
Coherent Functional Modules Improve Transcription Factor Target Identification, Cooperativity Prediction, and Disease Association
Transcription factors (TFs) are fundamental controllers of cellular regulation that function in a complex and combinatorial manner. Accurate identification of a transcription factor's targets is essential to understanding the role that factors play in disease biology. However, due to a high false positive rate, identifying coherent functional target sets is difficult. We have created an improved mapping of targets by integrating ChIP-Seq data with 423 functional modules derived from 9,395 human expression experiments. We identified 5,002 TF-module relationships, significantly improved TF target prediction, and found 30 high-confidence TF-TF associations, of which 14 are known. Importantly, we also connected TFs to diseases through these functional modules and identified 3,859 significant TF-disease relationships. As an example, we found a link between MEF2A and Crohn's disease, which we validated in an independent expression dataset. These results show the power of combining expression data and ChIP-Seq data to remove noise and better extract the associations between TFs, functional modules, and disease.
Published in the journal:
. PLoS Genet 10(2): e32767. doi:10.1371/journal.pgen.1004122
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004122
Summary
Transcription factors (TFs) are fundamental controllers of cellular regulation that function in a complex and combinatorial manner. Accurate identification of a transcription factor's targets is essential to understanding the role that factors play in disease biology. However, due to a high false positive rate, identifying coherent functional target sets is difficult. We have created an improved mapping of targets by integrating ChIP-Seq data with 423 functional modules derived from 9,395 human expression experiments. We identified 5,002 TF-module relationships, significantly improved TF target prediction, and found 30 high-confidence TF-TF associations, of which 14 are known. Importantly, we also connected TFs to diseases through these functional modules and identified 3,859 significant TF-disease relationships. As an example, we found a link between MEF2A and Crohn's disease, which we validated in an independent expression dataset. These results show the power of combining expression data and ChIP-Seq data to remove noise and better extract the associations between TFs, functional modules, and disease.
Introduction
Transcriptional networks are fundamental to many aspects of biology and disease. Gene expression is a carefully controlled process orchestrated by the activities of transcription factors (TFs) which regulate the transcription of each gene. TFs usually do not work in isolation, but instead multiple factors combine in different ways to regulate groups of genes in a concerted, often cooperative fashion [1]–[6]. The ENCODE project has begun to determine the binding locations of many transcription factors using chromatin immunoprecipitation (ChIP) followed by high-throughput sequencing (ChIP-Seq) [7], [8].
Despite the abundance of data about the genomic binding sites for transcription factors, determining transcription factor targets and when factors are active remains challenging. ChIP-Seq measurements can be noisy and reflect the particular condition in which the experiments are performed. Collecting more data alone will not solve this problem. As additional experiments are performed for each factor, critical and frequently used binding regions do become apparent, but it is often difficult to determine a signal threshold to distinguish common sites from condition-specific sites and general non-thematic associations from interesting biology. For example, NFκB binds to over 15,000 regions of the genome covering all possible regulatory targets of the factor. But in any given biological context, such as a local cooperative interaction with another transcription factor such as Stat1, only a handful of these genes are actively regulated by NFκB at any one time [4]. This property of TF function gives the illusion that TFs are operating broadly when in fact they perform specific context-dependent functions–in many cases with specific partners. These difficulties conspire to make the regulome challenging to study at a global level.
Thus, to understand transcription factor function, there is a need for computationally-efficient methods to (i) improve TF-target identification, (ii) identify small functional modules that represent context-specific biology, regulated by transcription factors, and (iii) annotate those modules with their functional implications (e.g. the role of the module in human disease).
Recently, a number of methods were developed to derive a network structure to connect sets of genes (modules) to the factors that control their expression [9]–[11]. These methods use gene expression data to derive the most parsimonious regulatory structure. However, because of their computational complexity, they can only account for a limited number of factors, require a specific type of input datasets, and, in their current form, cannot integrate other experimental data (e.g. ChIP-Seq). Thus, these methods may not be sufficient to capture the scale and complexity of the human regulome. Additionally, their usefulness is hampered by an assumption that the activity of the TF can be estimated by its expression, which, the authors of these methods acknowledge is not true in many cases [9]. Efficient approaches with the capability to integrate multiple data modalities are needed in order to properly leverage high-throughput experiments in the study of disease.
In this paper, we use factor analysis as a computationally-efficient method to (i) improve the identification of transcription factor targets, (ii) identify functional modules from gene expression data, and (iii) use these modules to annotate transcription factors and connect them to diseases. There are several methods for decomposing expression data to find groups of genes that work together. Network Component Analysis (NCA) is a method for inferring transcription factor activity from expression data [12] and has been used to build regulatory networks for model organisms [13]. However, NCA requires a priori knowledge of the regulatory structure which is often not available, and introduces bias in the associations between TFs and functional components. On the other hand, independent component analysis (ICA) is an unbiased and efficient method for deconvolving the signal from a fixed set of sources measured by a set of sensors (Figure 1). In essence, ICA is a computational method for extracting a set of signals from noisy data. When applied to gene expression data – like those recorded by microarrays – ICA can identify coherent functional modules (we refer to each ICA component as a module). Importantly, ICA allows genes to participate in multiple modules and thus has some ability to capture different biological contexts. A set of 423 data-driven modules derived from an ICA of 9,395 human expression microarrays covering a wide diversity of human biology was recently reported [14].

Here, we hypothesize that regulation of each of these ICA-derived modules is controlled by a small set of TFs. Using our method (which we call TFICA), we associated transcription factors to modules and then analyzed the genes contained within each module. Intersecting these target modules with ChIP-Seq binding sites improves target identification and elucidates the functional roles of the factors–both individually and in combination. We compare our approach to traditional methods in three areas: the identification of (i) transcription factor targets, (ii) TF-TF cooperativity, (iii) and the functional roles in the context of various diseases. In each of these cases, we found that our approach significantly outperforms the traditional methods. Further, we found improved performance when our approach is used in combination with traditional methods, implying that we are capturing an independent modality of transcription factor activity. Our data-driven approach is unbiased and computationally efficient enabling systematic identification of novel TF-disease relationships. Finally, we validate one such association between MEF2A and Crohn's disease.
Results
Functional modules improve the identification of transcription factor targets
We used a set of functional modules derived using ICA [14]. We then used ENCODE ChIP-Seq experimental data to connect transcription factors to individual modules if the factor bound a significant number of genes in that module (Figure 2A; see Materials and Methods). For 143 transcription factors and 379 modules, we identified 5,002 significant TF-module associations (with adjusted p<0.01, Fisher's exact test; Figure S1 and Table S1) for an overall FDR of 16.6%. We hypothesized that the components from ICA represent a single regulatory signal analogous to a single voice recorded by set of microphones (as in Figure 1). Thus, modules that associate with only one or a few factors correspond to cleaner signals than those associated with many factors. We identify 31 associations which we called “high-confidence” as there was only one TF significantly associated with the the module and another 142 “medium-confidence” associations, where the module was associated with three or fewer TFs.

We found that many of the modules nearly or fully overlap with targets of only one or a few transcription factors (Figure S3A–C). We found that for 171 modules the top associated TF could account for 80% of the targets in the module (Figure S3D). Additionally, the modules that explain the most variance across the compendium of 9,395 gene expression experiments are significantly associated with a larger number of TFs (Figure S4), and may represent large transcriptional programs.
Validation of target identification using shared functional annotations
Used in combination with the ChIP-Seq data, we hypothesized that these modules can improve identification of transcription factor targets. Specifically, we believe that putative targets (as determined by ChIP-Seq) which are also contained within significantly associated modules will be more likely to be “true” targets of the TF. To test this hypothesis, we used a set of specific GO terms [15] and we considered shared functional annotation as a proxy for a high quality TF-target association. This strategy has been used successfully in computational evaluation previously [15]. As expected, we found that TF-target pairs, particularly ones with high ChIP-Seq scores, were enriched for pairs with shared functional annotations (p<0.001; Figure 2B). When considering only the targets in the 5,002 modules associated with TFs, we find a significantly higher enrichment for shared annotations (Figure 2B). This enrichment is maintained across all ChIP-Seq peak scores (p<0.05). Additionally, if we only consider targets in the 142 medium-confidence or the 31 high-confidence modules, this enrichment is increased further at all peak score thresholds (p<0.001).
Validation of target identification using expression correlation
In an analogous fashion to the shared functional annotation approach, we used expression analysis to validate our TF-target associations under the assumption that factor expression can be used as a proxy for factor activity. We hypothesized that for high-confidence modules (i.e. those associated with just one TF) the genes within this module should be controlled predominantly by that single factor. To test this hypothesis we examined the correlation between the expression of the module (see Methods) and the expression of the factor across the compendium of 9,395 gene expression experiments. For example, AP-2γ is the sole significant association for module 360 (OR = 1.66; adjusted p = 0.006) and we found a significant correlation between the expression correlation between module 360 and AP-2γ (Spearman ρ = 0.38, p<0.001).
We systematically evaluated all 5,002 TF-module pairs in this manner (Table S2). We compared our method to two “naive” approaches for generating TF modules: (i) a “best-module” constructed from only the best ChIP-Seq hits for each TF according to their peak scores and (ii) a “matched-module” constructed from a random sample of the TF's ChIP-Seq targets with the same distribution of peak scores as was found in the ICA module. For high-confidence and medium-confidence modules TFICA outperforms the best-module method 60% of the time (binomial p = 0.011), and the matched-module method 77% of the time (binomial p = 3.2e-11; Figure S5). In fact, TFICA outperforms both naive approaches even for those modules associated with many transcription factors (>3). This holds until modules are associated with 40 or more transcription factors, at which point the individual factor expression signal can no longer be observed (Figure S5) and best-module begins to outperform TFICA. TFICA outperforms matched-modules regardless of the number of factor associated to the module (Figure S5). Additionally, six of the modules that are most enriched for a TF's targets are also the most correlated module for that TF (OR = 25.4, Fisher's exact P = 2.8e-7), and 15 are in the top 5% of all modules (OR = 5.6, Fisher's exact P = 1.9e-10). Finally, we found that the top co-expressed module is significantly enriched (Materials and Methods) for ChIP-Seq binding sites for 37 TFs (OR = 4.9, Fisher's exact P = 2.2e-12). We exhaustively evaluated the expression correlation between all TFs and all modules to estimate a null distribution, and found that our TFICA TF-module pairs were significantly more correlated than expected by chance (ρ = 0.05 vs. −0.06; t-test p = 1e-204). Additionally, in all, 327 TF-module pairs remained significantly correlated when compared to an empirically derived TF-specific null distribution (p<0.05; Figure S5).
TFICA modules are enriched for known transcription factor functional annotations
Many modules connected to TFs were significantly enriched for functional annotations known to be associated with the factor (Table 1 and Figure S2). For instance, sterol regulatory element-binding protein 2 (SREBP2) and module 158 was our most significant TF-module association (Figure 2A; OR = 45.2; 95% CI = (27.8, 71.6); adjusted p = 1e-31). SREBP2 is essential for cholesterol and fatty-acid biosynthesis, and module 158 is significantly enriched for GO terms related to lipid, sterol, cholesterol, and steroid synthesis (adjusted p<0.05; Table 1). In addition, SREBP2 shares many of the same target modules as SREBP1 (Figure S2), which are known regulatory partners.

Another example is the association between the transcription factor ZNF274 and module 111. Module 111 includes many zinc finger proteins of which ZNF274 is a known regulator [16]. In addition, ZNF274 clusters near SETDB1 and KAP1 (Figure S2A) and has been shown to recruit both of these transcription factors to repress the expression of other zinc finger proteins [17], [18] (Table 1).
Module 57 was associated with the greatest number of transcription factors (121 different factors; Table 1, Figure S2). This module also contains the greatest number of transcription factors as targets (14 factors). We found this module to be significantly enriched for DNA binding, regulation of transcription, and transcription regulator activity (among other regulatory terms; Table 1), and may represent a master regulatory module of other TFs.
Transcription factors that share associated modules show evidence of potential interactions
As we have demonstrated, we can significantly improve the identification of TF targets using TFICA modules. Therefore, we hypothesized that TFs that target overlapping modules may function together to regulate gene expression. We found 3,696 transcription factor pairs (comprising 135 individual TFs) that share a significant proportion of target modules (adjusted p<0.01, Fisher's exact test; Table S3). We assessed the putative TF-TF interactions predicted by TFICA using expression correlation, literature co-occurrences, and shared functional annotation. We compared the predictions of two TFICA similarity metrics (simple Tanimoto and a weighted approach which places more emphasis on higher confidence TF-module pairs) to those from a naive method of simply intersecting ChIP-Seq targets. We evaluated using multivariate linear models and assessed significance with an ANOVA (Figure 3A). Both TFICA approaches outperform the naive method in all 3 evaluations (Figure 3A) with weighted TFICA exhibiting the best performance. In addition, the combined model of both TFICA-similarity plus shared targets significantly outperforms the naive approach alone in all three of these metrics (Figure 3A and Figure S6). TF pairs from TFICA are significantly (1) more correlated in their overall gene expression across the compendium (simple: F = 28.6, p = 1.03e-7; weighted: F = 41.2, p = 1.75e-10), (2) more likely to co-occur in Pubmed abstracts (simple: F = 22.0, p = 2.92e-6; weighted: F = 57.2, p = 6.51e-14), and (3) more likely to share functional annotations (simple: F = 67.0 p = 5.24e-16; weighted: F = 119.4, p = 6.50e-27).

Of the top 30 pairs ranked TF-TF pairs according to module similarity, 14 have been previously reported, such as NF-YA and NF-YB, as well as Pol2 with a number of other initiating factors (Figure 3B). Many of the unreported results may be due to sparse annotation of individual genes (e.g. CHD2, CCNT2, and HEY1), and may indicate new biological links. For example, CHD2 clusters with CCNT2 and Sin3a, which are known cell cycle regulators. CHD2 has previously been proposed as involved in the cell cycle [19] consistent with its role as a DNA damage signaling protein.
Modules associate transcription factors with disease
We used enrichment analysis to associate ICA functional modules to diseases from the Gene Association Database [20]. Combined with the TFICA analysis, these two datasets allow us to create a transcription factor-disease network. We created a network of 143 transcription factors connected by their targeted functional modules (note that this network is naive to any disease associations). TFs clustered together according to the diseases with which they are significantly associated (Figure 4). In total, we found 7,808 significant associations between 141 transcription factors and 253 diseases. The average number of diseases we associate to a transcription factor is 36 (Figure S8A) with four transcription factors having just one significant disease association (e.g. BAF170 is associated with macular degeneration) and p300 associated with the most (204) diseases. The number of diseases associated with a transcription factor was significantly related to both the number of targets (Figure S8B; Spearman ρ = 0.47, P = 1.2e-7) and the number of GO annotations for the factor (Figure S8C; Spearman ρ = 0.39, P = 1.4e-5). The complete list of significant transcription factor-disease associations is available in Table S4.

Transcription factors with known relationships to disease clustered into distinct groups (Table 2). For example, we found a significant association between module 320 and acquired immunodeficiency syndrome (AIDS) (OR = 12.4; 95% CI = 4.7–28.7; adjusted p = 5.2E-4). Three of the TFs associated with this module (NFKB, IRF4, and BATF) are known to be involved in the transcriptional regulation of human immunodeficiency virus [21]–[23] and cluster together in the interaction network (Figure 4A).
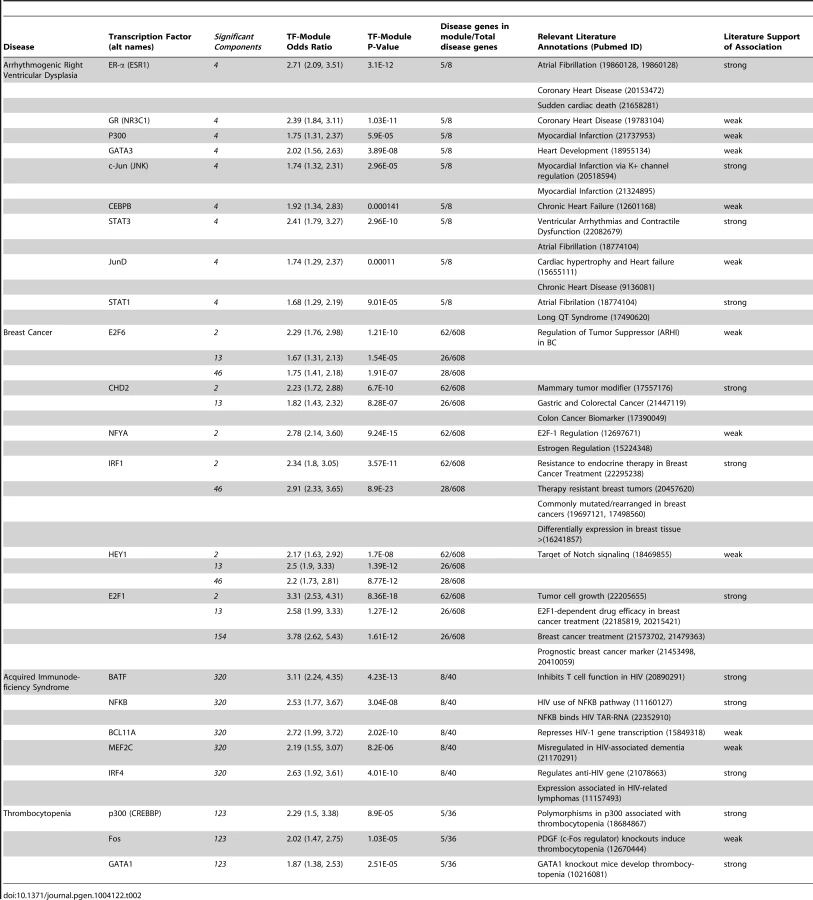
We found a significant association between module 4 and arrhythmogenic right ventricular dysplasia (OR = 82.2, 95% CI: 15.5–531.4, adjusted p = 1.1e-5). ER-α, c-Jun, STAT3, and STAT1 are associated with module 4, and all have known relationships with arrhythmias [24]–[28] and clustered together (Figure 4B). Thus, our network supports the previous suggestion that ER-α may be a promising prognostic marker for the development of atrial fibrillation [24]. In addition, we found that module 123 was significantly enriched for genes associated with thrombocytopenia (OR = 9.9; 95% CI = 2.89–27; adjusted p = 0.022). A number of TFs independently associated with thrombocytopenia, including p300 and GATA1,cluster together in the interaction network and are associated with module 123 (Figure 4C).
Finally, for breast cancer, we found significant associations with modules 2, 13, 46, and 154 with odds ratios of 8.6 (95% CI 5.6–13.4, adjusted p = 2.3e-19), 3.1 (1.8–5.2, adjusted p = 0.004), 3.1 (1.8–5.1, adjusted p = 0.002), and 4.2 (2.4–7.3, adjusted p = 2.1e-4), respectively. Based on their connectivity to these modules, the transcription factors E2F6, CHD2, NFYA, IRF1, HEY1, and E2F1 all cluster together in the TF-TF network (Figure 4D).
Evaluation on independent data sets shows improved transcription factor disease associations
We performed an evaluation of our TF-disease associations by comparing our derived associations to an independent standard created by combining (1) 37 transcription factor-disease associations from the GWAS Catalog, and (2) 46 associations from OMIM (see Methods). We assessed overall performance using the Area Under the Receiver Operating Characteristic Curve (AUROC). TFICA achieved an AUROC of 0.712 on this test dataset (Figure S7). For comparison, we also evaluated two control strategies: (1) a simple enrichment analysis on the ChIP-Seq targets associated with each transcription factor, and (2) the GREAT tool for annotation cis-regulatory elements in the genome [29]. The simple approach achieved an AUROC of 0.612, whereas GREAT achieved 0.687 (Figure S7). When combined together in a logistic regression model, TFICA significantly improved the performance of GREAT, increasing the AUROC from 0.687 to 0.761 (+10.7%, Chi-Squared = 19, P = 1.1E-5), suggesting the two approaches are complementary. Finally, we repeated this analysis using the AUROC50 which is a common measure used to evaluate performance at low false positive rates (FPR<0.5). We found an AUROC50 value of 0.185 for the naive metric, 0.248 for GREAT, 0.253 for TFICA, and 0.292 for the combined metric, indicating again that TFICA is adding an independent source of information for TF binding.
A regulatory network of human disease
Using the TFICA disease annotations, we visualized the highest confidence transcription factor-disease associations (see Materials and Methods) in a regulatory network connecting 62 transcription factors to 253 human diseases (Figure 5). As expected, substantial parts of this network reflect known biology. For example, TFICA associates HNF4G with metabolic disorders, which corresponds to its KEGG annotation. STAT3's role in fibrotic diseases is well-studied, as it is implicated in the proliferation of fibroblasts and excess ECM proteins [30]. In total, the network visualization describes 491 relationships, 33 are known associations according GAD, OMIM, and GWAS Catalog and thus the remaining 458 are potentially novel transcription factor-disease relationships. Transcription factors are connected with an average of 7.9 diseases and each disease was associated with an average of 1.9 transcription factors. The high confidence associations visualized in Figure 5 and all of the significant ICA-derived associations are available in Table S4.

Expression of MEF2A and its target genes differentiates patients with Crohn's disease from healthy controls
Using our network analysis, we identified MEF2A as the factor with the highest association with Crohn's disease. MEF2A has been previously associated with cardiovascular disease [31], but is not recognized to have a role in inflammatory bowel disease. MEF2A was associated with Crohn's disease through three modules: 69, 262, and 320. We validated this association using an independent expression dataset (not used in the training set) of 59 patients with Crohn's disease and 42 controls [32]. For each of these modules, the genes comprising the module showed significantly higher levels of differential expression between the two groups compared to genes not in one of the modules (P = 0.017, 1.4e-06, and 0.0084, for modules 69, 262, and 320, respectively). We used permutation testing to correct for a potential bias towards higher differential expression for genes contained within functional modules, after which module 262 remained significant (P = 0.025), which includes genes such as STAT4, CCR5, and SMAD3. We found that expression of MEF2A itself was significantly higher in Crohn's disease patients (Fig. 4A, P = 0.0013, Wilcoxon rank-sum test). Additionally, among genes targeted by MEF2A, genes in module 262 also exhibited a higher level of differential expression among patients with Crohn's disease (P = 0.0019).
To investigate the role of module 262 and MEF2A in classification of Crohn's disease, we projected the expression values in the Crohn's dataset to generate an expression value of the module (see Materials and Methods) and found an overall higher expression of the module in patients with Crohn's disease (Fig. 4A, P = 4.6e-09, Wilcoxon rank-sum test). We evaluated MEF2A and the module expression for their performance in a disease classifier using area under the receiver operating characteristic curve (AUROC). Both MEF2A and the aggregated expression of module 262 were significantly predictive (P = 0.0012 and P = 4.5e-06, logistic regression) with AUROCs of 0.77 and 0.86, respectively (Fig. 4B). Finally, we combined MEF2A expression and aggregated module expression into a single model and found that this combined statistic outperformed the other two classifiers (F-test p = 5.8e-11, AUROC = 0.90, Fig. 4B).
Discussion
We present a computationally efficient and conceptually simple method that is useful in linking transcription factors to their targets and to disease as well as derive several novel such relationships. Using current approaches, such an analysis is challenging as TFs in the ENCODE dataset bind near an average of 6,050 genes. Simple enrichment analysis on the full target set often does not reveal coherent functional groups. Factors may exhibit multifaceted functional roles and target genes in very different cellular contexts, and when all of a TF's targets are grouped together, it becomes difficult to isolate these individual contexts. Our method overlays data-driven functional module information – from a large compendium of human gene expression data – on top of TF binding data from ChIP-Seq. We demonstrate that our method (1) significantly improves TF target identification, (2) accurately identifies the functional roles of factors both independently and in combination with another factor, and (3) discovers new disease associations through these functional modules.
We show that TFICA identifies targets that are significantly more functionally coherent than targets identified by naive (peak-based) methods. Importantly, TFICA can identify these targets even in cases that lack strong support from ChIP-Seq binding data (i.e. sites that are not among the strongest bound peaks). We hypothesized that TFICA would be better able to identify targets that, despite lower binding levels, are biologically important. Our “matched” analyses tests this hypothesis and we observe that TF-target functional annotation sharing (Figure 2B) and expression correlation (Figure S5) is higher for TFICA targets than naively identified targets. In fact, despite that stronger binding more tightly couples TF and target expression, the expression correlation among modules identified by TFICA are consistent with those of the “best” module (genes with highest peak scores) until the modules are associated with more than 40 factors. Additionally, by linking TFs to established modules of gene expression we identify genes where binding of the factor is not observed, but instead, the TF is exerting indirect genetic control. In these cases, we hypothesize that the TF may be controlling expression of a module through its direct targets, some of which may be in the module.
Our factor interaction analysis is useful for suggesting the functional roles of TF through guilt by association, particularly for poorly described factors. For example, CHD2 is a helicase whose function remains to be fully understood. In our TF-TF network, we found that CHD2 is connected with the cyclin CCNT2 (Figure 3B), supporting the hypothesis that CHD2 plays a role in cell cycle [19]. It is important to acknowledge that our method for identifying TF-TF interactions, which uses all of the shared modules between two transcription factors may miss those that are unique to particular biological contexts. Future work will be required to model this type of interaction. In spite of this limitation, we identify many known relationships and outperform traditional approaches (Figure 3A).
Further, we were able to recapitulate many known TF-disease associations without any prior knowledge linking the factor to the disease. For example, our factor-disease network (Figure 5) links ER-α to arrhythmogenic right ventricular dysplasia, supporting recent findings that this protein may be used as a prognostic marker [24]. STAT1 and STAT3, which we also associate with arrhythmogenic right ventricular dysplasia, were recently found to be elevated in mice with sustained atrial fibrillation [27]. In fact, we find too many known associations in this network to enumerate here (refer to Table 2 for an annotated sampling of these associations, and Table S4 for the complete list). Furthermore, we found that the number of diseases associated with a given transcription factor varied widely from just one to hundreds. We hypothesize that this is related to the roles that a particular factor may play in different cellular contexts, with more general factors (e.g. p300, GR, and Pol2) associated with more diseases than more specific factors. We tested this by examining the relationship between the number of diseases associated with a TF and two measures of functional diversity: (1) the number of targets found for that TF by ChIP-Seq and (2) the number of unique GO annotations. In both cases, we found significant positive relationships (Figure S8B–C). In addition, our factor-disease network suggests a novel role for MEF2A in Crohn's disease – an association that would not have been found using the naive method (naive adj. p = 1) and that we validated using an independent data set and analysis (Figure 5A–B). Our module analysis suggests MEF2A is promoting inflammatory response via module 262, which includes STAT4, CCR5, and SMAD3.
It is important to note, however, that this approach is dependent on the quality of the functional networks used. Other methods for generating cohesive functional networks, including data-driven approaches (e.g. PPI networks) and knowledge-based (e.g. functional annotations from ontologies), may complement the approach and improve performance. Further, we derived our modules using a set of 9,395 gene expression experiments without regard to the particular context in which the experiment was performed. Focusing this analysis using a contextually specific set of experiments (e.g. only data focused on cardiovascular disease) could provide further specificity to the disease associations that are derived.
Our work dissects transcription factor function by examining associations with specific gene modules derived from a large compendium of human expression experiments under a wide variety of conditions. This approach is generally applicable in cases where the biological function can neither be described by a single gene nor the entire genome, but instead operates at an intermediate level – groups of genes or groups of functional pathways. We demonstrate improved identification of TF targets and construct a regulatory network of human disease. Finally, we find and validate a novel transcription factor-disease association. We make three databases publicly available to the community: (1) a database of 5,002 transcription factor-module associations, (2) a database of 3,696 putative transcription factor interacting pairs, and (3) a database of 7,808 transcription factor-disease relationships. These resources should further enable researchers to explore TF interactions as well as their roles in human disease.
Materials and Methods
Data sources
We obtained transcription factor binding data from UCSC, which included 2,750,490 reproducible binding sites from the ENCODE project [7], [8] and 41,972 gene annotations from RefGene (build hg19). 423 gene expression modules (a.k.a. “components”) determined previously from independent component analysis (ICA) are available at https://simtk.org/home/fcanalysis [14]. These modules were derived from a compendium of human gene expression data downloaded from the Gene Expression Omnibus (GEO). All 9,395 Affymetrix Human Genome U133 Plus 2.0 array deposited in GEO as of May 28, 2008 were downloaded. The ICA analysis was performed and published previously [14]. We obtained gene-disease associations from the Gene Association Database [20] and filtered for positive genome-wide and curated associations, as well as diseases with greater than five genes. We then used the NCBO Annotator [33] service to map the disease terms to terms in the Disease Ontology, resulting in 34,392 distinct associations. 4,267 and 5,279 validation associations were downloaded from the NHGRI GWAS catalog [34] (accessed on 3/31/12) and OMIM (http://omim.org), respectively. For both of these datasets, we mapped local disease terms to standardized terms in the Disease Ontology using the annotator, resulting in 1,842 and 9,866 annotations (of which 35 and 46 map to the transcription factors in our dataset), respectively.
Generating the transcription factor-ICA module network
We mapped transcription factor binding sites to the nearest gene within 100 kb (gene boundaries 100 kb upstream or downstream of the boundaries of the binding site) in the RefGene annotation to determine putative binding targets of each factor (Figure S1). We linked each transcription factor to each module using an enrichment analysis between the target genes of the transcription factor and the genes contained within each module. We used the hypergeometric distribution to model the expected amount of shared genes and a Fisher's Exact test to test the significance of any deviations. Finally, we corrected for multiple hypothesis testing and filtered for transcription factor-module pairs that were significant with an adjusted p-value less than 0.01 and an odds ratio greater than 1. We clustered TFs using complete-linkage clustering and Euclidean distance (Figure S2). In each module, TFs were ranked by number of genes in the module bound, and a greedy approach was used to determine how many TFs were necessary to account for genes in the module (Figure S3).
We associated each module with between 1 and 121 transcription factors. Our hypothesis is that those that are associated with fewer TFs will be of higher confidence than those associated with many, which may contain more noise. We therefore identified two subsets of TF-module pairs: (i) a “high-confidence” set of TF-module pairs where the module was only associated with one TF and (ii) a set of “medium-confidence” TF-module pairs where the module was associated to at most three TFs. We use these subsets in our evaluation of the TFICA TF-target predictions we make through the associated modules. Additionally, we computed the (Spearman) expression correlations between all TFs and all modules (projections) and used this distribution of correlations as an empirical null distribution. The correlations from the 5,002 significantly overlapping TF-module pairs were z-transformed to this distribution to determine the number of pairs that were empirically significantly correlated at the expression level.
For each of the 9,395 gene expression datasets, we projected the (log-transformed) gene expression measurements of each gene onto the loadings for each module. In cases where the module contained the transcription factor itself, we removed the transcription factor prior to computing the prediction. For modules with significant associations with at most three transcription factors, we tested the (Spearman) correlation between the projection and the logged gene expression value of the transcription factor.
We evaluated our TFICA approach by comparing to two different naive approaches: a “best” naive approach consisting of a set of equal size to the module and comprised of the top bound genes (i.e. those with the highest ChIP-Seq scores), and a “matched” naive approach, which is an equal-sized set with randomly chosen targets matched to the score distribution of the targets in the component. We then assessed whether TF expression is better correlated with projections from modules from TFICA (from above) compared to the average expression of the genes in these naive modules. We segregated by number of transcription factors associated with each module and presented how often TFICA outperforms the naive method for that “number of TFs” threshold. Additionally, we performed a paired t-test at each threshold comparing the r2 values between TF-module correlations from TFICA and the naive methods. Finally, we examined the proportion of TF-target pairs that share functional annotations based on the hypothesis that true regulatory targets are more likely to share these annotations. We split the TFICA associations into three groups: (i) high confidence associations, (ii) medium confidence associations, and (iii) all associations. We compared each of these three groups to the naive approach.
Building the transcription factor interaction network
We used the Tanimoto coefficient as a measure of similarity between each pair of transcription factors. The Tanimoto coefficient is a common set similarity comparison technique used to compare two sets. It is defined as the size of the intersection of the two sets divided by the size of the union of the two sets. In this case, the sets were the modules that were significantly associated with each transcription factor. We used these similarities to build two transcription factor interaction networks. The first was a small network of the top 30 interactions based on the tanimoto coefficient (Figure 3B). We annotated this network with curated information on the function (activator or repressor) and biological interactions between these factors. This network also visualizes the pairwise Pearson's correlation between the expression pattern across the 9,395 gene expression experiments in the compendium.
The second network contains 140 factors and was generated by connecting transcription factors whose pairwise tanimoto score was greater than 0.2. We used Cytoscape [35] (version 2.8.2) and the force-directed weighted layout visualize this network. To test the significance between the number of shared modules between each pair of transcription factors, we used enrichment analysis using the hypergeometric distribution and correction (adjusted p<0.01 and odds ratio >1).
Evaluation of TFICA TF-TF network versus other approaches
We evaluated our TFICA TF-TF network against a naive approach which linked TFs simply by their shared ChIP-Seq targets. In the naive approach, we used Tanimoto similarity to quantify the relationship between each pair of TFs. For TFICA TF similarity we used two approaches: (i) the Tanimoto similarity between the TFICA modules associated with each TF and (ii) a weighted version of the Tanimoto similarity between the TFICA modules associated with each TF. In the weighted version we upweighted high confidence TF-module associations over low-confidence. We believe that the fewer TFs a given module is associated with the higher the confidence. We used the following equation to generate a weighted Tanimoto similarity score for each TF-TF pair:

Construction of the transcription factor-disease network
We associated the derived 423 ICA modules to the 632 diseases reported in GAD (Genetic Association Database) by analyzing the overlap of the genes associated with a given disease with the genes in a given module. We used enrichment analysis with correction (FDR) for multiplicity to determine significant overlaps. Previously we linked factors to modules to produce TF-module pairs. From this analysis, we have module-disease pairs and thus, we can link TFs to diseases through their shared modules. For example, if a TF significantly targets a module and that module has a significant proportion of a disease's known genes, then we predict a relationship between the TF and the disease.
We generated two lists of transcription factor disease associations. The first contains all significant associations (adjusted p<0.01 and odds ratio >1), which was used for validation (below). The second is a small set of high confidence associations, which included a stringent set of associations (odds ratio >3, adjusted p<0.001), as well as the best association with adjusted p<0.01 and odds ratio greater than 1 for those diseases that had no “stringent” associations used for visualization. We used Cytoscape [35] (version 2.8.2) and the spring embedded layout to visualize the resulting network.
In order to assess the predictive performance of the algorithm we needed to optimize the alpha level for determining significance between modules and diseases. We used a subset of 632 associations from the GAD that are for transcriptions factors directly to calibrate this parameter. We computed the AUROC at a range of thresholds (Figure S7A) to determine the optimal value (determined to be adjusted FDR = 0.15).
We tested the performance of our method (using module-disease p value) against an independent validation dataset of known disease to gene (and thus, disease to TF) associations, a combination of the NHGRI GWAS catalog and OMIM (Figure S7B). We compared the performance to a naive method, where we evaluated the enrichment between the raw transcription factor targets and known disease genes (Fisher's test p value was used as a predictor). Additionally, we compared TFICA to an established method for determining enrichment for particular genomic annotations, GREAT (binomial p value was used as a predictor) [29]. We used the AUROC and AUROC50 values as summary statistics of the predictive performance of the methods. We combined the predictors from our method and GREAT using logistic regression and used an ANOVA to assess the additive contribution of our approach to GREAT testing significance with a Chi-Squared test. Comparisons of ROC curves were performed using DeLong's method in the pROC package for R.
Validation of association between MEF2A and Crohn's disease
We obtained a publicly-available expression dataset for 59 Crohn's patients and 42 healthy controls (GEO Accession Number: GSE3365) [32]. Gene expression measurements were determined using the median measurement of all probes in the gene. For each gene, differential expression between cases and controls was determined using a Student's t-test. The distributions of absolute values of these t-statistics were tested using a Student's t-test/Wilcoxon rank-sum test to determine significant differences in differential expression of gene sets.
To generate an expression value for the module, a projection was calculated, using the dot product of the expression values of each gene in the module and their individual loadings derived from ICA. Expression of MEF2A and the projection of the module were normalized to a z-score and summed to create a “combined” metric. These three metrics were compared among cases and controls using a Wilcoxon rank-sum test and fit using a logistic regression (binomial).
Statistical analysis
All statistical analyses were performed with the R statistical computing package (version 2.14.1). Enrichment analyses were computed using Fisher's exact test, with p-values corrected using the Benjamini-Hochberg FDR correction for multiple hypotheses. All expression correlations were performed using Spearman correlations. ROC curves were generated using the ROCR package for R [36] and comparisons of ROC curves were performed using DeLong's method in the pROC package for R.
Supporting Information
Zdroje
1. ZinzenRP, GirardotC, GagneurJ, BraunM, FurlongEEM (2009) Combinatorial binding predicts spatio-temporal cis-regulatory activity. Nature 462 : 65–70 doi:10.1038/nature08531
2. MacArthurS, LiX-Y, LiJ, BrownJB, ChuHC, et al. (2009) Developmental roles of 21 Drosophila transcription factors are determined by quantitative differences in binding to an overlapping set of thousands of genomic regions. Genome Biol 10: R80 doi:10.1186/gb-2009-10-7-r80
3. MirnyLA (2010) Nucleosome-mediated cooperativity between transcription factors. Proc Natl Acad Sci USA 107 : 22534–22539 doi:10.1073/pnas.0913805107
4. KarczewskiKJ, TatonettiNP, LandtSG, YangX, SliferT, et al. (2011) Cooperative transcription factor associations discovered using regulatory variation. Proc Natl Acad Sci USA 108 : 13353–13358 doi:10.1073/pnas.1103105108
5. ZhengW, ZhaoH, ManceraE, SteinmetzLM, SnyderM (2010) Genetic analysis of variation in transcription factor binding in yeast. Nature 464 : 1187–1191 doi:10.1038/nature08934
6. KasowskiM, GrubertF, HeffelfingerC, HariharanM, AsabereA, et al. (2010) Variation in transcription factor binding among humans. Science 328 : 232–235 doi:10.1126/science.1183621
7. BirneyE, StamatoyannopoulosJA, DuttaA, GuigóR, GingerasTR, et al. (2007) Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447 : 799–816 doi:10.1038/nature05874
8. ENCODE Project Consortium (2011) A user's guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol 9: e1001046 doi:10.1371/journal.pbio.1001046
9. SegalE, ShapiraM, RegevA, Pe'erD, BotsteinD, et al. (2003) Module networks: identifying regulatory modules and their condition-specific regulators from gene expression data. Nat Genet 34 : 166–176 doi:10.1038/ng1165
10. Pe'erD, TanayA, RegevA (2006) MinReg: A Scalable Algorithm for Learning Parsimonious Regulatory Networks in Yeast and Mammals. The Journal of Machine Learning Research 7 : 167–189.
11. Pe'erD, RegevA, TanayA (2002) Minreg: inferring an active regulator set. Bioinformatics 18 Suppl 1: S258–S267.
12. LiaoJC, BoscoloR, YangY-L, TranLM, SabattiC, et al. (2003) Network component analysis: reconstruction of regulatory signals in biological systems. Proc Natl Acad Sci USA 100 : 15522–15527 doi:10.1073/pnas.2136632100
13. HugheyJJ, LeeTK, CovertMW (2010) Computational modeling of mammalian signaling networks. Wiley Interdiscip Rev Syst Biol Med 2 : 194–209 doi:10.1002/wsbm.52
14. EngreitzJM, DaigleBJ, MarshallJJ, AltmanRB (2010) Independent component analysis: mining microarray data for fundamental human gene expression modules. J Biomed Inform 43 : 932–944 doi:10.1016/j.jbi.2010.07.001
15. TongAHY, LesageG, BaderGD, DingH, XuH, et al. (2004) Global mapping of the yeast genetic interaction network. Science 303 : 808–813 doi:10.1126/science.1091317
16. YanoK, UekiN, OdaT, SekiN, MasuhoY, et al. (2000) Identification and characterization of human ZNF274 cDNA, which encodes a novel kruppel-type zinc-finger protein having nucleolar targeting ability. Genomics 65 : 75–80 doi:10.1006/geno.2000.6140
17. FrietzeS, O'GeenH, BlahnikKR, JinVX, FarnhamPJ (2010) ZNF274 recruits the histone methyltransferase SETDB1 to the 3′ ends of ZNF genes. PLoS ONE 5: e15082 doi:10.1371/journal.pone.0015082
18. SchultzDC, AyyanathanK, NegorevD, MaulGG, RauscherFJ (2002) SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev 16 : 919–932 doi:10.1101/gad.973302
19. MarfellaCGA, OhkawaY, ColesAH, GarlickDS, JonesSN, et al. (2006) Mutation of the SNF2 family member Chd2 affects mouse development and survival. J Cell Physiol 209 : 162–171 doi:10.1002/jcp.20718
20. BeckerKG, BarnesKC, BrightTJ, WangSA (2004) The genetic association database. Nat Genet 36 : 431–432 doi:10.1038/ng0504-431
21. HiscottJ, KwonH, GéninP (2001) Hostile takeovers: viral appropriation of the NF-kappaB pathway. J Clin Invest 107 : 143–151 doi:10.1172/JCI11918
22. FarrowMA, KimE-Y, WolinskySM, SheehyAM (2011) NFAT and IRF proteins regulate transcription of the anti-HIV gene, APOBEC3G. J Biol Chem 286 : 2567–2577 doi:10.1074/jbc.M110.154377
23. QuigleyM, PereyraF, NilssonB, PorichisF, FonsecaC, et al. (2010) Transcriptional analysis of HIV-specific CD8+ T cells shows that PD-1 inhibits T cell function by upregulating BATF. Nature Medicine 16 : 1147–1151 doi:10.1038/nm.2232
24. SmalceljA, SertićJ, GolubićK, JurcićL, BanfićL, et al. (2009) Interactions of MinK and e-NOS gene polymorphisms appear to be inconsistent predictors of atrial fibrillation propensity, but long alleles of ESR1 promoter TA repeat may be a promising marker. Coll Antropol 33 : 933–937.
25. TangK, LiX, ZhengM-Q, RozanskiGJ (2011) Role of apoptosis signal-regulating kinase-1-c-Jun NH2-terminal kinase-p38 signaling in voltage-gated K+ channel remodeling of the failing heart: regulation by thioredoxin. Antioxid Redox Signal 14 : 25–35 doi:10.1089/ars.2010.3095
26. YajimaT, MurofushiY, ZhouH, ParkS, HousmanJ, et al. (2011) Absence of SOCS3 in the cardiomyocyte increases mortality in a gp130-dependent manner accompanied by contractile dysfunction and ventricular arrhythmias. Circulation 124 : 2690–2701 doi:10.1161/CIRCULATIONAHA.111.028498
27. TsaiC-T, LinJ-L, LaiL-P, LinC-S, HuangSKS (2008) Membrane translocation of small GTPase Rac1 and activation of STAT1 and STAT3 in pacing-induced sustained atrial fibrillation. Heart Rhythm 5 : 1285–1293 doi:10.1016/j.hrthm.2008.05.012
28. WuL, ArchackiSR, ZhangT, WangQK (2007) Induction of high STAT1 expression in transgenic mice with LQTS and heart failure. Biochem Biophys Res Commun 358 : 449–454 doi:10.1016/j.bbrc.2007.04.119
29. McLeanCY, BristorD, HillerM, ClarkeSL, SchaarBT, et al. (2010) GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 28 : 495–501 doi:10.1038/nbt.1630
30. MaL, ZhuangS (2011) The Role of STAT 3 in Tissue Fibrosis. curr chem biol 5 : 44–51 doi:10.2174/187231311793564342
31. Visvikis-SiestS, MarteauJ-B (2006) Genetic variants predisposing to cardiovascular disease. Curr Opin Lipidol 17 : 139–151 doi:10.1097/01.mol.0000217895.67444.de
32. BurczynskiME, PetersonRL, TwineNC, ZuberekKA, BrodeurBJ, et al. (2006) Molecular classification of Crohn's disease and ulcerative colitis patients using transcriptional profiles in peripheral blood mononuclear cells. J Mol Diagn 8 : 51–61 Available: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=16436634&retmode=ref&cmd=prlinks.
33. NoyNF, ShahNH, WhetzelPL, DaiB, DorfM, et al. (2009) BioPortal: ontologies and integrated data resources at the click of a mouse. Nucleic Acids Research 37: W170–W173 doi:10.1093/nar/gkp440
34. HindorffLA, SethupathyP, JunkinsHA, RamosEM, MehtaJP, et al. (2009) Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA 106 : 9362–9367 doi:10.1073/pnas.0903103106
35. SmootME, OnoK, RuscheinskiJ, WangP-L, IdekerT (2011) Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27 : 431–432 doi:10.1093/bioinformatics/btq675
36. SingT, SanderO, BeerenwinkelN, LengauerT (2005) ROCR: visualizing classifier performance in R. Bioinformatics 21 : 3940–3941 doi:10.1093/bioinformatics/bti623
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 2
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Genome-Wide Association Study of Metabolic Traits Reveals Novel Gene-Metabolite-Disease Links
- A Cohesin-Independent Role for NIPBL at Promoters Provides Insights in CdLS
- Classic Selective Sweeps Revealed by Massive Sequencing in Cattle
- Arf4 Is Required for Mammalian Development but Dispensable for Ciliary Assembly
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy