
Evaluating Human T-Cell Therapy of Cytomegalovirus Organ Disease in HLA-Transgenic Mice
Pre-emptive CD8 T-cell therapy of human cytomegalovirus (HCMV) disease in immunocompromised recipients of hematopoietic stem cell transplantation gave promising results in clinical trials, but limited efficacy and the need of HCMV-seropositive memory cell donors has so far prevented adoptive cell transfer from becoming clinical routine. Further development is currently hampered by the lack of experimental animal models that allow preclinical testing of the protective efficacy of human T cells in functional organs. While humanized mouse models with human tissue implants are technically and statistically demanding, and are limited to studying human T-cell activation and local virus control in the implants, a more feasible model for control of systemic infection and prevention of multiple-organ CMV disease is regrettably missing. Here we introduce such a model based on infection of genetically immunocompromised, HLA-A2.1-transgenic NOD/SCID/IL-2rg-/ - mice with a chimeric murine CMV engineered to express the HCMV NLV-peptide epitope. Mimicking the scenario of HCMV-unexperienced donors, human T cells transduced with a human T-cell receptor specific for HLA-A.2.1-presented NLV peptide controlled systemic infection and moderated organ disease resulting in a survival benefit. The model promises to become instrumental in defining T-cell properties that determine their protective efficacy for a further development of adoptive immunotherapy of post-transplantation CMV infection.
Published in the journal:
. PLoS Pathog 11(7): e32767. doi:10.1371/journal.ppat.1005049
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1005049
Summary
Pre-emptive CD8 T-cell therapy of human cytomegalovirus (HCMV) disease in immunocompromised recipients of hematopoietic stem cell transplantation gave promising results in clinical trials, but limited efficacy and the need of HCMV-seropositive memory cell donors has so far prevented adoptive cell transfer from becoming clinical routine. Further development is currently hampered by the lack of experimental animal models that allow preclinical testing of the protective efficacy of human T cells in functional organs. While humanized mouse models with human tissue implants are technically and statistically demanding, and are limited to studying human T-cell activation and local virus control in the implants, a more feasible model for control of systemic infection and prevention of multiple-organ CMV disease is regrettably missing. Here we introduce such a model based on infection of genetically immunocompromised, HLA-A2.1-transgenic NOD/SCID/IL-2rg-/ - mice with a chimeric murine CMV engineered to express the HCMV NLV-peptide epitope. Mimicking the scenario of HCMV-unexperienced donors, human T cells transduced with a human T-cell receptor specific for HLA-A.2.1-presented NLV peptide controlled systemic infection and moderated organ disease resulting in a survival benefit. The model promises to become instrumental in defining T-cell properties that determine their protective efficacy for a further development of adoptive immunotherapy of post-transplantation CMV infection.
Introduction
Reactivation of latent human cytomegalovirus (HCMV) infection is a frequent complication in patients after allogeneic hematopoietic stem cell transplantation (HSCT). Although potent antiviral drugs are available, their usage, however, is often limited by hematotoxicity and nephrotoxicity. In addition, the broad application of these drugs during pre-emptive treatment strategies is associated with a higher frequency of late-onset HCMV disease [1,2]. Preclinical research in murine models ([3–6], reviewed in [7–9]) as well as clinical phase I/II trials ([10–12], reviewed in [13,14]) have shown that the adoptive transfer of virus-specific CD8 T cells is a promising therapeutic option for preventing and treating CMV disease. However, the feasibility of HCMV-specific immunotherapy is currently impeded in clinical routine due to technical restrictions. It has also limitations in case the donor is HCMV-seronegative or carries only low frequencies of HCMV-specific memory T cells. In this situation, transduction of non-cognate T cells with virus specific T-cell receptors (TCR) may be an alternative means to transfer HCMV-specific T-cell function into HSCT recipients [15,16]. In any case, clinical protocols need to be improved before HCMV-specific cell therapy can be implemented in general clinical practice. To allow for a more reliable analysis of HCMV immunotherapies (e.g. adoptive T-cell therapy, therapeutic vaccination) animal models that mimic HCMV infections are needed.
Since HCMV replication is strictly restricted to cells and tissues of human origin ([17], reviewed in [18]), previous animal models utilized murine CMV (mCMV) as surrogate virus (reviewed in [7–9]) or mice infected with HCMV after implantation with human cells or tissues, for instance with tumor cell lines, fetal thymus, and liver biopsies ([19–23], reviewed in [24]). The implantation approach has shown that HCMV strains replicate locally with differences in pathogenicity, but fail to spread between tissue implants. To support systemic infection, Smith et al. [25] infected human CD34+ hematopoietic stem cell-engrafted mice with HCMV to establish latency and to induce virus reactivation in tissue-migrated monocytes and macrophages by granulocyte-colony stimulating factor (G-CSF) treatment. By model design, however, viral dissemination to functional organs relevant for viral pathogenesis (e.g. spleen, lungs, and liver) and transmission (salivary glands) cannot be assessed even in these advanced humanized mouse models.
We herein present a novel preclinical mouse model that allows the direct testing of HCMV-specific human T-cell products. In this, we combined the well-described murine model of mCMV infection of the immunocompromised host (reviewed in [7–9]) with the strong T-cell immunogenicity of the HLA-A*0201 (HLA-A2.1) restricted HCMV epitope pp65495-503 NLVPMVATV (briefly, NLV) [26]. We generated a chimeric recombinant mCMV expressing the NLV epitope (mCMV-NLV) during the infectious cycle to allow organ manifestation of the infection in the natural host similar to that seen in immunocompromised patients. After infection of HLA-A2.1 transgenic, constitutively combined-immunodeficient NOD/SCID/IL-2rg-/- (NSG/HHD) mice, which lack cells of adaptive immunity and are additionally deficient in natural killer (NK) cells [27], mCMV-NLV resulted in a rapid systemic infection that could be effectively combated by adoptively transferred human NLV-specific CD8 T cells as well as by human T cells transduced with an NLV-specific TCR. After migration into murine organs, these T cells not only reduced mCMV-NLV titers in an epitope-specific manner but also prolonged survival of infected mice, despite continued combined immunodeficiency in absence of hematopoietic reconstitution.
For the clinical correlate of reactivated HCMV infection associated with HSCT, our findings predict that pre-emptive CD8 T-cell immunotherapy, even though controlling CMV only transiently, can bridge the critical time of an immunocompromised state until immune reconstitution by HSCT takes over. Altogether, we provide a novel animal infection model that allows the direct evaluation and preclinical testing of HCMV-specific human T cells and potentially other HCMV immunotherapy approaches, including vaccination of HSCT recipients in parallel to hematopoietic reconstitution.
Results
Integration of the HCMV-NLV epitope into mCMV protein IE2 does not alter mCMV growth
The key strategy of our model was to antigenically ‘humanize’ both the murine virus and the murine host tissue for HLA class I-restricted recognition of infected cells by human T cells in functional host organs. For construction of the chimeric virus mCMV-NLV, we integrated the coding gene sequence of the HLA-A2.1-restricted pp65 (UL83)495-503 peptide epitope NLVPMVATV (abbreviated: NLV) of HCMV, with its natural flanking amino acids for authentic proteasomal cleavage and precursor peptide sequence, into the immediate-early (IE)2/m128 gene of mCMV-Δm157 (Fig 1A). Although NSG/HHD mice are deficient in NK cell activity due to lack of the interleukin-2 receptor (IL-2R) common γ chain (CD132) causing deficient IL-2/4/7/9/15/21 signaling [28,29], mCMV-Δm157 was chosen as parental virus to also formally exclude activation of the Ly49H+ subset of murine NK cells by m157-Ly49H interaction, a feature that is only valid in the C57BL/6 genetic background and might thus interfere with a broader application of the model (for a review, see [30]). For control experiments, a lack-of-epitope mCMV-NLV derivative was generated, in which a single amino acid replacement at the C-terminal HLA-A2.1 anchor position valine (V503) by alanine (mCMV-NLVAla) prevents epitope generation in the proteasome and MHC/HLA class-I (MHC/HLA-I) presentation (Fig 1A) (for the concept, see [31,32]).

The IE2/m128 gene encodes a regulatory but non-essential protein expressed in the IE phase of viral replication [33,34] and sporadically also during viral latency [35]. To verify, in the first place, that integration of the NLV epitope into the IE2 protein does not attenuate the virus, which otherwise would render the model inapplicable for studying viral pathogenesis, we compared the in vivo growth kinetics of parental virus mCMV-Δm157 and its antigenicity variant mCMV-NLV in NSG/HHD mice. In selected organs relevant for viral pathogenesis, such as liver, spleen, and lungs (Fig 1B), the two viruses were found to replicate in a log-linear fashion with comparable doubling times within each organ (for the principle, see [36,37]), though with organ-typic numerical differences, with most aggressive growth in the lungs. In conclusion, insertion of the NLV epitope did not impair in vivo viral growth. The chimeric virus thus fulfills the model’s first prerequisite of an unaltered replicative fitness.
The HCMV-NLV epitope is successfully processed and presented by HLA-A2.1 transgenic murine cells
The second prerequisite of the model is that the NLV peptide is actually generated by processing of the chimeric IE2-NLV protein in mCMV-NLV infected cells and that it is presented in association with HLA-A2.1 at the cell surface for recognition by TCR. To directly assess presentation, an endpoint which includes the preceding processing events, we chose two independent NLV peptide-specific, cytolytic CD8 T cell lines (CTLL) as probes: (i) a published, peptide-selected murine long-term CTLL, mCD8-NLV, expressing a murine TCR [38] for serving as a murine reference line, and (ii) a freshly generated peptide-selected human short-term polyclonal CTLL, hCD8-NLV, expressing human TCRs for modeling the situation of an HCMV-experienced, seropositive cell transfer donor as the clinical correlate. While mCD8-NLV consisted of cells with effector phenotype (CD44+CD62Llow TE/EM), including effector-memory cells, early effector cells, and short-lived effector cells ([39,40], reviewed in [41,42]) the short-term hCD8-NLV line was composed of cells with central memory and effector memory phenotype, TCM and TEM, respectively (S1 Fig). The functional avidities of these two CTLL were then tested in an IFN-γ secretion-based ELISpot assay for their sensitization by HLA-A2.1 transgenic NSG/HHD mouse embryonic fibroblasts (HLA-A2.1-MEF) exogenously loaded with graded concentrations of synthetic NLV peptide (Fig 2A). Notably, when corrected for background response measured with an unrelated peptide, both CTLL were similarly sensitive with an endpoint NLV peptide concentration of 10−9 M. Pretreatment of the HLA-A2.1-MEF stimulator cells with IFN-γ, which is known to enhance MHC class-I expression [43], only modestly increased the numbers of CTLL cells responding to exogenously peptide-loaded HLA-A2.1 molecules. Contrasting with the comparable functional avidities of the two CTLL, an assay based on their physical capacity to bind NLV peptide-folded HLA-A2.1 tetramers [44,45] revealed a higher structural avidity of mCD8-NLV (Fig 2B), which predicts more efficient in vivo function [8,46]. After this basal characterization of the two CTLL, we tested their capacity to detect naturally processed NLV peptide presented by HLA-A2.1 on the surface of infected NSG/HHD HLA-A2.1-MEF (Fig 2C). In both lines, a significant, though low, number of cells had an avidity sufficient for recognizing NLV-HLA-A2.1 complexes presented on infected cells. This number, however, was significantly enhanced when NSG/HHD HLA-A2.1-MEF were treated with IFN-γ prior to infection, which contrasts to the only modest effect of IFN-γ on exogenous peptide loading. The explanation likely is the overriding of the function of viral immune evasion molecules (viral regulators of antigen presentation, vRAPs) by IFN-γ, as described previously to occur in infected cells in vitro as well as in infected tissues in vivo ([47,48], reviewed in [49]).

In this context it is worth noting that the immune evasion molecules of mCMV indeed also target the transgenic HLA-A2.1 (S2 Fig). Specifically, as shown by flow cytometry, immune evasion mechanisms of mCMV caused cell surface down-modulation of both murine H2-Kd and the transgenic human HLA-A2.1 molecules in infected (gp36.5/m164+) but not in uninfected (gp36.5/m164-) cells of the same infected cell cultures when compared to uninfected control cultures. Accordingly, H2-Kd and HLA-A2.1 expression levels remained largely unaffected upon infection with the immunoevasin gene deletion mutant mCMV-ΔvRAP (S2 Fig) [48,50,51].
Importantly, both the normal and the IFN-γ-enhanced recognition of infected cells proved to be strictly NLV epitope-specific, as shown by baseline values of response to NSG/HHD HLA-A2.1-MEF infected with virus mCMV-NLVAla (Fig 2C).
NLV peptide-specific human CD8 T cells are protective by confining the infection of HLA-A2.1 transgenic murine tissues to nodular inflammatory foci (NIF) in an epitope-specific manner
After fulfillment of the model’s prerequisites of unaltered virulence and of successful transgenic antigen presentation, both NLV peptide-specific CTLL, characterized as detailed above, were tested for their capacity to control the replication of chimeric virus mCMV-NLV in tissues of HLA-A2.1 transgenic NSG/HHD mice, which can be engrafted with components of the human immune system due to a lack of an adaptive immune system and innate immune defects [27]. Since NSG/HHD mice are devoid of endogenous murine T cells, tissue infiltration by T cells and antiviral control can be attributed exclusively to the transferred cells.
Specifically, sublethally (2 Gy) γ-irradiated NSG/HHD mice were infected by intraplantar injection with 1x105 PFU of mCMV-NLV, a dose that would be lethal after approximately 12 to 17 days. Shortly thereafter, NLV-specific CTLL were intravenously transferred in order to mimic the clinical setting of pre-emptive therapy, and viral titers in spleen and lungs as well as numbers of infected cells in livers of individual mice were determined on day 11 (for the protocol scheme, see Fig 3A). In the case of transfer of human NLV-specific CTLL, single doses of IL-2 and IL-7 were given along with the cells for a short-term growth factor supply. In the beginning of the project, a key concern had been that a cross-species adoptive transfer of CD8 T cells might fail because of xenogeneic constraints in the chemokine-driven extravasion and tissue infiltration of intravenously transferred CD8 T cells. Not unexpectedly, based on extended literature to adoptive immunotherapy by CD8 T cells in fully murine models (reviewed in [7–9]), transfer of mCD8-NLV cells controlled the infection of spleen, lungs, and liver in a dose-dependent and epitope-specific fashion (Fig 3B). As shown for the liver, dose-dependent antiviral protection correlated with dose-dependent tissue infiltration by the adoptively transferred CD8 T cells. Likewise, lack of epitope after infection with mCMV-NLVAla abolished both tissue infiltration and control of virus replication. Importantly, as a new information, the results were qualitatively identical for the human CD8 T cells, hCD8-NLV, although higher cell numbers were needed (Fig 3C). Whether this relates to the lower structural avidity (recall Fig 2B) or to xenogeneic differences remains to be investigated. The more relevant conclusion, however, is that the functionality of antiviral human CD8 T cells can, in principle, be studied in murine tissues.
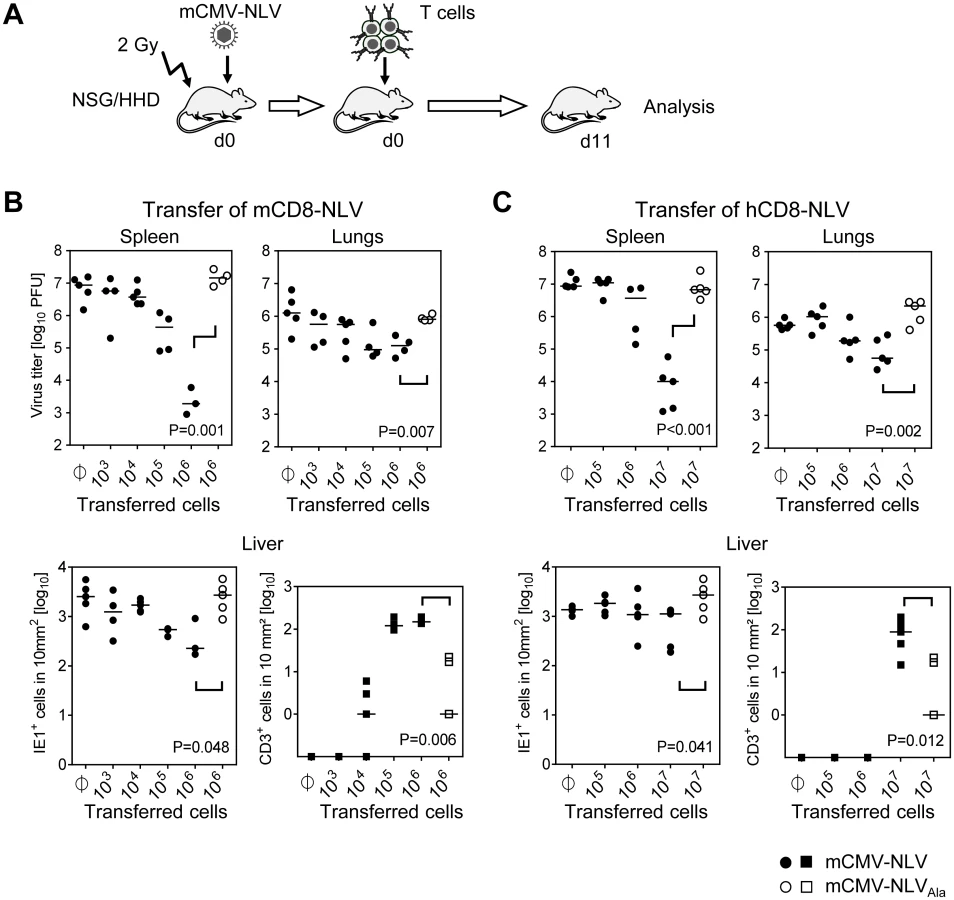
Protection against disseminated CMV organ infection has a microanatomical correlate in the formation of nodular inflammatory foci (NIF) [52–57]. As shown previously in a related murine model [54] and reproduced here for the example of mCD8-NLV cells, infection by a virus expressing the cognate epitope is confined to NIF where control takes place and where infected tissue cells and infiltrating CD8 T cells co-localize. In contrast, in the absence of the cognate epitope [54] after infection with virus mCMV-NLVAla, CD8 T cells do not infiltrate infected tissue at all, can thus not form NIF, and, accordingly, allow more widespread infection visible as many foci of infection (IF), with the consequence of virally-caused tissue pathology (Fig 4A). Importantly, these rules were found to apply, in principle, also to the human CD8 T cells, hCD8-NLV, although NIF appeared as being somewhat less condensed (Fig 4B). This may relate to the lower structural avidity (recall Fig 2B) and the higher cell numbers needed for antiviral protection (recall Fig 3C). Again, although the syngeneic cell transfer system is more efficient, which is not surprising, a xenogeneic system also allows NIF formation and control of intra-tissue virus spread, despite the species barrier.

TCR-transduced human CD8 T cells control the infection of HLA-A2.1 transgenic NSG/HHD mice
The need of an HCMV-experienced (HCMV antibody positive) donor as a source for virus-specific memory CD8 T cells is a limitation for an adoptive immunotherapy of HCMV reactivation in the highest-risk constellation of an HCMV-seronegative donor and—seropositive recipient (D-R+) [58–61], which can currently only be overcome by an HLA-matched third party memory T-cell donor [12,62,63]. An alternative approach, already successfully applied in tumor models [64,65], is the transduction of T cells derived from the HLA-matched HSCT donor with a virus-epitope specific TCR.
To model this clinical need, a first application of the here newly described HLA-A2.1 transgenic mouse CMV infection model was to test the in vivo antiviral function of human CD8 as well as CD4 T cells that were transduced by retroviral gene transfer of a modified high-affinity human T-cell receptor α/β (TCRNLV) previously shown to reprogram CD8 and CD4 T cells derived from HCMV-seronegative donors [16]. As a basal characterization of the TCRNLV transduced T cells enriched by drug selection, cytofluorometric analysis with NLV peptide-folded HLA-A2.1 tetramers for TCRNLV staining shows high transduction efficacies (Fig 5A), and Fig 5B and 5C reveal structural avidity as well as NLV epitope-specific cytolytic potential of CD8-TCRNLV and CD4-TCRNLV cells, respectively. Using NSG/HHD HLA2.1-MEF exogenously loaded with a high dose of synthetic NLV peptide as a positive standard, both transduced T-cell lines specifically lysed IFN-γ pre-treated mCMV-NLV-infected NSG/HHD cells, overcoming the presence of the mCMV immune evasion molecules (see above, [48]), and this lysis was TCR/epitope-specific as indicated by its absence after mock-transduction of the T cells as well as after infection of the target cells with the antigenicity-loss variant mCMV-NLVAla. Notably, CD8-TCRNLV CTL, but not mock-transduced CD8 T cells, lysed HLA-A2.1-expressing human primary foreskin fibroblasts infected with the HCMV immune evasion gene (US2,3,6,11) deletion mutant RV-KB6 expressing the NLV peptide [66], but not those infected with the combined US2,3,6,11 and pp65/UL83 deletion mutant HCMV-RV-KB15 lacking NLV peptide [16] (S3 Fig). This, again, showed the specificity in terms of the need for the specific TCR and the cognate epitope presented by HLA-A2.1. For completing the basal characterization, S4A and S4B Fig show the phenotyping of the human CD8 and CD4 T cells, respectively, each before (upper row) and after (lower row) TCRNLV transduction and subsequent lymphokine-driven expansion triggered by ligation of CD3 and CD28. In essence, and in accordance with previous studies on TCR transduction, the resulting CD8-TCRNLV population consisted primarily of TEM [67–70], whereas CD4-TCRNLV cells were predominantly TCM.

These cells, and the corresponding empty-vector transduction controls lacking cognate TCR, were then adoptively transferred into mCMV-NLV-infected NSG/HHD mice to test their in vivo antiviral efficacy (Fig 6). With the exception of the liver, where a tendential reduction in day 11 median virus titer did not reach statistical significance due to high variance in the therapy group, infectious virus load was found to be significantly reduced in spleen (P < 0.001) and lungs (P = 0.014) by 107 CD8-TCRNLV cells but not by 107 CD4-TCRNLV cells (P >0.05 throughout). This is important new information, as it contrasts with the cytolytic activities of both cell populations (recall Fig 5B and 5C), indicating that in vitro cytolytic activity against IFN-γ pre-treated, infected cells is not reliably predictive for an antiviral function in vivo.

TCR-transduced CD4 T cells enhance the antiviral efficacy of TCR-transduced CD8 T cells
Interestingly, CD4-TCRNLV cells, though not antiviral effectors on their own (see above), enhanced the antiviral function of CD8-TCRNLV cells when co-administered as a 1 : 4 mixture of 2 x 106 non-protective CD4-TCRNLV and 8 x 106 protective CD8-TCRNLV cells (spleen, P < 0.001; lungs, P = 0.009; liver, P = 0.049). Specifically, at least in spleen and lungs, this mixture turned out to be more efficient than the higher number of 107 protective CD8 T cells (P = 0.037 and P = 0.005, respectively), which indicates synergism in the sense of a helper effect of the CD4 T cells. Protection by the mixture was clearly NLV epitope-specific, as any antiviral function was abolished when the NSG/HHD mice were infected with the antigenicity-loss mutant mCMV-NLVAla.
Antiviral control, determined on day 11, should be preceded by migration of transferred cells into the tissues. As shown for spleen and liver (S5 Fig), CD8 and CD4 T cells could be recovered on day 3 after intravenous cell transfer from spleen leukocyte and liver non-parenchymal cell populations. Notably, after transfer of 107 CD8-TCRNLV cells in absence of CD4-TCRNLV cells, recovery of CD8-TCRNLV cells from the tissues was poor but was enhanced after transfer of the mixture. Thus, apparently, the CD4 T cells’ helper effect is by facilitating tissue infiltration, as limited longevity of transferred CD8-TCRNLV cells should not yet be an issue in the first 3 days.
NLV-specific human T cells prolong the survival of mCMV-NLV-infected HLA-A2.1 transgenic NSG/HHD mice by delaying viral pathology
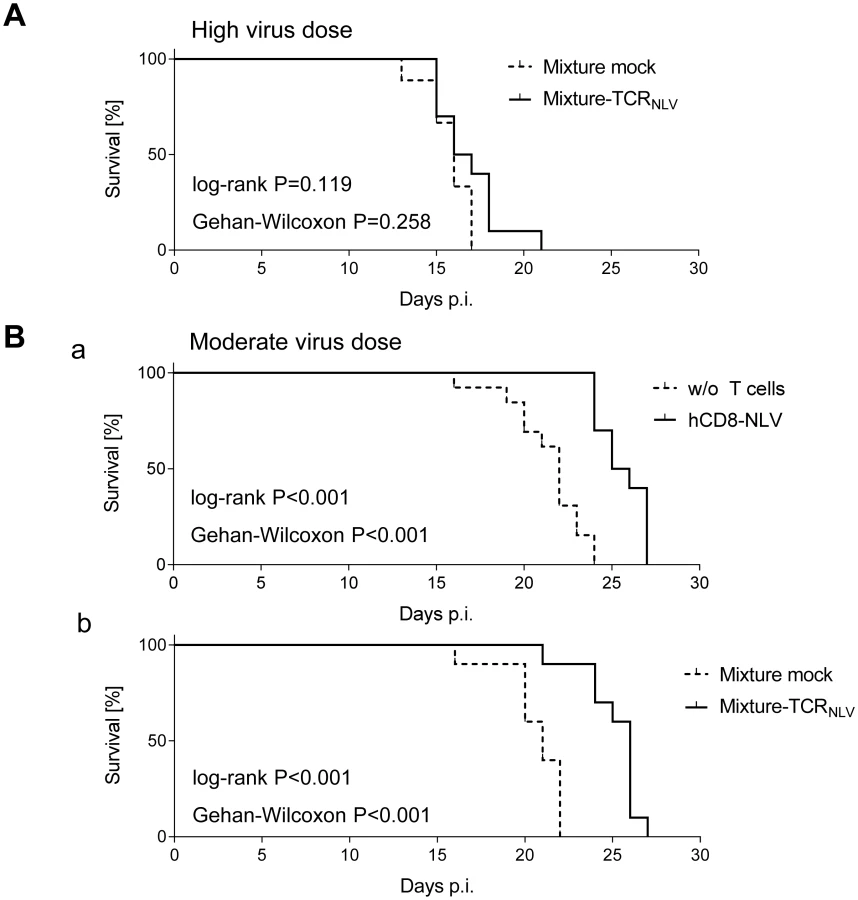
By design, the model’s strength is to test immediate effector functions of the transferred T cells, as their longevity in the NSG/HHD recipient is limited by the constitutive absence of IL-2/4/7/9/15/21 signaling and cytokine heterology, except for single doses of IL-2 and IL-7 coadministered with the human T cells. Furthermore, the model of a genetically, and thus enduringly, immunodeficient recipient is highly demanding for completeness of virus eradication at an early stage, as it is established experience that, in the long run, a few infectious units (just 1–5 PFU) of ‘therapy escapee virus’ will eventually cause death in NSG/HHD and related, immunodeficient mouse strains based on tissue pathology from exponential cytopathogenic viral spread over time [71].
Despite these inherent constraints, transfer of human NLV-specific T cells led to prolonged survival, which was modest at very high initial virus burden and did not reach statistical significance (Fig 7A) but was highly significant (P < 0.001 by log-rank test as well as by Gehan-Wilcoxon test) at a moderate, more realistic, initial virus dose (Fig 7B; see Discussion). Specifically, hCD8-NLV cells delayed mortality when compared to the no transfer group (Fig 7B and 7a), and the 1 : 4 mixture of TCRNLV-transduced CD4 and CD8 T cells delayed mortality when compared to the mock-transduced mixture lacking an NLV-specific TCR (Fig 7B and 7b).
To demonstrate a histopathological correlate of mortality, we performed immunohistochemical (IHC) imaging for livers from the mock-transduction control group and the therapy group (corresponding to Fig 7B and 7b) at the time of onset of mortality (day 20) in the control group (Fig 8). Overview sections, identifying infected liver cells by their expression of intranuclear viral IE1 protein (Fig 8a1 and 8b1; red staining), reveal extended plaque-like lesions from the cytopathogenic effect of virus replication in the control livers, whereas foci of infection were less frequent and less extended in the therapy group, though, as predictable from exponential (log-linear) growth ([36,37] and references therein) tissue infection is likely to close up to the control group with a few days delay explaining delayed onset of mortality in the therapy group. Higher-magnification 2-color IHC images show infected hepatocytes (iHc, red staining of IE1), which are distinctive by cytomorphology, infected endothelial cells (iEC, red staining of IE1 and black staining of CD31 antigen) (Fig 8a2 and 8b2), and infected macrophages (iMΦ, red staining of IE1 and turquoise-green staining of F4/80 antigen) (Fig 8a3 and 8b3). Quantification by cell counting reveals significant reductions in the numbers of infected cells in the therapy group, with no notable cell-type specific preferences (Fig 9A).


Although therapeutic cells equipped with a transgenic virus epitope-specific TCR are not expected to cause an immunopathology, there was residual concern that xenogeneic graft-versus-host interactions, possibly by the endogenous TCRs or by TCR-independent mechanisms, might cause immunopathology and thus obscure the results of the model, in particular the survival data. We have therefore compared general histopathology and caspase 3-dependent apoptosis in liver tissue sections from the control group and the therapy group at the time of onset of death in the control group (day 20 in this specific experiment) by 2-color IHC staining of infected cells (IE1+, red staining) and apoptotic cells (caspase-3+, brown staining) (Fig 10). Notably, histopathology proved to be confined to the foci of infection, as no lesions or necrotic areas were observed in tissue regions not yet reached by the infection. Interestingly, apoptotic cells were predominantly uninfected Hc (IE1-caspase-3+), though localizing to the foci of infection. This suggests an apoptosis induction in trans, but apoptotic iHc (IE1+caspase-3+) can also be found occasionally (IHC image in S6 Fig and quantitation in Fig 9B). The relatively low number of apoptotic infected cells relates to the known fact of cell death-inhibiting viral genes being expressed in CMV-infected cells (for reviews, see [72,73]. Most relevantly, however, the number of apoptotic cells, even that of uninfected apoptotic cells, was definitively not increased but rather tended toward being reduced in the therapy group (Fig 9B), a finding that rules out any relevant immunopathology by the xenogeneic cell transfer in this immunotherapy model. As reported recently, mCMV infection as such does not induce immunopathology mediated by CD8 T cells, except if the host is combined-deficient in NK cells and perforin [74]. Finally, IHC specific for CD8 did no longer reveal presence of transferred CD8 T cells in the liver on day 16 (Fig 9C), reflecting their limited longevity due to the multiple interleukin-signaling deficiency in NSG/HHD mice. In conclusion, histopathology is undoubtedly of viral etiology and is clearly reduced by antiviral human CD8 T-cell immunotherapy.

Discussion
We herein describe a new mouse model mimicking pre-emptive adoptive T-cell therapy of systemic HCMV infection in immunocompromised patients. The model is based on HLA-A2.1 transgenic NSG/HHD mice that are first infected with chimeric virus mCMV-NLV and are subsequently infused with human T cells specific for the immunodominant HLA-A2.1-restricted NLV epitope of HCMV pp65 (UL83). Despite the species barrier, transferred human T cells formed protective NIF, controlled virus spread, and limited viral pathology in classical organs of CMV disease manifestations without mediating immunopathology, thereby conveying a significant survival benefit to infected mice.
Previous mouse models for HCMV infection utilized transplantation of HCMV-infected human cells, e.g. fibroblasts embedded in agarose plugs [75], glioblastoma cell lines [76], and hepatocytes [23] or, alternatively, human fetal thymus/liver and retinal tissues infected with HCMV [19–21]. Although having contributed important information, implant models, however, are not designed for studying virus dissemination to functional organs and virus spread in these organs that causes histopathology resulting in morbidity and mortality. Therefore, life-saving therapeutic interventions could not be evaluated in such models. In contrast, systemic infection of NSG/HHD mice with the herein described chimeric virus mCMV-NLV provides a model that—in a reasonably good first approximation—resembles relevant aspects of HCMV disease in HSCT recipients, including the infection of vital organs involved in virus-associated morbidity and cause of death. It should be emphasized that the NLV peptide was genetically engineered into the non-essential IE2/m128 protein of mCMV. Thus, its synthesis and presentation occurs according to the regular gene expression kinetics of the viral IE2 carrier protein.
The NLV peptide was here just chosen as a paradigm. The model is meant as a modular system that can easily be adapted to scientific and medical needs. Specifically, by analogous strategy, the NLV peptide can be replaced with any other HLA-presented HCMV epitope of interest or might be combined with additional epitopes integrated at other positions of the mCMV genome [31]. Such chimeric viruses, together with various HLA transgenic NSG mouse strains, promise to allow analysis of the broad repertoire of HLA restricted T-cell responses against HCMV ([77], reviewed in [78,79]).
Importantly, like for HCMV infection of human cells, HLA-A2.1/NLV-antigen presentation was shown here to be susceptible to immune evasion mediated by virally encoded proteins that inhibit the trafficking of peptide-loaded MHC-I molecules to the cell surface [49,80–84]. Although the proteins involved and the precise molecular mechanisms by which they mediate immune evasion differ between individual CMV species, the common biological outcome is inhibition of antigen presentation, which has been shown to reduce but not to preclude a protective effect of adoptively transferred murine CD8 T cells in immunocompromised mice ([85,86], reviewed in [7–9,49,84]). Likewise, contrasting with earlier antigen presentation studies in transfected cells, HCMV ‘immune evasion’ genes gpUS2-11 also fail to completely prevent antigen presentation in infected cells [38,66]. In this context it is important to note that in the here presented model the species origin of the transferred CD8 T cells, secreting murine or human IFN-γ, appeared not to be qualitatively critical for controlling mCMV-NLV infection despite immune evasion mechanisms of mCMV being active that are relieved by murine IFN-γ [47,48]. For the future, the modular model is open for the option to further ‘humanize’ the conditions by replacing immune evasion genes of mCMV with those of HCMV in a next generation of chimeric mCMV-NLV viruses.
A concern affecting the validity of the model has been a potential species barrier for an efficient infiltration of infected murine tissues by human CD8 T cells. It is therefore important to recall that we observed tissue infiltration and epitope-specific formation of protective NIF by NLV-specific human CD8 T cells, reduction of viral load in representative murine target organs of CMV disease, and prolonged survival. The data in the model thus agree with previous work in humans showing that adoptive transfer of NLV-specific CD8 T cells is beneficial in patients undergoing allogeneic HSCT [87–89]. Importantly, a concern regarding a putative immunopathology by transferred xenogenic TCR-transduced T cells could be invalidated in the liver by absence of tissue necrosis and of apoptotic cells in tissue areas outside of the foci of infection.
Although previous studies have highlighted the importance of CD4 T-helper cell support in HSCT patients [87,90–93], antiviral T-cell immunotherapies so far mainly focused on cytolytic CD8 T cells. A helper contribution by CD4 T cells was proposed to be needed primarily for longevity of transferred CD8 T cells mediating enduring protection. Applying the here described new model for testing the requirements of immunotherapy by TCRNLV-transduced human CD8 T cells as a model for an HCMV seronegative HSCT donor, CD4 T cells indeed supported CD8 T cell-mediated antiviral control in mCMV-NLV-infected NSG/HHD mice, while not exerting antiviral effector functions on their own. As an interesting new information, this ‘helper’ effect corresponded to an enhanced early tissue infiltration by the CD8 T cells, indicating that CD4 T cell help is beneficial already at an early stage of immunotherapy and not only at late stages.
In the current version of the new model, the adoptive transfer of a single dose of human T cells resulted in epitope-specific tissue infiltration and tissue persistence of T cells within NIF structures for at least 11 days, and in a reduction of virus spread associated with confinement of the infection within NIF. At a very high dose of intraplantar infection, however, the survival benefit was insignificant, whereas at a moderate initial virus dose the survival benefit, though now statistically highly significant, did not result in cured long-term survivors, which corresponded to absence of tissue-infiltrating progeny of transferred T cells and relapse of virus spread at the time of onset of mortality.
For the interpretation of the relevance of only delayed but not prevented mortality, several aspects in which the model by design differs from the clinical correlate of HSCT-associated HCMV reactivation need to be considered: (i) in the model the virus doses used are much higher than to be expected in a clinical setting where acute primary infection of a seronegative HSCT recipient (R-) is a rare event, while reactivation in a seropositive, latently infected recipient (R+) likely originates from very few cells, if not just from one single cell. mCMV latency models indicated rare and stochastic reactivation events under immunocompromised conditions in vivo [94,95] as well as after tissue explantation ([96,97], reviewed in [41,42]). Indeed, it is clinical routine to start antiviral therapy, not just immunotherapy but also therapy with antiviral drugs, upon first detection of viral DNA by highly-sensitive PCR monitoring when the load of infectious virions is still minimal and mostly undetectable. This strategy is known as pre-emptive therapy, and has recently been reviewed in a clinical article by Seo and Boeckh [98]. Along the same line of argument, in approved HCMV vaccine trials, minimal doses of challenge virus (as low as 10 infectious units) were actually used to clinically evaluate vaccination efficacy (reviewed in [99]), a fact that not many are aware of. (ii) the model, by design, measures immediate antiviral effector functions of the transferred cells, as the NSG/HHD host does not support longevity and expansion of transferred cells due to multiple genetic deficiency in common γ-chain-dependent interleukin signaling, and (iii) in the clinical situation of pre-emptive immunotherapy, and likewise also of pre-emptive antiviral drug therapy, of HSCT-associated HCMV reactivation, the medical demand is only to bridge the critical phase of transient immunodeficiency until reconstitution of intrinsic host immunity in consequence of HSCT takes over. Such a temporary, transferred protection likely requires much lower cell numbers [63]. In contrast, in the model, as it currently stands, the NSG/HHD host is constitutively, and thus enduringly, immunodeficient so that even a single infectious unit of a ‘therapy escapee virus’ eventually causes death by viral tissue pathology. With these arguments in mind, the model provides a rigorous ‘stress test’ for the quality of adoptively transferred antiviral T cells, and the prolongation of host survival observed in this rigorous model predicts a good functionality of the tested human T cells in the less demanding clinical reality of low-dose HCMV reactivation followed by reconstitution of intrinsic antiviral immunity.
We wish to emphasize that the here introduced model should be understood as a basic module of a modular model system that is open to be developed further by ourselves and, hopefully, joined by other investigators. An obvious next step could be to improve the longevity and promote clonal expansion of adoptively transferred T cells by prolonged substitution with IL-2 and IL-7, and to include also IL-15 [100]. Another option for adaptation of the model to clinical needs could be the transfer of defined T-cell subsets with stem cell properties [101]. Finally, and most obviously, NSG/HHD adoptive transfer recipients could be further ‘humanized’ by human CD34+ stem-cell transplantation to reconstitute them with human immune system for the priming of intrinsic antiviral effector and helper cells [102,103] that take over for enduring viral control after the transient protection by adoptive immunotherapy has prevented life-threatening early-onset CMV disease.
In conclusion, we introduce here a promising mouse model of systemic CMV infection that allows the direct analysis of antiviral activity of HCMV-specific T-cell products in terms of infection control in functional organs and survival benefit. The model has best prospects to stimulate future research aimed at optimizing it for clinical needs, and has great potential to expedite the development of improved adoptive cell transfer as well as vaccine strategies against HCMV infection.
Material and Methods
Mice, cells, and viruses
C57BL/6 and NOD.Cg-Prkdcscid Il2rgtm1Wjl Tg(HLA-A/H2-D/B2M)1Dvs/SzJ (NSG/HHD) [27] mice were bred and maintained under SPF conditions in the Central Laboratory Animal Facilities at the University Medical Center Mainz and at the University Hospital of Regensburg. NSG/HHD were purchased from Jackson Laboratory (Bar Harbor, ME, USA). Mice were sacrificed by CO2 inhalation or cervical dislocation. Primary mouse embryonic fibroblasts (MEF) from C57BL/6 and NSG/HHD mice were generated by standard methods [104] and maintained in minimal essential medium (MEM) supplemented with 10% fetal calf serum. HLA-A2.1+ primary human foreskin fibroblasts (HFF) were grown in MEM supplemented with 10% fetal calf serum, 2 mM l-glutamine, 50 mg/L gentamicin and 0.5 ng/ml basic fibroblast growth factor (Life Technologies, Darmstadt, Germany) [66]. Human CD4 and CD8 T cells were isolated from buffy coat products of either HLA-A2.1+ CMV-seronegative or—seropositive healthy donors using immunomagnetic bead technology (Miltenyi Biotec, Bergisch Gladbach, Germany). Bacterial artificial chromosome (BAC)-derived virus mCMV-Δm157 [105] as used as non-chimeric reference virus. Virus stocks of mCMV-Δm157, mCMV-WT.BAC [106], and mCMV-Δm04/m06/m152 (mCMV-ΔvRAP) [50,51] were prepared from infected C57BL/6 MEF by sucrose-gradient ultracentrifugation as described [107]. HCMV immune evasion gene (US2,3,6,11) deletion mutant RV-KB6 and the combined US2,3,6,11, and pp65/UL83 deletion mutant RV-KB15 were described previously ([66] and [16], respectively).
Ethics statement
Animal research protocols of the University Medical Center Mainz were approved by the ethics committee of the Landesuntersuchungsamt Rheinland-Pfalz, permission numbers 23177-07/G09-1-004 and 23177-07/G11-1-004, according to German Federal Law §8 Abs. 1 TierSchG (animal protection law). Human blood cells were isolated from buffy coat products of healthy donors after written informed consent and approval by the ethics committee of the Landesärztekammer Rheinland-Pfalz and University Hospital of Regensburg (permission number 837.149.10 and 13-101-0240, respectively) and performed according to the Declaration of Helsinki.
Insertion of the HCMV epitope pp65495-503 NLVPMVATV into the mCMV genome
For generating mCMV-NLV, 69 nucleotides of HCMV ORF UL83 (n119,567-n119,499; GenBank accession no. X17403) were integrated at nucleotide position n186,511 into ORFm128 of the mCMV genome (GenBank accession no. NC_004065). These nucleotides code for 23 amino acids including the NLV epitope and its natural flanking regions. The mutagenesis was performed essentially as previously described [50]. A linear PCR fragment containing a kanamycin resistance gene flanked by two FRT sites and viral homologies of ORFm128 was generated by PCR with primers m128_NLV_for (5′-acg tcg ggc aga aag ctg ggt tat ctc gac gtg gcg gag aag atc ctg gcc cgc aac ctg gtg ccc atg gtg gct acg gtt cag ggt cag aat ctg aag tac cag gaa ttc agg acg acg acg aca agt aa-3`) and m128_NLV_rev (5′-gga tca cgc cga gaa cct cga ggg gac cgt tgc aca tgg ggt att cct tgc gca gca gga aca ctt aac ggc tga-3′) and plasmid pKD46 [108]. This fragment was inserted into the BAC plasmid pΔm157 [105] by homologous recombination in E. coli. After subsequent FLP-mediated excision of the resistance gene, the correct nucleotide insertion was verified by sequencing (GATC, Freiburg, Germany). The generation of mCMV-NLVAla was performed as described above except using primer m128_NLVAla_for (5′-acg tcg ggc aga aag ctg ggt tat ctc gac gtg gcg gag aag atc ctg gcc cgc aac ctg gtg ccc atg gtg gct acg gca cag ggt cag aat ctg aag tac cag gaa ttc agg acg acg acg aca agt aa-3`) instead of m128_NLV_for. Recombinant CMVs were reconstituted by transfection of purified BAC DNA into C57BL/6 MEF, and high titer virus stocks were purified as described [107].
Flow cytometry, antibodies, and peptides
Flow cytometry was performed on FACS Canto II or FACS LSR II (BD Biosciences, Heidelberg, Germany). Fluorochrome-labeled monoclonal Antibodies (mAb) were anti-mouse H-2Kb (clone AF6-88.5), CD62L-PE-Cy7 (clone MEL-14), CD8-FITC (clone 53–6.7), anti-human CD8-PerCP (clone SK1), CD8-APC (clone RPA-T8), CD4-APC (clone RPA-T4), CD28-FITC (clone CD28.2), CD95-PE (clone DX2), CD45RA-PE (clone HI100), CD45RO-PE (clone UCHL1), CD62L-FITC (clone DREG-56), HLA-A2.1-PE (clone BB7.2) (all BD Biosciences), anti-mouse CD44-PB (clone IM7) (BioLegend, San Diego, CA, USA), and anti-human CCR7-FITC (clone 150503) (R&D Systems, Minneapolis, MN, USA). Intracellular staining of MEF cells was performed with m164/gp36.5 antiserum [51], followed by Alexa Fluor 488-conjugated mAb goat anti-rabbit IgG (Life Technologies, Darmstadt, Germany). Fluorochrome-labeled HLA-A2.1/NLV tetramer was synthesized by Beckman Coulter (Krefeld, Germany). Peptides were synthesized by PSL (Heidelberg, Germany). Analyses were performed with software FlowJo 7.6.5 (Tree Star, Ashland, OR, USA).
T-cell stimulation and assays
Human NLV-specific CD8 T-cell lines were expanded from purified CD8 T cells of HLA-A2.1+ HCMV-seropositive healthy donors by stimulation with pp65 (UL83)495-503 NLV peptide (10−6 M)-loaded and irradiated (35 Gy) autologous peripheral blood mononuclear cells (PBMC) over 2 weeks at the T cell-to-PBMC ratio of 1 : 1 in AIM-V medium (Life Technologies). AIM-V was supplemented with 10% human serum, recombinant human interleukin (rhIL)-2 (50 IU/mL; Proleukin, San Diego, CA, USA), rhIL-7, and rhIL-15 (each 5 ng/mL; R&D Systems) (AIM-Vcytokine). A murine NLV-specific CD8 T-cell line was generated and weekly restimulated as previously described [38]. MEF were infected with mCMV under conditions of centrifugal enhancement of infectivity [104]. HFF were infected with HCMV-RVKB6 and—RVKB15 for 24h at an MOI of 5. Standard 4h [51Cr]-release and 20h interferon (IFN)-γ ELISpot-assays were performed in duplicates as reported [109,110]. Dose-escalating equilibrium tetramer binding data were plotted in Scatchard analysis of mean fluorescence intensity (MFI)/concentration of tetramer against MFI. The dissociation constant KD equals -1/slope [44,45].
TCR genes and retroviral transduction
TCRNLV is a codon-optimized and affinity fine-tuned variant of the previously described TCR AV18/BV13 [16,111]. A short self-cleaving F2A sequence [112] was used to link the N-terminus of TCRα chain to the C-terminus of TCRβ chain by ligation PCR technology [113]. The antisense oligonucleotide sequence was 5’-atg gct atg gtg aag cgg aag gac ttc gtg aaa caa acg ttg aat ttt gac ctt ctc aag ttg gcc gga gac gtg gag tcc aac ccc ggg cct atg gag aag aac ccc ctg gcc gcc ccc-3’. The coding sequence of the TCRβ-F2A-TCRα gene construct was inserted into the multiple-cloning-site of the drug-selectable retroviral vector pMX (BioCat, Heidelberg, Germany). Purified CD4 and CD8 T cells from HCMV-seronegative donors were pre-stimulated with anti-CD3/CD28 Dynabeads (Life Technologies) in AIM-V medium (supplemented with 100 IU/mL rhIL-2) and then retrovirally transduced with TCRNLV as previously described [113]. Following drug-selection on TCRNLV+ cells, T cells were expanded in vitro for a period of 7-12d, using anti-CD3/CD28 beads (3–5 μL/106 cells) in AIM-Vcytokine, prior to their adoptive transfer.
Quantitation of tissue infiltration and in vivo antiviral function of HCMV epitope-specific T cells
Eight - to 10-week-old NSG/HHD mice were sublethally (2 Gy) γ-irradiated for cell homing conditioning, followed by intraplantar infection with 1x105 PFU of the indicated viruses. Subsequently, a single dose of up to 1x107 human or murine T cells was injected intravenously. Human T cells were co-injected with rhIL-2 (1000 IU/mouse) and FcIL-7 (20 μg/mouse; Merck, Darmstadt, Germany). At indicated times post-infection, virus replication in spleen, lungs, and liver was assessed by quantitation of infectivity from the respective organ homogenates in a virus plaque assay (PFU assay) [107]. Infected cells and T cells in liver tissue sections were visualized simultaneously in their microanatomical context, specifically in nodular inflammatory foci (NIF), and quantitated by two-color immunohistochemistry (2C-IHC) specific for the intranuclear viral IE1 protein and a conserved CD3ε epitope [107]. For testing maintenance of transferred human T cells, flow cytometric analysis was performed on splenocytes retrieved by standard methods and on non-parenchymal liver cells isolated as described [97].
2-color immunohistochemical (2C-IHC) analyses of infection, differentiated by cell type, and of apoptosis
To detect any type of mCMV-infected cells, one-color IHC specific for the intranuclear viral protein IE1 was performed on liver tissue sections as described in detail elsewhere [107]. For differentiating infected cells by cell type, 2C-IHCs were performed by combining IE1-specific labeling with the labeling of cell type-specific markers, such as CD31 for the identification of endothelial cells (EC) or F4/80 (Ly71) antigen for the identification of macrophages (MΦ), essentially as described recently for a 3C-IHC analysis that has included the same markers [36]. 2C-IHC specific for IE1 and active caspase 3 was described previously [114] and applied with modifications. In brief, intranuclear IE1 protein was labeled with monoclonal Ab CROMA 101 and stained in red with alkaline phosphatase-1-conjugated polyclonal goat anti-mouse IgG (AbD Serotec, Puchheim, Germany) and fuchsin substrate-chromogen kit 2 (Dako-Cytomation, Hamburg, Germany). Active caspase 3 was detected with rabbit anti-active caspase 3 IgG (Biovision, Milpitas, CA, USA) and stained in brown by using the ImmPRESS reagent: anti-rabbit-Ig-peroxidase (Vector Laboratories, Peterborough, UK) with 3,3’-diaminobenzidine (DAB) as substrate. A light blue counterstaining was achieved with hematoxylin.
Statistical analyses and calculation of viral doubling times in host tissues
Statistical significance of differences between two independent data sets was evaluated by two-sided unpaired t test with Welch’s correction of unequal variances. Comparison of survival curves was performed with log-rank test and Gehan-Wilcoxon test. Differences were considered statistically significant for P values of <0.05. Viral doubling times (vDT = log2/a) and the corresponding 95% confidence intervals were calculated by linear regression analysis from the slopes a of log-linear growth curves [36]. All analyses were performed with Graphpad Prism 6.04 (GraphPad Software, San Diego, CA, USA).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Boeckh M, Leisenring W, Riddell SR, Bowden RA, Huang M, Myerson D et al. (2003) Late cytomegalovirus disease and mortality in recipients of allogeneic hematopoietic stem cell transplants: importance of viral load and T-cell immunity. Blood 101 : 407–414. 12393659
2. Ozdemir E, Saliba RM, Champlin RE, Couriel DR, Giralt SA, de Lima M et al. (2007) Risk factors associated with late cytomegalovirus reactivation after allogeneic stem cell transplantation for hematological malignancies. Bone Marrow Transplant 40 : 125–136. 17530009
3. Reddehase MJ, Weiland F, Munch K, Jonjic S, Luske A, Koszinowski UH (1985) Interstitial murine cytomegalovirus pneumonia after irradiation: characterization of cells that limit viral replication during established infection of the lungs. J Virol 55 : 264–273. 2991554
4. Steffens HP, Kurz S, Holtappels R, Reddehase MJ (1998) Preemptive CD8 T-cell immunotherapy of acute cytomegalovirus infection prevents lethal disease, limits the burden of latent viral genomes, and reduces the risk of virus recurrence. J Virol 72 : 1797–1804. 9499030
5. Holtappels R, Podlech J, Grzimek NK, Thomas D, Pahl-Seibert MF, Reddehase MJ (2001) Experimental preemptive immunotherapy of murine cytomegalovirus disease with CD8 T-cell lines specific for ppM83 and pM84, the two homologs of human cytomegalovirus tegument protein ppUL83 (pp65). J Virol 75 : 6584–6600. 11413326
6. Pahl-Seibert M, Juelch M, Podlech J, Thomas D, Deegen P, Reddehase MJ et al. (2005) Highly protective in vivo function of cytomegalovirus IE1 epitope-specific memory CD8 T cells purified by T-cell receptor-based cell sorting. J Virol 79 : 5400–5413. 15827154
7. Holtappels R, Böhm V, Podlech J, Reddehase MJ (2008) CD8 T-cell-based immunotherapy of cytomegalovirus infection: "proof of concept" provided by the murine model. Med Microbiol Immunol 197 : 125–134. doi: 10.1007/s00430-008-0093-2 18343947
8. Ebert S, Podlech J, Gillert-Marien D, Gergely KM, Büttner JK, Fink A et al. (2012) Parameters determining the efficacy of adoptive CD8 T-cell therapy of cytomegalovirus infection. Med Microbiol Immunol 201 : 527–539. doi: 10.1007/s00430-012-0258-x 22972232
9. Holtappels R, Ebert S, Podlech J, Fink A, Böhm V, Lemmermann NA et al. (2013) Murine model for cytoimmunotherapy of CMV disease after haematopoietic cell transplantation. In: Reddehase MJ, ed. Cytomegaloviruses: From Molecular Pathogenesis to Intervention. Vol 2. Norfolk, UK: Caister Academic Press. pp. 354–381.
10. Riddell SR, Watanabe KS, Goodrich JM, Li CR, Agha ME, Greenberg PD (1992) Restoration of viral immunity in immunodeficient humans by the adoptive transfer of T cell clones. Science 257 : 238–241. 1352912
11. Cobbold M, Khan N, Pourgheysari B, Tauro S, McDonald D, Osman H et al. (2005) Adoptive transfer of cytomegalovirus-specific CTL to stem cell transplant patients after selection by HLA-peptide tetramers. J Exp Med 202 : 379–386. 16061727
12. Feuchtinger T, Opherk K, Bethge WA, Topp MS, Schuster FR, Weissinger EM et al. (2010) Adoptive transfer of pp65-specific T cells for the treatment of chemorefractory cytomegalovirus disease or reactivation after haploidentical and matched unrelated stem cell transplantation. Blood 116 : 4360–4367. doi: 10.1182/blood-2010-01-262089 20625005
13. Thomas S, Herr W (2011) Natural and adoptive T-cell immunity against herpes family viruses after allogeneic hematopoietic stem cell transplantation. Immunotherapy 3 : 771–788. doi: 10.2217/imt.11.47 21668314
14. Sellar RS, Peggs KS (2012) Therapeutic strategies for the prevention and treatment of cytomegalovirus infection. Expert Opin Biol Ther 12 : 1161–1172. doi: 10.1517/14712598.2012.693471 22650422
15. Schub A, Schuster IG, Hammerschmidt W, Moosmann A (2009) CMV-specific TCR-transgenic T cells for immunotherapy. J Immunol 183 : 6819–6830. doi: 10.4049/jimmunol.0902233 19864595
16. Thomas S, Klobuch S, Besold K, Plachter B, Dorrie J, Schaft N et al. (2012) Strong and sustained effector function of memory - versus naive-derived T cells upon T-cell receptor RNA transfer: implications for cellular therapy. Eur J Immunol 42 : 3442–3453. doi: 10.1002/eji.201242666 22930221
17. Smith MG (1956) Propagation in tissue cultures of a cytopathogenic virus from human salivary gland virus (SGV) disease. Proc Soc Exp Biol Med 92 : 424–430. 13350368
18. Brune W (2013) Molecular basis of cytomegalovirus host species specificity. In: Reddehase MJ, ed. Cytomegaloviruses: from molecular pathogenesis to intervention. Vol 1. Norfolk, UK: Caister Academic Press. pp. 322–329.
19. Mocarski ES, Bonyhadi M, Salimi S, McCune JM, Kaneshima H (1993) Human cytomegalovirus in a SCID-hu mouse: thymic epithelial cells are prominent targets of viral replication. Proc Natl Acad Sci USA 90 : 104–108. 7678330
20. Brown JM, Kaneshima H, Mocarski ES (1995) Dramatic interstrain differences in the replication of human cytomegalovirus in SCID-hu mice. J Infect Dis 171 : 1599–1603. 7769298
21. Bidanset DJ, Rybak RJ, Hartline CB, Kern ER (2001) Replication of human cytomegalovirus in severe combined immunodeficient mice implanted with human retinal tissue. J Infect Dis 184 : 192–195. 11424017
22. Kern ER (2006) Pivotal role of animal models in the development of new therapies for cytomegalovirus infections. Antiviral Res 71 : 164–171. 16828175
23. Kawahara T, Lisboa LF, Cader S, Douglas DN, Nourbakhsh M, Pu CH et al. (2013) Human cytomegalovirus infection in humanized liver chimeric mice. Hepatol Res 43 : 679–684. doi: 10.1111/j.1872-034X.2012.01116.x 23442000
24. Smith MS, Streblow DN, Caposio P, Nelson JA (2013) Humanized mouse models of cytomegalovirus pathogenesis and latency. In Reddehase MJ, ed. Cytomegaloviruses: from molecular pathogenesis to intervention. Vol 1. Norfolk, UK: Caister Academic Press. pp. 417–436.
25. Smith MS, Goldman DC, Bailey AS, Pfaffle DL, Kreklywich CN, Spencer DB et al. (2010) Granulocyte-colony stimulating factor reactivates human cytomegalovirus in a latently infected humanized mouse model. Cell Host Microbe 8 : 284–291. doi: 10.1016/j.chom.2010.08.001 20833379
26. Wills MR, Carmichael AJ, Mynard K, Jin X, Weekes MP, Plachter B et al. (1996) The human cytotoxic T-lymphocyte (CTL) response to cytomegalovirus is dominated by structural protein pp65: frequency, specificity, and T-cell receptor usage of pp65-specific CTL. J Virol 70 : 7569–7579. 8892876
27. Shultz LD, Saito Y, Najima Y, Tanaka S, Ochi T, Tomizawa M et al. (2010) Generation of functional human T-cell subsets with HLA-restricted immune responses in HLA class I expressing NOD/SCID/IL2r gamma(null) humanized mice. Proc Natl Acad Sci USA 107 : 13022–13027. doi: 10.1073/pnas.1000475107 20615947
28. Giri JG, Ahdieh M, Eisenman J, Shanebeck K, Grabstein K, Kumaki S et al. (1994) Utilization of the beta and gamma chains of the IL-2 receptor by the novel cytokine IL-15. EMBO J 13 : 2822–2830. 8026467
29. Anderson DM, Kumaki S, Ahdieh M, Bertles J, Tometsko M, Loomis A et al. (1995) Functional characterization of the human interleukin-15 receptor alpha chain and close linkage of IL15RA and IL2RA genes. J Biol Chem 270 : 29862–29869. 8530383
30. Vidal S, Krmpotić A, Pyzik M, Jonjić S (2013) Innate immunity to cytomegalovirus in the murine model. In: Reddehase MJ, ed. Cytomegaloviruses: from molecular pathogenesis to intervention. Vol 2. Norfolk, UK: Caister Academic Press. pp. 192–214.
31. Lemmermann NA, Kropp KA, Seckert CK, Grzimek, Natascha K A, Reddehase MJ (2011) Reverse genetics modification of cytomegalovirus antigenicity and immunogenicity by CD8 T-cell epitope deletion and insertion. J Biomed Biotechnol 2011 : 812742. doi: 10.1155/2011/812742 21253509
32. Ebert S, Lemmermann NA, Thomas D, Renzaho A, Reddehase MJ, Holtappels R (2012) Immune control in the absence of immunodominant epitopes: implications for immunotherapy of cytomegalovirus infection with antiviral CD8 T cells. Med Microbiol Immunol 201 : 541–550. doi: 10.1007/s00430-012-0268-8 22976556
33. Messerle M, Keil GM, Koszinowski UH (1991) Structure and expression of murine cytomegalovirus immediate-early gene 2. J Virol 65 : 1638–1643. 1847480
34. Cardin RD, Abenes GB, Stoddart CA, Mocarski ES (1995) Murine cytomegalovirus IE2, an activator of gene expression, is dispensable for growth and latency in mice. Virology 209 : 236–241. 7747475
35. Grzimek NK, Dreis D, Schmalz S, Reddehase MJ (2001) Random, asynchronous, and asymmetric transcriptional activity of enhancer-flanking major immediate-early genes ie1/3 and ie2 during murine cytomegalovirus latency in the lungs. J Virol 75 : 2692–2705. 11222693
36. Lemmermann NA, Krmpotic A, Podlech J, Brizic I, Prager A, Adler H et al. (2015) Non-redundant and redundant roles of cytomegalovirus gH/gL complexes in host organ entry and intra-tissue spread. PLoS Pathog 11: e1004640. doi: 10.1371/journal.ppat.1004640 25659098
37. Podlech J, Reddehase MJ, Adler B, Lemmermann NA (2015) Principles for studying in vivo attenuation of virus mutants: defining the role of the cytomegalovirus gH/gL/gO complex as a paradigm. Med Microbiol Immunol. 2015 Mar 18. [Epub ahead of print]
38. Besold K, Frankenberg N, Pepperl-Klindworth S, Kuball J, Theobald M, Hahn G et al. (2007) Processing and MHC class I presentation of human cytomegalovirus pp65-derived peptides persist despite gpUS2-11-mediated immune evasion. J Gen Virol 88 : 1429–1439. 17412970
39. Obar JJ, Lefrancois L (2010) Memory CD8+ T cell differentiation. Ann N Y Acad Sci 1183 : 251–266. doi: 10.1111/j.1749-6632.2009.05126.x 20146720
40. Obar JJ, Lefrancois L (2010) Early events governing memory CD8+ T-cell differentiation. Int Immunol 22 : 619–625. doi: 10.1093/intimm/dxq053 20504887
41. Seckert CK, Griessl M, Büttner JK, Scheller S, Simon CO, Kropp KA et al. (2012) Viral latency drives 'memory inflation': a unifying hypothesis linking two hallmarks of cytomegalovirus infection. Med Microbiol Immunol 201 : 551–566. doi: 10.1007/s00430-012-0273-y 22991040
42. Seckert CK, Griessl M, Büttner JK, Freitag K, Lemmermann NA, Hummel MA et al. (2013) Immune surveillance of cytomegalovirus latency and reactivation in murine models: link to ‘memory inflation’. In: Reddehase MJ, ed. Cytomegaloviruses: from molecular pathogenesis to intervention. Vol 1. Norfolk, UK: Caister Academic Press. pp. 374–416.
43. Zhou F (2009) Molecular mechanisms of IFN-gamma to up-regulate MHC class I antigen processing and presentation. Int Rev Immunol 28 : 239–260. doi: 10.1080/08830180902978120 19811323
44. Holmberg K, Mariathasan S, Ohteki T, Ohashi PS, Gascoigne, Nicholas R J (2003) TCR binding kinetics measured with MHC class I tetramers reveal a positive selecting peptide with relatively high affinity for TCR. J Immunol 171 : 2427–2434. 12928390
45. Kuball J, Schmitz FW, Voss R, Ferreira EA, Engel R, Guillaume P et al. (2005) Cooperation of human tumor-reactive CD4+ and CD8+ T cells after redirection of their specificity by a high-affinity p53A2.1-specific TCR. Immunity 22 : 117–129. 15664164
46. Nauerth M, Weissbrich B, Knall R, Franz T, Dossinger G, Bet J et al. (2013) TCR-ligand koff rate correlates with the protective capacity of antigen-specific CD8+ T cells for adoptive transfer. Sci Transl Med 5 : 192ra87. doi: 10.1126/scitranslmed.3005958 23825303
47. Hengel H, Lucin P, Jonjic S, Ruppert T, Koszinowski UH (1994) Restoration of cytomegalovirus antigen presentation by gamma interferon combats viral escape. J Virol 68 : 289–297. 8254740
48. Fink A, Lemmermann NA, Gillert-Marien D, Thomas D, Freitag K, Böhm V et al. (2012) Antigen presentation under the influence of 'immune evasion' proteins and its modulation by interferon-gamma: implications for immunotherapy of cytomegalovirus infection with antiviral CD8 T cells. Med Microbiol Immunol 201 : 513–525. doi: 10.1007/s00430-012-0256-z 22961126
49. Lemmermann NA, Fink A, Podlech J, Ebert S, Wilhelmi V, Böhm V et al. (2012) Murine cytomegalovirus immune evasion proteins operative in the MHC class I pathway of antigen processing and presentation: state of knowledge, revisions, and questions. Med Microbiol Immunol 201 : 497–512. doi: 10.1007/s00430-012-0257-y 22961127
50. Wagner M, Gutermann A, Podlech J, Reddehase MJ, Koszinowski UH (2002) Major histocompatibility complex class I allele-specific cooperative and competitive interactions between immune evasion proteins of cytomegalovirus. J Exp Med 196 : 805–816. 12235213
51. Holtappels R, Gillert-Marien D, Thomas D, Podlech J, Deegen P, Herter S et al. (2006) Cytomegalovirus encodes a positive regulator of antigen presentation. J Virol 80 : 7613–7624. 16840340
52. Alterio de Goss M, Holtappels R, Steffens HP, Podlech J, Angele P, Dreher L et al. (1998) Control of cytomegalovirus in bone marrow transplantation chimeras lacking the prevailing antigen-presenting molecule in recipient tissues rests primarily on recipient-derived CD8 T cells. J Virol 72 : 7733–7744. 9733809
53. Podlech J, Holtappels R, Pahl-Seibert MF, Steffens HP, Reddehase MJ (2000) Murine model of interstitial cytomegalovirus pneumonia in syngeneic bone marrow transplantation: persistence of protective pulmonary CD8-T-cell infiltrates after clearance of acute infection. J Virol 74 : 7496–7507. 10906203
54. Böhm V, Podlech J, Thomas D, Deegen P, Pahl-Seibert M, Lemmermann NA et al. (2008) Epitope-specific in vivo protection against cytomegalovirus disease by CD8 T cells in the murine model of preemptive immunotherapy. Med Microbiol Immunol 197 : 135–144. doi: 10.1007/s00430-008-0092-3 18340461
55. Sacher T, Podlech J, Mohr CA, Jordan S, Ruzsics Z, Reddehase MJ et al. (2008) The major virus-producing cell type during murine cytomegalovirus infection, the hepatocyte, is not the source of virus dissemination in the host. Cell Host Microbe 3 : 263–272. doi: 10.1016/j.chom.2008.02.014 18407069
56. Stahl FR, Heller K, Halle S, Keyser KA, Busche A, Marquardt A et al. (2013) Nodular inflammatory foci are sites of T cell priming and control of murine cytomegalovirus infection in the neonatal lung. PLoS Pathog 9: e1003828. doi: 10.1371/journal.ppat.1003828 24348257
57. Ebert S, Becker M, Lemmermann NA, Büttner JK, Michel A, Taube C et al. (2014) Mast cells expedite control of pulmonary murine cytomegalovirus infection by enhancing the recruitment of protective CD8 T cells to the lungs. PLoS Pathog 10: e1004100. doi: 10.1371/journal.ppat.1004100 24763809
58. Boeckh M, Nichols WG (2004) The impact of cytomegalovirus serostatus of donor and recipient before hematopoietic stem cell transplantation in the era of antiviral prophylaxis and preemptive therapy. Blood 103 : 2003–2008. 14644993
59. Zhou W, Longmate J, Lacey SF, Palmer JM, Gallez-Hawkins G, Thao L et al. (2009) Impact of donor CMV status on viral infection and reconstitution of multifunction CMV-specific T cells in CMV-positive transplant recipients. Blood 113 : 6465–6476. doi: 10.1182/blood-2009-02-203307 19369230
60. Borchers S, Luther S, Lips U, Hahn N, Kontsendorn J, Stadler M et al. (2011) Tetramer monitoring to assess risk factors for recurrent cytomegalovirus reactivation and reconstitution of antiviral immunity post allogeneic hematopoietic stem cell transplantation. Transpl Infect Dis 13 : 222–236. doi: 10.1111/j.1399-3062.2011.00626.x 21585633
61. Ugarte-Torres A, Hoegh-Petersen M, Liu Y, Zhou F, Williamson TS, Quinlan D et al. (2011) Donor serostatus has an impact on cytomegalovirus-specific immunity, cytomegaloviral disease incidence, and survival in seropositive hematopoietic cell transplant recipients. Biol Blood Marrow Transplant 17 : 574–585. doi: 10.1016/j.bbmt.2010.07.020 20688181
62. Eiz-Vesper B, Maecker-Kolhoff B, Blasczyk R (2012) Adoptive T-cell immunotherapy from third-party donors: characterization of donors and set up of a T-cell donor registry. Front Immunol 3 : 410. doi: 10.3389/fimmu.2012.00410 23372567
63. Stemberger C, Graef P, Odendahl M, Albrecht J, Dossinger G, Anderl F et al. (2014) Lowest numbers of primary CD8(+) T cells can reconstitute protective immunity upon adoptive immunotherapy. Blood 124 : 628–637. doi: 10.1182/blood-2013-12-547349 24855206
64. Kershaw MH, Westwood JA, Darcy PK (2013) Gene-engineered T cells for cancer therapy. Nat Rev Cancer 13 : 525–541. doi: 10.1038/nrc3565 23880905
65. Rosenberg SA, Restifo NP (2015) Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348 : 62–68. doi: 10.1126/science.aaa4967 25838374
66. Besold K, Wills M, Plachter B (2009) Immune evasion proteins gpUS2 and gpUS11 of human cytomegalovirus incompletely protect infected cells from CD8 T cell recognition. Virology 391 : 5–19. doi: 10.1016/j.virol.2009.06.004 19570562
67. Li Y, Kurlander RJ (2010) Comparison of anti-CD3 and anti-CD28-coated beads with soluble anti-CD3 for expanding human T cells: differing impact on CD8 T cell phenotype and responsiveness to restimulation. J Transl Med 8 : 104. doi: 10.1186/1479-5876-8-104 20977748
68. Kunert A, Straetemans T, Govers C, Lamers C, Mathijssen R, Sleijfer S et al. (2013) TCR-Engineered T Cells Meet New Challenges to Treat Solid Tumors: Choice of Antigen, T Cell Fitness, and Sensitization of Tumor Milieu. Front Immunol 4 : 363. doi: 10.3389/fimmu.2013.00363 24265631
69. Barrett DM, Singh N, Liu X, Jiang S, June CH, Grupp SA et al. (2014) Relation of clinical culture method to T-cell memory status and efficacy in xenograft models of adoptive immunotherapy. Cytotherapy 16 : 619–630. doi: 10.1016/j.jcyt.2013.10.013 24439255
70. Cieri N, Mastaglio S, Oliveira G, Casucci M, Bondanza A, Bonini C (2014) Adoptive immunotherapy with genetically modified lymphocytes in allogeneic stem cell transplantation. Immunol Rev 257 : 165–180. doi: 10.1111/imr.12130 24329796
71. Ghazal P, Messerle M, Osborn K, Angulo A (2003) An essential role of the enhancer for murine cytomegalovirus in vivo growth and pathogenesis. J Virol 77 : 3217–3228. 12584345
72. Handke W, Krause E, Brune W (2012) Live or let die: manipulation of cellular suicide programs by murine cytomegalovirus. Med Microbiol Immunol 201 : 475–486. doi: 10.1007/s00430-012-0264-z 22965170
73. McCormick AL, Macarski ES (2013) Cell death pathways controlled by cytomegalovirus. In: Reddehase MJ, ed. Cytomegaloviruses: from molecular pathogenesis to intervention. Vol 1. Norfolk, UK: Caister Academic Press. pp. 264–277.
74. Arapovic J, Arapovic M, Golemac M, Traven L, Tomac J, Rumora D et al. (2015) The specific NK cell response in concert with perforin prevents CD8 T cell-mediated immunopathology after mouse cytomegalovirus infection. Med Microbiol Immunol. 2015 Mar 26. [Epub ahead of print]
75. Allen LB, Li SX, Arnett G, Toyer B, Shannon WM, Hollingshead MG (1992) Novel method for evaluating antiviral drugs against human cytomegalovirus in mice. Antimicrob Agents Chemother 36 : 206–208. 1317146
76. Pari GS, Netski D, St Jeor S, McCarthy D, Smith J, Georgio D et al. (1998) Generation of a nude mouse tumor model for in vivo replication of human cytomegalovirus. J Infect Dis 177 : 523–528. 9498427
77. Sylwester AW, Mitchell BL, Edgar JB, Taormina C, Pelte C, Ruchti F et al. (2005) Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J Exp Med 202 : 673–685. 16147978
78. Wills MR, Mason GM, Sissons JG (2013) Adaptive cellular immunity to human cytomegalovirus. In: Reddehase MJ, ed. Cytomegalovirus: from molecular pathogenesis to intervention. Vol 2. Norfolk, UK: Caister Academic Press. pp. 142–176.
79. Sissons JG, Wills MR (2015) How understanding immunology contributes to managing CMV disease in immunosuppressed patients: now and in future. Med Microbiol Immunol. 2015 Apr 21. [Epub ahead of print]
80. Reddehase MJ (2002) Antigens and immunoevasins: opponents in cytomegalovirus immune surveillance. Nat Rev Immunol 2 : 831–844. 12415307
81. Powers C, DeFilippis V, Malouli D, Fruh K (2008) Cytomegalovirus immune evasion. Curr Top Microbiol Immunol 325 : 333–359. 18637515
82. Lemmermann NA, Gergely K, Böhm V, Deegen P, Daubner T, Reddehase MJ (2010) Immune evasion proteins of murine cytomegalovirus preferentially affect cell surface display of recently generated peptide presentation complexes. J Virol 84 : 1221–1236. doi: 10.1128/JVI.02087-09 19906905
83. Jackson SE, Mason GM, Wills MR (2011) Human cytomegalovirus immunity and immune evasion. Virus Res 157 : 151–160. doi: 10.1016/j.virusres.2010.10.031 21056604
84. Lemmermann NA, Böhm V, Holtappels R, Reddehase MJ (2011) In vivo impact of cytomegalovirus evasion of CD8 T-cell immunity: facts and thoughts based on murine models. Virus Res 157 : 161–174. doi: 10.1016/j.virusres.2010.09.022 20933556
85. Holtappels R, Grzimek, Natascha K A, Simon CO, Thomas D, Dreis D, Reddehase MJ (2002) Processing and presentation of murine cytomegalovirus pORFm164-derived peptide in fibroblasts in the face of all viral immunosubversive early gene functions. J Virol 76 : 6044–6053. 12021337
86. Holtappels R, Thomas D, Reddehase MJ (2009) The efficacy of antigen processing is critical for protection against cytomegalovirus disease in the presence of viral immune evasion proteins. J Virol 83 : 9611–9615. doi: 10.1128/JVI.00936-09 19553308
87. Walter EA, Greenberg PD, Gilbert MJ, Finch RJ, Watanabe KS, Thomas ED et al. (1995) Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N Engl J Med 333 : 1038–1044. 7675046
88. Micklethwaite K, Hansen A, Foster A, Snape E, Antonenas V, Sartor M et al. (2007) Ex vivo expansion and prophylactic infusion of CMV-pp65 peptide-specific cytotoxic T-lymphocytes following allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 13 : 707–714. 17531781
89. Meij P, Jedema I, Zandvliet ML, van der Heiden, Pim L J, van de Meent, Marian, van Egmond, Esther H M et al. (2012) Effective treatment of refractory CMV reactivation after allogeneic stem cell transplantation with in vitro-generated CMV pp65-specific CD8+ T-cell lines. J Immunother 35 : 621–628. 22996368
90. Aubert G, Hassan-Walker AF, Madrigal JA, Emery VC, Morte C, Grace S et al. (2001) Cytomegalovirus-specific cellular immune responses and viremia in recipients of allogeneic stem cell transplants. J Infect Dis 184 : 955–963. 11574909
91. Einsele H, Roosnek E, Rufer N, Sinzger C, Riegler S, Loffler J et al. (2002) Infusion of cytomegalovirus (CMV)-specific T cells for the treatment of CMV infection not responding to antiviral chemotherapy. Blood 99 : 3916–3922. 12010789
92. Tu W, Chen S, Sharp M, Dekker C, Manganello AM, Tongson EC et al. (2004) Persistent and selective deficiency of CD4+ T cell immunity to cytomegalovirus in immunocompetent young children. J Immunol 172 : 3260–3267. 14978134
93. Peggs KS, Thomson K, Samuel E, Dyer G, Armoogum J, Chakraverty R et al. (2011) Directly selected cytomegalovirus-reactive donor T cells confer rapid and safe systemic reconstitution of virus-specific immunity following stem cell transplantation. Clin Infect Dis 52 : 49–57. doi: 10.1093/cid/ciq042 21148519
94. Reddehase MJ, Balthesen M, Rapp M, Jonjic S, Pavic I, Koszinowski UH (1994) The conditions of primary infection define the load of latent viral genome in organs and the risk of recurrent cytomegalovirus disease. J Exp Med 179 : 185–193. 8270864
95. Kurz SK, Reddehase MJ (1999) Patchwork pattern of transcriptional reactivation in the lungs indicates sequential checkpoints in the transition from murine cytomegalovirus latency to recurrence. J Virol 73 : 8612–8622. 10482614
96. Marquardt A, Halle S, Seckert CK, Lemmermann NA, Veres TZ, Braun A et al. (2011) Single cell detection of latent cytomegalovirus reactivation in host tissue. J Gen Virol 92 : 1279–1291. doi: 10.1099/vir.0.029827-0 21325477
97. Seckert CK, Renzaho A, Tervo H, Krause C, Deegen P, Kuhnapfel B et al. (2009) Liver sinusoidal endothelial cells are a site of murine cytomegalovirus latency and reactivation. J Virol 83 : 8869–8884. doi: 10.1128/JVI.00870-09 19535440
98. Seo S, Boeckh M (2013) Clinical Cytomegalovirus Research: Haematopoietic Cell Transplantation. In: Reddehase MJ, ed. Cytomegalovirus: From Molecular Pathogenesis to Intervention. Vol 2. Norfolk, UK: Caister Academic Press. pp. 337–353.
99. Plotkin S (2015) The history of vaccination against cytomegalovirus. Med Microbiol Immunol. 2015 Mar 20. [Epub ahead of print]
100. Wang X, Berger C, Wong CW, Forman SJ, Riddell SR, Jensen MC (2011) Engraftment of human central memory-derived effector CD8+ T cells in immunodeficient mice. Blood 117 : 1888–1898. doi: 10.1182/blood-2010-10-310599 21123821
101. Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF et al. (2011) A human memory T cell subset with stem cell-like properties. Nat Med 17 : 1290–1297. doi: 10.1038/nm.2446 21926977
102. Strowig T, Gurer C, Ploss A, Liu Y, Arrey F, Sashihara J et al. (2009) Priming of protective T cell responses against virus-induced tumors in mice with human immune system components. J Exp Med 206 : 1423–1434. doi: 10.1084/jem.20081720 19487422
103. Thomas S, Klobuch S, Sommer M, van Ewijk R, Theobald M, Meyer RG et al. (2014) Human CD8+ memory and EBV-specific T cells show low alloreactivity in vitro and in CD34+ stem cell-engrafted NOD/SCID/IL-2Rgammac null mice. Exp Hematol 42 : 28–38.e1–2. doi: 10.1016/j.exphem.2013.09.013 24120693
104. Podlech J, Holtappels R, Grzimek NK, Reddehase MJ (2002) Animal models: murine cytomegalovirus. In: Kaufmann SHE and Kabelitz D, eds. Methods in microbiology: immunology of infection. Vol. 32. Oxford, UK: Academic Press. pp. 493–525.
105. Bubic I, Wagner M, Krmpotic A, Saulig T, Kim S, Yokoyama WM et al. (2004) Gain of virulence caused by loss of a gene in murine cytomegalovirus. J Virol 78 : 7536–7544. 15220428
106. Wagner M, Jonjic S, Koszinowski UH, Messerle M (1999) Systematic excision of vector sequences from the BAC-cloned herpesvirus genome during virus reconstitution. J Virol 73 : 7056–7060. 10400809
107. Lemmermann NA, Podlech J, Seckert CK, Kropp KA, Grzimek NK, et al. (2010) CD8 T-Cell immunotherapy of cytomegalovirus disease in the murine model. In: Kaufmann SHE and Kabelitz D, eds. Methods in microbiology: immunology of infection. 2nd ed, Vol. 37. Oxford, UK: Academic Press. pp. 369–429.
108. Datsenko KA, Wanner BL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA 97 : 6640–6645. 10829079
109. Wehler TC, Nonn M, Brandt B, Britten CM, Grone M, Todorova M et al. (2007) Targeting the activation-induced antigen CD137 can selectively deplete alloreactive T cells from antileukemic and antitumor donor T-cell lines. Blood 109 : 365–373. 16931626
110. Dörrschuck A, Schmidt A, Schnurer E, Gluckmann M, Albrecht C, Wolfel C et al. (2004) CD8+ cytotoxic T lymphocytes isolated from allogeneic healthy donors recognize HLA class Ia/Ib-associated renal carcinoma antigens with ubiquitous or restricted tissue expression. Blood 104 : 2591–2599. 15231579
111. Heemskerk MH, Hagedoorn RS, van der Hoorn MA, van der Veken LT, Hoogeboom M, Kester MG et al. (2007) Efficiency of T-cell receptor expression in dual-specific T cells is controlled by the intrinsic qualities of the TCR chains within the TCR-CD3 complex. Blood 109 : 235–243. 16968899
112. Szymczak AL, Workman CJ, Wang Y, Vignali KM, Dilioglou S, Vanin EF et al. (2004) Correction of multi-gene deficiency in vivo using a single 'self-cleaving' 2A peptide-based retroviral vector. Nat Biotechnol 22 : 589–594. 15064769
113. Voss RH, Thomas S, Pfirschke C, Hauptrock B, Klobuch S, Kuball J et al. (2010) Coexpression of the T-cell receptor constant alpha domain triggers tumor reactivity of single-chain TCR-transduced human T cells. Blood 115 : 5154–5163. doi: 10.1182/blood-2009-11-254078 20378753
114. Cicin-Sain L, Ruzsics Z, Podlech J, Bubic I, Menard C, Jonjic S et al. (2008) Dominant-negative FADD rescues the in vivo fitness of a cytomegalovirus lacking an antiapoptotic viral gene. J Virol 82 : 2056–2064. 18094168
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 7
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- RNA Virus Reassortment: An Evolutionary Mechanism for Host Jumps and Immune Evasion
- Activation of TLR2 and TLR6 by Dengue NS1 Protein and Its Implications in the Immunopathogenesis of Dengue Virus Infection
- N-acetylglucosamine Regulates Virulence Properties in Microbial Pathogens
- Characterization of a Prefusion-Specific Antibody That Recognizes a Quaternary, Cleavage-Dependent Epitope on the RSV Fusion Glycoprotein
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy