of each gene (for example, innate, unstimulated expression and TLR-stimulated expression levels). Variables from the same gene were therefore grouped together, as follows. All variables were first log+1 transformed and then standardized by subtracting the mean and dividing by the standard deviation. Where two positively correlated measurements were made on the same gene, these were summed. Where more than two positively correlated measures were made, a principal components analysis (PCA) was carried out and the first principal component extracted [28]. By combining correlated measurements into a single variable for each gene, we aimed to reduce the number of statistical tests performed and also simplify interpretation by providing single variables incorporating aspects of both constitutive and potential maximal expression. In addition, as grouped measurements are likely to dilute the effect of any given single measurement, this should lead to stronger inference and more robust genetic associations. Gata3 expression values could not be combined as the two quantitative measurements taken were poorly correlated. Therefore, each Gata3 response variable was analysed separately. A summary of the immune response variables derived from standardized scores or PCA can be found in Table S12. For each of these response variables, linear models were used to construct non-genetic models containing the following terms as main effects along with their two way interactions: site (two levels: SQC and BLB); sex (2 levels: male and female); season [five levels, designated as: spring 2008 (March to May 2008), summer 2008 (June to August 2008), autumn 2008 (September to November 2008), winter 2008 (December 2008 to February 2009) and spring 2009 (March 2009)]; body weight (continuous) and eye-lens weight (continuous). Model simplification was then performed by deletion testing, to remove non-significant terms and create a base (‘minimal’) non-genetic model. Non-genetic models for Gata3 were constructed as above, except that a Box-Cox transformation [80] was performed on Gata3 mitogen-stimulated data, and a generalized linear model (GLM) with a binomial error distribution was performed on Gata3 unstimulated (detectable response/no detectable response) data. Haplotypes at each locus were tested for association with each response variable by fitting these genetic terms to the minimal non-genetic models as either (i) an additive model, where the effect, αi, of a haplotype Ai relative to haplotype A0 is assumed to be 0, αi and 2αi for genotypes A0A0, A0Ai and AiAi, respectively, or (ii) a heterozygosity model, where, for each gene, values of heterozygotes were compared to homozygotes. In some instances, both additive and heterozygote models returned significant results for the same locus, in which case the p-values for both terms are reported but the model with the lowest p-value is presented fully.
Pathogen variables from the cross-sectional data were analysed by GLMs using either a binomial error structure with a logit-link function (for binary, presence/absence variables) or quasi-Poisson errors with a log-link function (for count data which exhibited overdispersion). Non-genetic models were constructed as above. For the longitudinal data, pathogen variables were analysed using generalized linear mixed models (GLMMs) with a binomial error distribution, and fitted using restricted estimate maximum likelihood (REML) by the lmer function in R, for the following variables: site; sex; season [spring 2008 (March to May 2008), summer 2008 (June to August 2008), autumn 2008 (September to November 2008), winter 2008 (December 2008 to February 2009)]; maturity status (two levels, immature and currently/previously reproductively active); body weight (standardized by subtraction of the mean weight from each individual value); and recapture status (whether or not the animal had been caught previously). Two-way interactions were considered for all terms except recapture status. As animals caught at the same site in the same trap session experienced the same environmental conditions, site*trap session was included as a random effect [81]. To account for the correlation amongst different observations of the same individual, individual was also added as a random effect to the non-genetic model. However, due to problems with model convergence, individual could not be added to the model of Babesia microti infection. The Akaike Information Criterion (AIC) index [82] was used to select the most parsimonious non-genetic minimal model such that terms were discarded sequentially until no term could be removed without causing an increase in AIC of greater than two [83]. Genetic terms were tested under heterozygote and additive models, with significance assessed using either likelihood ratio tests (GLMs) or AIC (GLMMs). Additionally, for GLMMs, we also constructed equivalent GLMs as a check for consistency (i.e. to check that a significant association observed in a mixed model was also observed in a GLM) and to ensure that the use of mixed models and addition of random effects had not led to problems with model convergence.
Infection data were left out of the non-genetic models for the sake of parsimony. However, current infection status is likely to impact both on host immunological parameters and resistance to other pathogen species. We therefore performed post hoc tests to examine whether observed genetic associations remained after addition of pathogen data as explanatory variables. The addition of infection status had no effect on the significance of genetic associations with either immunological parameters or resistance to other pathogens.
Supporting Information
Zdroje
1. SommerS 2005 The importance of immune gene variability (MHC) in evolutionary ecology and conservation. Frontiers in Zoology 2 16
2. PiertneySBOliverMK 2006 The evolutionary ecology of the major histocompatibility complex. Heredity 96 7 21
3. JepsonABanyaWSisay-JoofFHassan-KingMNunesC 1997 Quantification of the relative contribution of major histocompatibility complex (MHC) and non-MHC genes to human immune responses to foreign antigens. Infection and Immunity 65 872 876
4. Acevedo-WhitehouseKCunninghamAA 2006 Is MHC enough for understanding wildlife immunogenetics? Trends in Ecology and Evolution 21 433 438
5. HillAVS 2001 The genomics and genetics of human infectious disease susceptibility. Annual Review of Genomics and Human Genetics 2 373 400
6. HollegaardMVBidwellJL 2006 Cytokine gene polymorphism in human disease: On-line databases, Supplement 3. Genes and Immunity 7 269 276
7. OllierWER 2004 Cytokine genes and disease susceptibility. Cytokine 28 174 178
8. de KoningD-JArchibaldAHaleyCS 2007 Livestock genomics: bridging the gap between mice and men. Trends in Biotechnology 25 483 489
9. GrenfellBTAmosWArnebergPBjørnstadONGreenmanJV 2002 Visions for future research in wildlife epidemiology. HudsonPJRizzoliAGrenfellBTHeesterbeekHDobsonAP The Ecology of Wildlife Diseases Oxford Oxford University Press 151 164
10. TelferSLambinXBirtlesRBeldomenicoPBurtheS 2010 Species interactions in a parasite community drive infection risk in a wildlife population. Science 330 243 246
11. CleavelandSHessGRDobsonAPLaurensonMKMcCallumHI 2002 The role of pathogens in biological conservation. HudsonPJRizzoliAGrenfellBTHeesterbeekHDobsonAP The Ecology of Wildlife Diseases Oxford Oxford University Press 139 150
12. GrahamALCattadoriIMLloyd-SmithJOFerrariMJBjørnstadON 2007 Transmission consequences of coinfection: cytokines writ large? Trends in Parasitology 23 284 291
13. KourilskyPTruffa-BachiP 2001 Cytokine fields and the polarization of the immune response. Trends in Immunology 22 502 509
14. GomezLMCamargoJFCastiblancoJRuiz-NarváezEACadenaJ 2006 Analysis of IL1B, TAP1, TAP2 and IKBL polymorphisms on susceptibility to tuberculosis. Tissue Antigens 67 290 296
15. Van DeventerSJH 2000 Cytokine and cytokine receptor polymorphisms in infectious disease. Intensive Care Medicine 26 Supplement 1 S98 S102
16. RomaniLPuccettiPBistoniF 1997 Interleukin-12 in infectious diseases. Clinical Microbiology Reviews 10 611 636
17. SmithAJPHumphriesSE 2009 Cytokine and cytokine receptor gene polymorphisms and their functionality. Cytokine & Growth Factor Reviews 20 43 59
18. ReedSG 1999 TGF-β in infections and infectious diseases. Microbes and Infection 1 1313 1325
19. BayleyJPOttenhoffTHMVerweijCL 2004 Is there a future for TNF promoter polymorphisms? Genes and Immunity 5 315 329
20. KelsoA 1998 Cytokines: Principles and prospects. Immunology & Cell Biology 76 300 317
21. AllenJEMaizelsRM 2011 Diversity and dialogue in immunity to helminths. Nature Reviews Immunology 11 375 388
22. VineyMERileyEMBuchananKL 2005 Optimal immune responses: Immunocompetence revisited. Trends in Ecology and Evolution 20 665 669
23. CharbonnelNGoüy de BellocqJMorandS 2006 Immunogenetics of micromammalmacroparasite interactions Micromammals and Macroparasites 401 442
24. AbbasAKMurphyKMSherA 1996 Functional diversity of helper T lymphocytes. Nature 383 787 793
25. BradleyJEJacksonJA 2008 Measuring immune system variation to help understand host-pathogen community dynamics. Parasitology 135 807 823
26. ColtmanDWWilsonKPilkingtonJGStearMJPembertonJM 2001 A microsatellite polymorphism in the gamma interferon gene is associated with resistance to gastrointestinal nematodes in a naturally-parasitized population of Soay sheep. Parasitology 122 571 582
27. EzenwaVOEtienneRSLuikartGBeja-PereiraAJollesAE 2010 Hidden consequences of living in a wormy world: Nematode-induced immune suppression facilitates tuberculosis invasion in African buffalo. American Naturalist 176 613 624
28. JacksonJABegonMBirtlesRPatersonSFribergIM 2011 The analysis of immunological profiles in wild animals: a case study on immunodynamics in the field vole, Microtus agrestis. Molecular Ecology 20 893 909
29. MarquetSDoumboOCabantousSPoudiougouBArgiroL 2008 A functional promoter variant in IL12B predisposes to cerebral malaria. Human Molecular Genetics 17 2190 2195
30. ComingsDEMacMurrayJP 2000 Molecular heterosis: A review. Molecular Genetics and Metabolism 71 19 31
31. RidruechaiCMahasirimongkolSPhromjaiJYanaiHNishidaN 2010 Association analysis of susceptibility candidate region on chromosome 5q31 for tuberculosis. Genes and Immunity 11 416 422
32. BorishLCSteinkeJW 2003 2. Cytokines and chemokines. Journal of Allergy and Clinical Immunology 111 S460 S475
33. SteinkeJWBorishL 2006 3. Cytokines and chemokines. Journal of Allergy and Clinical Immunology 117 S441 S445
34. TurnerAK 2010 Immunogenetics and polymorphism in a natural population of field voles. PhD Thesis: University of Liverpool, UK. Accessible at http://research-archive.liv.ac.uk/1468/
35. KasteleinRAHunterCACuaDJ 2007 Discovery and Biology of IL-23 and IL-27: Related but Functionally Distinct Regulators of Inflammation. Annual Review of Immunology 25 221 242
36. WeaverCTHarringtonLEManganPRGavrieliMMurphyKM 2006 Th17: An Effector CD4 T Cell Lineage with Regulatory T Cell Ties. Immunity 24 677 688
37. BlackWCBaerCFAntolinMFDuTeauNM 2001 Population genomics: Genome-wide sampling of insect populations Annual Review of Entomology 441 469
38. RoffDABenediktHBrianKH 2005 Variation and Life-History Evolution. Variation Burlington Academic Press 333 357
39. RoseMR 1982 Antagonistic pleiotropy, dominance, and genetic variation1. Heredity 48 63 78
40. CorwinEJ 2000 Understanding cytokines. Part I: Physiology and mechanism of action. Biological research for nursing 2 30 40
41. DowningTLloydATO'FarrellyCBradleyDG 2010 The Differential Evolutionary Dynamics of Avian Cytokine and TLR Gene Classes. Journal of Immunology 184 6993 7000
42. DeanMCarringtonMO'BrienSJ 2002 Balanced polymorphism selected by genetic versus infectious human disease. Annual Review of Genomics and Human Genetics 3 263 292
43. GrahamALAllenJEReadAF 2005 Evolutionary causes and consequences of immunopathology Annual Review of Ecology, Evolution, and Systematics 373 397
44. CouperKNBlountDGRileyEM 2008 IL-10: The master regulator of immunity to infection. Journal of Immunology 180 5771 5777
45. HamiltonWDZukM 1982 Heritable true fitness and bright birds: A role for parasites? Science 218 384 387
46. FolstadIKarterAJ 1992 Parasites, bright males, and the immunocompetence handicap. American Naturalist 139 603 622
47. PedersenABBabayanSA 2011 Wild immunology. Molecular Ecology 20 872 880
48. AbolinsSRPocockMJOHafallaJCRRileyEMVineyME 2011 Measures of immune function of wild mice, Mus musculus. Molecular Ecology 20 881 892
49. HawleyDMAltizerSM 2011 Disease ecology meets ecological immunology: understanding the links between organismal immunity and infection dynamics in natural populations. Functional Ecology 25 48 60
50. DemasGEZyslingDABeechlerBRMuehlenbeinMPFrenchSS 2011 Beyond phytohaemagglutinin: Assessing vertebrate immune function across ecological contexts. Journal of Animal Ecology DOI:10.1111/j.1365-2656.2011.01813.x. Epub March 14 2011
51. MackeyKSteinkampAChomczynskiP 1998 DNA extraction from small blood volumes and the processing of cellulose blood cards for use in polymerase chain reaction. Applied Biochemistry and Biotechnology - Part B Molecular Biotechnology 9 1 5
52. RaymondMRoussetF 1995 GENEPOP (Version 1.2): Population Genetics Software for Exact Tests and Ecumenicism. Journal of Heredity 86 248 249
53. RoussetF 2008 GENEPOP'007: a complete re-implementation of the genepop software for Windows and Linux. Molecular Ecology Resources 8 103 106
54. GuoSWThompsonEA 1992 Performing the exact test of Hardy-Weinberg proportion for multiple alleles. Biometrics 48 361 372
55. Garnier-GerePDillmannC 1992 A computer program for testing pairwise linkage disequilibria in subdivided populations. Journal of Heredity 83 239
56. WeirBS 1996 Genetic Data Analysis II Sunderland, MA Sinaur Associates, Inc
57. BlackWCKrafsurES 1985 A FORTRAN program for the calculation and analysis of two-locus linkage disequilibrium coefficients. TAG Theoretical and Applied Genetics 70 491 496
58. WeirBS 1979 Inferences about Linkage Disequilibrium. Biometrics 35 235 254
59. RiceWR 1989 Analyzing tables of statistical tests. Evolution 43 223 225
60. HolmS 1979 A Simple Sequentially Rejective Multiple Test Procedure. Scandinavian Journal of Statistics 6 65 70
61. StephensMScheetP 2005 Accounting for decay of linkage disequilibrium in haplotype inference and missing-data imputation. American Journal of Human Genetics 76 449 462
62. StephensMSmithNJDonnellyP 2001 A new statistical method for haplotype reconstruction from population data. American Journal of Human Genetics 68 978 989
63. LibradoPRozasJ 2009 DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25 1451 1452
64. PaunAReinertJTJiangZMedinCBalkhiMY 2008 Functional characterization of murine interferon regulatory factor 5 (IRF-5) and its role in the innate antiviral response. Journal of Biological Chemistry 283 14295 14308
65. MullenACHighFAHutchinsASLeeHWVillarinoAV 2001 Role of T-bet in commitment of Th1 cells before IL-12-dependent selection. Science 292 1907 1910
66. ZhuJYamaneHCote-SierraJGuoLPaulWE 2006 GATA-3 promotes Th2 responses through three different mechanisms: Induction of Th2 cytokine production, selective growth of Th2 cells and inhibition of Th1 cell-specific factors. Cell Research 16 3 10
67. HoriSNomuraTSakaguchiS 2003 Control of regulatory T cell development by the transcription factor Foxp3. Science 299 1057 1061
68. O'DonovanMRJohnsSWilcoxP 1995 The effect of PHA stimulation on lymphocyte sub-populations in whole-blood cultures. Mutagenesis 10 371 374
69. LivakKJSchmittgenTD 2001 Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25 402 408
70. BownKJLambinXTelfordGROgdenNHTelferS 2008 Relative importance of Ixodes ricinus and Ixodes trianguliceps as vectors of Anaplasma phagocytophilum and Babesia microti in field vole (Microtus agrestis) populations. Applied and Environmental Microbiology 74 7118 7125
71. TelferSBownKJSekulesRBegonMHaydenT 2005 Disruption of a host-parasite system following the introduction of an exotic host species. Parasitology 130 661 668
72. TelferSBegonMBennettMBownKJBurtheS 2007 Contrasting dynamics of Bartonella spp. in cyclic field vole populations: The impact of vector and host dynamics. Parasitology 134 413 425
73. TelferSCloughHEBirtlesRJBennettMCarslakeD 2007 Ecological differences and coexistence in a guild of microparasites: Bartonella in wild rodents. Ecology 88 1841 1849
74. BirtlesRJHazelSMBennettMBownKRaoultD 2001 Longitudinal monitoring of the dynamics of infections due to Bartonella species in UK woodland rodents. Epidemiology and Infection 126 323 329
75. OliverMKTelferSPiertneySB 2009 Major histocompatibility complex (MHC) heterozygote superiority to natural multi-parasite infections in the water vole (Arvicola terrestris). Proceedings of the Royal Society B: Biological Sciences 276 1119 1128
76. R Development Core Team 2009 R: A language and environment for statistical computing Vienna, Austria R Foundation for Statistical Computing
77. ClarkAG 2004 The role of haplotypes in candidate gene studies. Genetic Epidemiology 27 321 333
78. YangYLiSSChienJWAndriesenJZhaoLP 2008 A systematic search for SNPs/haplotypes associated with disease phenotypes using a haplotype-based stepwise procedure. BMC Genetics 9
79. VasemägiAPrimmerCR 2005 Challenges for identifying functionally important genetic variation: the promise of combining complementary research strategies. Molecular Ecology 14 3623 3642
80. BoxGEPCoxDR 1964 An analysis of transformations (with discussion). Journal of the Royal Statistical Society B 26 211 252
81. TelferSBirtlesRBennettMLambinXPatersonS 2008 Parasite interactions in natural populations: insights from longitudinal data. Parasitology 135 767 781
82. AkaikeH 1973 Information theory and an extension of the maximum likelihood principle. PetranBNCsakiF International Symposium on Information Theory Budapest Akademiai Kiadi 267 281
83. JohnsonJBOmlandKS 2004 Model selection in ecology and evolution. Trends in Ecology and Evolution 19 101 108
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 10
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- The Glycobiome Reveals Mechanisms of Pentose and Hexose Co-Utilization in Bacteria
- Global Mapping of Cell Type–Specific Open Chromatin by FAIRE-seq Reveals the Regulatory Role of the NFI Family in Adipocyte Differentiation
- Genetic Determinants of Serum Testosterone Concentrations in Men
- MicroRNA Expression and Regulation in Human, Chimpanzee, and Macaque Brains
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy