
A Barcode Screen for Epigenetic Regulators Reveals a Role for the NuB4/HAT-B Histone Acetyltransferase Complex in Histone Turnover
Dynamic modification of histone proteins plays a key role in regulating gene expression. However, histones themselves can also be dynamic, which potentially affects the stability of histone modifications. To determine the molecular mechanisms of histone turnover, we developed a parallel screening method for epigenetic regulators by analyzing chromatin states on DNA barcodes. Histone turnover was quantified by employing a genetic pulse-chase technique called RITE, which was combined with chromatin immunoprecipitation and high-throughput sequencing. In this screen, the NuB4/HAT-B complex, containing the conserved type B histone acetyltransferase Hat1, was found to promote histone turnover. Unexpectedly, the three members of this complex could be functionally separated from each other as well as from the known interacting factor and histone chaperone Asf1. Thus, systematic and direct interrogation of chromatin structure on DNA barcodes can lead to the discovery of genes and pathways involved in chromatin modification and dynamics.
Published in the journal:
. PLoS Genet 7(10): e32767. doi:10.1371/journal.pgen.1002284
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002284
Summary
Dynamic modification of histone proteins plays a key role in regulating gene expression. However, histones themselves can also be dynamic, which potentially affects the stability of histone modifications. To determine the molecular mechanisms of histone turnover, we developed a parallel screening method for epigenetic regulators by analyzing chromatin states on DNA barcodes. Histone turnover was quantified by employing a genetic pulse-chase technique called RITE, which was combined with chromatin immunoprecipitation and high-throughput sequencing. In this screen, the NuB4/HAT-B complex, containing the conserved type B histone acetyltransferase Hat1, was found to promote histone turnover. Unexpectedly, the three members of this complex could be functionally separated from each other as well as from the known interacting factor and histone chaperone Asf1. Thus, systematic and direct interrogation of chromatin structure on DNA barcodes can lead to the discovery of genes and pathways involved in chromatin modification and dynamics.
Introduction
The epigenetic landscape in the cell is dynamic and shaped by histone modifying and demodifying enzymes. In addition, histones themselves can also be dynamic; they can be moved along the DNA through the action of ATP-dependent nucleosome remodeling enzymes or can be evicted and replaced by new histones. Many histone modifying and remodeling enzymes have been identified and several factors have been found to be involved in changing nucleosome occupancy during gene activation and repression [1]–[3]. Recent studies indicate that histones can also be replaced by replication-independent mechanisms that do not involve obvious changes in nucleosome occupancy [3]–[9]. The replacement of existing chromatin-bound histones by newly synthesized histones most likely affects the stability of chromatin marks and thereby epigenetic mechanisms of gene regulation.
Histone replacement or turnover requires assembly and disassembly of nucleosomes, processes that most likely involve the action of histone chaperones. Chaperones are acidic proteins that bind the highly basic soluble histone proteins and thereby prevent non-specific interactions of histones with other proteins and DNA [10]–[12]. The HAT-B complex is one of the factors that binds newly synthesized histones H3 and H4 in the cytoplasm [13]. This evolutionary conserved complex, composed of the chaperone Hat2 and the acetyltransferase Hat1 (also known as Kat1), acetylates newly synthesized soluble histone H4 on lysine 12 (H4K12) and lysine 5 (H4K5) [14]–[17]. Hat1 specifically acts on soluble histones because it is inactive towards chromatin-bound nucleosomal histones [13]. Hat1 is the founding (and still only known) member of the family of type B HATs, which are cytoplasmic and specific for free histones [13], [14]. Whether the HAT-B complex or its acetyltransferase activity towards the H4 tail has a role in subsequent steps of histone trafficking or chromatin assembly is not well understood [14]. Cells lacking the HAT-B complex show no growth defect, indicating that acetylation of newly synthesized histones by Hat1 is not essential for replication-dependent histone deposition [14]. In addition, the acetylation marks introduced by HAT-B are removed upon deposition of new histones in chromatin [14]. However, several studies have indicated connections between Hat1 and chromatin [15], [18]–[24]. In addition, recent biochemical studies suggest that HAT-B guides newly synthesized histones from the cytoplasm to the nucleus, where it binds to the histone chaperone Hif1 to form the NuB4 complex and hand over the histones to other chaperones such as Asf1 [25], [26]. Asf1 is involved in the stimulation of H3K56 acetylation on soluble histones prior to their deposition [11], [12]. By binding to the chromatin assembly factor complex (CAF1) and chaperone Rtt106, Asf1 can subsequently deliver histones for deposition at the replication fork [27]–[30]. In addition, Asf1 can bind to the HIR complex and thereby deliver histones for replication-independent histone deposition [11], [12], [27]–[29], [31], [32]. How chaperones affect histone assembly and disassembly is still largely unknown but recent studies are starting to reveal some of the underlying mechanisms [30], [33]–[36].
We recently developed Recombination-Induced Tag Exchange (RITE) as an assay to measure histone turnover under physiological conditions [7]. RITE is a genetic pulse-chase method in which replacement of old by new histones can be examined by immunoblots or chromatin immunoprecipitation (ChIP). To unravel the significance of the high rate of histone turnover that we and others observed in yeast [4]–[9], [37], the underlying mechanisms will need to be identified. However, identification of genes involved in histone turnover is not straightforward. Screening for mutants that affect epigenetic processes is usually carried out using indirect read-outs such as activity of reporter genes or developmental phenotypes. Mutants that affect histone post-translational modifications have also been identified by global proteome analysis [38]. However, it is not clear whether and how histone turnover affects gene expression, reporter genes, or developmental phenotypes. As a consequence, no indirect reporter assays are available to screen for histone turnover genes by mutant hunts. The alternative, direct assessment of chromatin changes in mutant clones is typically laborious (involving ChIP-sequencing or ChIP-on-chip) and is usually not suitable for genetic screening. To speed up the discovery of histone turnover pathways, we directly interrogated chromatin structure using RITE combined with methods that have been developed for parallel analysis of fitness phenotypes in yeast [39], [40]. Using this strategy we identified mutants that either positively or negatively affected histone turnover and we provide the first in vivo evidence for a function of the NuB4 complex in histone exchange.
Results
Outline of a barcode screen for histone turnover mutants
The collection of gene-deletion mutants in Saccharomyces cerevisiae enables the systematic analysis of gene function. A pair of unique DNA barcodes (UpTag and DownTag) is present in each yeast deletion strain, flanking a common selectable marker gene used to knock out the respective genes (Figure 1). Molecular counting of the barcodes by DNA microarrays or digital counting by next-generation sequencing allows parallel analysis of the relative abundance of yeast clones in pooled cultures [40], [41]. The fitness of each yeast deletion mutant can be inferred from the changes in the relative abundance of the barcodes after exposure to the condition of interest. Using these same principles, we reasoned that in a pool of yeast deletion mutants the relative abundance of each barcode in a ChIP experiment might report on the abundance of a particular chromatin mark in that region in each mutant. Here we refer to the identification of epigenetic regulators by a barcode-ChIP-Seq approach as Epi-ID (Figure 1).

To explore the possibilities of Epi-ID and to search for genes involved in histone turnover we used the genetic pulse-chase method RITE to allow the detection of old and new histone H3 proteins in yeast [7] (Figure 1). Briefly, following deletion of one histone H3 gene copy, the sole remaining H3 gene was tagged with an HA tag flanked by LoxP sites, and a downstream orphan T7 tag. Initially all H3 proteins are tagged with an HA tag. Upon induction of a hormone-dependent Cre recombinase by the addition of estradiol, the HA tag in the genome is replaced by the T7 tag and from then on all newly synthesized H3 will be T7 tagged. Histone turnover results in replacement of H3-HA by H3-T7, which can be detected and quantified by immunoblot and ChIP (Figure 2). We note that histone turnover measurements obtained using RITE correlate well with measurements obtained using the previously used inducible pGAL-system to ectopically overexpress a tagged copy of histone H3 [42]. One of the advantages of RITE is that the tagged histone gene is expressed from its endogenous promoter, and old and newly synthesized histone H3 can be simultaneously detected and followed under any (physiological) condition of interest, independent of changes in nutrients to induce ectopic promoters [7], [43].
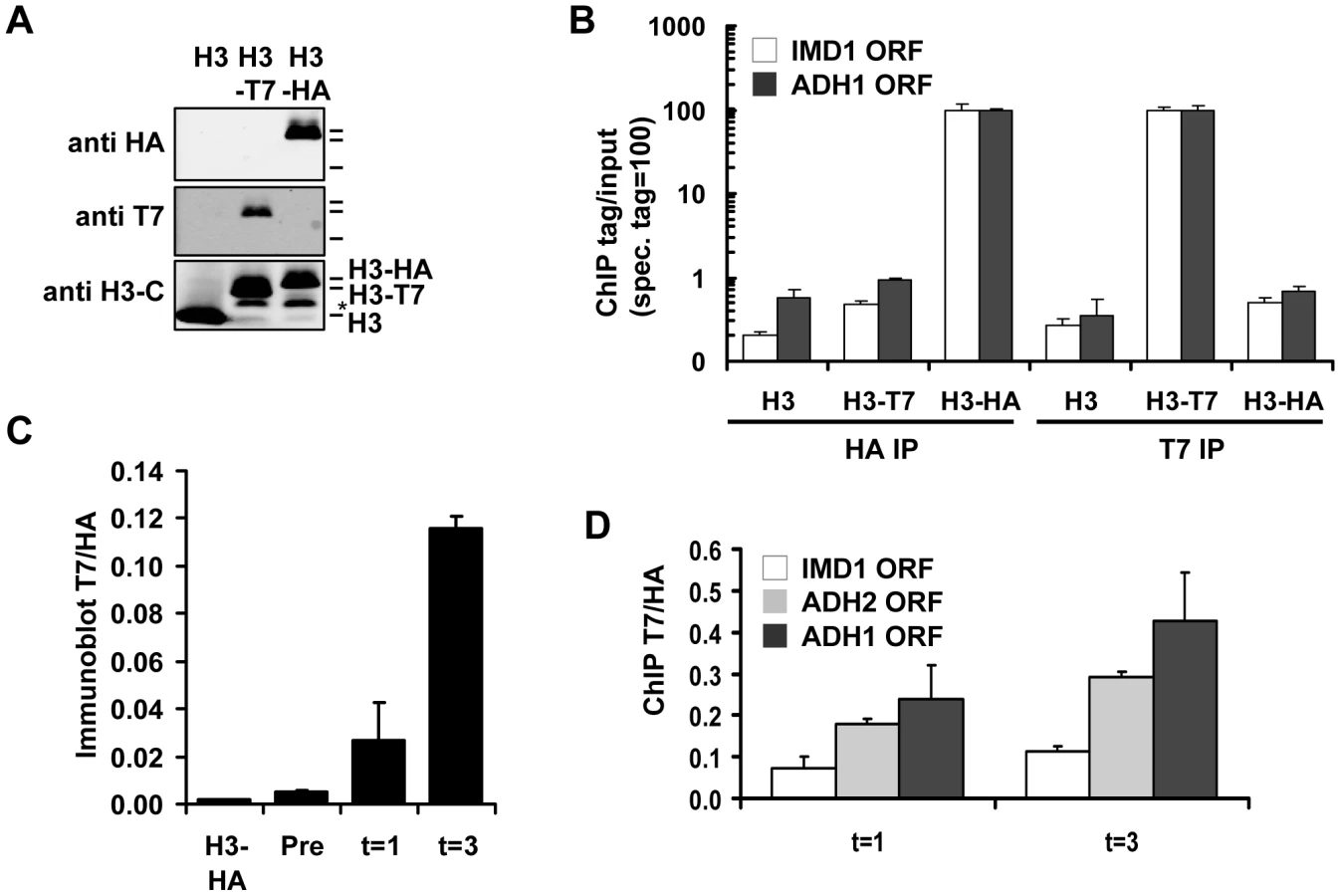
We introduced the RITE elements into 92 clones of the yeast deletion collection using Synthetic Genetic Array (SGA) analysis [44] (Figure 1). The deletions in this library represented genes known or suspected to be involved in epigenetic processes and a set of non-chromatin genes (Table S1). The clones of this new library of RITE deletion mutants were first grown separately in liquid cultures, then pooled, and subsequently arrested by starvation (Figure 3A and Figure S1). Recombination was induced to switch the epitope tags and chromatin samples were taken before and one and three days after induction of the tag switch. We previously found that yeast cells arrested by starvation (which we here refer to as G0) undergo replication-independent turnover of chromatin-bound histones [7]. In addition, we found a substantial amount of new bulk histone synthesis during three days of starvation by immunoblot analysis and ChIP (Figure 2C-2D and Figure S2). Arresting cells by starvation allows for efficient switching of the epitope tags by the induced Cre recombinase. Moreover, replication-dependent histone deposition and cell-cycle or growth rate differences between different mutants are eliminated. To measure histone turnover ChIP was performed on old (H3-HA) and new (H3-T7) histone H3. The barcode regions in the bound DNA were amplified using common primer sequences and adapters to allow parallel sequencing on the Illumina platform. Four base pair index tags were introduced in each sample to allow multiplex analysis (Figure S1). After digital barcode counting (see Materials and Methods) the relative ratio of new/old H3 was calculated as a value for replication-independent histone turnover in the pool of gene deletion mutants for each UpTag and DownTag barcode and for each of two time points after induction of the tag-switch (Figure 1, Figure 3A).

Validation of Epi-ID and candidate mutants
We performed three analyses to test the validity of the concept of Epi-ID. First, we verified that the independent measurements of the two time points (day 1 and 3) showed similar trends (Figure 3B–3C). Second, we compared UpTags with DownTags (U and D). The overall correlation between UpTag and DownTag barcodes suggests that position effects are not a major confounder in this assay (Figure 3D–3E; but also see Discussion). The few clones that did not correlate well between different time points or between UpTag and DownTag barcodes were eliminated from further analysis (see below). Third, the barcodes of the SIR3 and SIR4 deletion mutants (which do not mate and cannot be used for genetic crosses such as SGA), were integrated in the genome of strains constitutively expressing only H3–HA or only H3-T7. These clones were combined with the RITE library pool as internal negative and positive controls, respectively. The two control strains could be separated from each other at all three time points, both at the UpTag and DownTag barcodes. They also provided an indication of the dynamic range of the turnover measurements in this assay. For further analysis, clones for which severe growth defects were observed after the tag switch (and after release of the arrest by re-feeding) were excluded to eliminate mutants in which the new H3-T7 tagged histone may not be fully functional or causes tag-specific rather than true turnover effects (see Materials and Methods). Only those clones were included that showed low variation between the two time points and between UpTag and DownTag. The two control strains are shown as a reference (Figure 3F).
Of the resulting set of deletion mutants that passed the selection criteria, two clones with the lowest and two clones with the highest turnover signal were selected to examine whether the mutants affected turnover at loci independent of the barcode sequences. Each clone was grown individually and arrested by starvation. After induction of the epitope tag switch histone turnover was examined by ChIP-qPCR at four independent loci unrelated to the barcoded region analyzed in the parallel screen (promoter regions of IMD1, ADH2, HHT2, and ADH1) (Figure 3G). The changes in histone turnover at these four loci was similar to the changes measured at the barcodes, confirming that the chromatin changes of the barcodes reflected overall changes in the genome (Figure 3G). Nhp10 and Gis1 were found to be negative regulators of histone turnover. Hat1 positively regulated histone turnover. For every turnover experiment, the efficiency of the tag switch (percent of cells that had undergone a Cre-mediated recombination event) was determined (Table S2). By a colony plating assay we noticed that cells lacking HAP2 showed very poor Cre-mediated recombination, which was most likely the cause of the low ratio of new/old H3 in this clone (Figure S3). This clone was excluded from further analysis. Given the high conservation of Hat1 and its known activity towards new histones, we focused our further studies on Hat1.
The role of Hat1 in histone turnover
The histone acetyltransferase Hat1 together with the histone chaperone Hat2 forms the evolutionary conserved HAT-B complex that acetylates soluble histones. The functional consequences of Hat1's activity are not well understood. Hat1 plays a role in gene silencing [19] and DNA repair [14], suggesting that it affects chromatin structure. However, many of these phenotypes require additional mutations in the N-terminal tail of histone H3 and how chromatin is affected by Hat1 is not known. Our findings provide direct evidence that the Hat1 protein is important for efficient histone turnover in vivo (Figure 3F-3G). We first examined the role of Hat1's enzymatic activity. A strain containing a catalytically compromised (but not completely inactive) Hat1 protein (HAT1-E255Q) [19] showed a decrease in histone turnover similar to a hat1Δ strain (Figure 4A), suggesting that the acetyltransferase activity is important for efficient histone turnover. Hat1's primary known targets are lysines 5 and 12 of histone H4 (H4K5 and H4K12). Mutating the target lysines to arginine (H4K5,12R) did not substantially affect histone H3 turnover, whereas alanine or glutamine mutants (H4K5,12A and H4K5,12Q) showed enhanced turnover of histone H3 at most loci tested (Figure 4B and Figure S4). Arginine and lysine both contain a long hydrophobic side chain and a positive charge. Therefore, arginine might mimic the constitutively unacetylated (positively charged) state of lysine. Our results suggest that loss of the positive charge of H4K5,12 by acetylation is not sufficient to explain the role of Hat1 in histone turnover. However, changing the positively charged residues to neutral amino acids enhanced turnover. These results suggest that H4K5/K12 play a role in histone turnover but that loss of acetylation of these sites is not sufficient to cause a histone turnover defect.

Several lines of evidence suggest that H4K5 and H4K12 have evolutionary conserved roles in replication-dependent chromatin assembly [13], [26], [45]–[49] or nuclear import of histone H4 [50], [51]. However, in yeast, mutation of these lysines does not lead to growth defects and no changes in global chromatin organization have been observed [51]–[53]. To investigate the role of H4K5,12 in replicating cells we performed the tag switch in starved cells, released the switched population into fresh media, and then measured histone turnover in cells arrested in G2/M after one round of replication (monitored by FACS analysis). Cells expressing H4K5,12Q showed increased turnover at two of the three promoter regions analyzed, whereas cells expressing H4K5,12R showed decreased histone turnover (Figure 4C). In contrast, in two long coding sequences, the two H4K5,12 mutants affected turnover in a similar manner. In both H4K5,12 mutants turnover at the 3′ end was reduced relative to turnover at 5′ regions (Figure 4C). This is consistent with results we obtained previously with the H4K5,12R mutant in replicating cells and may indicate a role of these residues in 3′ to 5′ retrograde movement of old histones by passage of RNA Polymerase II [54].
All members of the NuB4 complex promote histone turnover
Hat1 in yeast and other organisms was initially identified as a cytoplasmic histone acetyltransferase [13], [14]. More recently, Hat1 was also found to be (predominantly) localized in the nucleus [14], [19], [26], [49], [55]. To investigate whether the role of Hat1 in histone turnover is mediated by a cytoplasmic or nuclear activity, we next examined the consequences of fusion of Hat1 to a nuclear export signal (Hat1-NES), which excludes Hat1 from the nucleus [19]. The NES fusion resulted in a modest decrease of histone turnover (Figure 5A), indicating that the cytoplasmic activity of Hat1 is not sufficient for Hat1's function in histone turnover and that at least part of Hat1's effect on histone turnover is mediated by a nuclear activity. To further investigate whether Hat1's role in histone turnover is indeed linked to its nuclear location, we analyzed the nuclear binding partners of Hat1. In the nucleus the members of the HAT-B complex, Hat1 and Hat2, interact with Hif1 (Hat1 Interacting Factor-1) and form the nuclear NuB4 complex [49], [55]. Hif1 belongs to the evolutionary conserved family of SHNi-TPR family of histone chaperones, which also includes Hs_NASP, Xl_N1/N2 and Sp_Sim3 [26], [56]. To examine the role of the NuB4 complex in histone turnover, we deleted Hif1 and compared this to independent deletions of Hat1 and Hat2. In this strain background, deletion of Hat1 by homologous recombination (which was confirmed by standard PCR analysis and by microarray analysis [e.g. see Figure S9]) did not affect histone turnover as much as in the mutant derived from the genetic cross with the yeast deletion collection or the catalytic mutant. The cause of this difference is unknown, but may involve differences in the genetic strain backgrounds.. However, cells lacking Hat2 or Hif1 showed reduced histone turnover, supporting the idea that the nuclear NuB4 complex plays a role in histone turnover (Figure 5B). To genetically test whether Hif1 and Hat-B affect turnover by means of a common pathway or protein complex (NuB4), we generated double mutant strains for epistasis analysis. Previous studies have shown that Hat2 is a central component of the NuB4 complex; deletion of HAT2 disrupts the nuclear localization of Hat1 and interactions between Hif1, Hat1, and histones [19], [49], [55]. Unexpectedly, deleting either HAT1 or HAT2 in combination with HIF1 resulted in a more severe decrease in histone turnover than in either one of the single mutants, suggesting that Hif1 and Hat1/Hat2 act at least in part by independent mechanisms (Figure 5C).

Histone turnover is strongly correlated with and induced by transcription by RNA Polymerase II [4]–[7] (and Figure S2). To investigate whether the observed G0 histone turnover defects in mutants of the NuB4 complex were caused by transcription defects we performed expression profiling and measured RNA Polymerase II occupancy. In mutant cells arrested in G0, no significant changes were found in the expression of the target genes analyzed in the turnover experiments when compared to wild-type cells (Figure 6A–6B). In addition, no significant changes (fold change >1.7, p<0.01) were found in the expression of the single H3 and H4 genes, with the exception of the H4K5,12 mutants, which showed a slight upregulation of the histone H3 gene. Thus, in G0 cells, reduced histone H3 turnover was not caused by reduced expression of (new) histones or by reduced expression of the loci at which histone turnover was measured. To compare transcriptional changes in the NuB4 mutants to other mutants, we also performed microarray analyses of NuB4 mutants made in the genetic background of the yeast deletion collection. These mutants were grown under standard mid-log conditions [57], [58]. We note that under these conditions the members of the NuB4 complex play the same role in histone turnover as in G0 (Figure S5). In general, no significant transcriptional changes were found in any of the NuB4 mutants compared to WT (fold change >1.7, p<0.01) in mid-log cultures. However, when examined in more detail, the expression profiles of mutants that contain a deletion of HIF1 and to a lesser extent HAT2, showed upregulation of the genes encoding histone H3 and H4 in mid-log cultures (Figure 6C). Regulation of histone gene expression seems to be a common property of nucleosome assembly factors [27], [59], providing further support for a link between the NuB4 complex and histone turnover.

Biochemical studies suggest that the NuB4 complex interacts with Asf1, which led to the suggestion that NuB4 might hand over newly synthesized histones to Asf1 for subsequent transfer to nucleosome assembly factors [25], [26], [60]. However, the histone genes clearly respond differently to deletion of ASF1 than to deletion of genes encoding members of the NuB4 complex [27], [59] (Figure 6C), suggesting a more complex relationship. Unfortunately, we could not test the genetic relationship between Asf1 and Hat1 because deletion of Asf1 in the strain background used for the RITE assay is lethal, similar to what has been reported previously [61]. To investigate the connection between Hat1 and Asf1 by alternative means, we used RITE as a genetic pulse-chase tool to examine the nature of the histone molecules bound to each protein. Rather than indirectly inferring the origin of the histones (new or chromatin derived) from the pattern of post-translational modifications, the epitope tag-switch pulse-chase allows for a direct distinction between resident and newly synthesized histones. In cells that had recently undergone a tag switch on H3 and therefore contained a mix of new and old histone H3, affinity purified Hat1 bound both new and old histones with a preference for new histones (Figure 7A–7B and Figure S6). Asf1 also bound both new and old histones but without a preference for new histones. (Figure 7A–7B and Figure S6). The binding of Hat1 and Asf1 to a different subset of the pool of soluble histones suggests that they affect different steps of chromatin assembly and disassembly.

Discussion
Using RITE as a biochemical-genetic pulse-chase tool, we previously observed rapid exchange of histone H3 in chromatin in yeast cells outside S-phase [7]. Similar results have been reported using an inducible pGAL-system to overexpress an ectopic tagged histone H3 copy [4]–[6], [37], [62], [63]. By using RITE, in contrast to the pGAL system, the tagged old and new histone H3 species are expressed by the endogenous H3 promoter from the endogenous chromosomal location [43]. Therefore, the high levels of histone exchange observed with RITE were not caused by misregulation of histone H3 expression. Indeed, qRT-PCR and microarray analyses showed that RITE strains containing tagged H3 express very similar H3 mRNA levels as wild-type cells containing untagged H3 at different phases of the cell cycle [7] (Figure S7). Interestingly, although histone mRNAs are cell cycle regulated and peak in S-phase when the demand for new histones is highest [64], [65], histone H3 mRNA expression is still relatively high outside S-phase, providing an explanation for the abundant synthesis of new histones outside S-phase [7]. To investigate the biological function of histone turnover and its consequences for chromatin structure and function, we developed the Epi-ID barcode screen for chromatin regulators and combined it with RITE. In this screen we found that Hat1 and subsequently also the other members of the NuB4 complex positively regulate histone turnover. To our knowledge, our data provide the first evidence that a Type B histone acetyltransferase complex regulates histone assembly in vivo. Hat1 was the first histone acetyltransferase identified [13], [66]. It is part of a multi-subunit complex that interacts with histone chaperones and acetylates free histones but is inactive towards nucleosomal histones [14], [26]. The biological significance of these biochemical activities of the Hat1 complexes remained elusive [14] although in genetic tests Hat1 was found to play a role in gene silencing and DNA damage response [14]. However, manifestation of these phenotypes required additional mutations in the N-terminal tail of histone H3 and whether these chromatin-related phenotypes are related to histone deposition defects remained unknown.
The known and conserved substrates of HAT-B/NuB4 are lysines 5 and 12 of histone H4 [14]. Mutation of these residues has revealed functions in histone H4 nuclear import and chromatin assembly [48], [50], [51]. However, H4K5,12 mutants generally show no major growth phenotypes or global changes in chromatin organization [24], [48], [52], [53]. Here we found a positive effect of H4K5,12A and H4K5,12Q mutants on histone turnover in promoters, suggesting that NuB4 may exert its turnover function via H4K5/K12 acetylation. However, H4K5,12R, mimicking the hypo-acetylated state of these lysines, did not cause a turnover defect (Figure 4). One possible explanation of these results is that NuB4 has additional substrates that contribute to its role in histone turnover [67]. We do not know whether other substrate lysines on histones or perhaps non-histone proteins are also involved and play roles redundant with the acetylated histone H4 tail.
The nuclear function for HAT-B in histone turnover (Figure 5) indicates that HAT-B's role in histone metabolism may be more complex than previously anticipated and extends beyond the acetylation of newly synthesized histones. This is in line with observations that Hat1 can be recruited to chromatin at origins of replication and DNA double strand breaks [20], [21] and with the role of members of the NuB4 complex in depositing histones following repair of a DNA double strand break [18]. Unexpectedly, our studies revealed that Hat1 and Hat2 act in parallel with Hif1, and that Hat1 and Asf1 bind a different subset of the soluble histone pool. In previous studies Hat1/Hat2, Hif1, and Asf1 have been shown to bind to each other [26], which led to the suggestion that Asf1 acts downstream of Hat1/Hat2/Hif1 and passes on new histones acetylated on H4K5/K12 (and H3K56) to chromatin assembly factors CAF-I, HIR, and Rtt106 [11], [12]. Our results suggest that Hat1/Hat2, Hif1 and Asf1 act, at least in part, via distinct pathways of chromatin assembly and/or disassembly (Figure 6C and Figure 7). The equal binding of Asf1 to new and old histones suggests that Asf1 may be involved in depositing as well as escorting histones evicted from chromatin (Figure 7C), which is in concordance with the finding that H3K56 acetylation (mediated by Rtt109/Asf1) is a mark of new histones, yet is important for histone eviction and nucleosome destabilization [11], [33]. Indeed, histone chaperones may not exclusively function in chromatin assembly [68]. For example Nap1, which can escort H3/H4 and H2A/H2B and assemble histone octamers into nucleosomes, but may orchestrate this by promoting nucleosome disassembly [33]. Another example is CAF1, which is involved in replication-coupled assembly of new histones into chromatin, yet histone H3 bound to this complex (or to Rtt106 or Asf1) contains methylated H3K79 [30], which is a mark of chromatin-bound histones [69], [70].
What are the functional consequences of altering histone turnover? Histone turnover might affect several aspects of the epigenome, such as nucleosome occupancy, DNA accessibility, or dynamics of histone modifications. No changes in growth or cell cycle progression were observed for single, double, or triple hat1Δ, hat2Δ, hif1Δ mutants (Figure S8) and no significant transcriptional changes were observed (see Figure 6 and Material and Methods). Apparently, slowing down turnover of histone H3 by loss of the NuB4 complex has no profound consequences under these conditions. Deletion of HAT2 or HIF1 resulted in a moderate increase in expression of the genes encoding histone H3 and H4 in mid-log cultures (Figure 6C). We expect that this may be a response to the histone turnover defects caused by deletion of Hat2 and Hif1, since deletion of Hat1, which overall has a lower impact on histone turnover, did not affect histone gene expression. It is possible that the phenotypes of the hat1Δ strain are relatively weak because of compensation of Hat1's activity by other HATs, such as Gcn5, which acetylates newly synthesized histone H3 [71]. In our microarray analyses, we did not observe significant upregulation of mRNA levels of other (putative) HATs in G0 cultures (Figure S9). Overall, our results indicate that loss of NuB4 function alone has no major consequences for global chromatin organization.
What is the function of histone turnover? Histone turnover leads to turnover of histone modifications and can thereby affect the pattern as well as dynamics of the epigenome. When a chromatin state is controlled by two opposing activities (e.g. modification and demodification by turnover) this could lead to a more rapid establishment a new equilibrium after perturbation of the epigenome, such as during DNA replication or after exposure to stress (e.g. see [72]). Based on models proposed for histone acetylation one could also envision that dynamic turnover (cycles of modification and demodification) rather than the steady state may be relevant for chromatin function [73]. Alternatively, histone turnover could counteract the accumulation of histone modifications that are less susceptible to demodification. For example, methylation of histone H3K79, which accumulates in a non-processive manner on aging histones [72] is enriched in genomic regions that show low histone turnover and retain old histone H3 molecules, suggesting that histone inheritance and dynamics help shape the epigenome [54]. The identification of additional mutants in future screens will help to further deconstruct the pathways of histone turnover and to discover their biological significance.
In the Epi-ID screen we also identified Gis1 and Nhp10 as negative regulators of histone turnover. Gis1 is a zinc-finger transcription factor involved in regulation of stress genes [74] and contains a Jumonji domain, which has been associated with histone demethylase activity [75]. Gis1 has also been reported to bind to several factors involved in DNA metabolism [76]. It will be interesting to test whether any of these Gis1-binding proteins or its putative demethylase activity is involved in this novel function of Gis1. Nhp10 is a non-essential subunit of the essential INO80 chromatin remodeling complex that can move or mobilize nucleosomes. Two recent studies suggest a role for INO80 in redeposition of histones during induced transcription [77], [78]. That Nhp10 slows down histone turnover provides further support for the idea that the INO80 complex can help to preserve the chromatin architecture during transcription. In an Epi-ID screen using 1536 chromosome biology mutants in which the old and new tags on histone H3 were swapped (old-T7 and new-HA), NHP10 and GIS1 mutants also showed more histone turnover (data not shown), indicating that the phenotypes observed were not caused by tag-specific effects and that Epi-ID can be scaled up.
The application of Epi-ID is not restricted to histone turnover. In fact, in screens for other epigenetic marks such as histone modifications or nucleosome occupancy Epi-ID, can be applied without the elaborate genetic crosses and genetic switches that are required for screens based on the RITE pulse chase assay. Future applications in yeast may benefit from other barcoded mutant collections that are being developed [79]–[81]. Although our study suggests that position effects of the barcoded marker are not major confounders in Epi-ID and can be (at least in part) excluded by comparing UpTag with DownTag barcodes, DNA barcodes at a common genetic locus separated from the gene deletion would be preferable for epigenetic screens. The recently developed Yeast Barcoders Library represents such a collection in which barcoded markers are integrated at the common HO locus thereby providing opportunities to further expand and improve the application of Epi-ID in yeast [82]. Finally, the basic principles of this approach should also be applicable to barcoded mutant libraries in other organisms, such as barcoded episomes, or transposon or virus insertion libraries.
Materials and Methods
Yeast strains, plasmids, and media
Yeast strains used in this study are listed in Table S3. Yeast media were described previously [7]. The pilot set of mutants (see Table S1) was manually made from the MATa haploid gene knockout library (Open Biosystems). H3-RITE (strain NKI4114) was crossed in duplicate with 92 mutants by Synthetic Genetic Array analysis [44] with the following modifications. After mating, diploids were selected and kept on Hygromycin, G418 and CloNat triple selection on rich media for one night. After 13 days on sporulation media a series of selections followed to select for the proper MAT haploids: twice on haploid MATa selection (YC-His+Can+SAEC), twice on triple resistance selection (YC-His+Can+SAEC+MSG+ Hygromycin, G418 and CloNat), and then twice on YC-His-Leu to select for H3-RITE strains in which the second, untagged, copy of H3 was deleted by insertion of LEU2. NKI2178 and NKI4179 are derivatives of BY4733. Plasmid pTW087, which was used to make strain NKI2178, was made by inserting a 6xHis tag behind the HA tag into pFvL118 [7] by PCR mutagenesis. Plasmid pTW088, which was used to make strain NKI4197, was made by replacing the HA tag in pTW081 [7] by a HA-6xHIS tag generated by PCR amplification from pTW087. NKI4128 was derived from a cross between Y7092 and NKI4004 [7], [44]. NKI8013 and NKI4140 were derived from NKI4179 and NKI4128 after elimination of the first tag and HphMX marker by induction of recombination. BAR1 was deleted using pMPY-ZAP. NKI2176 was derived from BY4733 using reagents described previously [7]. NKI2215 and NKI2216 were derived from NKI2176 by targeting the RITE cassettes from pFvL118 and pTW081 to the HHT2 locus [7]. NKI2300 and NKI2301 were derived from NKI2215 and NKI2216, respectively, after elimination of the first tag and HphMX marker by induction of recombination.
Switch assay and follow-up
The tag switch assay was performed as described previously [7] with a few adjustments. Briefly, all strains were grown in 600 µl YPD containing Hygromycin (200 µg/ml, Invitrogen) in 96-well format for three nights at 30°C. Cells were then pooled in 50 ml of saturated media without Hygromycin containing 1 µM β-estradiol (E-8875, Sigma-Aldrich). Approximately 1x109 cells were fixed with 1% formaldehyde for 15 minutes before addition of β-estradiol (t = 0), after 16 hours (t = 1) and after 3 additional days (t = 3) for chromatin immunoprecipitation. In the follow-up analysis of candidate turnover mutants, we identified several possible confounders in our specific turnover screen. In certain mutants low turnover measurements were caused by lack of the Cre-recombinase mediated tag-switch. These mutants were excluded from the follow-up studies. Some mutants showed severe loss of viability after the tag-switch and when released into fresh media. These clones were also excluded from the follow-up studies to avoid clones in which the new H3-T7 tagged histone may not be fully functional and possibly causes tag-specific rather than a physiological turnover effects.
ChIP-Seq
ChIP was performed as described previously [7]. One tenth of each sample was taken as input. After DNA isolation all samples were amplified using different SeqiXU1 primers in combination with P7U2 for the amplification of the UpTag and SeqiXD1 primers with P7D2 for amplification of the DownTag (primers are listed in Table S4). PCR amplification was conducted in 50 µl reactions using Phusion DNA polymerase (Finnzymes) with the following conditions: 10 cycles of 98°C/15 s, 56°C/15 s, 72°C/20 s; 20 cycles of 98°C/15 s, 72°C/15 s, 72°C/20 s. The amplicons of different conditions were pooled per tag, size separated on a 2% gel and the correct sized amplicons were excised and extracted using a Qiagen gel purification column. In a subsequent PCR reaction equal amounts of DNA of the UpTag and DownTag were amplified with primers P5seq and either P7U2 or P7D2 to attach the adapter fragments necessary for cluster formation and sequencing on the Illumina genome analyzer. PCR amplification was conducted in 50 µl reactions using Phusion® DNA polymerase with the following conditions: 10 cycles of 98°C/15 s, 56°C/15 s, 72°C/20 s; 20 cycles of 98°C/15 s, 72°C/25 s. The indexed barcode libraries were analyzed on an Illumina GAII genome analyzer and processed as described below.
Mapping sequence reads
The indexed barcode libraries were analyzed on an Illumina GAII. Sequence reads were expected to have the following composition: 4 bp index (i), 18 bp common UpTag (U1) or 17 bp common DownTag (D1) primer sequence, up to 20 bp unique UpTag or DownTag barcode sequence. A database of expected sequence reads was generated by combining the barcode sequences originally designed (http://www-sequence.stanford.edu/group/yeast_deletion_project/deletions3.html) with corrected sequences based on re-sequencing of the barcodes of the yeast diploid heterozygous deletion collection [40], [83]. Multiplex indexed barcodes were identified at position 1 to 6 allowing no mismatches. Barcodes were identified starting at position 22, 21, or 23, respectively, initially allowing no mismatches over a length of 11 nt. Unidentified reads were further analyzed in a second round by FASTA using the optimal alignment of gene tags. FASTA alignments were only considered with a minimal alignment length of 10 bases and a minimal identity of 90%. Only alignments that start within 2 bases from position 22 were allowed and alignments were not allowed to stop more than 5 bases from the end of the barcode. A set of unused barcodes [39], [41] was used to verify that allowing mismatches did not lead to a high false discovery rate and to determine cut-offs for P-values (see below). Out of a total number of 7446311 reads, 6249225 could be assigned to an indexed barcode amplicon. The mapped sequence reads were binned in UpTag and DownTag barcode fractions, further binned in sample fractions using the 4 bp indexes, and then the relative abundance of each barcode within each specific bin was determined using reads per million counts for each bin. Based on the behavior of the unused barcodes, to avoid false positive assignments clones with outlying up / down ratio counts (P-value <0.01) in any of the indexed samples were excluded from further analysis. Histone turnover was determined by calculating the ratio of T7 ChIP over HA ChIP for t = 1 and t = 3 days (t = 1, t = 3) and for the UpTag and DownTag barcodes. Only clones with a low variation between these four samples (SD <0.17; and thereby only clones for which both the UpTag and DownTag barcode were identified) were included for further analysis. Cut offs for variation were set such that all false positive identifications of the unused barcode set were excluded. Of the 92 clones in the screen, 53 were included in the final dataset. Drop-outs were caused by the genetic crossing or by the stringent selection criteria.
Follow-up analysis of individual mutants
Strains were grown individually to saturation in 50 ml of YPD; ChIP was performed only on samples after three days of saturation. ChIP DNA was quantified in real-time PCR using the SYBR Green PCR Master Mix (Applied Biosystems) and the ABI PRISM 7500 as described previously. An input sample was used to make a standard curve, which was then used to calculate the IP samples, all performed in the 7500 fast system software. As a measurement for turnover, the amount DNA of the T7-IP was divided over the HA-IP. The antibodies used for ChIP and immunoblots are HA (12CA5), T7 (A190-117A, Bethyl or 69522-3, Novagen), H3 C-terminus [7], RITE-spacer+LoxP [7], RNA PolII/Rpb1 (8WG16). Primers used for qPCR are listed in Table S5.
TAP-IP
The equivalent of 1x109 cells was washed with cold TBS, resuspended in 1ml cold TBS with a protease inhibitor cocktail. All steps were performed cold at 4°C unless otherwise stated. Cells were briefly spun and the pellet was frozen at −80°C. The pellet was dissolved in 400 µl lysis buffer (25 mM Hepes pH 7.9, 50 mM NaCl, 0.1% NP-40, 1 mM EDTA, 10% glycerol) containing a protease inhibitor cocktail. Cells were lysed by the addition of 400 µl glass beads and vortexing for 15 min on a multivortex. The total lysate was spun at maximum speed for 5 min, the soluble fraction was transferred to a new tube and 1 ml of lysis buffer was added. The lysate was then spun for 5 min 14K, transferred to a new tube, then spun for 15 min 14K and again transferred to a new tube. Of this fraction 50 µl was used as input, the rest was incubated with 30 µl IgG beads (Invitrogen) for 2 hrs. The beads were washed three times with cold lysis buffer for 5 min and once with TEV buffer (50 mM Tris pH 8, 0.5 mM EDTA, 50 mM NaCl, and 1 mM DTT). The beads were resuspended in 100 µl TEV buffer to which 175 µg recombinant TEV protease is added and kept overnight. The soluble fraction contains the immunoprecipitated fraction and was analyzed by quantitative immunoblotting. Lysates were separated on a 16% polyacrylamide gel and blotted onto 0.45 µm nitrocellulose membrane. Membranes were blocked with 2% Nutrilon (Nutricia) in PBS. Primary antibody incubations were performed overnight in Tris-buffered saline-Tween with 2% Nutrilon, anti-HA (mouse 12CA5), anti-T7 (Novagen, 1∶1000) or a polyclonal antibody obtained against the LoxP peptide (1∶2500) [7]. Secondary antibody incubations were performed for 45 minutes using LI-COR Odyssey IRDye 800CW (1∶12.000). Immunoblots were subsequently scanned on a LI-COR Odyssey IR Imager (Biosciences) using the 800 channel. Signal intensities were determined using Odyssey LI-COR software version 3.0.
FACS analysis
To monitor cell cycle progression and cell cycle arrests the DNA content of the cells was measured by flow cytometry as described previously [7], using SYTOX Green and a 530/30 filter (Becton-Dickinson). Analysis was performed using FCSexpress2.
Expression profiling
Each mutant strain was profiled four times from two independently inoculated cultures and harvested in early mid-log phase in synthetic complete medium with 2% glucose or harvested in starvation conditions in rich media as described above for the turnover experiments. Sets of mutants were grown alongside corresponding WT cultures and processed in parallel. Dual-channel 70-mer oligonucleotide arrays were employed with a common reference WT RNA. All steps after RNA isolation were automated using robotic liquid handlers. These procedures were first optimized for accuracy (correct FC) and precision (reproducible result), using spiked-in RNA for calibration [84]. After quality control, normalization, and dye-bias correction [85], statistical analysis for mid-log cultures was performed for each mutant versus the collection of 200 WT cultures as described by Lenstra et al [57]. The reported FC is an average of the four replicate mutant profiles versus the average of all WTs. HAT1, HAT2, and HIF1 single, double, and triple mutants in the BY4742 background were not different from wild type (less than three genes changed p<0.01, FC >1.7 after removal of WT variable genes). Mutants in G0 were compared to replicates of the corresponding wild-type RITE strain. Due to variability under conditions of starvation [86] we did not perform genome-wide statistical analyses of expression changes in G0 cultures.
Accession numbers
Microarray data have been deposited in ArrayExpress under accession numbers E-TABM-1175 (mutants) and E-TABM-773/E-TABM-984 (200 WT replicates), as well as in GEO under accession number GSE30168.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. RandoOJAhmadK 2007 Rules and regulation in the primary structure of chromatin. Curr Opin Cell Biol 19 250 256
2. BonasioRTuSReinbergD 2010 Molecular signals of epigenetic states. Science 330 612 616
3. HenikoffS 2008 Nucleosome destabilization in the epigenetic regulation of gene expression. Nat Rev Genet 9 15 26
4. DionMFKaplanTKimMBuratowskiSFriedmanN 2007 Dynamics of replication-independent histone turnover in budding yeast. Science 315 1405 1408
5. JamaiAImoberdorfRMStrubinM 2007 Continuous histone H2B and transcription-dependent histone H3 exchange in yeast cells outside of replication. Mol Cell 25 345 355
6. RufiangeAJacquesPEBhatWRobertFNouraniA 2007 Genome-wide replication-independent histone h3 exchange occurs predominantly at promoters and implicates h3 k56 acetylation and asf1. Mol Cell 27 393 405
7. VerzijlbergenKFMenendez-BenitoVvan WelsemTvan DeventerSJLindstromDL 2010 Recombination-induced tag exchange to track old and new proteins. Proc Natl Acad Sci U S A 107 64 68
8. KimHJSeolJHHanJWYounHDChoEJ 2007 Histone chaperones regulate histone exchange during transcription. EMBO J 26 4467 4474
9. DealRBHenikoffJGHenikoffS 2010 Genome-wide kinetics of nucleosome turnover determined by metabolic labeling of histones. Science 328 1161 1164
10. ParkYJLugerK 2008 Histone chaperones in nucleosome eviction and histone exchange. Curr Opin Struct Biol 18 282 289
11. DasCTylerJKChurchillME 2010 The histone shuffle: histone chaperones in an energetic dance. Trends Biochem Sci 35 476 489
12. De KoningLCorpetAHaberJEAlmouzniG 2007 Histone chaperones: an escort network regulating histone traffic. Nat Struct Mol Biol 14 997 1007
13. ParthunMRWidomJGottschlingDE 1996 The major cytoplasmic histone acetyltransferase in yeast: links to chromatin replication and histone metabolism. Cell 87 85 94
14. ParthunMR 2007 Hat1: the emerging cellular roles of a type B histone acetyltransferase. Oncogene 26 5319 5328
15. BensonLJPhillipsJAGuYParthunMRHoffmanCS 2007 Properties of the type B histone acetyltransferase Hat1: H4 tail interaction, site preference, and involvement in DNA repair. J Biol Chem 282 836 842
16. ChangLLorangerSSMizzenCErnstSGAllisCD 1997 Histones in transit: cytosolic histone complexes and diacetylation of H4 during nucleosome assembly in human cells. Biochemistry 36 469 480
17. PovedaASendraR 2008 Site specificity of yeast histone acetyltransferase B complex in vivo. FEBS J 275 2122 2136
18. GeZWangHParthunMR 2011 Nuclear Hat1p Complex (NuB4) Components Participate in DNA Repair-linked Chromatin Reassembly. J Biol Chem 286 16790 16799
19. MersfelderELParthunMR 2008 Involvement of Hat1p (Kat1p) catalytic activity and subcellular localization in telomeric silencing. J Biol Chem 283 29060 29068
20. SuterBPogoutseOGuoXKroganNLewisP 2007 Association with the origin recognition complex suggests a novel role for histone acetyltransferase Hat1p/Hat2p. BMC Biol 5 38
21. QinSParthunMR 2006 Recruitment of the type B histone acetyltransferase Hat1p to chromatin is linked to DNA double-strand breaks. Mol Cell Biol 26 3649 3658
22. RosalenyLEAntunezORuiz-GarciaABPerez-OrtinJETorderaV 2005 Yeast HAT1 and HAT2 deletions have different life-span and transcriptome phenotypes. FEBS Lett 579 4063 4068
23. QinSParthunMR 2002 Histone H3 and the histone acetyltransferase Hat1p contribute to DNA double-strand break repair. Mol Cell Biol 22 8353 8365
24. KellyTJQinSGottschlingDEParthunMR 2000 Type B histone acetyltransferase Hat1p participates in telomeric silencing. Mol Cell Biol 20 7051 7058
25. FillinghamJRechtJSilvaACSuterBEmiliA 2008 Chaperone control of the activity and specificity of the histone H3 acetyltransferase Rtt109. Mol Cell Biol 28 4342 4353
26. CamposEIFillinghamJLiGZhengHVoigtP 2010 The program for processing newly synthesized histones H3.1 and H4. Nat Struct Mol Biol 17 1343 1351
27. FillinghamJKainthPLambertJPvan BakelHTsuiK 2009 Two-color cell array screen reveals interdependent roles for histone chaperones and a chromatin boundary regulator in histone gene repression. Mol Cell 35 340 351
28. SuttonABucariaJOsleyMASternglanzR 2001 Yeast ASF1 protein is required for cell cycle regulation of histone gene transcription 9. Genetics 158 587 596
29. LambertJPFillinghamJSiahbaziMGreenblattJBaetzK 2010 Defining the budding yeast chromatin-associated interactome. Mol Syst Biol 6 448
30. LiQZhouHWurteleHDaviesBHorazdovskyB 2008 Acetylation of histone H3 lysine 56 regulates replication-coupled nucleosome assembly. Cell 134 244 255
31. SharpJAFoutsETKrawitzDCKaufmanPD 2001 Yeast histone deposition protein Asf1p requires Hir proteins and PCNA for heterochromatic silencing 5. Curr Biol 11 463 473
32. GreenEMAntczakAJBaileyAOFrancoAAWuKJ 2005 Replication-Independent Histone Deposition by the HIR Complex and Asf1. Curr Biol 15 2044 2049
33. AndrewsAJChenXZevinAStargellLALugerK 2010 The histone chaperone Nap1 promotes nucleosome assembly by eliminating nonnucleosomal histone DNA interactions. Mol Cell 37 834 842
34. BurgessRJZhouHHanJZhangZ 2010 A role for Gcn5 in replication-coupled nucleosome assembly. Mol Cell 37 469 480
35. LukERanjanAFitzgeraldPCMizuguchiGHuangY 2010 Stepwise histone replacement by SWR1 requires dual activation with histone H2A.Z and canonical nucleosome. Cell 143 725 736
36. Papamichos-ChronakisMWatanabeSRandoOJPetersonCL 2011 Global regulation of H2A.Z localization by the INO80 chromatin-remodeling enzyme is essential for genome integrity 1. Cell 144 200 213
37. LingerJTylerJK 2006 Global replication-independent histone H4 exchange in budding yeast. Eukaryot Cell 5 1780 1787
38. KroganNJDoverJWoodASchneiderJHeidtJ 2003 The Paf1 complex is required for histone H3 methylation by COMPASS and Dot1p: linking transcriptional elongation to histone methylation. Mol Cell 11 721 729
39. SmithAMHeislerLESt OngeRPFarias-HessonEWallaceIM 2010 Highly-multiplexed barcode sequencing: an efficient method for parallel analysis of pooled samples. Nucleic Acids Res 38 e142
40. SmithAMHeislerLEMellorJKaperFThompsonMJ 2009 Quantitative phenotyping via deep barcode sequencing. Genome Res 19 1836 1842
41. PierceSEDavisRWNislowCGiaeverG 2007 Genome-wide analysis of barcoded Saccharomyces cerevisiae gene-deletion mutants in pooled cultures. Nat Protoc 2 2958 2974
42. Radman-LivajaMRandoOJ 2010 Nucleosome positioning: how is it established, and why does it matter? Dev Biol 339 258 266
43. DealRBHenikoffS 2010 Capturing the dynamic epigenome. Genome Biol 11 218
44. TongAHBooneC 2006 Synthetic genetic array analysis in Saccharomyces cerevisiae. Methods Mol Biol 313 171 192
45. VerreaultAKaufmanPDKobayashiRStillmanB 1996 Nucleosome assembly by a complex of CAF-1 and acetylated histones H3/H4. Cell 87 95 104
46. BensonLJGuYYakovlevaTTongKBarrowsC 2006 Modifications of H3 and H4 during Chromatin Replication, Nucleosome Assembly, and Histone Exchange. J Biol Chem 281 9287 9296
47. SobelRECookRGPerryCAAnnunziatoATAllisCD 1995 Conservation of deposition-related acetylation sites in newly synthesized histones H3 and H4. Proc Natl Acad Sci U S A 92 1237 1241
48. Ejlassi-LassalletteAMocquardEArnaudMCThirietC 2011 H4 replication-dependent diacetylation and Hat1 promote S-phase chromatin assembly in vivo. Mol Biol Cell 22 245 255
49. AiXParthunMR 2004 The nuclear Hat1p/Hat2p complex: a molecular link between type B histone acetyltransferases and chromatin assembly. Mol Cell 14 195 205
50. BlackwellJSJrWilkinsonSTMosammaparastNPembertonLF 2007 Mutational analysis of H3 and H4 N termini reveals distinct roles in nuclear import. J Biol Chem 282 20142 20150
51. GlowczewskiLWaterborgJHBermanJG 2004 Yeast chromatin assembly complex 1 protein excludes nonacetylatable forms of histone H4 from chromatin and the nucleus. Mol Cell Biol 24 10180 10192
52. DionMFAltschulerSJWuLFRandoOJ 2005 Genomic characterization reveals a simple histone H4 acetylation code. Proc Natl Acad Sci U S A 102 5501 5506
53. MaXJWuJAltheimBASchultzMCGrunsteinM 1998 Deposition-related sites K5/K12 in histone H4 are not required for nucleosome deposition in yeast. Proc Natl Acad Sci U S A 95 6693 6698
54. Radman-LivajaMVerzijlbergenKFWeinerAvan WelsemTFriedmanN 2011 Patterns and mechanisms of ancestral histone protein inheritance in budding yeast. PLoS Biol 9 e1001075 doi:10.1371/journal.pbio.1001075
55. PovedaAPamblancoMTafrovSTorderaVSternglanzR 2004 Hif1 is a component of yeast histone acetyltransferase B, a complex mainly localized in the nucleus. J Biol Chem 279 16033 16043
56. DunleavyEMPidouxALMonetMBonillaCRichardsonW 2007 A NASP (N1/N2)-related protein, Sim3, binds CENP-A and is required for its deposition at fission yeast centromeres. Mol Cell 28 1029 1044
57. LenstraTLBenschopJJKimTSchulzeJMBrabersNA 2011 The specificity and topology of chromatin interaction pathways in yeast. Mol Cell 42 536 549
58. van WageningenSKemmerenPLijnzaadPMargaritisTBenschopJJ 2010 Functional overlap and regulatory links shape genetic interactions between signaling pathways. Cell 143 991 1004
59. FeserJTruongDDasCCarsonJJKieftJ 2010 Elevated histone expression promotes life span extension. Mol Cell 39 724 735
60. MosammaparastNGuoYShabanowitzJHuntDFPembertonLF 2002 Pathways mediating the nuclear import of histones H3 and H4 in yeast. J Biol Chem 277 862 868
61. SharpJARizkiGKaufmanPD 2005 Regulation of histone deposition proteins Asf1/Hir1 by multiple DNA damage checkpoint kinases in Saccharomyces cerevisiae. Genetics 171 885 899
62. KaplanTLiuCLErkmannJAHolikJGrunsteinM 2008 Cell cycle - and chaperone-mediated regulation of H3K56ac incorporation in yeast. PLoS Genet 4 e1000270 doi:10.1371/journal.pgen.1000270
63. Katan-KhaykovichYStruhlK 2011 Splitting of H3-H4 tetramers at transcriptionally active genes undergoing dynamic histone exchange. Proc Natl Acad Sci U S A 108 1296 1301
64. GradolattoARogersRSLavenderHTavernaSDAllisCD 2008 Saccharomyces cerevisiae Yta7 regulates histone gene expression. Genetics 179 291 304
65. GauthierNPJensenLJWernerssonRBrunakSJensenTS 2010 Cyclebase.org: version 2.0, an updated comprehensive, multi-species repository of cell cycle experiments and derived analysis results. Nucleic Acids Res 38 D699 D702
66. KleffSAndrulisEDAndersonCWSternglanzR 1995 Identification of a gene encoding a yeast histone H4 acetyltransferase. J Biol Chem 270 24674 24677
67. YeJAiXEugeniEEZhangLCarpenterLR 2005 Histone H4 lysine 91 acetylation a core domain modification associated with chromatin assembly. Mol Cell 18 123 130
68. JasencakovaZScharfANAskKCorpetAImhofA 2010 Replication stress interferes with histone recycling and predeposition marking of new histones. Mol Cell 37 736 743
69. van LeeuwenFGafkenPRGottschlingDE 2002 Dot1p modulates silencing in yeast by methylation of the nucleosome core. Cell 109 745 756
70. SawadaKYangZHortonJRCollinsREZhangX 2004 Structure of the conserved core of the yeast Dot1p, a nucleosomal histone H3 lysine 79 methyltransferase 2. J Biol Chem 279 43296 43306
71. AdkinsMWCarsonJJEnglishCMRameyCJTylerJK 2007 The histone chaperone anti-silencing function 1 stimulates the acetylation of newly synthesized histone H3 in S-phase. J Biol Chem 282 1334 1340
72. De VosDFrederiksFTewijMvan WelsemTVerzijlbergenKF 2011 Progressive methylation of aging histones by Dot1 acts as a timer. EMBO Rep in press
73. ClaytonALHazzalinCAMahadevanLC 2006 Enhanced histone acetylation and transcription: a dynamic perspective. Mol Cell 23 289 296
74. PedruzziIBurckertNEggerPDe VirgilioC 2000 Saccharomyces cerevisiae Ras/cAMP pathway controls post-diauxic shift element-dependent transcription through the zinc finger protein Gis1. EMBO J 19 2569 2579
75. TuSBullochEMYangLRenCHuangWC 2007 Identification of histone demethylases in Saccharomyces cerevisiae. J Biol Chem 282 14262 14271
76. TronnersjoSHanefalkCBalciunasDHuGZNordbergN 2007 The jmjN and jmjC domains of the yeast zinc finger protein Gis1 interact with 19 proteins involved in transcription, sumoylation and DNA repair. Mol Genet Genomics 277 57 70
77. KlopfEPaskovaLSoleCMasGPetryshynA 2009 Cooperation between the INO80 complex and histone chaperones determines adaptation of stress gene transcription in the yeast Saccharomyces cerevisiae. Mol Cell Biol 29 4994 5007
78. HannumGSrivasRGuenoleAvan AttikumHKroganNJ 2009 Genome-wide association data reveal a global map of genetic interactions among protein complexes. PLoS Genet 5 e1000782 doi:10.1371/journal.pgen.1000782
79. Ben AroyaSCoombesCKwokTO'DonnellKABoekeJD 2008 Toward a comprehensive temperature-sensitive mutant repository of the essential genes of Saccharomyces cerevisiae. Mol Cell 30 248 258
80. DaiJHylandEMYuanDSHuangHBaderJS 2008 Probing nucleosome function: a highly versatile library of synthetic histone H3 and H4 mutants. Cell 134 1066 1078
81. HoCHMagtanongLBarkerSLGreshamDNishimuraS 2009 A molecular barcoded yeast ORF library enables mode-of-action analysis of bioactive compounds. Nat Biotechnol 27 369 377
82. YanZCostanzoMHeislerLEPawJKaperF 2008 Yeast Barcoders: a chemogenomic application of a universal donor-strain collection carrying bar-code identifiers 36. Nat Methods 5 719 725
83. EasonRGPourmandNTongprasitWHermanZSAnthonyK 2004 Characterization of synthetic DNA bar codes in Saccharomyces cerevisiae gene-deletion strains. Proc Natl Acad Sci U S A 101 11046 11051
84. van BakelHHolstegeFC 2004 In control: systematic assessment of microarray performance. EMBO Rep 5 964 969
85. MargaritisTLijnzaadPvan LeenenDBouwmeesterDKemmerenP 2009 Adaptable gene-specific dye bias correction for two-channel DNA microarrays. Mol Syst Biol 5 266
86. RadonjicMAndrauJCLijnzaadPKemmerenPKockelkornTT 2005 Genome-wide analyses reveal RNA polymerase II located upstream of genes poised for rapid response upon S. cerevisiae stationary phase exit. Mol Cell 18 171 183
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 10
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- The Glycobiome Reveals Mechanisms of Pentose and Hexose Co-Utilization in Bacteria
- Global Mapping of Cell Type–Specific Open Chromatin by FAIRE-seq Reveals the Regulatory Role of the NFI Family in Adipocyte Differentiation
- Genetic Determinants of Serum Testosterone Concentrations in Men
- MicroRNA Expression and Regulation in Human, Chimpanzee, and Macaque Brains
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy