
Novinky v patogenezi dvou podtypů myelodysplastického syndromu: 5q minus syndromu a sideroblastické anémie, částečně platné i u jiných onkohematologických onemocnění
New findings in the pathogenesis of two myelodysplastic syndrome subtypes: 5q - syndrome and sideroblastic anaemia, partially relevant for other onco-haematological diseases
Myelodysplastic syndrome (MDS) is a pre-cancerosis, or rather a pre-leukaemic state. It is not a definitive disease but rather involves heterogeneous clinical manifestations ranging from cytopenias to conditions similar to acute myeloid leukaemia. This characteristic of MDS explains the difficulty of elucidating its pathogenesis. Admirable progress regarding cell structure and metabolism as well as cell growth regulation has extended our understanding of MDS pathogenesis. Recently, two interesting findings in the pathogenesis of 5q - syndrome and refractory anaemia with ringed sideroblasts (RARS) have been reported. In 5q - syndrome, ribosomal stress has been identified as the underlying mechanism of refractory anaemia. It has also been partially explained why ribosomal stress does not hamper effective megakaryopoiesis in this MDS subtype. In the majority of RARS patients, a mutation of the SF3B1 gene, which plays a role in pre-mRNA splicing, has been uncovered. The contribution of this SF3B1 mutation to the development of ringed sideroblasts and its role in iron metabolism within erythroblasts represent an impulse for further research. SF3B1 mutation in chronic lymphocytic leukaemia as opposed to RARS is associated with poor prognosis.
Key words:
pathogenesis of MDS, 5q - syndrome, ribosomal stress, RARS, mutation of SF3B1, defect RNA splicing
Autoři:
R. Neuwirtová 1; O. Fuchs 2; A. Jonášová 1
Působiště autorů:
I. interní klinika – klinika hematologie, Všeobecná fakultní nemocnice, Lékařská fakulta, Univerzita Karlova Praha
1; Ústav hematologie a krevní transfuze Praha
2
Vyšlo v časopise:
Transfuze Hematol. dnes,18, 2012, No. 4, p. 154-161.
Kategorie:
Souhrnné práce, původní práce, kazuistiky
Souhrn
Myelodysplastický syndrom (MDS) je prekanceróza, resp. preleukemie. Nejde tedy o definitivní podobu choroby, ale o heterogenní klinické projevy počínaje cytopeniemi až ke stavům málo se lišícím od akutní myeloidní leukemie. S charakterem tohoto syndromu souvisí i obtížnost objasňování patogeneze MDS. Díky obdivuhodnému pokroku ve znalosti struktury a metabolismu buňky a v regulaci růstu buněk se posunují naše znalosti i v patogenezi MDS kupředu. V poslední době došlo k zajímavým objevům v patogenezi dvou podtypů MDS a to 5q-syndromu a refrakterní anémie s prstenčitými sideroblasty (RARS). U 5q-syndromu se podařilo objasnit příčinu refrakterní anémie tzv. ribozomálním stresem a částečně odpovědět na otázku, proč u tohoto syndromu ribozomální stres nebrání efektivní megakaryopoéze. U RARS byla nalezena ve vysokém procentu mutace jednoho z genů, které participují na sestřihu RNA na mRNA. Účast mutace na vzniku prstenčitých sideroblastů a metabolismu železa v těchto buňkách je podnětem k dalšímu výzkumu. Tatáž mutace u chronické lymfatické leukemie na rozdíl od RARS je spojena se špatnou prognózou choroby.
Klíčová slova:
patogeneze MDS, 5q-syndrom, ribozomální stres, RARS, mutace SF3B1, defektní RNA sestřih
Úvod
Myelodysplastický syndrom (MDS) je poměrně mladý syndrom ve srovnání s jinými onkohematologickými chorobami. Byl popsán francouzsko-americko-britskou skupinou (FAB) hematologů v roce 1982 (1). FAB skupina vybrala příhodné jméno myelodysplastický syndrom podle základního společného znaku, což je morfologická dysplasie krvetvorných buněk. Jde ale o velmi heterogenní skupinu chorob. Zatím co tzv. MDS o vysokém riziku už má částečné rysy akutní myeloidní leukemie, onemocnění řazená do MDS o nízkém riziku nemají klinický charakter nádorového onemocnění, s kterým se neslučuje předčasná apoptóza buněk. Nicméně laboratorní vyšetření už v této fázi MDS odkrývá řadu vlastností, typických pro nádorovou buňku (obr. 1). Jde vlastně o prekancerózu, v tomto případě o preleukemii.

Složitost myelodysplastického syndromu se odráží i v obtížném odkrývání patogeneze procesů, vedoucích ke klinickým obrazům MDS. Hematologové, kteří bádají v patogenezi MDS, mohou jen závidět třeba těm kolegům, kteří se zabývají chronickou myeloidní leukemií, kde je jednotný nález filadelfského Ph+ chromozomu a konzistentní klinický obraz. Proto nezbývá v oblasti MDS než se smířit s tím, že zatím postupně odkrýváme nové etiopatogenetické procesy a sestavujeme je jako střípky do mozaiky. Ačkoli ještě mnoho kaménků zbývá do konečného obrazu (pokud se to vůbec někdy podaří), přece jen se nám už části obrazu vynořují.
Historicky první globální představu o patogeneze MDS nám umožnil sekundární MDS. Sekundární MDS vzniká po působení různých mutagenů, jako je záření nebo cytostatika po terapii nejčastěji maligních chorob, nebo u profesí, kde se člověk setkává s benzenem, organickými rozpouštědly nebo insekticidy (2, 3). I u primárního MDS předpokládáme poškození genomu pluripotentní kmenové buňky mutageny ze životního prostředí, které se u disponovaných osob během života sčítají. Vzniká patologický klon, který získává růstovou výhodu, takže postupně obsadí celou dřeň. U nemocných se sekundárním MDS téměř vždy nalézáme chromozomové aberace; např. po alkylačních cytostaticích a benzenu to jsou defekty 5. a 7. chromozomu. Tato skutečnost dala podnět k širokému cytogenetickému výzkumu u MDS. Mezi chromozomovými aberacemi převažují delece chromozomů. Cytogenetické vyšetřování je bezpodmínečně nutné pro stanovení diagnózy a prognózy, ale přináší i fakta významná pro výzkum patogeneze MDS (4).
Milníkem v porozumění klinického nálezu MDS a startem pro cílený výzkum patogeneze MDS byla úvaha o předčasné apoptóze krvetvorných buněk, která vysvětlila paradox hyper - nebo normocelulární dřeně a chudé, cytopenické periferie (5). Rozvinulo se vyšetřování pro - a anti-apoptotických faktorů. Nicméně předčasná apoptóza nevysvětlí beze zbytku diskrepantní nález dřeně a periferie. Uvažuje se také o předčasné senescenci krvetvorných buněk (6). Proto probíhalo a průběžně probíhá vyšetřování úlohy cytokinů, onkogenů resp. tumor supresorových genů a řady faktorů signálních soustav, účastnících se buněčného cyklu. V malém procentu MDS nemocných se předpokládá poškození kmenové buňky autoreaktivními cytotoxickými T lymfocyty (7, 8, 9). S cílem blíže objasnit etiopatogenezi MDS se studují různé negativní vlivy, jako je oxidativní poškození buněk, neschopnost reparovat poškozenou DNA, defekty imunity. K objasnění patogeneze MDS by mohly velmi pomoci experimentální modely, což se daří u MDS jen výjimečně a jen částečně odpovídá model na zvířeti MDS u pacientů (10).
Obrovským skokem kupředu v objasňování patogeneze MDS je zavedení molekulárně genetických metod. Podařilo se prokázat defekty v epigenetických procesech, zabraňující přístup transkripčních faktorů k DNA jako jsou hypermetylace DNA nebo aktivita histondeacetyláz. Mnoho výzkumných prací se zabývá expresí genů (11), úlohou mikroRNA (12). Zdokonalování vyšetřovacích metod (např. sekvenování) umožňuje odhalování bodových mutací genů. Modelem pro MDS mohou být některé vrozené mutace vedoucí k poruchám krvetvorby, jako je tomu u příbuznosti DBA a 5q - syndromu (13). V řadě případů MDS nemusí docházet k mutaci genů, ale stačí chybění genu na jedné alele chromozomu (haploinsuficience) (14), což je u MDS s častými delecemi chromozomů nebo jejich částí příčinou patologických klinických obrazů.
Dvě novinky v patogenezi 5q minus syndromu a sideroblastické anémie
Kaménky do mozaiky patogeneze MDS představují dva pozoruhodné objevy z velmi nedávné doby, týkající se 5q - syndromu a sideroblastické anémie, resp. refrakterní anémie s věnečkovými sideroblasty (RARS). Díky moderním laboratorním metodám a díky výpočetní technice se další doplňující významné nálezy objevují v neobyčejně krátké době. Ve spolupráci řady hematologických center jsou novinky v patogenezi MDS vzápětí prověřovány v klinické medicíně. Finálně lze pak doufat, že nové poznatky přinesou i tolik potřebné nové způsoby terapie. Navíc se rýsuje, že oba nové objevy v patogeneze MDS mají význam i v patogeneze a klinice jiných onkohematologických a onkologických chorob.
U nemocných s delecí dlouhého ramene 5. chromozomu se podařilo vysvětlit mechanismus anémie haploinsuficiencí genu pro protein malé subjednotky ribozomu (RPS14) (15). U 65 % nemocných se sideroblastickou anémií (MDS-RARS, RCMD-RS) byla nalezena mutace genu SF3B1, která vede k defektnímu sestřihu (splicingu) pre-mRNA na mRNA (16). Tento nález otvírá vrátka k poznání mechanismu hromadění železa v mitochondriích kolem jádra erytroblastů.
Haploinsuficience genu RPS14 vysvětluje patogenezi refrakterní anémie u 5q - syndromu
MDS s izolovanou delecí dlouhého ramene 5. chromozomu (del 5q) tvoří 15 % nemocných s MDS. Mezi nimi nejčastější podskupinou jsou nemocní s tzv. 5q minus syndromem. Ten už v roce l974 popsal významný cytogenetik van den Berghe (1). Jeho popis klinického obrazu tohoto typu MDS platí dodnes, jak také ukazuje kolektivní studie naší MDS skupiny (18). Je to vedle delece dlouhého ramene 5. chromozomu makrocytová anémie, normální nebo zvýšený počet destiček, megakaryocyty s nelobulizovaným jádrem, nezvýšený počet blastů a většinou početně chudší červená řada ve dřeni. Mimochodem navíc platí van den Bergheho pozorování, že převážně jde o ženy (v naší sestavě v 80 %), což se zatím neví proč. Ostatní pacienti s izolovaným del(5q) v naší sestavě, kteří nesplňují kritéria 5q - syndromu, mají často trombocytopenii, bohatou erytropoézu, jen malé procento nelobulizovaných megakaryocytů a horší prognózu.
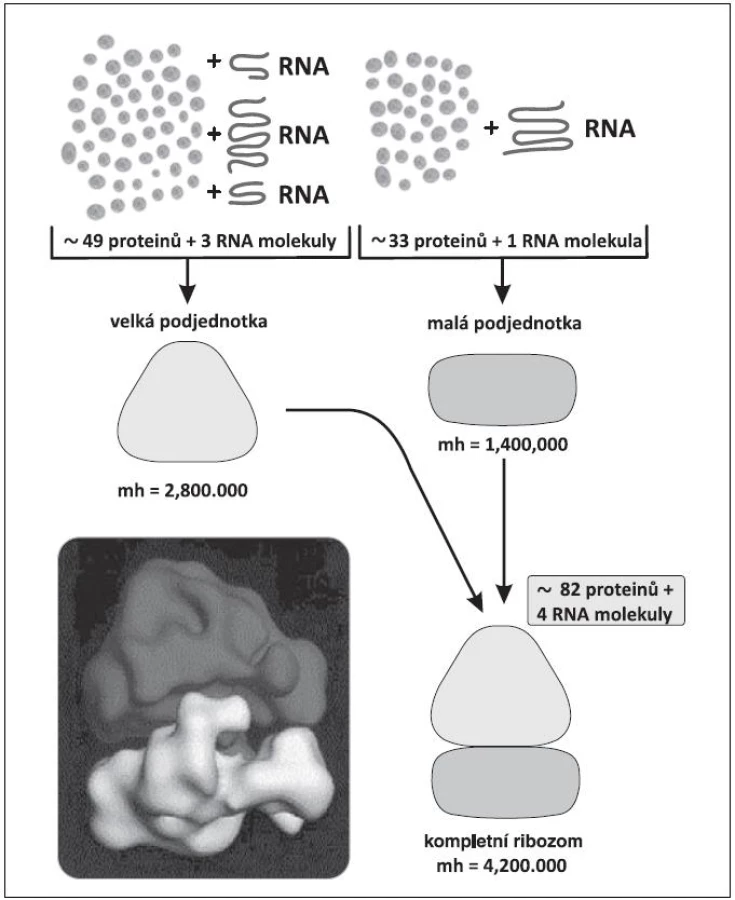
Všem pacientům s del(5q) je společné chybění úseku q31-32 (dnes dle některých autorů až 33) tzv. common deleted region – CDR. Boultwoodová a spol. odvedli skvělou práci před 10 lety, když stanovili 40 genů, lokalizovaných na tomto úseku (19). Při pátrání po jejich funkci byl zpočátku přičítán patogenetický význam genu SPARC (Secreted Protein, Acidic and Rich in Cystein), o kterém se později zjistilo, že má sice pro MDS pacienty s del(5q) také význam, nikoli však klíčový, protože hraje úlohu hlavně v adhezivitě buněk. S objevem zásadního významu přišel Ebert a spol. v roce 2008 (15). Sofistikovanou metodou pomocí manipulace s shRNA (short hairpin RNA=krátká RNA obsahující vlásenku) postupně vyřazoval geny na CDR až prokázal, že haploinsuficience genu RPS14 pro jeden z proteinů malé podjednotky ribozomu (obr. 2) s následnou defektní tvorbou ribozomů má význam pro erytropoézu. Při chybění RPS14 se in vitro snižuje tvorba buněk červené řady, které mají fenotyp erytroidních buněk u 5q - syndromu. Práce byla přijata pro publikaci v Nature také proto, že přispěla k popisu významu tzv. haploinsuficience genů. Nejdříve se anémie na podkladě haploinsuficience genu RPS14 vysvětlovala nespecificky tím, že ze tří krvetvorných řad je zejména erytropoéza nejvíce závislá na intenzivní proteosyntéze. Brzo však byla patogeneze anémie elegantně upřesněna (20, 21). Při nedostatečné tvorbě byť jen jednoho z 33 proteinů malé podjednotky ribozomu se omezí tvorba ribozomů a některé z nekonzumovaných ribozomálních proteinů se v buňce hromadí a vyvolávají tzv. ribozomální stres (21). Navážou se na faktor HDM2 (u myší MDM2 - mouse double minute 2) a inhibují jeho aktivitu. HDM2 je ubiquitin-ligáza v ubiquitin-proteasomovém systému (obr. 3) (22, 23). Jeden z proteinů, jehož hladinu ligáza HDM2 reguluje ubiquitinylací a degradací v proteasomu, je p53, nejdůležitější proapoptotický faktor buňky. HDM2 se také váže k transaktivační oblasti p53 a inhibuje transkripční aktivitu p53 (24). Potlačená aktivita HDM2 v rámci ribozomálního stresu vede k zvýšení p53 i k zvýšení transkripční aktivity p53 v erytroidních buňkách, tím k jejich apoptóze a k refrakterní anémii (14, 20). Tento mechanismus anémie u 5q - syndromu je patrně obdobný i u Diamond-Blackfanovy anémie (DBA). U DBA dochází k ribozomálnímu stresu na podkladě vrozené mutace některých genů pro ribozomální proteiny (13, 25) s podobnými následky pro patogenezi anémie.

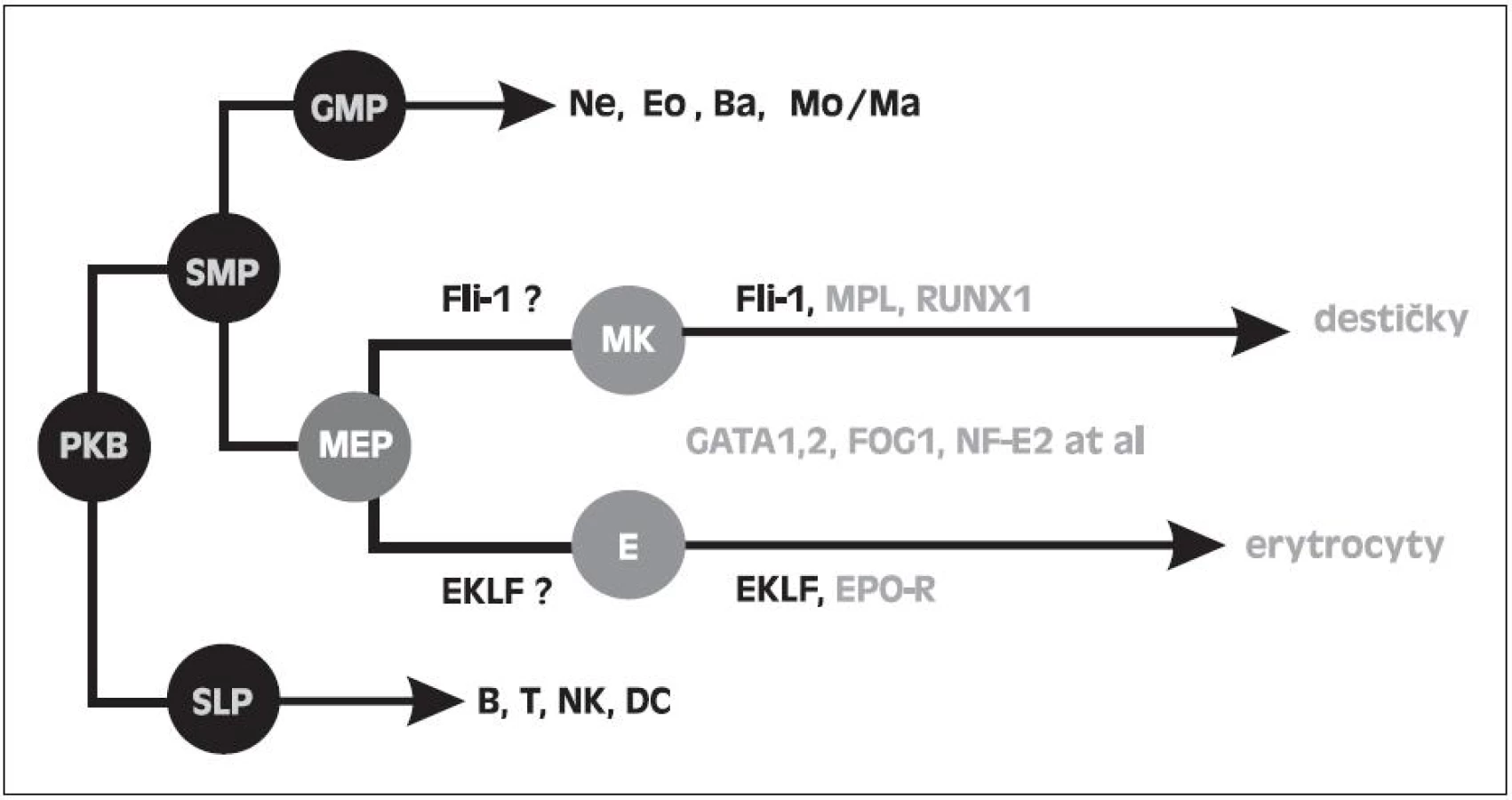
Jak je však možné, že u 5q - syndromu s refrakterní anémií při inefektivní erytropoéze je současně možná efektivní megakaryopoéza, která produkuje destičky při horní hranici normy nebo často ve zvýšeném počtu, ačkoli v buňkách obou řad je přítomna haploinsuficience RPS14 a tudíž i ribozomální stres. Tuto otázku jsme se pokusili vysvětlit naším nálezem zvýšené hladiny transkripčního faktoru Fli1 (Friend leukemia virus integration 1) u nemocných s 5q - syndromem. Na obrázku 4 je uvedeno schéma vývoje krevních buněk ze společné kmenové buňky, kde destičková a erytroidní řada má dočasně společný progenitor MEP (megakaryocyto-erytroidní progenitor). Kromě řady dalších faktorů se podílí dle prací in vitro na diferenciaci MEPu do erytroidní řady faktor EKLF (Erythroid Krüppel like factor) a do destičkové řady Fli1 (26, 27). Naše výsledky prokazující zvýšení mRNA FLi1 v mononukleárech krve a dřeně u nemocných s 5q - syndromem jsou uvedeny na obrázku 5. U těchto nemocných jsme naopak nacházeli snížení EKLF ve srovnání s kontrolami. Předběžné výsledky byly předneseny na výroční konferenci Americké hematologické společnosti (ASH Annual Meeting) v roce 2009 (28), na Olomouckých hematologických dnech (OHD) v roce 2010 (29) a na rozšířeném počtu nemocných prezentovány na ASH Annual Meeting 2011 (30). Aktuálně byla práce přijata do tisku. Příčinu zvýšení Fli1 s největší pravděpodobností vysvětluje nález Kumara a spol. a to haploinsuficiencí mikroRNA 145, jejíž gen je lokalizován na CDR 5q (31). Jedním z cílových genů pro tuto mikroRNA je právě gen pro Fli1. Snížená mikroRNA 145 tedy nebrání mediátorové RNA (mRNA) pro Fli1 dospět do translační fáze a úspěšně zajistit zvýšenou syntézu Fli1. Tento faktor podporuje megakaryopoézu u 5q - syndromu dvojím způsobem. Jednak je to vliv na diferenciaci MEP do destičkové řady (32), jednak se podle Truonga váže na promotor genu pro HDM2 a chrání tuto ligázu před ribozomálním stresem (33). V buňkách destičkové řady může HDM2 plnit svou funkci a to regulovat hladinu p53. Na rozdíl od erytroidních buněk se p53 nezvyšuje, buňky destičkové řady nepodléhají předčasné apoptóze a megakaryopoéza je efektivní. Toto je naše vysvětlení efektivní megakaryopoézy na rozdíl od inefektivní erytropoézy. V literatuře se uvádí spekulativní vysvětlení a to, že megakaryopoéza je méně závislá na proteosyntéze než erytropoéza (34). Mimochodem můžeme spekulovat, zda vliv Fli1 na HDM2 a hladinu p53 není vlastně součástí jeho úlohy jako onkogenu, který tímto mechanismem má bránit apoptóze nádorové buňky.

Jen krátkou poznámkou se vracíme k druhému faktoru a to EKLF v diferenciaci MEP do erytroidní řady. Prokázali jsme sníženou hladinu mRNA EKLF jako u 5q - syndromu, tak i u DBA (30). Zatím chybí experimentální důkazy účasti snížené hladiny EKLF na anémii u obou ribozomopatií. Jediné, co jsme zjistili je, že snížení EKLF je skutečné: u nemocných s 5q - syndromem není způsobeno hypermetylací genu pro EKLF, jak prokázala Hájková (30).
Mutace genu SF3B1 u MDS typu RARS nebo RCMD-RS
Zcela jiný systém je objeven v etiopatogeneze RARS. Jde o mutace genu SF3B1, který je angažovaný v tzv. mechanismu sestřihu RNA neboli RNA splicingu (16, 35, 36, 37). Čeští molekulární genetici vidí ve splicingu sestřih, zatímco anglosaský svět ve slově splicing rozumí navazování dvou konců např. provázku (podle slovníku). V obou odborných jazykových výrazech je zachycena obojí činnost: jde o vystřižení nekódujících (zatím s nepoznanou funkcí) intronů z pre-mRNA a pospojování pouze exonů, nositelů informace (obr. 6). Tak vzniká mediátorová RNA, která má donést informaci o syntéze určitého proteinu do translační fáze. Tento proces se odehrává v jádře a to v tzv. spliceosomu. Spliceosom je velký komplex složený z RNA a malých nukleárních ribonukleoproteinů (snRNA), které mají své specifické úlohy a dále z více než 100 asociovaných proteinů. Mutace genů pro komponenty spliceosomu vedou k poruchám proliferace buňky v pozitivním i negativním smyslu. Homozygotní mutace se nesnáší s životem buňky. U 45–85 % nemocných s MDS a CMML byla zatím nalezena somatická mutace jednoho z osmi genů: SF3B1, SRSF2, U2AF35, ZRSR2, SF3A1, PRP40B, SF1, U2AF65 (16).

Zatím nejvíce poznatků se týká mutace genu SF3B1 (16, 35, 36). Tato mutace byla nalezena ve dřeni u 20 až 45 % MDS nemocných. Gen pro SF3B1 je lokalizován na 2q33.1 (37). Bližší analýzou se ukázalo, že většinou byla mutace potvrzena u těch typů MDS, kde byly přítomny věnečkové sideroblasty (65 až 85 % případů) (35). Věnečkové nebo také prstenčité sideroblasty jsou, jak je známo, erytroblasty, které mají nahromaděné železo (navázané na speciálním feritinu, tzv. mitoferrin) v mitochondriích a ty jsou lokalizované v erytroidních buňkách kolem jádra.
Mutace SF3B1 byla nalezena i u jiných onemocnění s průměrnou frekvencí procenta výskytu, jak je uvedeno v tabulce 1. Je velmi vzácná u dětské MDS a JMML (38). Mutace SF3B1 byla popsána u ojedinělých nemocných s CMML a MDS/MPS (39, 40, 41) a u deseti ze 155 pacientů s primární myelofibrózou; u šesti z nich byly přítomny věnečkové sideroblasty (42).

Z klinického hlediska je překvapením, že v prvních klinických pracích při srovnání MDS s věnečkovými sideroblasty s mutací SF3B1 a s nemutovaným genem jsou parametry: délka života (OS), doba do transformace do AML (LFS) a přežívání bez vedlejších příznaků (EFS) příznivější u nemocných s prokázanou mutací (obr. 7) (39, 40, 41). Tito nemocní mají vyšší počet leukocytů a destiček a také bohatší buněčnost červené řady než MDS nemocní s nemutovaným SF3B1 (39). Mutace genů tedy nemusí vždy znamenat horší prognózu pro svého nositele. Nicméně další práce už neprokazovaly tak výrazný přínos mutace SF3B1 k životu erytroidní buňky. Buď našli lepší přežívání jen u RCMD-RS, ale ne u RARS, tj. u nemocných s dysplazií jen červené řady (obr. 8), nebo dokonce autoři uzavírají skepticky, že příznivá délka života souvisí s obecně příznivou prognózou diagnózy RARS nezávisle na mutaci (37, 41). Zatím asi musíme vyčkat dalších klinických studií.


Jakmile se seznámí hematolog s novinkou o častém výskytu mutace SF3B1 u RARS, musí ho napadnout otázka, jak souvisí mutace SF3B1 s poruchou hromadění železa v mitochondriích ve věnečkových sideroblastech. Zatím předběžnou odpověď přináší sdělení Boultwoodové a spol., že pro RARS je typické snížení exprese genu ABCB7 (ATP-binding cassette protein B7). Tento gen kóduje protein, který má zajišťovat transport železa z mitochondrií do cytoplazmy (43, 44). U sideroblastické anémie je časté spojení SF3B1 se sníženou expresí genu ABCB7. Lze tedy předpokládat, že vázne transport železa resp. shluků železa se sírou (Fe/S) z mitochondrií do cytosolu (44).
Další zajímavostí, jejíž význam zatím není jasný, je časté spojení mutace SF3B1 s mutací genu pro metyltransferázu DNMT3A (16). První klinické studie svědčí pro horší prognózu nemocných s RARS, kteří mají současně mutaci SF3B1 a DNMT3A (45, 46). Zřejmě přicházíme do náročné etapy analýzy současných mutací řady genů a nejen těch ze spliceosomu. Tyto mutované geny a jimi kódované faktory mezi sebou i s faktory normálními vzájemně interagují s různým klinickým dopadem. Anglosaská literatura má pro tuto situaci hezký výraz “cross talk“ mutací. Výskyt několika mutací se zdá zvláště častý u MDS (41, 46, 47, 48).
Opačný význam pro prognózu onemocnění než u MDS s věnečkovými sideroblasty má přítomnost mutace SF3B1 u nemocných s CLL (49, 50, 51) (tab. 1). Prognóza je špatná zejména u těch pacientů, kteří současně mají del(11q). Rossi zjistil u nemocných s CLL s mutací SF3B1 rezistenci na fludarabin (51).
Pro kliniky lze vyvodit z těchto zbrusu nových objevů několik podnětů. V určení prognózy MDS s věnečkovými sideroblasty bude vhodné vyšetřit kromě IPSS přítomnost mutace SF3B1. Ještě významnější bude vyšetření genu SF3B1 u nemocných s CLL. Dále bude vhodné revidovat WHO klasifikaci MDS z roku 2008. Do této klasifikace vrátit RCMD-RS (refractory cytopenia with multilineage dysplasia and ring sideroblasts) jako samostatný podtyp MDS. Zajímavé budou výsledky studií, zda mutace SF3B1 má prognostický význam u nemocných, kteří mají méně než 15 % sideroblastů (hranice pro oprávněnost určení sideroblastické anémie). Také je otázka, zda přítomnost SF3B1 zlepšuje prognózu u nemocných s MDS o vysokém riziku. Dále by mohlo být zajímavé zjistit, zda sideroblastická krvetvorba, častá u sekundárních MDS nebo u MDS se současnou jinou malignitou, je také spojena s přítomností mutace SF3B1. Specielně u MDS pak očekáváme přínos nejen pro upřesnění diagnózy a prognózy, ale i pro stále dosti omezenou terapii.
Závěr
V posledních 2–3 letech se podařilo obohatit poznatky o patogenezi MDS o dva objevy ze zcela nových oblastí. U 5q - syndromu byl prokázán mechanismus anémie ve snížené tvorbě ribozomů, vedoucí posléze k vysoké hladině proapoptotického faktoru p53 v erytroblastech a k refrakterní anémii. Na rozdíl od erytropoézy je u 5q-syndromu efektivní megakaryopoéza. Čeští autoři prokazují v buňkách magakaryocytové řady zvýšenou hladinu faktoru Fli1, který je součástí mechanismu chránícího tyto buňky před ribozomálním stresem. Zcela nový pohled na patogenezi MDS typu sidroblastické anémie přináší nález mutace genu SF3B1, který je složkou mechanismu provádějícího sestřih RNA na mRNA. Zatímco u MDS je mutace SF3B1 prognosticky příznivá, u CLL (výskyt mutace SF3B1 asi u l5 % nemocných) znamená horší prognózu onemocnění a rezistenci na fludarabin.
Podíl autorů na rukopisu
R.N. , O.F. , A.J.
Poděkování
Práce byla podpořena grantem NT/13836-4/2012 IGA MZ ČR a grantem PRVOUK-P27/LF1/2.
Doc. MUDr. Radana Neuwirtová CSc.
I. interní klinika, klinika hematologie
Všeobecná fakultní nemocnice
Univerzita Karlova, 1. Lékařská fakulta
U Nemocnice 2
128 08 Praha 2
Doručeno do redakce: 10. 7. 2012.
Přijato po recenzi: 18. 9. 2012
Zdroje
1. Bennett JM, Catovsky D, Daniel MT, et al. Proposals for the classification of the myelodysplastic syndromes. Br J Haematol 1982; 51 : 189-199.
2. Pedersen-Bjergaard J. Therapy related myelodysplasia. Leukemia Res 2007; 31: S1-S3.
3. Bělohlávková P, Neuwirtová R, Čermák J, et al. Sekundární myelodysplastický syndrom: retrospektivní analýza dat z registru České pracovní MDS skupiny. Transfuze Hematol dnes 2009; 15 : 237-243.
4. Michalová K, Zemanová Z. Molekulární cytogenetika v diagnostice nádorových onemocnění. Cas Lek Cesk 2006; 145 : 532-537.
5. Yoshida Y, Mufti G. Apoptosis and its significance in MDS: controversies revisited. Leukemia Res 1999; 23 : 777-785.
6. Wang Y, Cen J, He J, et al. Accelerated cellular senescence in myelodysplastic syndrome. Exp Hematol 2009; 37 : 1310-1317.
7. Jonášová A, Neuwirtová R, Čermák J. Cyclosporin A therapy in hypoplastic MDS patiensts and certain refractory anaemias without hypoplastic bone marrow. Br J Haematol 1998; 100 : 304-309.
8. Cukrová V, Neuwirtová R, Bartůňková J, et al. Defective cytotoxicity of T lymphocytes in myelodysplastic syndrome (MDS). Exp Hematol 2009; 37 : 386-394.
9. Dobbelstein C, Ganser A. Immunosuppresive therapy for myelodysplastic syndromes. Curr Pharm Des 2012; 18 : 3184-3189.
10. Barlow JL, Drynan LF, Hewett DR, et al. A p53-dependent mechanism underlies macrocytic anemia in a mouse model of human 5q - syndrome. Nat Med 2009; 15 : 59-66.
11. Vašíková A, Budinská E, Beličková M, et al. Differential gene expression of bone marrow CD34+ cells in early and advanced myelodysplastic syndrome. Neoplasma 2009; 56 : 335-342.
12. Dostalova-Merkerova M, Krejcik Z, Votavova H, et al. Distinctive microRNA expression in CD34+ in bone marrow cells from patients with myelodysplastic syndrome. Eur J Hum Genet 2011; 19 : 311-319.
13. Cmejla R, Cmejlova J, Handrkova H, et al. Ribosomal protein S17 gene (RPS17) is mutated in Diamond-Blackfan anemia. Hum Mutat 2007; 28 : 1178 - 1182.
14. Ebert BL. Molecular dissection of the 5q deletion in MDS. Semin Oncol 2011; 38 : 621-626.
15. Ebert BL, Pretz J, Bosco J, et al. Identification of RPS14 as a 5q - syndrome gene by interference screen. Nature 2008; 451 : 335-339.
16. Steensma DP. Surprising splicing: the new most frequent class of genetic alteration in MDS. Hematologist 2012; 9 : 5 and 7.
17. Van den Berghe H, Cassiman JJ, David G, et al. Dicstinct haematological disorder with deletion of long arm of no. 5 chromosome. Nature 1974; 251 : 437-438.
18. Neuwirtová R, Jonášová A, Čermák J. Analýza nemocných s myelodysplastickým syndromem (MDS) s delecí dlouhého ramene 5. chromosomu (del(5q)), sledovaných Českou MDS pracovní skupinou. Význam pro diagnostické zařazení a určení prognózy. Transfuze hematol dnes 2009; 15 : 204-209.
19. Boultwood J, Fidler C, Strickson AJ, et al. Narrowing and genomic annotation of the commonly deleted region of the 5q - syndrome. Blood 2002; 99 : 4638-4641.
20. Pellagatti A, Marafioti T, Paterson JC, et al. Induction of p53 and up-regulation of the p53 pathway in the human 5q - syndrome. Blood 2010; 115 : 2721-2723.
21. Zhang Y, Lu H. Signaling to p53: Ribosomal proteins find their way. Cancer Cell 2009; 16 : 369-377.
22. Glickman MH, Ciechanover A. The ubiquitin-proteasom proteolytic pathway: destruction for the sake of construction. Physiol Rev 2002; 82 : 373-428.
23. Fuchs O, Neuwirtova R. Ubikvitiny, proteasomy, sumoylace a použití dnes a zítra v terapii nádorů i jiných chorob. I. Ubikvitin - proteasomový systém a transkripční faktor NF-κB. Vnitř Lék 2006; 52 : 371-378.
24. Lai Z, Yang T, Kim YB, et al. Differentiation of Hdm2-mediated p53 ubiquitination and Hdm2 autoubiquitination activity by small molecular weight inhibitors. Proc Natl Acad Sci USA 2002; 99 : 14734-14739.
25. Cmejla R, Cmejlova J, Handrkova H, et al. Identification of mutations in the ribosomal protein L5 (RPL5) and ribosomal protein L11 (RPL11) genes in Czech patients with Diamond-Blackfan anemia. Hum Mutat 2009; 30 : 321-327.
26. Frontelo P, Manwani D, Galdass M, et al. Novel role for EKLF in megakaryocyte lineage commitment. Blood 2007; 110 : 3871-3880.
27. Builloux F, Juban G, Cohet N, et al. EKLF restricts megakaryocytic differentiation at the benefit of erythrocytic differentiation. Blood 2008; 112 : 576-584.
28. Newirtova R, Fuchs O, Provaznikova D, et al. Fli-1 and EKLF gene expression in patients with MDS 5q - syndrome. Blood 2009; 114 : 1090-1091(Abstract 2788).
29. Neuwirtová R, Fuchs O, Jonášová A, et al. Transkripční faktory EKLF a Fli1 u 5q minus syndromu. Srovnání s MDS o nízkém riziku s normálním 5. chromosomem. Transfuze Hematol dnes 2010; 16 : 32-33(abstrakt 1754).
30. Neuwirtová R, Fuchs O, Pospíšilová D, et al. The significance of megakaryoytic transcription factor Fli1 and erythroid transcription factor EKLF in the ribosomopathies. 5q - syndrome and Diamon-Blackfan anemia, the role of Fli1 in p53 regulation and in 5q - syndrome megakaryopoiesis. Blood 2011; 118 : 1634 (Abstract 3825).
31. Kumar MS, Narla A, Nonami A, et al. Coordinate loss of a microRNA and protein-coding gene cooperate in the pathogenesis of 5q - syndrome. Blood 2011; 118 : 4066-4073.
32. Kawada H, Ito T, Pharr PN, et al. Defective megakaryopoiesis and abnormal erythroid development in Fli-1 gene-targeted mice. Int J Hematol 2001; 73 : 463-468.
33. Truong AH, Cervi D, Lee J, et al. Direct transcriptional regulation of MDM2 by Fli-1. Oncogene 2005; 24 : 962-969.
34. Galili N, Cruz R, Stratton J, et al. A pluralistic approach to the study of myelodysplastic syndromes: evolving pathology of the seed via the soil. In: Saba HI and Mufti GJ Advances in Malignant Hematology. 1st Ed. Oxford, UK, Wiley-Blackwell, 2011. Publikováno elektronicky DOI 10.1002/9781444394016.ch9.
35. Yoshida K, Sanada M, Shiraishi YS, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011; 478 : 64-69.
36. Visconte V, Makishima H, Jankowska A, et al. SF3B1, a splicing factor is frequently mutated in refractory anemia with ring sideroblasts. Leukemia 2012; 26 : 542-545.
37. Patnaik MM, Lasho TR, Hodnefield JM, et al. SF3B1 mutations are prevalent in MDS with ring sideroblasts but do not hold independent prognostic value. Blood 2012; 119 : 569-572.
38. Hirabayashi S, Flotho C, Moetter J, et al. Spliceosomal gene aberrations are rare, coexist with oncogenic mutations, and are unlikely to exert a driver effect in childhood MDS and JMML. Blood 2012; 119 : 3578-3584.
39. Papaemmanuil E, Cazzola M, Boultwoud J, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med 2011; 365 : 1384-1395.
40. Malcovati L, Papaemmanuil E, Bowen D, et al. Clinical significance of SF3B1 mutations in MDS and myeodysplastic/myeloproliferative neoplasms. Blood 2011; 118 : 6239-3246.
41. Makishima H, Visconte V, Sakaguchi H, et al. Mutations in the spliceosome machinery, a novel and ubiquitous pathway. Blood 2012; 116 : 3203-3210.
42. Lasho TL, Finke CM, Hanson CA, et al. SF3B1 mutations in primary myelofibrosis: clinical, histopathology and genetic correlates among 155 patients. Leukemia 2012; 26 : 1135-1137.
43. Boultwood J, Pellagatti A, Nikpour M, et al. The role of the iron transporter ABCB7 in refractory anemia with ring sideroblasts. PLoS ONE 2008; 3 : 1-5.
44. Sheftel AD, Richardson DR, Prchal J, Ponka P. Mitochondrial iron metabolism and sideroblastic anemia. Acta Haematol 2009; 122 : 120-133.
45. Bejar R, Stevenson K, Caughey B, et al. Validation of a prognostic model and the impact of SF3B1, DNMT3A and other mutations in 289 genetically characterized lower risk MDS patient samples. Blood 2011; 118 : 969(Abstract 969).
46. Damm F, Kosmider O, Geisi-Boyer V, et al. Mutations affecting mRNA splicing define distinct clinical phenotypes and correlate with patients outcome in myelodysplastic syndromes. Blood 2012; 116 : 3211-3218.
47. Bejar R, Stevenson K, Abdel-Wahab O, et al. Clinical effect of point mutations in MDS. N Engl J Med 2011; 364 : 2496-2506.
48. Maciejewski JP, Padgett RA. Defect in spliceosomal machinery: new pathway of leukaemogenesis. Brit J Haematol 2012; 158; 165-173.
49. Wang L, Lawrenze MS, Wan Y, et al. SF3B1 and other novel cancer genes in chronic lymhocytic leukemia. N Engl J Med 2011; 365 : 2497-2506.
50. Quesada V, Conde L, Ordonez CZ, et al. Exone sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in CLL. Nat Genet 2011; 44 : 47-52.
51. Rossi D, Bruscagin A, Spina V, et al. Mutations of the SF3B1 splicing factor in chronic lymphocytic leukemia: association with progression and fludarabine – refractoriness. Blood 2011; 118 : 6904-6908.
Štítky
Hematologie a transfuzní lékařství Interní lékařství OnkologieČlánek vyšel v časopise
Transfuze a hematologie dnes

2012 Číslo 4
- Srovnání vlivu omeprazolu a pantoprazolu na antiagregační účinek klopidogrelu
- Současné postavení a přínos sartanů v klinické praxi
- Prognostický význam hladiny natriuretických peptidů při léčbě empagliflozinem
- Hypertrofická obstrukční kardiomyopatie ve světle moderní farmakoterapie – kazuistika
- Management hypertenze v těhotenství
Nejčtenější v tomto čísle
- První zkušenosti jednoho centra s použitím přípravku Octaplas® v léčbě pacientky s vrozenou formou trombotické trombocytopenické purpury
- Pokyny autorům Transfuze a hematologie dnes
- Zajištění operačních výkonů u pacientů s von Willebrandovou chorobou preparátem Wilate® – první klinické zkušenosti jednoho centra v ČR
- Podpůrná terapie megestrol acetátem u myeloproliferativních neoplazií
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy