
Small RNAs Prevent Transcription-Coupled Loss of Histone H3 Lysine 9 Methylation in
In eukaryotes, histone H3 lysine 9 methylation (H3K9me) mediates silencing of invasive sequences to prevent deleterious consequences including the expression of aberrant gene products and mobilization of transposons. In Arabidopsis thaliana, H3K9me maintained by SUVH histone methyltransferases (MTases) is associated with cytosine methylation (5meC) maintained by the CMT3 cytosine MTase. The SUVHs contain a 5meC binding domain and CMT3 contains an H3K9me binding domain, suggesting that the SUVH/CMT3 pathway involves an amplification loop between H3K9me and 5meC. However, at loci subject to read-through transcription, the stability of the H3K9me/5meC loop requires a mechanism to counteract transcription-coupled loss of H3K9me. Here we use the duplicated PAI genes, which stably maintain SUVH-dependent H3K9me and CMT3-dependent 5meC despite read-through transcription, to show that when PAI sRNAs are depleted by dicer ribonuclease mutations, PAI H3K9me and 5meC levels are reduced and remaining PAI 5meC is destabilized upon inbreeding. The dicer mutations confer weaker reductions in PAI 5meC levels but similar or stronger reductions in PAI H3K9me levels compared to a cmt3 mutation. This comparison indicates a connection between sRNAs and maintenance of H3K9me independent of CMT3 function. The dicer mutations reduce PAI H3K9me and 5meC levels through a distinct mechanism from the known role of dicer-dependent sRNAs in guiding the DRM2 cytosine MTase because the PAI genes maintain H3K9me and 5meC at levels similar to wild type in a drm2 mutant. Our results support a new role for sRNAs in plants to prevent transcription-coupled loss of H3K9me.
Published in the journal:
. PLoS Genet 7(10): e32767. doi:10.1371/journal.pgen.1002350
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002350
Summary
In eukaryotes, histone H3 lysine 9 methylation (H3K9me) mediates silencing of invasive sequences to prevent deleterious consequences including the expression of aberrant gene products and mobilization of transposons. In Arabidopsis thaliana, H3K9me maintained by SUVH histone methyltransferases (MTases) is associated with cytosine methylation (5meC) maintained by the CMT3 cytosine MTase. The SUVHs contain a 5meC binding domain and CMT3 contains an H3K9me binding domain, suggesting that the SUVH/CMT3 pathway involves an amplification loop between H3K9me and 5meC. However, at loci subject to read-through transcription, the stability of the H3K9me/5meC loop requires a mechanism to counteract transcription-coupled loss of H3K9me. Here we use the duplicated PAI genes, which stably maintain SUVH-dependent H3K9me and CMT3-dependent 5meC despite read-through transcription, to show that when PAI sRNAs are depleted by dicer ribonuclease mutations, PAI H3K9me and 5meC levels are reduced and remaining PAI 5meC is destabilized upon inbreeding. The dicer mutations confer weaker reductions in PAI 5meC levels but similar or stronger reductions in PAI H3K9me levels compared to a cmt3 mutation. This comparison indicates a connection between sRNAs and maintenance of H3K9me independent of CMT3 function. The dicer mutations reduce PAI H3K9me and 5meC levels through a distinct mechanism from the known role of dicer-dependent sRNAs in guiding the DRM2 cytosine MTase because the PAI genes maintain H3K9me and 5meC at levels similar to wild type in a drm2 mutant. Our results support a new role for sRNAs in plants to prevent transcription-coupled loss of H3K9me.
Introduction
The eukaryotic cell is under constant threat from transposons and other invasive sequences. Transposons can drain cellular resources for RNA and protein synthesis and can damage the cell through expression of aberrant gene products or activation of transposon movement. A major mechanism to protect against these deleterious effects is to target transposons and other repetitive sequences for silencing mediated through chromatin modifications. In most eukaryotes, transposon chromatin is marked by methylation of histone H3 at the lysine 9 position (H3K9me). In some eukaryotes including mammals and plants transposon chromatin is also marked by cytosine methylation (5meC). An important question is how H3K9me and 5meC are accurately maintained on transposons but not on host genes.
A conserved strategy to maintain H3K9me and 5meC is to use the modifications as methyltransferase (MTase) binding recognition motifs. For example, in Arabidopsis thaliana, dimethylation of H3K9 (H3K9me2) maintained by three partially redundant histone MTases—SUVH4 (also known as KYP, At5g13960), SUVH5 (At2g35160), and SUVH6 (At2g22740)—is associated with 5meC maintained by the CMT3 cytosine MTase (At1g69770) [1]–[4]. The SUVH MTases contain a 5meC binding domain and CMT3 contains an H3K9me binding domain, suggesting that the SUVH/CMT3 pathway involves an amplification loop that can perpetuate both H3K9me and 5meC [5], [6]. Consistent with this model, mutations in the CMT3 or MET1 (At5g49160) cytosine MTases, which act to maintain 5meC in non-CG and CG sequence contexts respectively, result in reduced H3K9me2 levels on transposons and repetitive sequences [3], [7]–[10]. In addition, a suvh4 suvh5 suvh6 triple H3 K9 MTase mutant displays similar reduced non-CG methylation patterns to a cmt3 mutant [4]. Although the H3K9me/5meC amplification loop provides a mechanism to stably maintain both modifications in untranscribed regions of the genome, at junctions where modified sequences are transcribed through from nearby unmodified promoters, H3K9me can be removed by transcription-associated histone replacement or histone demethylation [7], [11]. What prevents transcriptional destabilization of H3K9me patterns?
Duplicated Arabidopsis genes encoding the tryptophan synthesis enzyme phosphoribosylanthranilate isomerase (PAI) provide an ideal system to understand the balance between transcription and SUVH-mediated H3K9me2/CMT3-mediated 5meC. In most Arabidopsis strains there are three unlinked PAI gene duplications that lack 5meC [12]. However, in the Wassilewskija (Ws) strain one of the PAI loci is rearranged as a tail-to-tail inverted repeat (IR) of two genes PAI1–PAI4 (At1g07780), which triggers the recognition of PAI sequences as invaders. The PAI1–PAI4 IR as well as two unlinked singlet genes PAI2 (At5g05590) and PAI3 (At1g29410) are modified by H3K9me2 and 5meC, coextensive with their regions of shared sequence identity [3], [13] (see Figure S1 for PAI gene maps). The PAI1–PAI4 IR is fused to a heterologous promoter with a transcription start site approximately 500 base pairs (bp) upstream of the PAI1 5meC boundary, which drives constitutive expression of PAI1 transcripts [14]. The polyadenylated transcripts that accumulate from this locus consist of a majority class that terminates normally in the PAI1 3′ untranslated region at the center of the IR and a minority class that extends through PAI1 into palindromic PAI4 sequences to provide a source of fold-back double-stranded RNA (dsRNA). Therefore the PAI1–PAI4 locus is able to stably maintain H3K9me2 and 5meC on the IR sequences even in the face of substantial read-through transcription. The PAI2 and PAI3 singlet genes also stably maintain H3K9me2 and 5meC even though they are likely to be only partially silenced by limited upstream modifications: at PAI2 5meC extends only 250 bp upstream of the predicted transcription start site, and at PAI3 5meC extends only as far as the predicted transcription start site [12], [13].
Arabidopsis uses three cytosine MTase pathways to control 5meC: the CMT3 pathway maintains 5meC mainly in non-CG contexts in conjunction with the SUVH H3K9 MTases, the MET1 pathway maintains 5meC mainly in CG contexts, and the DRM2 (At5g14620) pathway initiates 5meC on new invasive sequences under the guidance of small RNAs (sRNAs), as well as contributing to maintenance of non-CG methylation at some loci [15]. In a cmt3 or a suvh4 suvh5 suvh6 mutant, the Ws PAI genes are depleted for 5meC in non-CG contexts [4], [16]. In addition, in a cmt3 met1 double mutant the PAI genes are depleted for 5meC in all contexts [3]. Therefore, the DRM2 pathway plays a minimal role in the maintenance of PAI 5meC patterns. However, genetic or epigenetic changes that impair the production of transcripts that read through from PAI1 into palindromic PAI4 sequences at the PAI1–PAI4 IR cause reduced levels of PAI 5meC in non-CG contexts [13], [14], [17], [18]. In light of these results, we hypothesized that sRNAs processed from dsRNAs might underlie a mechanism to prevent the loss of SUVH/CMT3-mediated modifications due to read-through transcription, independently of the role for sRNAs in guiding DRM2.
To test the hypothesis that sRNAs control the SUVH/CMT3 pathway, we used mutations in Arabidopsis dicer-like (DCL) ribonucleases to block processing of sRNAs from dsRNAs, and monitored the effects on Ws PAI gene H3K9me2 and 5meC levels. Arabidopsis encodes four DCLs (reviewed in [19]). DCL1 (At1g01040) is specialized for processing 21 nucleotide (nt) microRNAs (miRNAs) needed for developmental gene regulation, whereas DCL2 (At3g03300), DCL3 (At3g43920), and DCL4 (At5g20320) have partially redundant roles in processing sRNAs used in other silencing pathways. For example, DCL3 processes 24 nt sRNAs used to guide DRM2 to matching target sequences such as transgene insertions and transposons [20], [21]. In a dcl3 mutant DCL2 and DCL4 can partially compensate by processing 22 nt and 21 nt sRNAs respectively corresponding to the same genomic target sequences [22], [23].
Here we show that the dcl2 dcl3 dcl4 mutant has reduced levels of H3K9me2 and non-CG methylation on PAI sequences relative to wild type, corresponding to loss of PAI sRNAs. We also show that a drm2 mutant maintains similar levels of PAI H3K9me2 and 5meC relative to wild type. Therefore the PAI genes illustrate that DCL-dependent sRNAs help maintain SUVH/CMT3-mediated modifications through a distinct mechanism from their role in guiding DRM2. In the dcl mutant there is a weaker reduction in PAI 5meC levels but a similar or stronger reduction in PAI H3K9me2 levels compared to a cmt3 mutant, indicating a connection between sRNAs and maintenance of H3K9me2 patterns independent of CMT3 function. We also show that upon inbreeding in the absence of DCL function, remaining PAI 5meC is destabilized. Our results reveal a new pathway for sRNA control of H3K9me2 and associated 5meC patterns in plants. This pathway provides a homeostatic mechanism to use a product of read-through transcription—sRNAs—as a means to counteract transcription-coupled loss of H3K9me2 on transposons and repeats.
Results
Dicer mutations cause reduced levels of PAI non-CG methylation
To test whether dicer-dependent sRNAs contribute to maintenance of PAI 5meC patterns, we generated strains where dicer mutations were combined with the three methylated PAI loci from Ws and assayed PAI 5meC patterns using both DNA gel blot and bisulfite sequencing assays. For PAI DNA gel blot analysis we cleaved genomic DNA with each of three 5meC-sensitive restriction enzymes that have cleavage sites within methylated PAI sequences: HincII (sensitive to methylation of the outermost non-CG cytosines in 5′ atGTCAACag 3′, where the recognition sequence is shown in uppercase), MspI (sensitive to methylation of the outer non-CG cytosines in 5′ CCGG 3′, and HpaII (sensitive to methylation of either the inner CG or outer non-CG cytosines in 5′ CCGG 3′). HincII cleaves at the translational start codons of PAI1, PAI2 and PAI4 but not PAI3, and the MspI/HpaII isoschizomers cleave in the second introns of PAI2, PAI3, and PAI4 but not PAI1 (Figure S1).
We found that genomic DNA prepared from dcl2, dcl3, and dcl4 single insertional null mutants and the dcl2 dcl4 and dcl3 dcl4 double mutants had similar PAI cleavage patterns to wild type Ws genomic DNA when assessed by HincII, MspI, or HpaII DNA gel blot assays (Figure 1). In contrast, genomic DNA prepared from the dcl2 dcl3 and dcl2 dcl3 dcl4 mutants displayed increased cleavage with HincII at PAI1–PAI4 and PAI2, and with MspI at PAI1–PAI4, PAI2, and PAI3 relative to wild type Ws, diagnostic of partially reduced non-CG methylation levels at all three PAI loci. Bisulfite sequencing of PAI1 and PAI2 proximal promoter/first exon regions in the dcl2 dcl3 dcl4 mutant compared to wild type Ws and Ws cmt3 showed that there was a partial loss of 5meC in CHG and CHH contexts. Therefore, the bisulfite sequencing data are consistent with the DNA gel blot assays. The results indicate that DCL2 and DCL3 act redundantly to maintain PAI non-CG methylation patterns.
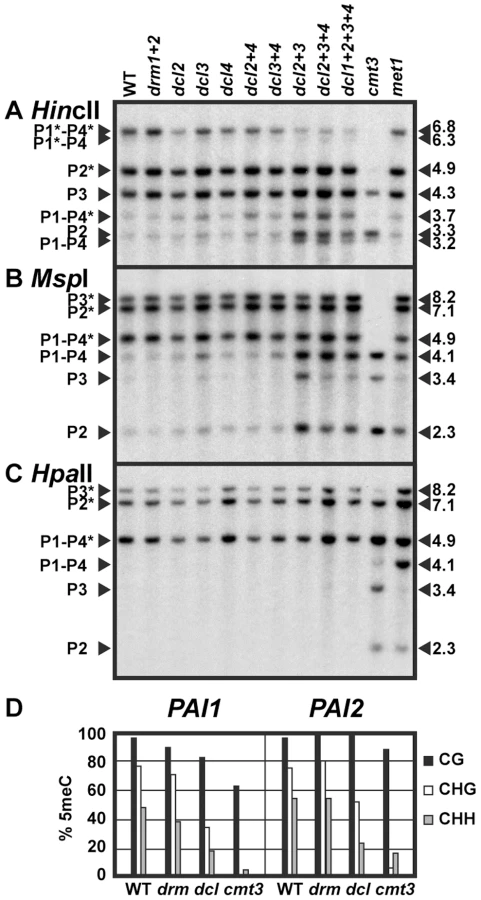
To determine whether the miRNA processing dicer DCL1 contributes to the remaining PAI non-CG methylation in the dcl2 dcl3 dcl4 triple mutant relative to cmt3, we included a dcl1 dcl2 dcl3 dcl4 quadruple mutant strain in the DNA gel blot analysis (Figure 1). Because dcl1 null alleles are embryo-lethal we used a partial-function dcl1-9 allele that is viable but female-sterile [24], [25]. The dcl1 dcl2 dcl3 dcl4 mutant displayed similar cleavage patterns to the dcl2 dcl3 dcl4 mutant, indicating that the dcl1-9 mutation does not enhance the partial loss of 5meC conferred by mutation of the other three DCL genes. In subsequent studies we focused on the dcl2 dcl3 dcl4 mutant, which has global depletion of sRNAs other than miRNAs [23].
For comparison to the dcl mutants we included genomic DNA prepared from cytosine MTase mutants in the Ws background (Figure 1). DRM2 is the major cytosine MTase controlling initiation of 5meC, but the related DRM1 MTase (At5g15380) could also contribute to this pathway [26]. Therefore we used a drm1 drm2 double null insertional mutant. DNA from the Ws drm1 drm2 mutant displayed similar PAI cleavage patterns to wild type Ws in all three DNA gel blot assays, and similar 5meC patterns to wild type Ws in bisulfite sequencing analysis of PAI1 and PAI2 proximal promoter regions. DNA from a Ws met1 mutant displayed increased cleavage at all three PAI loci with HpaII, and partially increased cleavage at PAI1–PAI4 and PAI2 with MspI, but no difference from wild type PAI cleavage patterns with HincII in DNA gel blot assays, diagnostic of a partial loss of 5meC in CG and CCG contexts. DNA from the cmt3 mutant displayed nearly complete cleavage with HincII and MspI, and partially increased cleavage with HpaII in DNA gel blot assays, diagnostic of strong loss of 5meC mainly in CHG and CHH contexts. Compared to the cytosine MTase mutants across the three DNA gel blot assays, the dcl mutant PAI demethylation phenotypes are consistent with a partial defect in the SUVH/CMT3 pathway rather than a defect in the DRM or MET1 pathways.
DCL3 and DRM2 are key factors in establishing new 5meC imprints. We used a previously developed genetic assay combining the Ws PAI IR dsRNA source locus with an unmethylated PAI2 target gene from another strain background to show that dcl3 and drm1 drm2 mutations impair the acquisition of new 5meC on PAI2 (Text S1, Figure S2). Therefore the PAI genes use the same DCL3/DRM pathway for establishing 5meC imprints as other characterized loci. However, once PAI 5meC patterns are established, the DCL3/DRM pathway plays a minimal role in long-term maintenance (Figure 1).
The dcl2 dcl3 dcl4 mutant has reduced levels of PAI H3K9me2
We used chromatin immunoprecipitation (ChIP) analysis with H3K9me2-specific antibodies on chromatin prepared from the dcl2 dcl3 dcl4 mutant compared to chromatin prepared from wild type, suvh4 suvh5 suvh6, cmt3, drm1 drm2, or cmt3 drm1 drm2 strains to determine whether the dcl mutations affect levels of H3K9me2 as well as non-CG methylation on PAI sequences. Chromatin was analyzed by quantitative PCR with primer pairs specific for the PAI1 arm of the PAI1–PAI4 IR locus or the PAI2 singlet gene.
At both PAI1–PAI4 and PAI2 the dcl2 dcl3 dcl4 mutant had reduced levels of H3K9me2 relative to wild type, although not as strongly as in the suvh4 suvh5 suvh6 H3K9 MTase mutant (Figure 2). Comparing the ChIP results to the assays for 5meC (Figure 1), the reduced PAI H3K9me2 levels in the dcl2 dcl3 dcl4 mutant are still sufficient to support substantial CMT3 activity. Therefore CMT3 might be able to use even sparsely distributed H3K9me2 as a localization signal.

At both PAI loci the cmt3 mutant also had partially reduced levels of H3K9me2, presumably because reduced 5meC levels impair SUVH localization to PAI sequences. At PAI2 increased transcription due to proximal promoter demethylation in a cmt3 mutant could also contribute to reduced H3K9me2 levels, perhaps accounting for a stronger relative reduction at PAI2 than at PAI1–PAI4 [3], [16]. The dcl2 dcl3 dcl4 mutant had similar reduction in H3K9me2 levels to the cmt3 mutant at PAI2, but a stronger reduction at PAI1–PAI4. In contrast, the dcl2 dcl3 dcl4 mutant had weaker reductions in PAI2 and PAI1–PAI4 non-CG methylation levels compared to cmt3 (Figure 1). This comparison indicates that the dcl2 dcl3 dcl4 mutations impair maintenance of H3K9me2 independently of effects on CMT3 function. If the dcl mutations acted by impairing CMT3 to cause a partial reduction in PAI 5meC levels as the primary consequence, then the resulting reduction in H3K9me2 levels would be expected to be less than in the cmt3 mutant.
At both PAI1–PAI4 and PAI2, the drm1 drm2 mutant displayed similar levels of H3K9me2 to wild type, and the drm1 drm2 cmt3 mutant displayed similar levels of H3K9me2 to cmt3 (Figure 2). Therefore, the DRM cytosine MTases do not contribute to maintenance of PAI H3K9me2 patterns.
The dcl2 dcl3 dcl4 mutant is depleted for PAI sRNAs
To determine whether reduced PAI non-CG methylation and H3K9me2 levels in dcl2 dcl3 dcl4 correlate with loss of PAI sRNAs, we used RNA gel blot analysis to detect PAI sRNAs (Figure 3). As a negative control we used a mutant derivative of Ws, Δpai1–pai4, where the PAI1–PAI4 IR source of dsRNA DCL substrates has been deleted by homologous recombination between flanking direct repeat sequences [17]. As a positive control we used the Δpai1–pai4 strain transformed with a PAIIR transgene consisting of an IR of approximately 700 bp of PAI cDNA sequences transcribed by the strong constitutive Cauliflower Mosaic Virus 35S promoter [14]. We previously determined that PAI sRNAs could be detected in the Δpai1–pai4(PAIIR) transgenic strain but not in wild type Ws using RNA gel blot analysis with a PAI cDNA riboprobe. Furthermore, high-throughput sRNA sequencing in the C24 strain that has a similar PAI1–PAI4 IR to Ws detected PAI sRNAs at low levels [27]. We therefore optimized detection of rare PAI sRNAs by designing a high-affinity locked nucleic acid (LNA) probe corresponding to the sense strand of a 35 nt sequence in the PAI fifth exon.

In wild type Ws the LNA probe detected low levels of PAI sRNA species of both shorter and longer sizes, between 21 and 24 nt relative to size markers, above the background signal in the Δpai1–pai4 negative control strain (Figure 3). This pattern is consistent with processing of PAI1–PAI4 palindromic transcripts into sRNAs by more than one dicer. Correspondingly, the C24 high-throughput sequencing analysis detected PAI sRNAs between 21 and 24 nt long covering the entire IR region [27]. The wild type Ws levels of endogenous PAI sRNAs were comparable to levels detected in a hundred-fold dilution of RNA prepared from the Δpai1–pai4(PAIIR) positive control transgenic strain (Figure 3). The transgenic strain produced mostly smaller PAI sRNAs, as previously observed for this strain using a PAI cDNA riboprobe [14], presumably due to differences in PAI IR expression patterns and structure.
PAI sRNA species were depleted in the dcl2 dcl3 dcl4 strain to similar background levels as detected in Δpai1–pai4 (Figure 3). This result supports the hypothesis that loss of PAI sRNAs underlies the reduction in SUVH-dependent H3K9me2 and CMT3-dependent 5meC on PAI sequences. The lack of residual PAI sRNAs in dcl2 dcl3 dcl4 indicates a minimal contribution of DCL1 to generating these species, although DCL1 is functional in processing microRNAs such as miR167.
The dcl2 dcl3 dcl4 mutant has destabilized PAI2 silencing and 5meC
To determine whether loss of sRNAs causes destabilization of remaining PAI silencing modifications upon inbreeding by self-pollination, we introduced the dcl2 dcl3 dcl4 mutations into a Ws pai1 reporter background where silencing of the PAI2 singlet gene can be monitored by visual inspection. In wild type Ws, PAI1 expressed from the heterologous upstream promoter is the major source of PAI enzyme; expression of PAI2 is impaired by H3K9me2/5meC on proximal promoter sequences and PAI3 and PAI4 do not encode functional enzyme due to polymorphisms [12]. In the Ws pai1 missense mutant, the impairment of PAI2 expression is revealed through tryptophan deficiency phenotypes including reduced size and blue fluorescence under ultraviolet (UV) light caused by accumulation of the tryptophan precursor anthranilate [28]. The stable maintenance of PAI2 H3K9me2/5meC in pai1 is reflected in stable maintenance of blue fluorescence across generations of inbreeding. Mutations that decrease PAI2 H3K9me2 and/or 5meC levels in the Ws pai1 background, including cmt3, met1, and suvh4, result in reduced fluorescence [2], [16], [28].
The initial pai1 dcl2 dcl3 dcl4 strain displayed partially reduced PAI 5meC patterns similar to the PAI1 dcl2 dcl3 dcl4 strain (Figure 1, Figure 4). The pai1 dcl2 dcl3 dcl4 plants were larger and less fluorescent than pai1 plants, reflecting the partial reduction of non-CG methylation levels on PAI2 (Figure 4).
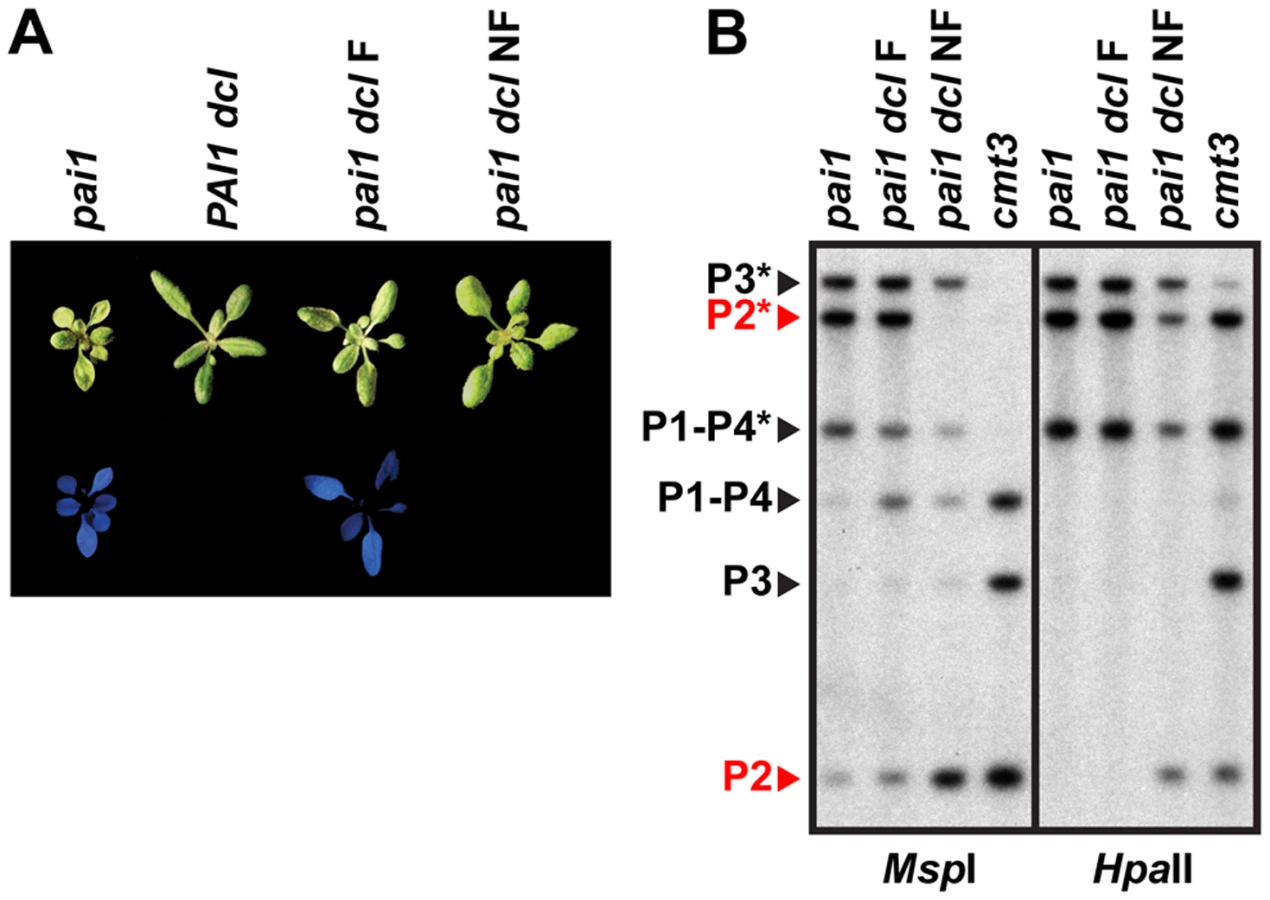
Examination of pai1 dcl2 dcl3 dcl4 inbred populations revealed that blue fluorescence diagnostic of PAI2 silencing was not stably maintained. In a population of 191 pai1 dcl2 dcl3 dcl4 plants we found two non-fluorescent segregants (1.0%). In contrast, no non-fluorescent individuals were found in control populations of thousands of pai1 plants, consistent with our previous results. Each of the non-fluorescent pai1 dcl2 dcl3 dcl4 plants yielded approximately 75% non-fluorescent and 25% fluorescent second-generation progeny (54 non-fluorescent out of 70 total progeny plants [77%] for one line and 31 non-fluorescent out of 42 total progeny plants [74%] for another line). Approximately one third of the second-generation non-fluorescent plants lacked remaining PAI2 5meC in a HincII DNA gel blot assay, and these individuals yielded 100% non-fluorescent third-generation progeny, whereas the remaining second-generation non-fluorescent plants had partial levels of PAI2 5meC and yielded approximately 75% non-fluorescent third-generation progeny. For example, 12 out of 28 [43%] non-fluorescent second-generation progeny from one line had fully demethylated PAI2 phenotypes in the HincII assay and each of these individuals yielded 100% non-fluorescent progeny; the pai1 dcl NF line shown in Figure 4 is derived from one of these individuals. The segregation patterns are consistent with reduced PAI2 5meC levels and increased expression occurring on just one of the two chromosomes in the parental non-fluorescent plant and being inherited in a Mendelian fashion.
DNA gel blot analysis of a non-fluorescent pai1 dcl2 dcl3 dcl4 line indicated a nearly complete loss of CCG methylation monitored by MspI cleavage and partially reduced CG methylation monitored by HpaII cleavage at PAI2 relative to the fluorescent parental line, consistent with the reversion of tryptophan deficiency phenotypes (Figure 4). However, PAI1–PAI4 and PAI3 maintained similar 5meC patterns to the fluorescent parental line. Therefore, in pai1 dcl2 dcl3 dcl4 the loss of PAI2 5meC and silencing detected by the blue fluorescence screen is not coupled to destabilization of 5meC at the other PAI loci.
Both the initial loss of PAI non-CG methylation and the stochastic further loss of PAI2 silencing and 5meC in dcl2 dcl3 dcl4 are similar to patterns we previously observed in the Δpai1–pai4 mutant [17]. This comparison indicates that regardless of whether PAI sRNAs are depleted by loss of DCL function or by loss of the source of PAI dsRNA substrates for DCL cleavage (Figure 3), PAI2 silencing is similarly destabilized. The destabilization could be due to a combination of effects at PAI2 including impairment of H3K9me2 maintenance, increased transcription, and impairment of the DRM2/DCL3 pathway in resetting 5meC imprints (Text S1, Figure S2).
The dcl2 dcl3 dcl4 mutant has reduced non-CG methylation levels at a subset of other SUVH/CMT3 target loci
To determine whether other SUVH/CMT3 target loci besides the PAI genes have reduced 5meC levels in the dcl2 dcl3 dcl4 mutant, we used a survey approach with MspI DNA gel blot assays (Figure 5). We monitored representative sequences of three types: a degenerate (86% identical) inverted repeat locus IR1074 [29], highly repetitive 5S rDNA and 180 bp centromeric sequences (CEN), or low-copy transposons Ta3 [7] and Mu1 [30]. At all of these sequences, there was no difference in MspI cleavage between wild type and the drm1 drm2 mutant, but greatly increased cleavage in the cmt3 mutant, indicating that CCG methylation at the monitored MspI sites is dependent on CMT3 with a minimal contribution from the DRM MTases. For each sequence, we tested both the pai1 dcl2 dcl3 dcl4 parental fluorescent strain and an isogenic non-fluorescent progeny line to determine whether these two strains had differences in 5meC patterns at loci other than PAI2 (Figure 4).

The IR1074, 5S rDNA, and CEN sequences showed partially increased MspI cleavage in both of the dcl mutant strains relative to wild type and cmt3 (Figure 5). For the highly repetitive sequences the increased cleavage was evident as a slight shift downwards in the peak intensity of the ladder of cleaved bands (Figure 5B). Therefore, similarly to the PAI genes, these sequences require DCL function for maintenance of CMT3-dependent 5meC. The sequences had similar 5meC patterns in the fluorescent versus non-fluorescent pai1 dcl2 dlc3 dcl4 lines, indicating that the loss of PAI2 5meC in the non-florescent line is not coupled to more general destabilization of 5meC.
In contrast, Ta3 and Mu1 showed no differences in MspI cleavage between the dcl mutant lines and wild type controls (Figure 5). The Mu1 transposon has a polymorphic arrangement between Ws and the Columbia (Col) strain in which the dcl alleles were originally isolated, and the dcl2 dcl3 dcl4 strain carries both Mu1 arrangements. Despite this complication, comparisons to wild type versus cmt3 controls in each strain background showed no evidence of demethylation in the dcl mutant lines. Therefore, not all SUVH/CMT3 targets display DCL-dependent maintenance of 5meC. In light of our hypothesis that sRNAs prevent loss of H3K9me2 due to read-through transcription, the different effects of the dcl mutations at different SUVH/CMT3 target loci could reflect the extent to which read-through transcription occurs across the modified sequences. For example, the dcl-sensitive locus IR1074 is likely to be transcribed across to make fold-back dsRNA because this locus produces sRNAs even in an RNA-dependent RNA polymerase rdr2 mutant background [29].
We also monitored H3K9me2 levels at the single-locus targets IR1074 and Ta3 by ChIP in the dcl2 dcl3 dcl4 mutant and the same control strains used for analysis of the PAI genes (Figure 6). At IR1074 H3K9me2 levels were reduced in the dcl2 dcl3 dcl4 mutant relative to wild type, although not as strongly as in the suvh4 suvh5 suvh6 mutant. In contrast, at Ta3 H3K9me2 was maintained at similar levels between the dcl2 dcl3 dcl4 mutant and wild type. The H3K9me2 ChIP results agree with the 5meC results indicating that full modification of IR1074 but not Ta3 depends on DCL function (Figure 5A, Figure 5C).

The cmt3 mutant had reduced levels of H3K9me2 at both IR1074 and Ta3, presumably due to impaired SUVH localization and/or increased transcription caused by loss of non-CG methylation (Figure 5, Figure 6). However, at both loci the drm1 drm2 mutant maintained H3K9me2 at similar levels to wild type, and the drm1 drm2 cmt3 mutant maintained H3K9me2 at similar levels to cmt3 (Figure 6). Therefore, CMT3 but not the DRM cytosine MTases contributes to maintenance of H3K9me2 patterns at IR1074 and Ta3.
At IR1074, the dcl2 dcl3 dcl4 mutant and the cmt3 mutant displayed similar reductions in H3K9me2 levels relative to wild type (Figure 6). However, dcl2 dcl3 dcl4 had a weaker reduction in IR1074 non-CG methylation levels than cmt3 (Figure 5). This relationship is similar to that observed for PAI1–PAI4 and PAI2 (Figure 1, Figure 2), and supports the view that loss of sRNAs in the dcl2 dcl3 dcl4 mutant impairs maintenance of H3K9me2 patterns independently of CMT3 function at loci subject to read-through transcription.
Discussion
In Arabidopsis, H3K9me2 maintained by the SUVH4, SUVH5, and SUVH6 histone MTases is used to guide 5meC in non-CG contexts maintained by the CMT3 cytosine MTase [1]–[4]. The SUVH MTases contain 5meC-binding domains, and CMT3 contains an H3K9me-binding domain, leading to the model that the SUVH/CMT3 pathway involves an amplification loop between 5meC and H3K9me2 [5], [6]. However, this amplification loop is not sufficient to maintain full levels of 5meC and H3K9me2 on the Ws PAI gene duplications, including a constitutively transcribed IR locus PAI1–PAI4 and partially silenced singlet genes PAI2 and PAI3. In previous work we showed that production of palindromic transcripts from the PAI1–PAI4 IR is also required for maintenance of PAI non-CG methylation [13], [14], [17], [18]. For example, in a Δpai1–pai4 mutant the PAI2 and PAI3 genes have reduced non-CG methylation, and the remaining 5meC on PAI2 is destabilized upon inbreeding [17]. Here we use mutations in the DCL dicer ribonucleases to show that PAI sRNAs processed from PAI dsRNAs are the key species that reinforce the SUVH/CMT3 amplification loop between H3K9me2 and non-CG methylation.
Arabidopsis uses DCL-dependent sRNAs incorporated into argonaute (AGO) effector proteins as nucleic acid sequence-specificity guides in a variety of pathways including miRNA control of development, RNA interference, and guidance of 5meC mediated by the DRM2 cytosine MTase 19,23. The DRM2 pathway contributes together with the SUVH/CMT3 pathway to maintenance of non-CG methylation at many 5meC target loci [15], [31]. This overlap has obscured whether sRNAs have an independent role in the SUVH/CMT3 pathway. However, the Ws PAI genes maintain 5meC in non-CG contexts almost entirely through the SUVH/CMT3 pathway once initial 5meC is established (Figure 1, Text S1, Figure S2). The reduction in PAI non-CG methylation and H3K9me2 levels in dcl mutant backgrounds therefore indicates a direct connection between DCL-dependent sRNAs and the SUVH/CMT3 pathway (Figure 1, Figure 2). DCL2 and DCL3 are the key dicers required for maintaining H3K9me2 and 5meC patterns on the PAI genes, suggesting a preference for longer 22 and 24 nt sRNAs in this pathway.
ChIP analysis shows that the dcl2 dcl3 dcl4 mutant has reduced H3K9me2 levels at PAI loci similar to or stronger than in the cmt3 mutant (Figure 2). However, the dcl2 dcl3 dcl4 mutant has weaker reductions in PAI 5meC levels than the cmt3 mutant (Figure 1). Therefore reduced H3K9me2 levels in the dcl mutant cannot be accounted for as a secondary effect of impaired CMT3 function. Instead, the ChIP results support the view that the dcl mutations directly impair maintenance of H3K9me2 patterns. In this view, the partial loss of PAI H3K9me2 in dcl2 dcl3 dcl4 reduces CMT3 localization, resulting in reduced PAI non-CG methylation as a secondary effect.
Similarly to the PAI genes, a subset of other SUVH/CMT3 target loci including highly repetitive 5S rDNA and CEN sequences have partially reduced non-CG methylation levels in the dcl2 dcl3 dcl4 mutant (Figure 5). However, some loci such as the low copy transposons Ta3 and Mu1 can maintain full 5meC levels relative to wild type despite the loss of DCL function. This variation could reflect the degree to which different SUVH/CMT3 target loci are transcribed across. This variation could also reflect which RNA polymerases are most active at different loci. Arabidopsis encodes five RNA polymerases: the conserved eukaryotic RNA polymerases POLI, POLII, and POLIII, and plant-specific POLIV and POLV implicated in targeting DRM2-dependent 5meC [32]. In particular, POLV is proposed to transcribe across target loci to make “scaffold” transcripts that recruit sRNA/AGO complexes and components of the DRM2 pathway [33], [34]. Because of its specialized role in making silencing-associated transcripts, POLV might be less disruptive of H3K9me2 than other RNA polymerases designed to express host genes. In this case, protein-encoding loci like the PAI genes that are transcribed by RNA POLII, and 5meC targets that depend on RNA POLII for scaffold transcript synthesis [35], might have a stronger dependence on an sRNA-based mechanism to maintain H3K9me2 than POLV-transcribed regions of the genome.
Our previous studies with allelic variants of the PAI1–PAI4 IR locus support the hypothesis that the level of transcription across the locus determines the extent to which sRNAs are needed for maintenance of PAI 5meC levels. In one study we used transgene-expressed sRNAs to direct 5meC and transcriptional silencing to the upstream promoter that drives transcription through PAI1–PAI4, thereby impairing production of PAI dsRNAs and sRNAs [14]. In this transgenic strain the PAI1–PAI4 locus was able to maintain full 5meC levels, whereas the PAI2 and PAI3 singlet genes had partially reduced non-CG methylation levels. These patterns are consistent with a model where the decreased transcription of PAI1–PAI4 specifically reduces its dependence on PAI sRNAs. In a second study we characterized a mutant derivative of Ws where a rearrangement in the center of the PAI1–PAI4 IR introduces a new polyadenylation site and reduces the levels of transcripts that extend into palindromic PAI4 sequences, without altering promoter sequences or the level of transcription across the locus [18]. In the rearrangement mutant the PAI1–PAI4 IR locus as well as the PAI2 and PAI3 singlet genes had partially reduced non-CG methylation levels. These patterns are consistent with a model where read-through transcription at all three loci together with reduced PAI sRNAs results in loss of H3K9me2 and 5meC at all three loci, similarly to the situation in the dcl2 dcl3 dcl4 mutant. Taken together, our results support a homeostatic mechanism where sRNAs produced from heterochromatic regions by read-through transcription feed back to counteract depletion of H3K9me2 and associated 5meC levels caused by read-through transcription.
The mechanistic relationship between sRNAs and maintenance of H3K9me2 patterns remains to be determined. In the fission yeast Schizosaccharomyces pombe, an sRNA-loaded AGO protein in the RITS effector complex interacts with nascent transcripts at centromeric repeats to recruit the Clr4 H3K9 MTase (reviewed in [36]). Plants could use an analogous effector complex interaction mechanism to target SUVH H3K9 MTases to specific regions of the genome. Consistent with this possibility, in a suvh4 suvh5 mutant background, the remaining SUVH6 MTase maintains levels of H3K9me2 and associated 5meC similar to wild type at the PAI1–PAI4 IR but not at the PAI2 singlet gene [4]; this locus-specific activity could reflect preferential interactions between SUVH6 and effector complexes that assemble near a site of dsRNA synthesis. Alternatively, sRNA-AGO complexes could recruit intermediate factors that then promote SUVH activity at specific targets. A third possibility is that sRNAs could guide a pathway that protects heterochromatic sequences from H3K9 demethylation. For example, the IBM1 JumonjiC domain H3K9 demethylase acts to prevent H3K9me2 and non-CG methylation from accumulating in transcribed genes 11,37,38. IBM1 could be excluded from also acting at heterochromatic sequences through a mechanism that involves sRNA-AGO complexes. Furthermore, sRNA-dependent mechanisms that promote addition of H3K9me2 or prevent removal of H3K9me2 could operate in concert.
Pathways where sRNA-AGO complexes guide H3K9me to appropriate regions of the genome have been identified in organisms ranging from fission yeast to the protozoan Tetrahymena thermophila to the insect Drosophila melanogaster, even though these organisms lack 5meC [39]–[41]. Our discovery that Arabidopsis also uses sRNAs to maintain H3K9me could represent a plant-specific variation on this fundamentally conserved strategy. In this case, the sRNA/SUVH/CMT3 pathway and the sRNA/DRM2 pathway could have both evolved from a basal mechanism involving sRNA-AGO guidance of H3K9 MTases. Consistent with this possibility, SUVH variants that lack catalytic activity but maintain methyl-DNA binding are required for DRM2-dependent 5meC [42].
The plant sRNA/H3K9me maintenance mechanism is interwoven with the SUVH/CMT3 chromatin binding amplification loop and partially redundant functions of the MET1 and DRM pathways to create a reinforced silencing network. However, loss of the sRNA/H3K9me maintenance mechanism cannot be completely buffered by the other pathways, and results in both immediate reductions and longer-term destabilization of H3K9me2 and 5meC. The unique properties of the PAI genes make them ideal reporters to further understand how sRNAs are harnessed to control maintenance of H3K9me2 on appropriate target sequences in plant genomes.
Materials and Methods
Plant strains
T-DNA insertional dcl alleles were obtained from the Arabidopsis Biological Resource Center (ABRC) or from the laboratory of James Carrington at Oregon State University. The dcl2-1, dcl3-1, and dcl4-2 mutations are likely null alleles originally isolated in the Col strain [21], [23]. The dcl1-9 mutation is a partial function allele originally isolated in Ws, but then crossed five times to the Landsberg erecta (Ler) strain 24,25. Each dcl mutant was crossed to Ws. PCR-based genotype markers were used to identify dcl mutant progeny homozygous for the three PAI loci from Ws (Table S1). Each dcl allele was crossed a second time with Ws to increase the proportion of the genome contributed by the Ws parent. The resulting dcl single mutant strains were then crossed with each other to generate double, triple, and quadruple mutant combinations. The dcl mutations were also crossed into the Ws pai1 reporter strain [28]. The Ws drm1 drm2 double T-DNA insertional null strain was obtained from the laboratory of Steven Jacobsen at UCLA [43]. The Col cmt3-11T T-DNA insertional null strain was obtained from the ABRC [44]. The Ws pai1, Ws Δpai1–pai4, Ws Δpai1–pai4(PAIIR), Ws cmt3i11a, Ws x met1-1, and pai1 suvh4R302* suvh5-1 suvh6-1 strains were previously described [4], [14], [16], [17], [28]. Ws cmt3illa and Ws drm1 drm2 mutants were crossed to make the Ws drm1 drm2 cmt3 strain.
Analysis of 5meC patterns
Plant genomic DNA preparation and DNA gel blot assays for 5meC were performed as previously described [12]. Bisulfite sequencing of the top strands of PAI1 and PAI2 proximal promoter regions was performed as previously described [14]. PAI bisulfite sequencing primers are listed in Table S1.
sRNA analysis
Total RNA was extracted with TRIzol reagent (Invitrogen) using the manufacturer's protocol. Low molecular weight (LMW) RNA was enriched by precipitating high molecular weight RNA out of solution with 0.5 M NaCl, 10% polyethylene glycol (MW 8000). The remaining LMW RNA was precipitated with 100% ethanol and resuspended in water treated with diethyl pyrocarbonate. LMW RNA was fractionated on a 17% acrylamide 7 M urea gel and transferred to a Hybond-N membrane (GE Healthcare). sRNA 5′ ends were chemically crosslinked to the membrane as previously described [45]. Membranes were hybridized in OligoHyb buffer (Ambion) overnight at 42°C with 32P 5′ end-labeled oligonucleotide probes. Probes were either an LNA modified PAI1 exon 5 sense 35-mer (Exiqon) or an miR167 antisense 21-mer (Table S1). Probed membranes were washed three times with a 2× SSC, 0.1% SDS solution. sRNA sizes were estimated from an ethidium bromide-stained low molecular weight DNA ladder (USB), and by comparison to the PAI sRNA species observed in the Δpai1–pai4(PAIIR) control strain [14].
ChIP analysis
Formaldehyde crosslinking and chromatin preparations were performed as previously described [46] starting with two grams of aerial tissue from three-week-old plants grown in soilless potting medium (Fafard mix 2) under continuous illumination. Chromatin was immunoprecipitated with anti-H3K9me2 monoclonal antibody [47] or carried through the protocol with no antibody added as a control (mock precipitation). Immunoprecipitations were performed as previously described [3]. Each ChIP assay was performed in at least three independent biological replicates. Quantitative PCR amplification of immunoprecipitated DNA was performed using the 7300 Real-Time PCR System (ABI), with three replicate reactions for each sample. ChIP primer sequences are listed in Table S1.
Supporting Information
Zdroje
1. JacksonJPLindrothAMCaoXJacobsenSE 2002 Control of CpNpG DNA methylation by the KRYPTONITE histone H3 methyltransferase. Nature 416 556 560
2. MalagnacFBarteeLBenderJ 2002 An Arabidopsis SET domain protein required for maintenance but not establishment of DNA methylation. EMBO J 21 6842 6852
3. EbbsMLBarteeLBenderJ 2005 H3 lysine 9 methylation is maintained on a transcribed inverted repeat by combined action of SUVH6 and SUVH4 methyltransferases. Mol Cell Biol 25 10507 10515
4. EbbsMLBenderJ 2006 Locus-specific control of DNA methylation by the Arabidopsis SUVH5 histone methyltransferase. Plant Cell 18 1166 1176
5. LindrothAMShultisDJasencakovaZFuchsJJohnsonL 2004 Dual histone H3 methylation marks at lysines 9 and 27 required for interaction with CHROMOMETHYLASE3. EMBO J 23 4286 4296
6. JohnsonLMBostickMZhangXKraftEHendersonI 2007 The SRA methyl-cytosine-binding domain links DNA and histone methylation. Curr Biol 17 379 384
7. JohnsonLCaoXJacobsenS 2002 Interplay between two epigenetic marks. DNA methylation and histone H3 lysine 9 methylation. Curr Biol 12 1360 1367
8. TariqMSazeHProbstAVLichotaJHabuY 2003 Erasure of CpG methylation in Arabidopsis alters patterns of histone H3 methylation in heterochromatin. Proc Natl Acad Sci U S A 100 8823 8827
9. MathieuOReindersJCaikovskiMSmathajittCPaszkowskiJ 2007 Transgenerational stability of the Arabidopsis epigenome is coordinated by CG methylation. Cell 130 851 862
10. PillotMBarouxCVazquezMAAutranDLeblancO 2010 Embryo and endosperm inherit distinct chromatin and transcriptional states from the female gametes in Arabidopsis. Plant Cell 22 307 320
11. InagakiSMiura-KamioANakamuraYLuFCuiX 2010 Autocatalytic differentiation of epigenetic modifications within the Arabidopsis genome. EMBO J 29 3496 3506
12. MelquistSLuffBBenderJ 1999 Arabidopsis PAI gene arrangements, cytosine methylation and expression. Genetics 153 401 413
13. LuffBPawlowskiLBenderJ 1999 An inverted repeat triggers cytosine methylation of identical sequences in Arabidopsis. Mol Cell 3 505 511
14. MelquistSBenderJ 2003 Transcription from an upstream promoter controls methylation signaling from an inverted repeat of endogenous genes in Arabidopsis. Genes Dev 17 2036 2047
15. ListerRO'MalleyRCTonti-FilippiniJGregoryBDBerryCC 2008 Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 133 523 536
16. BarteeLMalagnacFBenderJ 2001 Arabidopsis cmt3 chromomethylase mutations block non-CG methylation and silencing of an endogenous gene. Genes Dev 15 1753 1758
17. BenderJFinkGR 1995 Epigenetic control of an endogenous gene family is revealed by a novel blue fluorescent mutant of Arabidopsis. Cell 83 725 734
18. MelquistSBenderJ 2004 An internal rearrangement in an Arabidopsis inverted repeat locus impairs DNA methylation triggered by the locus. Genetics 166 437 448
19. RamachandranVChenX 2008 Small RNA metabolism in Arabidopsis. Trends Plant Sci 13 368 374
20. ChanSWZilbermanDXieZJohansenLKCarringtonJC 2004 RNA silencing genes control de novo DNA methylation. Science 303 1336
21. XieZJohansenLKGustafsonAMKasschauKDLellisAD 2004 Genetic and functional diversification of small RNA pathways in plants. PLoS Biol 2 E104
22. GasciolliVMalloryACBartelDPVaucheretH 2005 Partially redundant functions of Arabidopsis DICER-like enzymes and a role for DCL4 in producing trans-acting siRNAs. Curr Biol 15 1494 1500
23. HendersonIRZhangXLuCJohnsonLMeyersBC 2006 Dissecting Arabidopsis thaliana DICER function in small RNA processing, gene silencing and DNA methylation patterning. Nat Genet 38 721 725
24. JacobsenSERunningMPMeyerowitzEM 1999 Disruption of an RNA helicase/RNAse III gene in Arabidopsis causes unregulated cell division in floral meristems. Development 126 5231 5243
25. SchauerSEJacobsenSEMeinkeDWRayA 2002 DICER-LIKE1: blind men and elephants in Arabidopsis development. Trends Plant Sci 7 487 491
26. CaoXJacobsenSE 2002 Role of the Arabidopsis DRM methyltransferases in de novo DNA methylation and gene silencing. Curr Biol 12 1138 1144
27. MolnarAMelnykCWBassettAHardcastleTJDunnR 2010 Small silencing RNAs in plants are mobile and direct epigenetic modification in recipient cells. Science 328 872 875
28. BarteeLBenderJ 2001 Two Arabidopsis methylation-deficiency mutations confer only partial effects on a methylated endogenous gene family. Nucleic Acids Res 29 2127 2134
29. LuCKulkarniKSouretFFMuthuValliappanRTejSS 2006 MicroRNAs and other small RNAs enriched in the Arabidopsis RNA-dependent RNA polymerase-2 mutant. Genome Res 16 1276 1288
30. LippmanZMayBYordanCSingerTMartienssenR 2003 Distinct mechanisms determine transposon inheritance and methylation via small interfering RNA and histone modification. PLoS Biol 1 E67
31. CaoXJacobsenSE 2002 Locus-specific control of asymmetric and CpNpG methylation by the DRM and CMT3 methyltransferase genes. Proc Natl Acad Sci U S A 99 Suppl 4 16491 16498
32. PikaardCSHaagJRReamTWierzbickiAT 2008 Roles of RNA polymerase IV in gene silencing. Trends Plant Sci 13 390 397
33. WierzbickiATHaagJRPikaardCS 2008 Noncoding transcription by RNA polymerase Pol IVb/Pol V mediates transcriptional silencing of overlapping and adjacent genes. Cell 135 635 648
34. WierzbickiATReamTSHaagJRPikaardCS 2009 RNA polymerase V transcription guides ARGONAUTE4 to chromatin. Nat Genet 41 630 634
35. ZhengBWangZLiSYuBLiuJY 2009 Intergenic transcription by RNA polymerase II coordinates Pol IV and Pol V in siRNA-directed transcriptional gene silencing in Arabidopsis. Genes Dev 23 2850 2860
36. MoazedD 2009 Small RNAs in transcriptional gene silencing and genome defence. Nature 457 413 420
37. MiuraANakamuraMInagakiSKobayashiASazeH 2009 An Arabidopsis jmjC domain protein protects transcribed genes from DNA methylation at CHG sites. EMBO J 28 1078 1086
38. SazeHShiraishiAMiuraAKakutaniT 2008 Control of genic DNA methylation by a jmjC domain-containing protein in Arabidopsis thaliana. Science 319 462 465
39. VolpeTAKidnerCHallIMTengGGrewalSI 2002 Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi. Science 297 1833 1837
40. LiuYMochizukiKGorovskyMA 2004 Histone H3 lysine 9 methylation is required for DNA elimination in developing macronuclei in Tetrahymena. Proc Natl Acad Sci U S A 101 1679 1684
41. Pal-BhadraMLeibovitchBAGandhiSGRaoMBhadraU 2004 Heterochromatic silencing and HP1 localization in Drosophila are dependent on the RNAi machinery. Science 303 669 672
42. JohnsonLMLawJAKhattarAHendersonIRJacobsenSE 2008 SRA-domain proteins required for DRM2-mediated de novo DNA methylation. PLoS Genet 4 e1000280
43. CaoXSpringerNMMuszynskiMGPhillipsRLKaepplerS 2000 Conserved plant genes with similarity to mammalian de novo DNA methyltransferases. Proc Natl Acad Sci U S A 97 4979 4984
44. ChanSWHendersonIRZhangXShahGChienJS 2006 RNAi, DRD1, and histone methylation actively target developmentally important non-CG DNA methylation in Arabidopsis. PLoS Genet 2 e83
45. PallGSHamiltonAJ 2008 Improved northern blot method for enhanced detection of small RNA. Nat Protoc 3 1077 1084
46. SalehAAlvarez-VenegasRAvramovaZ 2008 An efficient chromatin immunoprecipitation (ChIP) protocol for studying histone modifications in Arabidopsis plants. Nat Protoc 3 1018 1025
47. KimuraHHayashi-TakanakaYGotoYTakizawaNNozakiN 2008 The organization of histone H3 modifications as revealed by a panel of specific monoclonal antibodies. Cell Struct Funct 33 61 73
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 10
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- The Glycobiome Reveals Mechanisms of Pentose and Hexose Co-Utilization in Bacteria
- Global Mapping of Cell Type–Specific Open Chromatin by FAIRE-seq Reveals the Regulatory Role of the NFI Family in Adipocyte Differentiation
- Genetic Determinants of Serum Testosterone Concentrations in Men
- MicroRNA Expression and Regulation in Human, Chimpanzee, and Macaque Brains
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy