
Transcriptome-Wide Analyses of 5′-Ends in RNase J Mutants of a Gram-Positive Pathogen Reveal a Role in RNA Maturation, Regulation and Degradation
RNA decay and maturation have in recent years been recognised as major regulatory mechanisms in bacteria. In contrast to Escherichia coli, the Firmicute (Gram-positive) bacteria often do not encode the well-studied endonuclease RNase E, but instead rely on the endonucleases RNase Y, RNase J1 and RNase J2, of which the latter two have additionally been shown to have 5′ to 3′ exonucleolytic activity. We have previously demonstrated that these RNases could be deleted individually in the pathogenic Firmicute Staphylococcus aureus; however, we here present that, outside a narrow permissive window of growth conditions, deleting one or both of the RNase J genes presents serious difficulties for the cell. Moreover, an active site mutant of RNase J1 behaved like a deletion, whereas no phenotypes were detected for the RNase J2 active site mutant. Furthermore, in order to study the in vivo enzymatic activity of RNase J1 and J2, a method was developed to map the exact 5′-ends of mature and processed RNA, on a global scale. An enrichment of 5′ RNA ends could be seen in the RNase J mutants, suggesting that their exonucleolytic activity is crucial for normal degradation of bulk RNA. Using the data to examine specific RNAs, we demonstrated that RNase J activity is needed for correct 5′ maturation of both the 16S rRNA and the RNase P ribozyme, and can also inactivate the latter, possibly as quality control. Additional examples show that RNase J perform initial cleavages, apparently competing with ribosomes for access to mRNAs. The novel 5′ mapping assay offers an exceptionally detailed view of RNase activity, and reveals that the roles of RNase J proteins are diverse, ranging from maturation and post-transcriptional regulation to degradation.
Published in the journal:
. PLoS Genet 10(2): e32767. doi:10.1371/journal.pgen.1004207
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004207
Summary
RNA decay and maturation have in recent years been recognised as major regulatory mechanisms in bacteria. In contrast to Escherichia coli, the Firmicute (Gram-positive) bacteria often do not encode the well-studied endonuclease RNase E, but instead rely on the endonucleases RNase Y, RNase J1 and RNase J2, of which the latter two have additionally been shown to have 5′ to 3′ exonucleolytic activity. We have previously demonstrated that these RNases could be deleted individually in the pathogenic Firmicute Staphylococcus aureus; however, we here present that, outside a narrow permissive window of growth conditions, deleting one or both of the RNase J genes presents serious difficulties for the cell. Moreover, an active site mutant of RNase J1 behaved like a deletion, whereas no phenotypes were detected for the RNase J2 active site mutant. Furthermore, in order to study the in vivo enzymatic activity of RNase J1 and J2, a method was developed to map the exact 5′-ends of mature and processed RNA, on a global scale. An enrichment of 5′ RNA ends could be seen in the RNase J mutants, suggesting that their exonucleolytic activity is crucial for normal degradation of bulk RNA. Using the data to examine specific RNAs, we demonstrated that RNase J activity is needed for correct 5′ maturation of both the 16S rRNA and the RNase P ribozyme, and can also inactivate the latter, possibly as quality control. Additional examples show that RNase J perform initial cleavages, apparently competing with ribosomes for access to mRNAs. The novel 5′ mapping assay offers an exceptionally detailed view of RNase activity, and reveals that the roles of RNase J proteins are diverse, ranging from maturation and post-transcriptional regulation to degradation.
Introduction
RNA decay is an important regulatory process in bacteria and is thought to be a major factor in post-transcriptional control [1]. Some bacterial mRNAs have halflives of only seconds, most mRNAs have halflives of minutes, and a few especially stable RNAs can last for hours [2], however the signals that distinguish a short-lived from a long-lived RNA are only poorly understood. The standard RNA decay paradigm, based on the model organism Escherichia coli, describes an initial endonucleolytic cut made by RNase E and regulated by largely unknown mechanism(s). Then 3′ to 5′ exonucleolytic degradation follows, carried out by polynucleotide phosphorylase (PNPase), which is normally inhibited by hairpin structures at the 3′-end of the RNA [3]–[5]. However, this model can rarely be applied to bacteria outside the beta and gamma-proteobacteria, since large families such as the Firmicutes often do not encode RNase E homologs, but instead encode RNase Y [6] and one or more RNase J paralog(s) [7]. A role for RNase J1 in general RNA decay was shown by depleting it in Bacillus subtilis and Streptococcus pyogenes, which greatly increases the half-lives of many mRNAs [8]–[10]. Moreover, B. subtilis and S. aureus RNase Y, RNase J1, and RNase J2 have been proposed to be a part of a larger degradosome-complex, identified via bacterial two-hybrid interactions [11], [12], which might coordinate and/or regulate the RNA decay.
The characterisation of the in vivo enzyme activity of B. subtilis RNase J1 revealed a 5′ to 3′ exoribonucleolytic activity, previously unheard of in bacteria [13], and structural studies have provided important clues to the molecular basis for this activity [14]–[16]. Removal of the final 38 nucleotides from the 5′-end of pre-16S rRNA in B. subtilis is dependent on this exonucleolytic activity, underlining its importance [13], [17].
In contrast, RNase J2 exhibits little to no exonucleolytic activity [18], which is accompanied by a virtual lack of phenotypes in a B. subtilis RNase J2 mutant [7], [8]. A hint to the role of RNase J2 may be found in bacterial two-hybrid experiments, where RNase J1 and J2 interact strongly with each other [11], [12], and several lines of evidence from both B. subtilis and S. aureus suggest that RNase J1 and J2 can form both homo - and hetero-dimers as well as hetero-tetramers, although reports do not agree on which form is predominant [12], [16], [18].
Moreover, individually both B. subtilis RNase J1 and J2 have been shown in vitro to endonucleolytically cut a number of RNAs, and that the interplay between the two proteins changes the specificity of this activity [7], [18]. However, the physiological role of this, and to what extent it complements the endonucleolytic activity of RNase Y, is not clear [9].
RNase J1 was originally identified as an essential gene in B. subtilis [19], and both RNase J1 and J2 were reported to be essential in S. pyogenes [10]. In S. aureus, a saturated transposon insertion screen of the genome failed to find insertions in either RNase J1 or RNase J2, suggesting that both were essential in this organism [20]. In contrast, using our recently developed system for generating allelic replacements in S. aureus, we were able to isolate deletion mutants of both RNase J1 and J2 [21].
In this work we examine the range of severe phenotypes caused by the loss of RNase J1 and RNase J2, however at the same time we show that a deletion of both is viable in S. aureus. This is in concert with results published during the preparation of this manuscript by Figaro and coworkers [22], where, in contrast to previous data, the two RNase J genes were shown to be non-essential in B. subtilis. In addition, we show that mutations in the RNase J1 active site are virtually equivalent to a complete deletion of the RNase J1 gene, whereas the RNase J2 active site appears to have no function under the tested conditions, even though a complete deletion of RNase J2 results in growth defects that approach the RNase J1 deletion. To study the details of RNase J1 and J2 mutations, we furthermore developed a new method for examining the 5′-ends of the entire mono-phosphorylated transcriptome. Using this assay, we show that the major role of RNase J (RNase J will be used as generic term, where we discuss J1 and J2, and not a specific one of them) in RNA decay is the 5′ to 3′ exonucleolytic activity, but examples are also given where the RNase J endonucleolytic activity might play an important role. Finally, we show that RNase J activity is responsible for the normal pathway of RNase P RNA and 16S rRNA 5′-maturation.
Results
RNase J1 and J2 are only essential under specific growth conditions in S. aureus
In order to consolidate the apparent contradiction between the findings of Chaudhuri and coworkers (2009), which indicated essentiality of RNase J1 and J2, and our own previous findings, in which the two genes are non-essential [21], we used total genome sequencing to confirm the deletions of the two RNase J genes (S0940 and SA1118, using the S. aureus N315 nomenclature, which will be used throughout this manuscript). Out of more than three million 100 bp reads from the Illumina machine, none mapped to the deleted regions, in contrast to the sequence of unrelated strains, where at least 1100 reads would map to each of the two RNase J genes (Table 1). This confirmed that RNase J1 and RNase J2 regions had indeed been deleted in our mutants, and additionally ruled out that the rnase genes had been moved to an alternative genomic location by an unfortunate recombination event. The sequencing data was also used to perform a search for single nucleotide polymorphisms (SNPs) and small inserts and deletions (indels) in our mutant strains, localising any potential second-site mutations which might aid the cell in surviving the loss of the RNase J genes. However, the only secondary mutation identified in the RNase J1 deletion strain (strain ΔJ1) was a silent valine to valine in the gluconate operon repressor (C to A at position 2578631 in SA2295), and no secondary mutations were detected in the RNase J2 deletion strain (strain ΔJ2).

Furthermore, to ensure that the viability of the RNase J1 deletion mutants was not a peculiarity of the SA564-strain used [21], the RNase J1 deletion was generated twice, independently, in an RN4220 background (strains PR02-03 and 06, Table 2).

The main difference between our method for generating mutants and that of Chaudhuri and coworkers, was the passages at 44°C used by the latter to eliminate their thermosensitive transposon-carrying plasmid. Indeed, the authors warn that their list of essential genes reflect conditions at 44°C in the specific medium used, and state: “Consequently, genes required for high temperature survival will be scored as putatively essential” [20]. Therefore, we tested whether high temperature was limiting growth of the RNase J mutants, by spotting a dilution series of RNase J1 and J2 mutants on Mueller-Hinton (MH) plates, incubated at a range of temperatures. Corresponding with the warning from Chaudhuri and coworkers, the RNase J deletion mutants did indeed grow extremely poorly at 42°C, but, surprisingly, also at 30°C and below, and it is therefore only at 37°C that the mutants will grow relatively unaffected (Figure 1).

The requirements for the medium were examined by using a defined medium (RH-medium, containing only glucose, amino acids, vitamins, buffer, and salts), along with LB-medium and MH-medium. The spot-tests revealed that RNase J deletion mutants require a complex medium for growth, and will not grow on RH-medium. Furthermore, even though LB medium contains a complex range of various salts and nutrients, it has a low Magnesium concentration (30 to 50 µM; [23]), and this deficiency apparently also inhibits growth of the RNase J mutants, unless additional MgCl2 is added, in which case the ΔJ2 mutant will grow (Figure 1A). Thus, RNase J mutants are only viable at a very restrictive temperature range and are highly susceptible to medium changes.
A double RNase J1/J2 mutant is viable
In the single RNase J1 and J2 mutants, it is still a possibility that the activity of the remaining RNase J partially compensates for the loss of the other RNase J. To examine the consequences of a complete absence of RNase J activity, a double mutant (ΔJ1ΔJ2) was generated, where the RNase J2 mutant was used as parent for a second round of mutagenesis, to substitute the RNase J1 gene with an erythromycin resistance cassette. The full genome of the double mutant was also sequenced, to confirm the deletions (Table 1). The double mutant does grow at 37°C, but even slower than the single mutants (Figure 1B). Both strains ΔJ1 and ΔJ1ΔJ2 generate visible aggregates when grown in liquid culture, making it difficult to obtain reproducible growth data by measuring OD600 or CFU-counts. We therefore used a semi-quantitative spot-assay to evaluate the growth of the various mutant strains, by comparing the size of individual colonies on the same petri-dish.
RNase J1 and RNase J2 are both needed for full functionality, but the active site of RNase J2 is dispensable
RNase J2 is not essential in B. subtilis, and a deletion mutant exhibits only very mild phenotypes [8]. In S. aureus the RNase J2 deletion does result in strong growth defects, however these defects are less severe than those observed for the ΔJ1 strain (Figure 1). This raised the possibility that the effects observed from deleting RNase J2 were in fact due to a partially malfunctioning RNase J1, in the absence of RNase J2. Alternatively, since the interaction of RNase J2 with the degradosome appears to go through RNase J1 [11], [12] it is possible that the stronger phenotypes observed for the ΔJ1 strain are due to removing RNase J2 from the degradosome in absence of RNase J1.
Active-site mutants were therefore generated, in order to maintain the possibility of correctly forming a J1+J2 complex, but eliminating the enzymatic activity of either one or the other of the ribonucleases. The active site of S. aureus RNase J1 has a 74-HGHEDH-motif, which matches the signature motif (HxHxDH) of the metallo-β-lactamase family [18], while the S. aureus RNase J2 active site sequence 76-HGHEHA exhibits several differences from the motif (Figure S1). A H76A mutation disrupts the unique active site of B. subtilis RNase J1 [13], [16], [17], and since both RNase J1 and J2 of S. aureus have the first two Zinc-coordinating histidines of the HxHxDH motif, these were chosen for substitution with alanines, generating the active site mutants RNase J1 H74A-G-H76A (J1AGA) and RNase J2 H76A-G-H78A (J2AGA) in place of the wild-type genes on the chromosome (Table 2; Figure S1).
The growth of J1AGA and J2AGA mutants were then examined, under the conditions where ΔJ1 and ΔJ2 are inhibited. The J2AGA mutant grew like wild-type under all tested conditions consistent with a proposed structural role of RNase J2, whereas the J1AGA strain exhibited defects that were very similar to the ΔJ1 mutant (Figure 1B), and it is therefore not dissociation of the degradosome that produces the phenotypes.
Overexpressing RNase J1 partially complements the loss of RNase J2
Using the growth-inhibitory conditions established above, it was tested whether overexpression of one RNase J could rescue the loss of the other. The genes for RNase J1 and RNase J2 were cloned into the multicopy plasmid pEB01 including upstream regions containing ribosome binding sites (RBSs) and putative promoters. In the case of RNase J1, this meant inclusion of a small upstream ORF (SA0941) (Table 3). The ΔJ1 mutant was completely rescued by introduction of the pJ1 plasmid, but could not be complemented by pJ2 or by pJ1AGA. In contrast, the ΔJ2 strain could be partially rescued by the pJ1 plasmid, and fully complemented by pJ2 (Figure 2). Interestingly, pJ1AGA was also able to partially complement the loss of RNase J2, supporting the hypothesis that the role of RNase J2 is mainly structural, and does not require enzymatic activity.


Mapping the 5′-ends of mono-phosphorylated RNAs
RNase J1, and to a lesser degree RNase J2, from B. subtilis were shown in several studies to have 5′ to 3′ exoribonucleolytic activity, and that mono-phosphorylated 5′ ends were strongly preferred over tri-phosphorylated ends. To examine the effects of RNase J1 and J2 on 5′-ends of RNAs in S. aureus, the Exact Mapping Of Transcriptome Ends (EMOTE) assay was developed to map the exact 5′ base of a large number of mono-phophorylated RNAs in the cell (see flowchart in Figure 3). Briefly, large excess of a custom-designed 18 nt RNA oligo (Rp5) was ligated to total RNA preparations from WT, ΔJ1, ΔJ2, ΔJ1ΔJ2, J1AGA, ΔY, and ΔcshA strains (Table 2). The enzyme used, T4 RNA ligase 1, will not ligate tri-phosphorylated ends to Rp5, and the assay is therefore specific for the 5′ mono-phosphorylated ends that are preferred and generated by RNase J. The cDNA was generated using a semi-random primer (DROAA), which also adds one of the two linkers needed for sequencing with Illumina HiSeq technology. In order to avoid cDNA generated from the excess of Rp5, the DROAA primer terminated in two deoxyadenosines, a combination which is extremely common in the S. aureus genome, but is unable to basepair with the uridine-lacking Rp5 oligo. Second strand synthesis and PCR amplification was carried out using a primer specific for the Illumina linker in DROAA, paired with a Rp5-specific primer which also added the second Illumina linker sequence to the DNA. 50 bp Illumina sequencing was then performed from the Rp5-end of the DNA, revealing the 24 first bases of each RNA, more than enough to map the reads precisely in a bacterial genome (Figure 3). The first of these 24 bases thus represents the 5′-end of an RNA molecule in the original sample, and the number of Illumina reads that correspond to each original 5′ position, are then added up and tabularised (see Tables S1, S2 and S3 for examples). In the tables, the position given is the number of the first (5′) base in the detected RNA, and a cleavage at position X is therefore defined as a cleavage between base X-1 and base X. Each column shows the number of reads that detect a specific 5′-end from a given strain.

Table 4 gives a general overview of the output from the EMOTE assay. The first step was to remove reads that do not have the Rp5 sequence (about 6%), and therefore cannot be used to determine 5′-ends. Next the barcodes were identified to assign each read to a specific bacterial strain. These pools of reads were then examined for the presence of the CC motif, which ensures that the read reflects a genuine RNA 5′-end. Aligning the 24 bases after the CC motif to the S. aureus N315 genome sequence resulted in either uniquely mapping reads, or reads that mapped to multiple locations on the genome (such as rRNA genes, IS elements, etc.). Uniquely mapping and multiply mapping reads were organised in separate tables for further analyses, as shown in Table S1 and S2.

Limitations in the EMOTE method
It is important to note that even though the RNA ligation step confers specificity to mono-phophorylated RNAs, the EMOTE data does not distinguish between mono-phosphorylated 5′-ends generated by endonucleases, exonucleases, or pyrophosphohydrolases. It is therefore often possible to detect transcription start-sites, because tri-phosphorylated ends are converted to mono-phosphorylated ends by one of the S. aureus homologues of the Nudix family of pyrophosphohydrolases. In B. subtilis, only one of several has so far been identified [24] (the homolog in S. aureus is SA1612). Furthermore, the RNA ligase exhibits some sequence specific bias in its activity, and although the ligation step is carried out with large excess of Rp5, it is still possible that some 5′-ends are under - or over-represented. This bias is however identical in all strains for 5′-ends at a given position, and the bias is therefore removed when the data from two strains are compared.
If an RNA fragment is shorter than about 100 nucleotides, then it will be under-represented in our EMOTE assay, since the chosen RNA purification method depletes small RNA from the sample. This is further accentuated by the paucity of potential priming sites for the DROAA oligo on very short RNAs, however, we are still able to detect 5S rRNA and some tRNAs in our EMOTE data (Table S1 and S2).
In addition to the potential biases and interpretation problems mentioned above, an additional problem presented itself, which was not directly related to the EMOTE method. We had great difficulty removing ribosomal RNA from our samples, and in our hands, the Ambion MicrobeExpress kit works very poorly for S. aureus rRNA, and we suspect that the pull-down oligos do not hybridise fully to the rRNA of this organism. This led to high percentage of reads mapping to the 5′-ends of 16S and 23S rRNA (Table 4), so it is certain that an improved rRNA depletion method/protocol would improve the depth of the EMOTE data. The data presented here are therefore limited to relatively high abundance RNAs, however we fully expect that a deeper sequencing would allow detection of even poorly expressed RNAs. A rough estimate of how many genes we are able to detect is illustrated in the final column of Table 4, where the number of genes with more than 10 reads are listed.
Validation of the EMOTE assay
The EMOTE method was evaluated by comparing our data to the results from the recent landmark paper on RNase III function in S. aureus [25]. The described RNase III cleavage of the 16 rRNA processing stem, the cut inside the rnc gene, and the double-stranded cut of a hairpin in the cspA gene were all detected by our method (Tables S3, S4 and S5). The cleavage of the 16S rRNA processing stem by RNase III was detected at the exact previously published site, and served as a basis for further analyses of the 16S rRNA gene (see below). In the rnc gene we detect a large number of RNA with a 5′ base at position 1216563 and a smaller sub-population at the adjacent base 1216564, and in addition, a weaker signal is seen for 5′-bases 10–16 bases downstream, corresponding to the cleavage detected in vivo by primer extension. In the cspA gene, we could detect a high number of 5′-ends at positions 1409120 and 1409083, which not only confirm the previously published results, but additionally our exact method serves to refine the positions of the RNase III cleavages to one base further towards the base of the cspA hairpin [25].
Lack of RNase J activity results in a transcriptome-wide accumulation of mono-phosphorylated 5′ ends
RNase J1 has been proposed to be a major player in bulk RNA decay [7], by processively digesting the RNAs from the 5′-end once they become mono-phosphorylated, either by an endonucleolytic cut, or by the action of a pyrophosphohydrolase. This hypothesis has been supported by the observed stabilisation of individual RNAs during depletion of RNase J1 in B. subtilis and S. pyogenes [8], [10]. However, until now it has been unclear to what extent it is a general mechanism that affects a majority of the transcriptome, or whether only a small subset of RNAs are dependent on RNase J1 for correct degradation.
To resolve this, the number of reads for a given position in the WT data-set was compared to the number of reads in the RNase J mutants. Only positions where at least two reads mapped in both the WT and mutant data-sets were included, reducing background noise, and potential bias caused by lack of sequencing depth. Table 5 shows the number of positions that fulfilled the criteria, and the percentage of these that are either 4-fold more abundant or 4-fold less abundant in the mutants. The ΔY and ΔcshA data are included as controls, and show neither strong enrichment nor strong reduction in the 5′-ends. The strong enrichment of 5′-ends in the RNase J mutants, signifies that under wild-type conditions, the bulk of the RNA is indeed degraded by the 5′ exonucleolytic activity of the RNase J complex. The assay does not reveal what happens to the 3′-ends of the RNA, however, the strains are viable, and it must therefore be presumed that the RNA is degraded via the 3′-ends in the RNase J mutants.

Possible outcomes caused by the loss of RNase J activity
When an RNase is analysed in an isolated in vitro system, the interpretation is often straight-forward since the presence/absence of the RNase will generate certain specific patterns of RNA products. In vivo however, many RNases function together, often with overlapping functions, to degrade or mature RNA, and it is therefore important to take this into account when interpreting the data. Several scenarios can be imagined when the RNase J genes are deleted, due to the potential combination of exo - and endo-nucleolytic activities.
-
If an RNA is degraded (or matured) directly from the original 5′-end, after a pyrophosphohydrolase has generated a mono-phosphate, then a loss of 5′ exonucleolytic activity will result in an accumulation of 5′-ends, which can be detected by the EMOTE assay.
-
Endonucleolytic cuts performed by a non-RNase J, which are then 5′ degraded by the RNase J1+J2 will be poorly detectable in the EMOTE data from the WT strain, since these RNA species are quickly removed, as soon as they are generated. However in the RNase J mutants, where the 5′ exonuclease activity is absent, such a cleavage site will be visible in the EMOTE data, since the generated 5′-ends will accumulate (for example at position +452 to +477 in the SA1279-rnpB transcript). Sites from i and ii will contribute to the accumulation observed for all the examined RNase J mutants (Table 5), which shows that RNase J2 is needed for exonucleolytic activity, but also that mutating the active site of RNase J1 is sufficient to see the same level of 5′-end accumulation as in the ΔJ1ΔJ2 strain (Table 5), strongly suggesting that in vivo, it is primarily the RNase J1+J2 complex which is the 5′ exonuclease.
-
A more complex situation arises when, for example, an endonucleolytic cut is made by RNase J1, and then 5′ exonucleolytically degraded. Then we would expect to observe an accumulation in the ΔJ2 strain, since the endonucleolytic cut can be made by RNase J1, and the exonucleolytic degradation is inhibited by the loss of RNase J2 from the RNase J1+J2 complex. Both the WT and the ΔJ1ΔJ2 strains will exhibit low signals in the EMOTE data, but for different reasons: In the WT, the 5′-end generated by the endonucleolytic activity of RNase J1 is removed by the exonucleolytic activity, whereas in the ΔJ1ΔJ2 strain, there is no RNase J to perform the endonucleolytic cleavage in first place. The latter can additionally be used to distinguish between scenario ii and iii, because a 5′-end generated by a non-RNase J will accumulate in the ΔJ1ΔJ2 strain, whereas it will be absent if the cut is made by an RNase J.
-
Finally, to complicate matters even further, it is almost certain that the in vivo 5′-exonucleolytic activity of RNase J can be blocked or paused by external factors such as ribosome binding or secondary structures. It is therefore not immediately possible to determine whether a 5′-end was generated by exo - or endo-nucleolytic cleavage, and consequently the interpretation of both EMOTE and other types of data is often not straight-forward.
Below, we present four examples where RNase J activity impacts on RNAs (Figures 4, 5, 6 and 7) and although they are certainly not exhaustive for all situations, they illustrate different outcomes of the absence of RNase J activity.
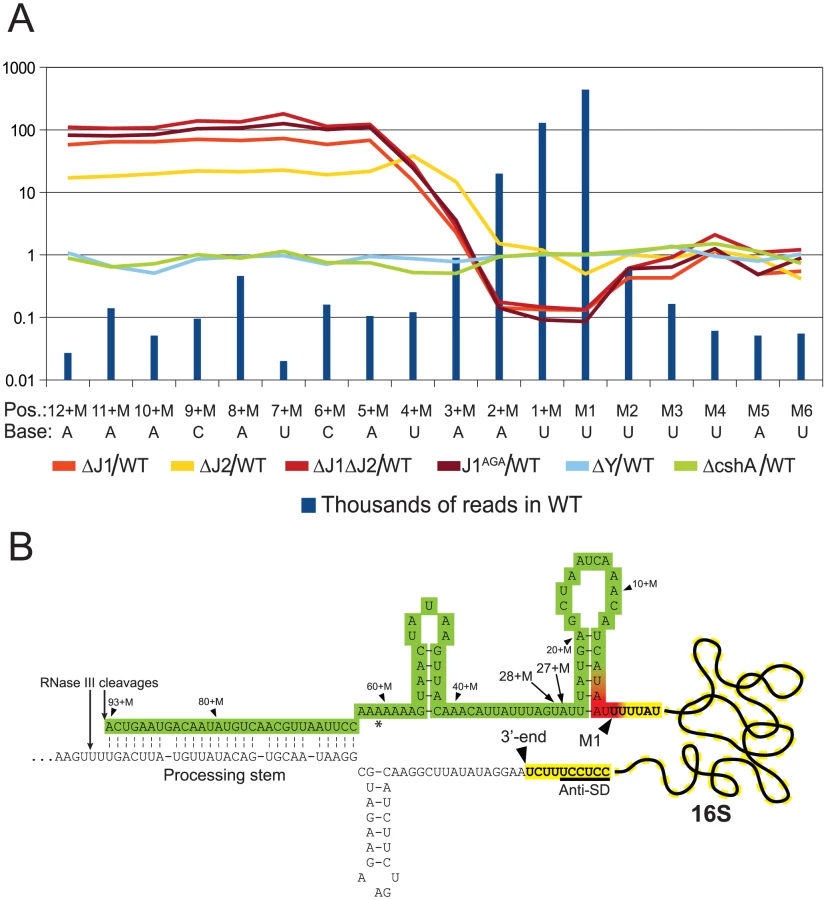



Example 1: The final step in 5′ maturation of 16S rRNA is carried out by RNase J1, aided by RNase J2
Not surprisingly, and despite the ribosome depletion step, the vast majority of reads from the EMOTE mapped to the 16S and 23S rRNAs (small RNAs like 5S rRNA and tRNAs were depleted during the RNA purification procedure). The rRNAs represent a large and stable population of mono-phosphorylated RNAs in the cell, whereas mono-phosphorylated mRNAs are often undergoing degradation and therefore have short half-lives and much lower steady-state levels. When comparing the number of reads for each position at the 5′-end of 16S rRNA, it became clear that the RNase J mutants were unable to properly finalise the 5′ maturation of this RNA. Instead, there was an accumulation of immature 16S rRNA (Figure 4, Table S5), showing that RNase J1 and J2 are responsible for cropping the final 4 nucleotides to form mature 16S rRNA. The J1AGA mutant exhibits identical deficiencies to ΔJ1 and ΔJ1ΔJ2, indicating that the active site of RNase J1 is required for 16S maturation in S. aureus. A similar analysis of the 5′-end of 23S rRNA revealed no enrichment in the +M region of any of our examined mutants (Table S2), but this is not surprising, since 5′ maturation of B. subtilis 23S rRNA was shown to be carried out by the Mini-III RNase, and RNase J activity is only important when Mini-III is missing [26].
Example 2: RNase J matures and degrades the 5′-end of RNase P RNA
The second example is the region containing SA1279, SA1278 and rnpB, the RNA component of the ribozyme RNase P. SA1279 encodes cell division protein GpsB, whereas the SA1278 ORF is entirely inside the rnpB gene, and might not be translated.
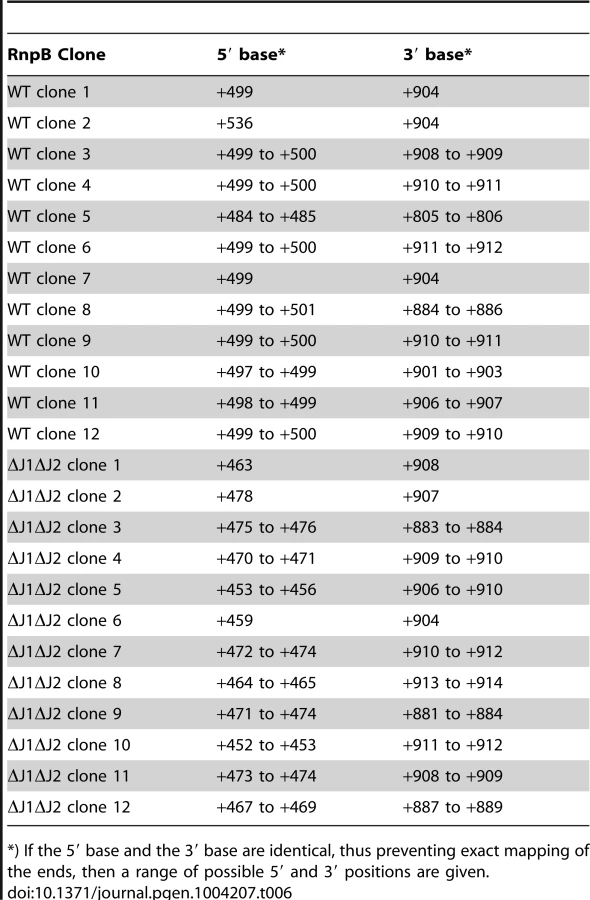
RNA species with three different 5′-ends are abundant in this region (Figure 5A, Table S6). Position +1 is within the SA1280 ORF, and has upstream −10 (TATA) and −35 (TTGAtt) motifs. It is probably the major transcription start site for the SA1279 gene, but there are no obvious upstream transcription termination sites, so transcription start sites even further upstream could also contribute to SA1279 expression. Furthermore, a strong hairpin can form at +1, protecting the RNA from 5′ degradation, without interfering with translation of SA1279. The +485 site corresponds to the 5′-end of the RNase P RNA (RnpB), and is also a possible transcription start site, although the −35 motif is very weak (cTGtCt). The +485 5′-end is protected by Helix P1, which forms between the beginning and the end of RnpB (Figure 5D and [27]). No obvious transcription termination site could be identified between +1 and +485, and it is likely that both transcription start sites contribute to rnpB expression. The +499 5′-end is more difficult to explain, since no convincing transcription start motifs are present, and the +499 RNA would be unable to form the conserved Helix P1 in RNase P. The +499 RNA species are much less abundant in the RNase J mutants, although the ΔJ2 strain still has a small population of this RNA (Figure 5A). Similarly, a smaller proportion of the +485 RNA was detected in the RNase J mutants, but this was counterbalanced by the appearance of several small populations of RNAs starting at positions +452 to +477 (Figure 5A and 5C).
RnpB molecules with a 5′-end at +499, where the P1 Helix cannot form a protecting secondary structure with the 3′-end, could potentially provide an entry-point for 3′ to 5′ exonucleases. To determine whether this was the case, total RNA was circularised with T4 RNA ligase 1, an rnpB-specific oligo was used to generate cDNA, and PCR products across the ligated 5′-3′ junction of RnpB were obtained using divergent rnpB-specific primers (Figure 5B). The average length of PCR-products amplified from the WT strain were visibly shorter than those from the ΔJ1 and ΔJ1ΔJ2 strains (Figure 5E). To obtain the exact 5′ and 3′-ends of individual RnpB molecules, the bands from WT and ΔJ1ΔJ2 were cut from the gel and ligated to a cloning vector, whereupon twelve clones from each strain were sequenced. The pairs of 5′-ends and 3′-ends shown in Table 6 qualitatively proves the existence of both the +485 and +499 RNA species in the WT strain, and reveal that 3′ heterogeneity of RnpB is limited, such that the size differences of the PCR-products in Figure 5E are due to lack of 5′ processing in the ΔJ1ΔJ2 strain.
To show that an RNA species really does start at +1 and extends into the rnpB gene, we first performed a Northern blot, were we could detect a faint band corresponding in size to this longer RNA (Figure S2A). This was then confirmed by using the cDNA generated from circularised RNA (described above) as template for a PCR where the reverse rnpB-primer was substituted with a reverse primer annealing inside the upstream SA1279 ORF (Figure 5B). The extremely faint bands (Figure S2B) were again cut from the gel, and six clones from each strain were sequenced. Table 7 shows the existence of RNA molecules with a 5′-end at +1, but also that some slightly (7 to 14 nt) longer RNAs can be found when no RNase J is present to trim the 5′-ends.

Example 3: RNase J removes the first 51 nucleotides from SA2322 mRNA
Newly transcribed full-length SA2322 mRNA, encoding a hypothetical membrane protein, starting at position +1 or +2, can form a predicted energetically favourable 9 bp hairpin structure (Helix II in Figure 6) between nucleotide +15 and +42, and this is possibly further stabilised by a secondary helix between nt +4 to +12 and +56 to +63 (Helix I, in Figure 6). However, a significant proportion of RNA is detected which starts at position +52 (Figure 6A, Table S7), a position that lack recognisable transcription initiation signals, and is not caused by cleavage by RNase Y, since the +52 position is easily detected in the ΔY strain. The RNase J mutants however, either lack the cut (ΔJ1, ΔJ1ΔJ2 and J1AGA) or have a severe paucity of it, compared to the WT strain (ΔJ2) (Figure 6A), and it is therefore likely that RNase J activity is responsible for the +52 cleavage (Figure 6, see discussion for details).
Example 4: Strain ΔJ1ΔJ2 has different RNA sub-populations than WT, ΔJ1 and ΔJ2
The fourth, and perhaps most intriguing, example is the SA1075 mRNA, encoding the acyl carrier protein hmrB, in which the RNase J single mutants behave different from the double RNase J deletion (Figure 7), as opposed to previous examples, where the four RNase J mutants follow each other closely (Figures 4, 5, and 6).
Newly formed SA1075 mRNA, is predicted to form a secondary structure that seems to block translation by sequestering the RBS and the start codon (Figure 7B and C). However, cleavage at +16, generating a new 5′-end, renders the RBS accessible, presumably allowing translation to occur. In the WT, ΔJ1, ΔJ1ΔJ2, and ΔY strains, a major population starting at +16 can be observed, however, in strains ΔJ2 and J1AGA, this RNA species is much less visible. Even though the overall abundance of detected SA1075 RNA is reduced in the RNase J mutants, the percentage with 5′-ends at +1 goes up in the RNase J mutants, (Figure 7A, Table S8). The cleavage at position +45 of the SA1075 is very noticeable in strains ΔJ1, ΔJ2 and J1AGA, but is only at background level in ΔJ1ΔJ2, WT, ΔY, and ΔcshA (Figure 7A). A Northern blot was used to confirm the observed data, using probe R2, which hybridises near the 3′-end (Figure 7B). The +1 and +16 bands are not separated, however the +45 band is visible in all RNase J mutant strains except ΔJ1ΔJ2 (Figure 7D). Either RNase J1 or RNase J2 must therefore be needed for this cleavage to occur, whether by exo - or endonucleolysis, and since the +45 position is also detected in the J1AGA strain, which encodes the entire RNase J1 protein, but only lacks an active site, it seems highly unlikely (albeit possible) that the cut is made by another RNase, requiring the presence of one of the RNase J proteins for its activity (Figure 7, see discussion for details).
Discussion
We have previously shown that RNase J1 and RNase J2 individually are not needed for S. aureus viability [21], and we have here shown that the double mutant is also viable, proving that it is not a case of one RNase J taking over the function of the other in the single mutants. Several growth conditions were examined, and it was found that growth of the RNase J mutants was strongly inhibited outside a narrow range of laboratory growth conditions. These pronounced growth defects reflect the importance of RNA degradation and are in accordance with the major defects in cell morphology observed in B. subtilis [22]. We believe that the previous failure to isolate RNase J mutants could at least partially be attributed to their reduced growth under various conditions and that our recently developed selection/counterselection system was a key element in overcoming the difficulties of isolating RNase J mutants [21]. Moreover, due to the controversy of our findings when comparing with earlier publications from both S. aureus and other Firmicutes [7], [10], [19], [20], we confirmed the deletion of the RNase J genes by full genome sequencing (Table 1), and only identified a single valine to valine point mutation on the ΔJ1 chromosome, which appears to be unrelated to the viability of ΔJ1.
RNase J1 and J2 are working together to correctly cleave and degrade RNA
RNase J1 and RNase J2 active-site mutants were examined, and surprisingly, the J2AGA mutant exhibited no growth defects, whereas the J1AGA mutant behaved almost exactly like the full RNase J1 deletion mutant, both in the 5′-mapping experiments and in terms of growth, with the only major difference being an ability to grow on magnesium complemented LB-medium (Figure 1). This seems to indicate that the only active RNase J in S. aureus is RNase J1, but that the RNase J2 protein is needed for RNase J1 to function correctly.
Taking into account data mainly from B. subtilis [11], , but also from S. aureus [12], RNase J1 and RNase J2 strongly interact, and will generate a population of hetero-dimers or tetramers. We therefore propose a model where RNase J1 provides the active site of the complex, accounting for the strong phenotype of the J1AGA mutant, and the lack of phenotypes for the J2AGA mutant. In contrast, RNase J2 serves mainly as a structural protein, ensuring the proper function of the RNase J1+J2 complex. The phenotypes seen for the RNase J2 deletion mutants are therefore caused by a disruption of the RNase J1+J2 complex, which can be partially overcome by over-expressing RNase J1 from a plasmid, however, the over-expressed RNase J1 does not need to be enzymatically active (Figure 2). This hypothesis is strengthened by the almost identical molecular data from ΔJ1 and J1AGA, and ΔJ2, found throughout our EMOTE and Northern blot data (Figures 4, 5, 6 and 7; Table S1 and S2). Nevertheless, the model does not completely exclude a minor enzymatic activity of RNase J2, perhaps when it is not complexed to RNase J1, an activity that would account for the +45 cut of SA1075 in ΔJ1 (Figure 7 and see below). Moreover, the interplay between RNase J1 and J2 may be different in organisms where RNase J2 was shown to be either essential or virtually dispensable, such as S. pyogenes and B. subtilis, respectively [10], [7].
5′ EMOTE analyses of RNase J mutants
RNase J enzymes were reported to have endonucleolytic and 5′ to 3′ exonucleolytic activities. To obtain a better picture of these enzymatic activities within the cell, it was necessary to develop an assay that allowed us to examine the 5′-ends of all cellular RNA in the mutants, and compare it to the wild-type (Figure 3). Two additional mutants of the RNA decay machinery (ΔY and ΔcshA) were included as controls, to ensure that observed effects were RNase J specific. Processed RNA and degradation intermediates possess a mono-phosphate 5′-end, and are readily detected in our assay, whereas primary 5′-ends are generally protected by their triphosphate ends, and are only detected due to the pyrophosphohydrolase activity of enzymes such as RppH [24]. The assay is therefore useful for studying enzymes like RNase J, that endo - or exonucleolytically cleave RNA and produce molecules with monophosphorylated 5′-ends.
RNase J1 and J2 are responsible for 5′ maturation of 16S rRNA
The EMOTE analysis shows a clear enrichment in the RNase J1 mutants at all positions upstream of the 4+M position (defined here as the mature rRNA with additional 4 nucleotides at the 5′-end), whereupon the abundance rapidly decreases to an extreme under-representation at position M1 (the first nucleotide of the mature rRNA). The proportion between the RNase J1 mutants and the WT stays at the same high plateau from position 13+M to 4+M (Figure 4), and our data indicate that this plateau extends all the way to the RNase III cleavage site (93+M) (Table S3).
Downstream of position 5+M, in the wild-type, 5′ ends are strongly enriched, presumably due to rapid exonucleolytic activity of RNase J trimming the 16S rRNA down to position 4+M, and then a somewhat slower final trimming to obtain the fully mature 16S rRNA at position M1. It is furthermore possible that the difference between the severity of phenotypes seen for ΔJ2 and ΔJ1 (Figure 1) can be explained by the higher proportion of mature 16S rRNA observed in ΔJ2 (Figure 4).
Our interpretation is that an unidentified endonuclease cleaves semi-randomly between 93+M and 5+M, whereupon the RNase J1+J2 complex exonucleolytically degrades the resulting 5′-end until it reaches the M1 position. The endonuclease responsible for the random cuts in S. aureus is not likely to be RNase Y, since the ΔY data follow the WT data closely (Figure 4). Even in the total absence of RNase J, there is however still a small but significant amount of 16S rRNA with a correctly matured 5′-end, but it is unclear whether this is caused by occasional cleavage at the M1 position by the above mentioned unidentified endonuclease, or by a backup-system for RNase J (Figure 4).
In B. subtilis, the 16S rRNA is specifically cut by the unidentified endonuclease at position 38+M, whereupon it is trimmed to maturity by RNase J1 [13], [17]. In S. aureus, a peak in the reads at positions 27+M and 28+M in both WT and all mutant strains (Figure 4, Table S3) suggests that these two positions are equivalent to 38+M in B. subtilis, but that the S. aureus enzyme is less specific.
Since RNase J is responsible for maturing the 5′ end of 16S rRNA, they must somehow limit their processive exonuclease activity, in order to avoid degrading the entire rRNA. Mathy and coworkers [13] observed that in vitro, the B. subtilis RNase J1 will digest the entire 16S rRNA molecule, but this is obviously not the case in vivo, where RNase J processivity is arrested at the M1 site, presumably by ribosomal proteins.
RnpB-maturation and possibly quality control by RNase J
We have shown that the +1 transcript of SA1279 continues into rnpB (Figure 5) and that this long RNA is processed to obtain a mature RNase P RNA (Table 7, Figure S2). We propose that the accumulation of RNA species with 5′-ends in the region downstream of the SA1279 ORF (+452 to +477), detected in the RNase J mutants, represent cleavage products by an unidentified endonuclease, which in the WT strain are trimmed by RNase J to form the mature RNase P RNA at position +485 (Figure 5C). This model thus explains the reduction in observed +485 RNA in the RNase J mutant strains, and the remaining, lower, level of mono-phosphorylated mature RNase P RNA is generated directly by transcription initiation and pyrophosphohydrolysis at this site (Figure 5B), despite the poor −35 motif. It is possible that it is RNase Y that performs the endonucleolytic cleavage of the +452 to +477 region. However, our data can neither confirm nor reject this, since the resulting 5′-ends are removed so quickly by RNase J in the WT strain that they do not appear in our data, and thus the absence of these species in the ΔY mutant is not conclusive (Figure 5).
The formation of +499 RNA population is less obvious, however our data clearly show that it is caused by RNase J. The evolutionary conserved P1 Helix, which presumably protects the 5′-end of the RnpB RNA from 5′ degradation cannot form during transcription, but only once the entire RNA has been made. One possibility is therefore that a 5′ exonucleolytic attack is made by RNase J on nascent RNA, which has not yet formed the P1 Helix (Figure 5D). This exonucleolytic digestion would then continue until blocked by one of the many secondary structures in RNase P RNA, or by the protein component (RnpA) of the ribozyme. The +499 end would in this model represent a degradation intermediate, where RNase J either is blocked or pauses long enough to accumulate a detectable population of +499 RNA. The EMOTE data, due to potential ligation and PCR biases, cannot be used quantitatively to compare the relative abundance of two different 5′-ends such as +485 and +499, and the same is the case for the circularised and cloned junctions shown in Table 6, which seem to indicate that the +499 5′-end is more abundant in the WT strain than the +485 end. Instead, Table 6 confirms qualitatively that the 5′-end of the RNA can be both +485 and +499 in WT.
Adding further complexity to the maturation of RNase P is the existence of a small anti-sense RNA, which can base-pair to the 3′-end of RnpB [2], [28]. One could imagine that this anti-sense RNA is in competition with the 5′-end of the RnpB to hybridise to the 3′-end, and if the competition is successful, then the P1 Helix will fail to form, and the 5′-end is free to be degraded by RNase J (Figure 5D).
SA2322 mRNA maturation by RNase J
Our data cannot directly determine whether the +52 cut of SA2322 is due exonucleolytic digestion of the primary transcript, or an endonucleolytic cut directly at +52. In either scenario, 5′ to 3′ exonucleolytic digestion beyond +52 is very likely inhibited by ribosomes binding the RBS, as seen for the hbs gene in B. subtilis [29]. Importantly, if Helix I, helped by Helix II, forms in vivo in full-length mRNA, the +52 cut may serve a regulatory function, since Helix I partially sequesters the RBS of SA2322. It is also worth noting that the transcription start site of SA2322 appears to be less precise in all the RNase J mutants (Figure 6A). This might be due to lessened fidelity of the transcription initiation complex, altered activity of pyrophosphohydrolases, which would reveal new products to our assay, or perhaps that RNAs starting at position +3 or +4 are so rapidly degraded by RNases J in the WT strain, that these RNA species simply are not observed.
RNase J1 and J2 are individually responsible for cleavage at position +45 of SA1075 mRNA
The essential [20] SA1075 gene, where a cut at position +16 liberates the RBS, serves as a second example of an RNase-mediated regulatory mechanism. This cleavage however, is not mediated by RNase J. Instead, a second cleavage at position +45, inside the SA1075 ORF, is performed by either RNase J1 or J2 individually, and reveals an intriguing difference in activity between the RNase J1+J2 complex, and RNase J1 or J2 alone.
It appears that three scenarios are possible for a newly transcribed SA1075 mRNA: i) Translation is initiated before Hairpin I can form, and successively loading ribosomes prevent the formation of Hairpin I. If ever a pause in translation initiation occurs, then Hairpin I will form, blocking further initiation, unless ii) an RNase cleaves at position +16, to destroy Hairpin I and allow translation initiation. However, if an SA1075 mRNA molecule is not shielded by ribosomes, then it is a potential target for iii) an RNase J cleavage at position +45, which removes the RBS and start codon, and initiates degradation of the mRNA (Figures 7C and E).
While our data cannot identify the enzyme that cleaves SA1075 mRNA at position +16, it is possible that it is RNase III, because the 5′ fragment of the SA1075 mRNA was recovered in a pull-down experiment, using an inactive RNase III mutant as bait, whereas a fragment starting at +16 was obtained when wild-type RNase III was used ([25]; P. Romby, personal communication). If so, then the fate of an SA1075 mRNA is decided by a competition between RNase J, RNase III and the ribosome (which itself is matured by RNase J and RNase III).
Several hypotheses can explain difference at position +45, between the ΔJ1, ΔJ2 and J1AGA mutants and ΔJ1ΔJ2, observed in both the Northern blot and EMOTE data (Figure 7). It is possible that the +45 cleavage is caused by an unrelated RNase, which is over-expressed in the ΔJ1, ΔJ2 and J1AGA mutants, but not in the ΔJ1ΔJ2 strain. It is also possible that RNase J1 or J2 individually still retain a small amount of exonucleolytic activity, even though no such activity has been shown for RNase J2 [18], and that this activity is able to digest until position +45, where it is blocked by an unknown mechanism. What this mechanism might be is uncertain, since the +45 RNA does not contain a ribosomal binding site, nor is any secondary structure predicted which could block RNase J exonucleolysis.
Our preferred hypothesis, is that the +45 is an endonucleolytic cleavage which can be performed by either of the two RNase J proteins individually, but that any subsequent 5′ to 3′ exonucleolytic degradation can only be carried out by a functional RNase J1+J2 complex. This would explain why 5′-ends at position +45 are not seen in the WT strain, since the RNAs are rapidly 5′ to 3′ digested, neither seen in the ΔJ1ΔJ2 strain, because the +45 cleavage is never made. In contrast, the +45 RNA is highly enriched in the ΔJ1, ΔJ2 and J1AGA mutants, because they are able to perform the +45 cut, but unable to exonucleolytically degrade the resulting product. This hypothesis would also correspond with the observation by Mäder and coworkers [8] that several genes had significantly altered abundance in their B. subtilis RNase J1 and/or RNase J2 mutants, but did not exhibit any change in the double mutant.
Supporting this hypothesis are the data presented in Table 5, where the severely inhibited or lacking in vivo exonuclease activity in strains ΔJ1, ΔJ2 and J1AGA results in a similar accumulation of 5′-ends to that found in strain ΔJ1ΔJ2.
Concluding remarks
As seen in Figure 1, the RNase J mutants exhibit severe growth defects when taken outside the narrow ‘comfort zone’ of 37°C and Mueller-Hinton medium. The exact cause(s) of these problems have not been identified, but since our 5′ data shows that RNase J exerts a global influence on mRNAs (as well as on maturation of stable RNAs), the growth defects are most likely due to mis-regulation of a number of genes, each of which probably not causing major phenotypes, but generating severe effects when combined. Possible candidate genes could be SA1075 (hmrB) and RNase P, which are both essential [20]. In all mutant studies, especially those where the growth of the cell is severely affected, there is always a risk of detecting secondary effects of the mutation. This is important to keep in mind when interpreting the RNase J mutant data, even though we have attempted to strengthen the observations by using a range of different RNase J mutants and including the ΔY and ΔcshA strains as controls.
It appears that the 5′ to 3′ exonucleolytic activity is crucial for normal growth, whereas the endonucleolytic activity only serves a secondary function. The normal growth of strain J2AGA supports this, if the S. aureus RNase J2 is similar to its B. subtilis counterpart, which almost exclusively exhibits endonucleolytic activity in vitro [18].
While we do not assume that our four presented examples are exhaustive for all the functions of RNase J, they reveal how RNase J activity remodels sub-populations of RNA (Figures 4, 5, 6 and 7), and show that the roles of RNase J are diverse, ranging from maturation and post-transcriptional regulation to degradation. Furthermore, our data clearly show that RNase J1 is the work-horse, while RNase J2 plays a supporting role, with no detected phenotypes of the J2AGA mutant. However, we also detect that an important RNase J-mediated cleavage takes place in the SA1075 gene in ΔJ1, which demonstrates that RNase J2 is capable of performing some tasks by itself. The implied interplay between RNase J1, RNase J2 and the RNase J1+J2 complex in vivo, which might correlate with switching between endo - and exo-nucleolytic activity, is likely to have fine-tuned regulatory functions, arising from the evolutionary event that duplicated the RNase J gene in an early Firmicute ancestor.
Materials and Methods
Growth mediums
Mueller-Hinton (Beckton-Dickinson, Sparks, MD, USA) and LB-plates (Merck, Darmstadt, Germany) were prepared with 13 g/l agar (Merck), and RH-plates were made by autoclaving 100 ml milliQ water with 7.5 g agar, cooling it to 55°C and mixing with 500 ml modified RPMI-medium (Sigma, R7388) prewarmed to 55°C. +U, +C, and +Mg, indicates addition of 20 mg/l uracil, 10 mg/l chloramphenicol, and 5 mM MgCl2, respectively.
Constructing allelic replacement mutants
Mutants were generated using the pRLY-vector series (Table 3), and following the protocol described in Redder and Linder, 2012. However, since the RNase J mutants are sensitive to both heat and cold, strains PR01-20, PR01-25, PR02-03, and PR02-06 were kept between 32°C and 39°C throughout their construction, and strain PR01-27 and PR01-37 were grown at 37°C at all times, since the lack of origin of replication in the pRLY-vectors used for these strains precluded the need for plasmid establishment and plasmid elimination phases (see also Redder and Linder, 2012).
Spotting dilutions to determine growth defects
Strains were cultured overnight in liquid MH+U with agitation. Dilution series were then made in MH medium, to obtain 10−5 and 10−6 dilutions, 5 µl of which were then spotted on MH and RH plates with added uracil (10 mg/l). The plates were then incubated at the indicated temperature(s) until the WT colonies reached appropriate size, typically 18 hours for 42°C and 37°C, 24 hours for 30°C and 48 hours for 25°. All strains compared in Figure 1 were spotted on the same plate, to eliminate variations between batches. The same was the case for the strains in Figure 2.
EMOTE assay of mono-phosphorylated 5′-ends
2 ml of mid-exponential phase cultures (OD600 between 0.40 and 0.45) were harvested by centrifugation, the supernatant removed, and 1 ml cold 1∶1 ethanol/acetone was immediately added to protect the RNA. After washing the bacterials pellets in 1×TE buffer (10 mM Tris, 1 mM EDTA, pH 8), the cells were lysed with 100 µg lysostaphin (AMBI Products LLC, Lawrence, NY, USA) in 200 µl TE with 1 U/µl Murine RNase Inhibitor (M0314S, New England Biolabs, Ipswich, MA, USA), and homogenised using RNeasy Shredder columns (Qiagen, Hombrechtikon, Switzerland), whereupon total RNA was prepared using the RNeasy mini kit (Qiagen) with an on-column DNase I treatment (Cat: 79254; Qiagen).
For each strain, 0.5 nmol of Rp5 oligo (Table 8), 5 µg of total RNA, and TE buffer to a total volume of 55 µl was heated for 120 seconds at 95°C and then flash-cooled in ice/water. A premix of 10 µl 10× T4 RNA ligase buffer, 10 µl 10 mM ATP, 30 U T4 RNA ligase 1, 1 µl Murine RNase inhibitor (all from New England Biolabs), and 21 µl water was added to each tube of denatured RNA, and incubated for 3 hours at 37°C. After ethanol precipitation and re-suspension in 20 µl TE, the protocol for the MicrobeExpress kit (Ambion, Life Technologies, Zug, Switzerland) was followed to deplete the ribosomal RNA, followed by re-suspension in 25 µl water.
cDNA with one Illumina-sequencing adaptor was generated from 4.5 µl Rp5-ligated and rRNA-depleted total RNA, with 100 pmol DROAA oligo, 6 µl water, 4 µl 5× Reverse Transcriptase buffer, 2 µl 100 mM DTT, 1 µl 10 mM dNTP, 0.5 µl Murine RNase inhibitor, and 1 µl M-MLV (-H) Reverse Transcriptase (New England Biolabs), which was incubated for 10 minutes at room temperature, 50 minutes at 42°C, and 30 minutes at 65°C to inactivate the enzymes. 180 µl water with 1 mg RNase I was added and the cDNA was purified using the Promega SV PCR-purification kit (Promega, Madison, WI, USA).
The second Illumina sequencing adaptor, together with barcodes for identifying from which strain the sequence originated, was added in a second-strand PCR: 32 µl H2O, 1.5 µl 10 µM oligo D5La (D5Lb, D5Lc, etc.), 1.5 µl 10 µM B-oligo (Table 8), 10 µl 5× Phusion HF buffer, 1.5 µl 10 mM dNTP, 0.5 µl Phusion enzyme, 3 µl cDNA. Program: 2 min @ 98°C, 20 sec @ 98°C, 15 sec @ 55°C, 30 sec @ 72°C (30 cycles), 3 min @ 72°C. 6 µl of each PCR product was loaded on an agarose gel, to verify that the yields were similar. 40 µl of the WT PCR product was mixed with 20 µl from each of the other strains, and the mixture was purified using the Promega SV PCR-purification kit, and eluted twice with 50 µl water. The purified mixture was loaded on an agarose gel, and the smear between 300 bp and 1400 bp was extracted using the Promega SV Gel extraction kit (Promega), according to protocol (Figure 3), and the DNA was sent for 50 bp Illumina sequencing (Fasteris, Plan-des-Ouates, Switzerland).
Two biological replicates were carried out for strains WT, ΔJ1, ΔJ2, ΔJ1ΔJ2, ΔY and ΔcshA, but only one for strain J1AGA. Data presented for J1AGA and in Figure 4 are from the second biological replicate, and the latter exhibit the same patterns as the first biological replicate.
Analysing the EMOTE data
Before mapping on the reference genome, reads were split into groups looking for the exact match with the corresponding barcode sequence. After sorting, only reads having the control sequence (CC) at the proper position were selected. Then, reads were trimmed in order to only keep the sequence corresponding to the RNA. Reads were mapped on the reference genome (Staphylococcus aureus N315, accession number: NC_002745.2) using Bowtie 0.12.7 with the standard parameters. A first mapping was performed, keeping reads that mapped to unique positions on the chromosome (Table S1). Reads mapping at multiple positions on the chromosome were used on a second step (Table S2). Both mappings were analysed separately.
Analysing the 5′-ends of rRNAs used data from the group of reads that mapped to multiple locations (the five rRNA operons for example). The percentage of detected RNAs with 5′-ends at a given position was determined by dividing the number of reads mapping to that position by the total number of reads with a 5′-end within the region from the 16S rRNA processing stem cleavage by RNase III (93+M) to the 3′-end of the mature 16S rRNA. The proportion of 5′-ends compared to the WT strain (shown in Figure 4) was then obtained by dividing the percentage from the mutant strain with the percentage from the WT strain. The full data-set for 16S rRNA is shown in Table S3.
5′-ends of mRNAs and RNase P RNA used data from the group of reads that mapped to unique positions on the chromosome. The percentages for a given position was calculated as described above, however, the proportion between mutant and WT percentages was not calculated, since several positions had a WT percentage of zero, which would lead to division by zero. The full data-sets for the examples given in the text are shown in Tables S6, S7, and S8.
Northern blots
Total RNA was prepared in the same way as for the EMOTE assay, but with an additional step of ethanol precipitation to concentrate the RNA. 4 µg total RNA from each strain was loaded on a 5% polyacryl amide gel with 8M urea, and afterwards transferred to a Hybond-N nitrocellulose membrane (Amersham) using a Biorad Protean Tetra-cell blotting system. The RNA was crosslinked to the membrane with a UV Stratalinker 2400 (Stratagene), and the marker was revealed using Methylene blue. Probes were hybridised over night at 37°C in ExpressHyb hybridization solution (Clontech, Mountain View, CA, USA), excess probe was washed away, and the signal was detected using a Typhoon FLA 7000 phosphorimager (General Electric). The membrane was stripped for 2 hours at 75°C with 0.2% SDS and 10 mM Tris pH 7.5, whereupon a new probe was hybridised. Loading control was done with a 5S rRNA probe, used last to ensure that the strong 5S signal did not interfere with the other experiments. Probes used for Northern blotting (Table S9) were 5′ labelled using 32P γ-ATP and T4 polynucleotide kinase (New England Biolabs).
Supporting Information
Zdroje
1. CondonC, BechhoferDH (2011) Regulated RNA stability in the Gram positives. Curr Opin Microbiol 14 : 148–154.
2. RobertsC, AndersonKL, MurphyE, ProjanSJ, MountsW, et al. (2006) Characterizing the effect of the Staphylococcus aureus virulence factor regulator, SarA, on log-phase mRNA half-lives. J Bacteriol 188 : 2593–2603.
3. McDowallKJ, KaberdinVR, WuSW, CohenSN, Lin-ChaoS (1995) Site-specific RNase E cleavage of oligonucleotides and inhibition by stem-loops. Nature 374 : 287–290.
4. CarpousisAJ (2007) The RNA degradosome of Escherichia coli: an mRNA-degrading machine assembled on RNase E. Annu Rev Microbiol 61 : 71–87.
5. AndersonKL, RouxCM, OlsonMW, LuongTT, LeeCY, et al. (2010) Characterizing the effects of inorganic acid and alkaline shock on the Staphylococcus aureus transcriptome and messenger RNA turnover. FEMS Immunol Med Microbiol 60 : 208–250.
6. ShahbabianK, JamalliA, ZigL, PutzerH (2009) RNase Y, a novel endoribonuclease, initiates riboswitch turnover in Bacillus subtilis. EMBO J 28 : 3523–3533.
7. EvenS, PellegriniO, ZigL, LabasV, VinhJ, et al. (2005) Ribonucleases J1 and J2: two novel endoribonucleases in B.subtilis with functional homology to E.coli RNase E. Nucleic Acids Res 33 : 2141–2152.
8. MaderU, ZigL, KretschmerJ, HomuthG, PutzerH (2008) mRNA processing by RNases J1 and J2 affects Bacillus subtilis gene expression on a global scale. Mol Microbiol 70 : 183–196.
9. DurandS, GiletL, BessieresP, NicolasP, CondonC (2012) Three essential ribonucleases-RNase Y, J1, and III-control the abundance of a majority of Bacillus subtilis mRNAs. PLoS Genet 8: e1002520.
10. BugryshevaJV, ScottJR (2010) The ribonucleases J1 and J2 are essential for growth and have independent roles in mRNA decay in Streptococcus pyogenes. Mol Microbiol 75 : 731–743.
11. Lehnik-HabrinkM, PfortnerH, RempetersL, PietackN, HerzbergC, et al. (2010) The RNA degradosome in Bacillus subtilis: identification of CshA as the major RNA helicase in the multiprotein complex. Mol Microbiol 77 : 958–971.
12. RouxCM, DemuthJP, DunmanPM (2011) Characterization of components of the Staphylococcus aureus messenger RNA degradosome holoenzyme-like complex. J Bacteriol 193 : 5520–6.
13. MathyN, BenardL, PellegriniO, DaouR, WenT, et al. (2007) 5′-to-3′ exoribonuclease activity in bacteria: role of RNase J1 in rRNA maturation and 5′ stability of mRNA. Cell 129 : 681–692.
14. Li de la Sierra-GallayI, ZigL, JamalliA, PutzerH (2008) Structural insights into the dual activity of RNase J. Nat Struct Mol Biol 15 : 206–212.
15. DorleansA, Li de la Sierra-GallayI, PitonJ, ZigL, GiletL, et al. (2011) Molecular basis for the recognition and cleavage of RNA by the bifunctional 5′-3′ exo/endoribonuclease RNase J. Structure 19 : 1252–1261.
16. NewmanJA, HewittL, RodriguesC, SolovyovaA, HarwoodCR, et al. (2011) Unusual, dual endo - and exonuclease activity in the degradosome explained by crystal structure analysis of RNase J1. Structure 19 : 1241–1251.
17. BrittonRA, WenT, SchaeferL, PellegriniO, UickerWC, et al. (2007) Maturation of the 5′ end of Bacillus subtilis 16S rRNA by the essential ribonuclease YkqC/RNase J1. Mol Microbiol 63 : 127–138.
18. MathyN, HebertA, MerveletP, BenardL, DorleansA, et al. (2010) Bacillus subtilis ribonucleases J1 and J2 form a complex with altered enzyme behaviour. Mol Microbiol 75 : 489–498.
19. KobayashiK, EhrlichSD, AlbertiniA, AmatiG, AndersenKK, et al. (2003) Essential Bacillus subtilis genes. Proc Natl Acad Sci U S A 100 : 4678–4683.
20. ChaudhuriRR, AllenAG, OwenPJ, ShalomG, StoneK, et al. (2009) Comprehensive identification of essential Staphylococcus aureus genes using Transposon-Mediated Differential Hybridisation (TMDH). BMC Genomics 10 : 291.
21. RedderP, LinderP (2012) New Range of Vectors with a Stringent 5-Fluoroorotic Acid-Based Counterselection System for Generating Mutants by Allelic Replacement in Staphylococcus aureus. Appl Environ Microbiol 78 : 3846–3854.
22. FigaroS, DurandS, GiletL, CayetN, SachseM, et al. (2013) Bacillus subtilis mutants with knockouts of the genes encoding ribonucleases RNase Y and RNase J1 are viable, with major defects in cell morphology, sporulation, and competence. J Bacteriol 195 : 2340–2348.
23. Papp-WallaceKM, MaguireME (2008) Regulation of CorA Mg2+ channel function affects the virulence of Salmonella enterica serovar typhimurium. J Bacteriol 190 : 6509–6516.
24. RichardsJ, LiuQ, PellegriniO, CelesnikH, YaoS, et al. (2011) An RNA pyrophosphohydrolase triggers 5′-exonucleolytic degradation of mRNA in Bacillus subtilis. Mol Cell 43 : 940–949.
25. LioliouE, SharmaCM, CaldelariI, HelferAC, FechterP, et al. (2012) Global regulatory functions of the Staphylococcus aureus endoribonuclease III in gene expression. PLoS Genet 8: e1002782.
26. RedkoY, CondonC (2010) Maturation of 23S rRNA in Bacillus subtilis in the absence of Mini-III. J Bacteriol 192 : 356–359.
27. ReiterNJ, OstermanA, Torres-LariosA, SwingerKK, PanT, et al. (2010) Structure of a bacterial ribonuclease P holoenzyme in complex with tRNA. Nature 468 : 784–789.
28. HowdenBP, BeaumeM, HarrisonPF, HernandezD, SchrenzelJ, et al. (2013) Analysis of the Small RNA Transcriptional Response in Multidrug-Resistant Staphylococcus aureus after Antimicrobial Exposure. Antimicrob Agents Chemother 57 : 3864–3874.
29. Daou-ChaboR, MathyN, BenardL, CondonC (2009) Ribosomes initiating translation of the hbs mRNA protect it from 5′-to-3′ exoribonucleolytic degradation by RNase J1. Mol Microbiol 71 : 1538–1550.
30. ZukerM (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31 : 3406–3415.
31. KazantsevAV, KrivenkoAA, HarringtonDJ, HolbrookSR, AdamsPD, et al. (2005) Crystal structure of a bacterial ribonuclease P RNA. Proc Natl Acad Sci U S A 102 : 13392–13397.
32. OunS, RedderP, DidierJP, FrancoisP, CorvagliaAR, et al. (2013) The CshA DEAD-box RNA helicase is important for quorum sensing control in Staphylococcus aureus. RNA Biol 10 : 157–165.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 2
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Genome-Wide Association Study of Metabolic Traits Reveals Novel Gene-Metabolite-Disease Links
- A Cohesin-Independent Role for NIPBL at Promoters Provides Insights in CdLS
- Classic Selective Sweeps Revealed by Massive Sequencing in Cattle
- Arf4 Is Required for Mammalian Development but Dispensable for Ciliary Assembly
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy