
Ubiquilin 1 Promotes IFN-γ-Induced Xenophagy of
More people die from Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis (TB), than any other bacterial pathogen. It has long been appreciated that Mtb can survive and divide within macrophages, white blood cells that normally kill bacteria. Macrophages are able to partially control Mtb through a degradative process called autophagy. Autophagy is activated by the cytokine interferon-gamma (IFN-γ), which promotes control of Mtb infection. How the tubercle bacilli are targeted to the autophagy pathway remains unclear. Here we show that the human protein ubiquilin 1 can interact with Mtb surface proteins and associate with Mtb that are present in the host cell cytosol. We propose a model in which activating autophagy with IFN-γ promotes UBQLN1 recruitment to Mtb, which in turn leads to recruitment of the autophagy machinery, autophagy-mediated degradation of the bacteria, and activation of effector T cells. Since IFN-γ is critical in human control of Mtb, our study suggests that polymorphisms in ubiquilins, known to influence susceptibility to neurodegenerative illnesses, might also play a role in host defense against Mtb.
Published in the journal:
. PLoS Pathog 11(7): e32767. doi:10.1371/journal.ppat.1005076
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1005076
Summary
More people die from Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis (TB), than any other bacterial pathogen. It has long been appreciated that Mtb can survive and divide within macrophages, white blood cells that normally kill bacteria. Macrophages are able to partially control Mtb through a degradative process called autophagy. Autophagy is activated by the cytokine interferon-gamma (IFN-γ), which promotes control of Mtb infection. How the tubercle bacilli are targeted to the autophagy pathway remains unclear. Here we show that the human protein ubiquilin 1 can interact with Mtb surface proteins and associate with Mtb that are present in the host cell cytosol. We propose a model in which activating autophagy with IFN-γ promotes UBQLN1 recruitment to Mtb, which in turn leads to recruitment of the autophagy machinery, autophagy-mediated degradation of the bacteria, and activation of effector T cells. Since IFN-γ is critical in human control of Mtb, our study suggests that polymorphisms in ubiquilins, known to influence susceptibility to neurodegenerative illnesses, might also play a role in host defense against Mtb.
Introduction
Mycobacterium tuberculosis (Mtb) infects one-third of the world’s population. It can remain dormant in its host for decades and ultimately kills more people than any other bacteria. Mtb survives within macrophages by preventing its own delivery to the degradative, phagolysosomal compartment [1]. Macrophages that are activated by IFN-γ partially overcome the arrest in phagosome maturation imposed by Mtb [2,3]. IFN-γ stimulates macroautophagy [4–6] (hereafter autophagy), a process by which double-membrane organelles termed autophagosomes capture and degrade cytoplasmic components. In non-selective autophagy, which occurs in response to nutrient limitation, a portion of the cytoplasm is engulfed. In a form of autophagy that is called xenophagy, invading microorganisms are targeted. Autophagosomes that sequester Mtb fuse with lysosomes and impair mycobacterial replication. Autophagy partially restricts Mtb replication, and conditions that activate autophagy, including exposure to IFN-γ, promote mycobacterial clearance [4,6–8].
A prevailing model for how autophagy contributes to antimicrobial host defense begins with bacteria damaging or escaping phagosomes [9,10]. In the case of Mtb, damage and/or escape depends upon the mycobacterial ESX-1 Type VII secretion system and the secreted effector EsxA (also known as ESAT-6) [11–15]. Phagosomal damage allows mycobacterial DNA and peptidoglycan to activate host cytosolic sensors [14,16]. Ubiquitinated proteins accumulate around the bacteria, which partially depends upon the E3 ligase parkin (encoded by the PARK2 gene) [17]. The host proteins p62 (also known as SQSTM1), NDP52 (nuclear dot protein 52 kD, also known as CALCOCO2), and NBR1 (next to BRCA1 gene 1) can all bind ubiquitin as well as the autophagy protein LC3; they are thought to serve as cargo adaptors that link ubiquitin-conjugated Mtb or phagosomal remnants to LC3 [8,9,17]. A major unanswered question in the field is how Mtb, or any other bacteria, are physically linked to the autophagy machinery. In mitophagy, an analogous process in which mitochondria are selectively cleared from the cytoplasm, certain outer mitochondrial membrane proteins directly bind LC3 or recruit parkin to damaged mitochondria [18]. How parkin and other E3 ligases are recruited to the invading tubercle bacilli is uncertain; no mechanism comparable to that described for mitophagy has been shown to link the bacterial surface to the autophagy machinery.
We found that host UBQLN1 (also known as PLIC-1) binds a subset of Mtb secreted proteins and recognizes Mtb during xenophagy. UBQLN1 is a member of a family of highly related proteins that contain a ubiquitin-like (UBL) domain, a ubiquitin-associated domain (UBA), and STI1 motifs that are found in the co-chaperone Sti-1 (also known as HOP) (Fig 1A). UBQLN1 and UBQLN2 are thought to facilitate degradation of ubiquitinated targets by the proteasome [19,20]. More recently, they have also been shown to play a role in autophagy. Ubiquilins associate with autophagosomes, participate in autophagosome formation, and protect against starvation-induced cell death [21,22]. They are implicated in clearing protein aggregates in neurodegenerative disorders, including Alzheimer’s disease, amyotrophic lateral sclerosis (ALS), and Huntington’s disease [23–25]. Here, we show that UBQLN1 recognizes Mtb, acts upstream of ubiquitination, and promotes autophagy-mediated clearance of Mtb. Therefore, we provide evidence that UBQLN1 serves as a link between the bacterial surface and the host autophagy pathway.
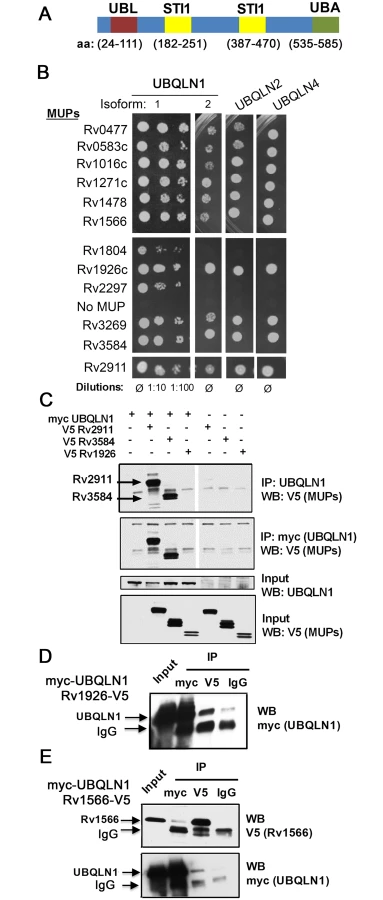
Results
UBQLN1 interacts with exported Mtb proteins
Previously, we used a stringent yeast two-hybrid (Y2H) system to map Mtb-human protein-protein interactions. UBQLN1 interacted with 12 Mtb proteins, which we call MUPs for mycobacterial ubiquilin-interacting proteins [26] (S1 Text, S1 Table). UBQLN1 exhibited selectivity in its interactions, as we identified it only 12 times in a screen of 339 secreted Mtb proteins. It did not interact with any of the 60 non-secreted Mtb proteins that were also screened or Antigen 85b, EsxA, and EsxB when directly tested [26]. Specificity was also indicated by the finding that MUPs interacted weakly or not at all with NDP52, an autophagy receptor that, like UBQLN1, contains a ubiquitin-binding domain (S1 Fig). To evaluate whether MUPs interact with other ubiquilin family members, we examined murine UBQLN1, UBQLN2, and UBQLN4, which are 88%, 67%, and 60% identical to human UBQLN1, respectively. We did not test UBQLN3 because its expression is restricted to the testes [27]. Most MUPs interacted with murine UBQLN1 (both splice isoforms), UBQLN2, and UBQLN4 (Fig 1B). The MUPs are largely uncharacterized; there is not a domain common to all of them, although two MUPs contain p60 domains (S1 Table). Most have a predicted signal peptide that targets them for secretion, and the majority have been found in culture filtrate in at least one study, suggesting they are accessible to host interactions. Few MUPs are found exclusively in the culture filtrate; most are also present in the cell membrane or whole cell lysate (S2 Fig)[28]. To conclude, we found that ubiquilin family members can interact with numerous Mtb secreted and surface proteins.
To determine whether MUPs interact with UBQLN1 in mammalian cells, we expressed V5-tagged MUPs in HEK293 cells. We could detect expression of eight MUPs in HEK293 cells, and we tested four in co-immunoprecipitation assays with human UBQLN1. We could not detect endogenous UBQLN1 in HEK293 cells, so we co-transfected myc-UBQLN1 along with V5-tagged MUPs. myc-UBQLN1 immunoprecipitated MUPs Rv2911 and Rv3584, but not Rv1926. MUPs were not detected in immunoprecipitates from cells lacking myc-UBQLN1 (Fig 1C). Although myc-UBQLN1 did not co-immunoprecipitate Rv1926c-V5, Rv1926c-V5 did co-immunoprecipitate UBQLN1 when using an antibody recognizing V5, whereas minimal myc-UBQLN1 was detected when an isotype control antibody was used (Fig 1D). The amount of UBQLN1 that co-immunoprecipitate with Rv1926 was 27% of that which could be precipitated directly using the myc antibody. In the case of Rv1566-V5 and myc-UBQLN1, we could co-immunoprecipitate the proteins in both directions (Fig 1E), and in both cases 7% of the amount directly precipitated was co-immunoprecipitated. Thus, all of the MUPs tested co-immunoprecipitated in at least one direction with UBQLN1.
UBQLN1 associates with Mtb in vitro
Since UBQLN1 can interact with Mtb surface proteins, we reasoned that it might associate with Mtb in a cell free system. We incubated Mtb with cytosol from HEK293 cells that had been transfected with plasmid encoding UBQLN1, truncated versions of UBQLN1, or vector control (Fig 2A, 2B and 2C). After four hours of incubation, we washed the bacteria to remove unbound host proteins. Actin was removed after the first wash, whereas UBQLN1 remained associated with Mtb after five washes. As additional controls for specificity, we tested cytosol from HEK293 cells transfected with GFP or the E3 ligase parkin. In contrast to UBQLN1, neither GFP nor parkin associated with Mtb (Fig 2B). To test if Ubqln1 binds Mtb directly, we purified GST-UBLQN1 and added it to Mtb in vitro. As we found with HEK293 cell lysate, UBQLN1 remained associated with Mtb after five washes (Fig 2D).

To further understand how UBQLN1 interacts with Mtb, we examined the roles of the UBL and UBA domains. A recombinant, truncated version of UBQLN1 lacking the UBL domain (UBQLN1-ΔUBL) failed to bind Mtb, whereas the UBA domain mutant had preserved binding (Fig 2C). Although recombinant UBQLN1-ΔUBL did not bind Mtb in vitro, when UBQLN1-ΔUBL was expressed in HEK293 cells or yeast, it interacted with intact Mtb as well as MUPs (Fig 2D and S3 Fig). This suggests that in co-immunoprecipitation and Y2H experiments, additional proteins, including UBQLN family members, may bridge or stabilize the UBQLN1-Mtb and MUP interactions. When we expressed UBQLN1 lacking the UBA domain (UBQLN1-ΔUBA) in HEK293 cells, there were numerous products both smaller and larger than the predicted protein, suggesting that UBQLN1 - ΔUBA was degraded and post-translationally modified. Degradation products associated with Mtb in the cell free system (Fig 2C) and also co-immunoprecipitated with MUPs (Rv1566 and Rv2911; S3 Fig). In the Y2H, UBQLN1-ΔUBA failed to interact with MUPs, perhaps related to its propensity for degradation (S3 Fig). The UBA domain alone failed to associate with Mtb (Fig 2C). In conclusion, Mtb associates with recombinant UBQLN1 in vitro and in a cell free system. The UBL domain appears important in vitro, although in the context of cytoplasm, it is dispensable, perhaps related to the ability of endogenous UBQLN family members or other adaptor proteins to multimerize and recruit the truncated protein. Determining the contribution of the UBA domain is confounded by the propensity for the truncated protein to be degraded, but the combined data suggest the UBA domain is not required.
UBQLN1 associates with Mtb, restricts its growth, and promotes T cell activation in IFN-γ activated macrophages
UBQLN1 interacted with Mtb proteins and was recruited to the bacteria from host cytosol. Therefore, it should co-localize with the bacteria during an infection. We used a UBQLN1-specific antibody to examine its localization by immunofluorescence microscopy. UBQLN1 was predominantly found in small cytoplasmic punctae (S4 Fig). When we examined the relationship of UBQLN1 to Mtb in unactivated macrophages, we found a low level of co-localization. The association was enhanced in IFN-γ activated macrophages, in which we found UBQLN1 co-localized with 13% of Mtb (Fig 3A and 3B). Although the association of UBQLN1 with Mtb was more prominent in IFN-γ activated macrophages, the protein was present at equivalent levels in untreated macrophages (Fig 3C) and the overall cellular distribution of UBQLN1 looked similar in activated and naïve cells (S4 Fig). In IFN-γ activated macrophages, there was little co-localization of UBQLN1 with an Mtb ΔesxA mutant, which does not permeabilize the phagosome (Fig 3B) [12,14], suggesting that the association of UBQLN1 with Mtb requires phagosomal damage to provide access to the bacteria.
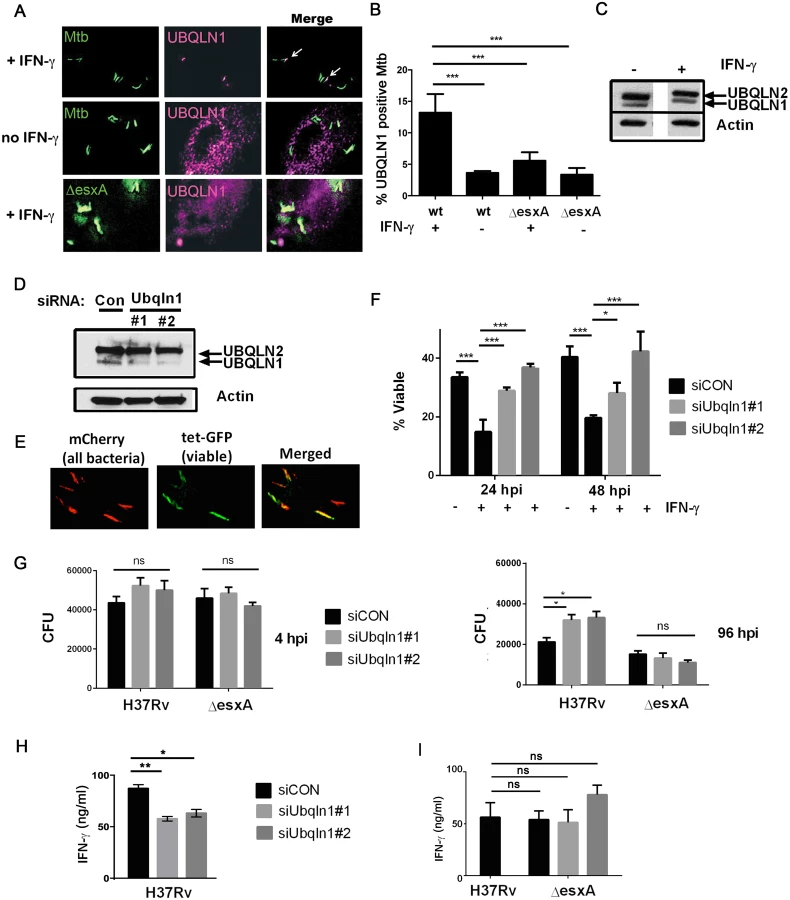
To determine whether UBQLN1 plays a role during infection, we examined macrophages in which UBQLN1 was depleted using RNAi. We used two different siRNAs which reduced UBQLN1 levels by 88% (siRNA #1) and 90% (siRNA #2; Fig 3D) based upon quantification using ImageJ and normalizing to actin levels. There was no significant effect on UBQLN2 levels. Following UBQLN1-depletion, bone marrow-derived macrophages (BMDMs) were infected with a live/dead Mtb reporter strain that expresses mCherry constitutively and GFP under control of a tetracycline-inducible promoter [29]. After treatment with anhydrotetracycline (AnTc), metabolically active bacteria express both GFP and mCherry, whereas dead bacteria only express mCherry (Fig 3E). As expected, control macrophages that were activated with IFN-γ restrained growth of Mtb. In contrast, UBQLN1-depleted macrophages were impaired in restricting Mtb growth (Fig 3F). We corroborated these results by plating for colony forming units (CFU). UBQLN1 silencing had no effect on bacterial uptake 4 hours post infection (hpi), but it rendered IFN-γ activated macrophages defective in their ability to control Mtb at 96 hpi (Fig 3G). UBQLN1 was also required to control Mtb in IFN-γ activated RAW264.7 (RAW) cells, a murine macrophage cell line (S5 Fig). In accordance with the localization data, there was no effect of UBQLN1 silencing in unactivated macrophages (S5 Fig) or on infections with the ΔesxA mutant (Fig 3G and S5 Fig). To determine whether UBQLN1 controls the general antimicrobial capacity of macrophages, we examined its effect on the intracellular growth of Staphylococcus aureus and Mycobacterium smegmatis. There was no effect of depleting UBQLN1 on S. aureus or M. smegmatis survival (S6 Fig). Thus, although UBQLN1 restricts growth of Mtb, it is not universally required for the antimicrobial capacity of macrophages towards all bacteria.
Because MUPs also bound UBQLN2 and UBQLN4, we examined whether these family members play a role in controlling Mtb replication in macrophages. We found that UBQLN2, but not UBQLN4, was present in macrophage lysate (S4 Fig). We attempted to silence UBQLN2 using siRNA, but we only achieved limited depletion, which did not result in any change in mycobacterial CFU (S4 Fig). Thus, UBQLN1 restricts Mtb growth in activated macrophages, and we were unable to draw definitive conclusions about UBQLN2.
Another important function of macrophages is to present antigen presentation to and activate T cells. We examined the ability of UBQLN1-silenced macrophages to activate Th1 polarized CD4+ T cells using P25TCR-Tg T cells which recognize the peptide 25 epitope of Mtb Antigen 85B. T cell production of IFN-γ was antigen specific, as IFN-γ was not detected when macrophages were infected with a strain lacking Antigen 85b (ΔfbpB). Notably, IFN-γ secretion was lower when P25TCR-Tg T cells were co-cultured with UBQLN1-depleted macrophages compared to co-culture with controls (Fig 3H). There was no effect of UBQLN1 silencing on MHCII surface expression (S7 Fig). UBQLN1 silencing did not impair the ability of macrophages infected with ΔesxA to activate T cells (Fig 3I), consistent with the idea that UBQLN1 acts on bacilli that access the cytosol. This also argues that impaired T cell activation is not caused by a non-specific effect such as decreased macrophage viability. In summary, UBQLN1 limits Mtb replication and enhances the ability of macrophages to activate effector T cells.
UBQLN1 co-localizes with Mtb that are cleared via autophagy
The above findings demonstrate that UBQLN1 recognition of Mtb and its role in controlling Mtb replication depends upon the bacterial ESX-1 system and host IFN-γ activation, both of which promote Mtb xenophagy [4,14]. We examined the accumulation of LC3-II by western blotting in uninfected macrophages, and found that UBQLN1 was required for basal autophagy in uninfected, IFN-γ activated BMDMs (S5 Fig). To evaluate whether UBQLN1 is involved in xenophagy, we examined the localization of UBQLN1 positive Mtb relative to autophagy components (Fig 4A). In activated macrophages, we found that more than 60% of the UBQLN1 positive Mtb co-localized with ubiquitinated proteins, which surround mycobacteria that damage or escape the phagosome [8,12,30]. 45% of the UBQLN1 positive bacteria also co-localized with p62, an autophagy adaptor that is recruited to ubiquitinated bacteria and binds LC3. Finally, more than 60% of the UBQLN1 positive bacteria co-localized with LC3. Likewise, a prominent fraction of the bacteria that associated with FK2, p62, or LC3 were UBQLN1 positive (Fig 4B), all of which is consistent with the idea that UBQLN1 functions in autophagy-mediated clearance of Mtb.

We next asked whether UBQLN1 positive Mtb are trafficked through the autophagy pathway. Initially, we examined whether IFN-γ-mediated control of Mtb depends upon Atg16L1, a component of the autophagy elongation complex that conjugates LC3 to phosphatidylethanolamine on the incipient autophagosome. IFN-γ restricted bacterial growth in wild type (wt) macrophages (Atg16L1flox/flox Cre-), but not in autophagy-deficient macrophages (Atg16L1flox/flox Lyz-Cre+) (Fig 4C), demonstrating that autophagy is required for the antibacterial properties of IFN-γ. In IFN-γ activated macrophages, there were more p62-positive Mtb in autophagy-deficient cells than controls (22.8 +/ - 2.2% versus 11.8 +/ - 5.3%; p<0.03), consistent with the idea that p62 associates with Mtb that are directed to the autolysosome where p62 is subsequently degraded. Similarly, IFN-γ-activated, Atg16L1-deficient macrophages contained more UBQLN1 positive Mtb compared to autophagy-competent cells (20.4 +/ - 4.7% vs 10.5 +/ - 2.7%, p<0.04), suggesting that UBQLN1-decorated Mtb are also targeted for xenophagy. Moreover, the fraction of UBQLN1 positive bacteria that were also positive for p62 increased from ~50% in wt macrophages to ~80% in the autophagy-deficient cells (Fig 4D and 4E), implying that double positive bacteria are eliminated by autophagy. Combined, these data support the idea that UBQLN1 associates with bacteria that are cleared through autophagy.
UBQLN1 promotes Mtb ubiquitination independently of parkin
UBQLN1 binds mono and polyubiquitin [31], so we thought that it might be recruited to ubiquitinated Mtb, like the autophagy adaptors p62 and human NDP52. Alternatively, since UBQLN1 can bind MUPs, we also thought that UBQLN1 might directly recognize Mtb and act upstream of ubiquitination. To distinguish these possibilities, we examined ubiquitination of Mtb in UBQLN1-silenced BMDMs. As expected, IFN-γ promoted the ubiquitination of Mtb. Notably, the recruitment of ubiquitin to Mtb in activated cells depended upon UBQLN1. Similarly, the enhanced co-localization of p62 and LC3 with Mtb seen in IFN-γ treated macrophages was blunted in cells lacking UBQLN1 (Fig 5A, 5B and 5C). Thus, we conclude that UBQLN1 promotes Mtb-associated ubiquitination and subsequent recruitment of adaptor proteins and the autophagy machinery during IFN-γ promoted xenophagy.

The E3 ligase parkin is required for ubiquitin recruitment to Mtb in naïve macrophages [17], and we found that it also played a role in IFN-γ activated macrophages. In activated macrophages, the parkin knockout macrophages had half as many ubiquitin positive Mtb as wt macrophages (Fig 6A). We asked whether parkin is also required for UBQLN1 recruitment to Mtb. While there was a slight trend towards decreased UBQLN1 positive Mtb in the parkin mutant, it was not statistically significant (Fig 6B). This suggests that UBQLN1 largely localizes independently of parkin and ubiquitination. In addition, in macrophages lacking parkin we found that UBQLN1 silencing diminished ubiquitin association with Mtb, much as it did in wt macrophages (Fig 6C). Combined, these results suggest that in activated macrophages, UBQLN1 and parkin act independently to recruit ubiquitinated proteins to Mtb.

Discussion
Although in vitro autophagy makes only a limited contribution to macrophage control of Mtb, in vivo mice are profoundly susceptible to Mtb infection if they lack parkin or autophagy proteins in myeloid cells [8,17,32]. In addition to controlling bacterial replication, xenophagy modulates cytokine responses and promotes MHCII antigen presentation [32,33]. Here, we provide multiple lines of evidence that UBQLN1 associates with Mtb and links them to the autophagy machinery: UBQLN1 binds MUPs, binds Mtb in vitro, and localizes to Mtb during infection. The association of UBQLN1 with Mtb depends upon EsxA, which is likely related to the role of EsxA in damaging the phagosomal membrane; however, it is also possible that EsxA plays some other role in recruiting UBQLN1 to the bacteria. Consistent with a role for UBQNL1 in recognizing Mtb, in the absence of UBQLN1 there is less ubiquitin, p62, and LC3 recruited to Mtb, which correlates with impaired control of bacterial replication and diminished CD4+ T cell activation. Overall, our data are consistent with a model in which UBQLN1 recognizes Mtb that become accessible to the cytosol upon phagosomal damage; UBQLN1 then assists in recruiting the autophagy machinery.
Why did we only detect a role for UBQLN1 in IFN-γ activated macrophages when the protein is also present in unactivated macrophages? UBQLN1 probably does not function in the IFN-γ signaling pathway, as there was no difference in surface MHCII, an IFN-γ induced gene, between control and UBQLN1-silenced BMDMs (S7 Fig). The apparent selectivity for activated macrophages may simply reflect that we see very low levels xenophagy in unactivated cells, as evidenced by little FK2, p62, and LC3 association with Mtb in naïve macrophages (Fig 5). It may be difficult to detect significant differences when we alter the trafficking of this minor bacterial population, particularly using RNAi, which generates hypomorphic effects rather than complete loss of function. In addition, autolysosomes may be less antimicrobial in naïve cells compared with activated macrophages. Other investigators have reported higher levels of ubiquitin and LC3 association in unactivated macrophages (for example see [8,17]), which might be due to strain or other experimental differences. When we activated autophagy chemically with rapamycin, it resulted in considerable toxicity in siRNA-transfected, Mtb-infected cells making it difficult to draw any conclusions about the role of UBQLN1 in this context. However, we suspect UBQLN1 would play a detectable role in naive macrophages if they had robust xenophagy.
It is surprising that UBQLN1 recognizes so many Mtb proteins that appear to have little sequence conservation. In addition, there are likely to be additional MUPs as our Y2H screen only examined 399 Mtb proteins [26]. The MUPs do not appear to share much in common (S1 Table), although one thing almost all of the MUPs have is a predicted signal peptide. However, not all proteins containing signal peptides interact with UBQLN1, since UBQLN1 did not interact with Ag85B or the other proteins with signal sequences in our original Y2H screen [26]. Domain mutants of the MUPs have so far not been revealing, as all fragments have failed to interact. One possibility is that MUPs are prone to misfolding and aggregation. UBQLN1 is proposed to have chaperone activity [34], which may involve the STI1 motifs. Our findings that UBQLN1 promotes ubiquitination and adaptor recruitment is consistent with it playing an early role in xenophagy; however, it is possible that we identified the MUP-UBQLN1 interaction fortuitously, and MUPs are not what is responsible for recruiting UBQLN1 to Mtb. To verify the UBQLN1-MUP interactions or identify the bona fide UBQLN1 interacting proteins during an infection is technically challenging and an area of ongoing effort.
How do we envision UBQLN1 might work to promote xenophagy? One possibility is that it recruits or activates an E3 ligase, which ubiquitinates bacterial or host phagosomal components. In several studies, UBQLN1 has been shown to promote ubiquitination of target proteins [35–37], although the E3 ligases were not identified. For Mtb xenophagy, parkin was a strong candidate, since it was recently shown to be required for ubiquitination around Mtb [17]. However, UBQLN1 localized to Mtb and promoted ubiquitination even in the absence of parkin (Fig 6). In addition, UBQLN1 and parkin did not interact in Y2H or co-immunoprecipitation experiments. Hence, a different E3 ligase likely acts downstream of UBQLN1 and parallel to parkin. Another possibility is that UBQLN1 is part of an amplification loop that fosters the association of ubiquitinated proteins with Mtb; UBQLN1 might localize to Mtb by virtue of binding MUPs, misfolded or aggregated proteins, or ubiquitinated proteins and then recruit additional ubiquitinated proteins. Whether UBQLN2 also plays a role in Mtb xenophagy warrants additional investigation. Although we did not detect a role for UBQLN2, this may have been due to insufficient silencing.
In conclusion, we show that UBQLN1 is required for autophagy-mediated clearance of Mtb in response to IFN-γ. UBQLN1 associates with Mtb and promotes recruitment of ubiquitinated proteins, autophagy adaptors, and the autophagy machinery. We speculate that UBQLN1 recognizes MUPs or other aggregation prone proteins generated by Mtb or present in the phagosome. In doing so, UBQNL1 promotes innate resistance to Mtb in the same way that it protects cells from cytotoxicity due to aggregation-prone cellular proteins, such as APP, TDP-43, and polyQ-expanded Huntington’s disease protein [24]. Thus, in addition to their role in Alzheimer’s disease, polymorphisms in UBQLN1 may influence susceptibility to tuberculosis, analogous to the dual role of PARK2 (which encodes parkin) in Parkinson’s and leprosy [17,38]. UBQLN2 mutations, which confer risk of ALS [23], are also worthy of further investigation. Overexpression of UBQLN1 ameliorates damage in murine models of stroke and Huntington’s disease [39,40]. Therefore, therapeutics that promote the activity of ubiquilins might have efficacy in neurodegenerative disorders and tuberculosis.
Materials and Methods
Details regarding Y2H, cell culture conditions, bacterial and mouse strains, flow cytometry, T cell activation assay, recombinant protein production, antibodies, siRNAs, and plasmids can be found in the S1 Text.
Co-immunoprecipitation and western blotting (WB)
For WB, cellular lysates were prepared in phosphate buffered saline (PBS) with 1% NP-40 and Halt Protease Inhibitor Cocktail (Thermo Scientific). For immunoprecipitations (IPs), HEK293 cells were lysed in PBS with 0.1% NP-40 and passed 25 times through a 25 gauge needle. Lysates were incubated with Sepharose G agarose beads (GE Healthcare) pre-bound with anti-Ubqln1 (Abcam) or anti-myc antibody (Fig 1C). Alternatively, Sepharose G Dynabeads coated with anti-myc, anti-V5, or control IgG antibodies were used (Fig 1D and 1E). Bound proteins were analyzed by WB.
Intracellular bacterial growth assays
RAW cells or BMDMs were transfected with siRNAs for 2d (RAW) or 3d (BMDM) prior to infection. 200 U/ml murine IFN-γ (Gibco) was added 24h before infection as indicated. For Mtb and M. smegmatis, macrophages were infected with a single cell suspension at an MOI of 3 with at least three replicates per experiment as previously described [26]. 4 hpi macrophages were extensively washed, lysed with 0.1% Triton X-100 at indicated time points, and serial dilutions were plated on 7H11. CFU were counted 15–21 days later for Mtb and 2–3 days later for M. smegmatis. For S. aureus infection, bacteria were opsonized with human serum for 1h prior to infection. Macrophages were infected at an MOI of 1, washed extensively 30 min post-infection, and lysed in 0.1% Triton-X-100 at indicated time points. S. aureus were plated on Tryptic Soy Agar, and CFU were quantified the following day.
Microscopic analysis of infected cells
RAW cells or BMDMs from C57BL/6, parkin KO, or LC3-GFP-expressing mice were plated in 8 well chamber slides. They were infected with a single cell suspension of Mtb expressing GFP, DsRed, or the live/dead plasmid at MOI of 5 followed by washing 4 hpi. At indicated time points, they were fixed in 1% paraformaldehyde (PFA)/PBS overnight. For live/dead analysis, 200 nM anhydrotetracycline (AnTc) was added 20–24 hours prior to fixation. % viable Mtb was calculated using the live/dead strain as a ratio of GFP-bright, metabolically active bacteria to total mCherry-positive bacteria. For immunofluorescence microscopy, macrophages were permeabilized with 0.1% Tween-20 prior to immunostaining with primary and corresponding secondary antibody. Images were acquired using the Nikon Eclipse TiE/B fluorescent microscope at 60x magnification and deconvoluted as previously described [26]. At least 100 bacteria from a minimum of three independent fields were examined per experiment. For reproduced images, in some cases background was subtracted by selecting an ROI (region of interest) where there were no cells. For reproduced images, contrast was altered equally for a given single channel image for all samples in an experiment. For example, the signal corresponding to UBLQN1 was contrast adjusted equally for all panels in Fig 3A and likewise for all panels in Fig 4A. In Fig 5, the Ub, p62, and LC3 channels are adjusted equivalently for the siCON panel and the siUBQNL1 panel. Similarly, the UBQLN1 and p62 channels were equally adjusted for the Cre+ and Cre - samples.
Bacterial binding assay
1 x 106 HEK293 were plated, transfected the following day, and lysed 2 days later in PBS with 0.1% NP-40 (lysis buffer) by passage through a 25 gauge needle 25 times in the same manner as co-immunoprecipitation experiments. Mtb were grown to between O.D. = 0.5 and 1, washed twice with PBS, and one O.D. of bacteria was mixed with HEK293 lysate and incubated at 4°C for 4 h. Bacteria were then pelleted, resuspended in 1 mL lysis buffer, transferred to a new tube, and washed 5 times with 1 mL lysis buffer. The resulting bacterial pellet was suspended in 100 μl lysis buffer, transferred to a new tube with SDS loading buffer, boiled, and analyzed by WB.
Ethics statement
This study was conducted in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The New York University School of Medicine Institutional Animal Care and Use Committee approved all work with mice (protocol #130707–02). Euthanasia was performed prior to bone marrow harvest in accordance with the 2013 AVMA Guidelines for the Euthanasia of Animals.
Supporting Information
Zdroje
1. Stanley SA, Cox JS (2013) Host-pathogen interactions during Mycobacterium tuberculosis infections. Curr Top Microbiol Immunol 374 : 211–241. doi: 10.1007/82_2013_332 23881288
2. Via LE, Fratti RA, McFalone M, Pagan-Ramos E, Deretic D, et al. (1998) Effects of cytokines on mycobacterial phagosome maturation. J Cell Sci 111 (Pt 7): 897–905. 9490634
3. MacMicking JD (2012) Interferon-inducible effector mechanisms in cell-autonomous immunity. Nat Rev Immunol 12 : 367–382. doi: 10.1038/nri3210 22531325
4. Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, et al. (2004) Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 119 : 753–766. 15607973
5. Matsuzawa T, Kim BH, Shenoy AR, Kamitani S, Miyake M, et al. (2012) IFN-γ elicits macrophage autophagy via the p38 MAPK signaling pathway. J Immunol 189 : 813–818. doi: 10.4049/jimmunol.1102041 22675202
6. Singh SB, Davis AS, Taylor GA, Deretic V (2006) Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science 313 : 1438–1441. 16888103
7. Yuk JM, Shin DM, Lee HM, Yang CS, Jin HS, et al. (2009) Vitamin D3 induces autophagy in human monocytes/macrophages via cathelicidin. Cell Host Microbe 6 : 231–243. doi: 10.1016/j.chom.2009.08.004 19748465
8. Watson RO, Manzanillo PS, Cox JS (2012) Extracellular M. tuberculosis DNA Targets Bacteria for Autophagy by Activating the Host DNA-Sensing Pathway. Cell 150 : 803–815. doi: 10.1016/j.cell.2012.06.040 22901810
9. Deretic V, Saitoh T, Akira S (2013) Autophagy in infection, inflammation and immunity. Nat Rev Immunol 13 : 722–737. doi: 10.1038/nri3532 24064518
10. Huang J, Brumell JH (2014) Bacteria-autophagy interplay: a battle for survival. Nat Rev Microbiol 12 : 101–114. doi: 10.1038/nrmicro3160 24384599
11. van der Wel N, Hava D, Houben D, Fluitsma D, van Zon M, et al. (2007) M. tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell 129 : 1287–1298. 17604718
12. Wong KW, Jacobs WR (2011) Critical role for NLRP3 in necrotic death triggered by Mycobacterium tuberculosis. Cell Microbiol 13 : 1371–1384. doi: 10.1111/j.1462-5822.2011.01625.x 21740493
13. Simeone R, Bobard A, Lippmann J, Bitter W, Majlessi L, et al. (2012) Phagosomal rupture by Mycobacterium tuberculosis results in toxicity and host cell death. PLoS Pathog 8: e1002507. doi: 10.1371/journal.ppat.1002507 22319448
14. Manzanillo PS, Shiloh MU, Portnoy DA, Cox JS (2012) Mycobacterium tuberculosis activates the DNA-dependent cytosolic surveillance pathway within macrophages. Cell Host Microbe 11 : 469–480. doi: 10.1016/j.chom.2012.03.007 22607800
15. Simeone R, Sayes F, Song O, Gröschel MI, Brodin P, et al. (2015) Cytosolic Access of Mycobacterium tuberculosis: Critical Impact of Phagosomal Acidification Control and Demonstration of Occurrence In Vivo. PLoS Pathog 11: e1004650. doi: 10.1371/journal.ppat.1004650 25658322
16. Pandey AK, Yang Y, Jiang Z, Fortune SM, Coulombe F, et al. (2009) NOD2, RIP2 and IRF5 play a critical role in the type I interferon response to Mycobacterium tuberculosis. PLoS Pathog 5: e1000500. doi: 10.1371/journal.ppat.1000500 19578435
17. Manzanillo PS, Ayres JS, Watson RO, Collins AC, Souza G, et al. (2013) The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature 501 : 512–516. doi: 10.1038/nature12566 24005326
18. Randow F, Youle RJ (2014) Self and nonself: how autophagy targets mitochondria and bacteria. Cell Host Microbe 15 : 403–411. doi: 10.1016/j.chom.2014.03.012 24721569
19. Ko HS, Uehara T, Tsuruma K, Nomura Y (2004) Ubiquilin interacts with ubiquitylated proteins and proteasome through its ubiquitin-associated and ubiquitin-like domains. FEBS letters 566 : 110–114. 15147878
20. Kleijnen MF, Shih AH, Zhou P, Kumar S, Soccio RE, et al. (2000) The hPLIC proteins may provide a link between the ubiquitination machinery and the proteasome. Mol Cell 6 : 409–419. 10983987
21. N'Diaye EN, Kajihara KK, Hsieh I, Morisaki H, Debnath J, et al. (2009) PLIC proteins or ubiquilins regulate autophagy-dependent cell survival during nutrient starvation. EMBO reports 10 : 173–179. doi: 10.1038/embor.2008.238 19148225
22. Rothenberg C, Srinivasan D, Mah L, Kaushik S, Peterhoff CM, et al. (2010) Ubiquilin functions in autophagy and is degraded by chaperone-mediated autophagy. Human molecular genetics 19 : 3219–3232. doi: 10.1093/hmg/ddq231 20529957
23. Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, et al. (2011) Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 477 : 211–215. doi: 10.1038/nature10353 21857683
24. Takalo M, Haapasalo A, Natunen T, Viswanathan J, Kurkinen KM, et al. (2013) Targeting ubiquilin-1 in Alzheimer's disease. Expert Opin Ther Targets 17 : 795–810. doi: 10.1517/14728222.2013.791284 23600477
25. Bertram L, Hiltunen M, Parkinson M, Ingelsson M, Lange C, et al. (2005) Family-based association between Alzheimer's disease and variants in UBQLN1. N Engl J Med 352 : 884–894. 15745979
26. Mehra A, Zahra A, Thompson V, Sirisaengtaksin N, Wells A, et al. (2013) Mycobacterium tuberculosis Type VII Secreted Effector EsxH Targets Host ESCRT to Impair Trafficking. PLoS Pathog 9: e1003734. doi: 10.1371/journal.ppat.1003734 24204276
27. Conklin D, Holderman S, Whitmore TE, Maurer M, Feldhaus AL (2000) Molecular cloning, chromosome mapping and characterization of UBQLN3 a testis-specific gene that contains an ubiquitin-like domain. Gene 249 : 91–98. 10831842
28. de Souza GA, Leversen NA, Målen H, Wiker HG (2011) Bacterial proteins with cleaved or uncleaved signal peptides of the general secretory pathway. J Proteomics 75 : 502–510. doi: 10.1016/j.jprot.2011.08.016 21920479
29. Martin CJ, Booty MG, Rosebrock TR, Nunes-Alves C, Desjardins DM, et al. (2012) Efferocytosis is an innate antibacterial mechanism. Cell Host Microbe 12 : 289–300. doi: 10.1016/j.chom.2012.06.010 22980326
30. Collins CA, De Mazière A, van Dijk S, Carlsson F, Klumperman J, et al. (2009) Atg5-independent sequestration of ubiquitinated mycobacteria. PLoS Pathog 5: e1000430. doi: 10.1371/journal.ppat.1000430 19436699
31. Raasi S, Varadan R, Fushman D, Pickart CM (2005) Diverse polyubiquitin interaction properties of ubiquitin-associated domains. Nat Struct Mol Biol 12 : 708–714. 16007098
32. Castillo EF, Dekonenko A, Arko-Mensah J, Mandell MA, Dupont N, et al. (2012) Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc Natl Acad Sci U S A 109: E3168–3176. doi: 10.1073/pnas.1210500109 23093667
33. Jagannath C, Lindsey DR, Dhandayuthapani S, Xu Y, Hunter RL, et al. (2009) Autophagy enhances the efficacy of BCG vaccine by increasing peptide presentation in mouse dendritic cells. Nat Med 15 : 267–276. doi: 10.1038/nm.1928 19252503
34. Kim SH, Shi Y, Hanson KA, Williams LM, Sakasai R, et al. (2009) Potentiation of amyotrophic lateral sclerosis (ALS)-associated TDP-43 aggregation by the proteasome-targeting factor, ubiquilin 1. J Biol Chem 284 : 8083–8092. doi: 10.1074/jbc.M808064200 19112176
35. Beverly LJ, Lockwood WW, Shah PP, Erdjument-Bromage H, Varmus H (2012) Ubiquitination, localization, and stability of an anti-apoptotic BCL2-like protein, BCL2L10/BCLb, are regulated by Ubiquilin1. Proc Natl Acad Sci U S A 109: E119–126. doi: 10.1073/pnas.1119167109 22233804
36. El Ayadi A, Stieren ES, Barral JM, Boehning D (2012) Ubiquilin-1 regulates amyloid precursor protein maturation and degradation by stimulating K63-linked polyubiquitination of lysine 688. Proc Natl Acad Sci U S A 109 : 13416–13421. doi: 10.1073/pnas.1206786109 22847417
37. Gao L, Tu H, Shi ST, Lee KJ, Asanaka M, et al. (2003) Interaction with a ubiquitin-like protein enhances the ubiquitination and degradation of hepatitis C virus RNA-dependent RNA polymerase. J Virol 77 : 4149–4159. 12634373
38. Mira MT, Alcaïs A, Nguyen VT, Moraes MO, Di Flumeri C, et al. (2004) Susceptibility to leprosy is associated with PARK2 and PACRG. Nature 427 : 636–640. 14737177
39. Liu Y, Lü L, Hettinger CL, Dong G, Zhang D, et al. (2014) Ubiquilin-1 protects cells from oxidative stress and ischemic stroke caused tissue injury in mice. J Neurosci 34 : 2813–2821. doi: 10.1523/JNEUROSCI.3541-13.2014 24553923
40. Safren N, El Ayadi A, Chang L, Terrillion CE, Gould TD, et al. (2014) Ubiquilin-1 overexpression increases the lifespan and delays accumulation of Huntingtin aggregates in the R6/2 mouse model of Huntington's disease. PLoS One 9: e87513. doi: 10.1371/journal.pone.0087513 24475300
41. Philips JA, Porto MC, Wang H, Rubin EJ, Perrimon N (2008) ESCRT factors restrict mycobacterial growth. Proc Natl Acad Sci U S A 105 : 3070–3075. doi: 10.1073/pnas.0707206105 18287038
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 7
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- RNA Virus Reassortment: An Evolutionary Mechanism for Host Jumps and Immune Evasion
- Activation of TLR2 and TLR6 by Dengue NS1 Protein and Its Implications in the Immunopathogenesis of Dengue Virus Infection
- N-acetylglucosamine Regulates Virulence Properties in Microbial Pathogens
- Characterization of a Prefusion-Specific Antibody That Recognizes a Quaternary, Cleavage-Dependent Epitope on the RSV Fusion Glycoprotein
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy