
A Novel Antiviral Target Structure Involved in the RNA Binding, Dimerization, and Nuclear Export Functions of the Influenza A Virus Nucleoprotein
Influenza A virus nucleoprotein (NP) is a highly conserved multifunctional protein that plays an essential role in infection by all subtypes of influenza A virus, making it an attractive target for new antiviral drugs. NP regulates viral polymerase activity and transport of the viral genome into/from the host cell nucleus by forming the viral ribonucleoprotein complex (vRNP). Because NP regulates replication and transcription of the viral genome in addition to its role in nuclear export (all of which are essential for the production of viral progeny), it is a promising drug target. Here, we used the antiviral compound RK424 to identify a novel pocket structure within NP. This structure encompassed three different functional domains that are involved in the above-mentioned replication steps. RK424 inhibits viral genome replication/transcription and nuclear export of NP by destabilizing the NP oligomer and inhibiting the binding of chromosome region maintenance 1 (CRM1) to NP via nuclear export signal (NES) 3, which is located in close proximity to the NP pocket. Taken together, these findings suggest that this small NP pocket is a novel antiviral target.
Published in the journal:
. PLoS Pathog 11(7): e32767. doi:10.1371/journal.ppat.1005062
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1005062
Summary
Influenza A virus nucleoprotein (NP) is a highly conserved multifunctional protein that plays an essential role in infection by all subtypes of influenza A virus, making it an attractive target for new antiviral drugs. NP regulates viral polymerase activity and transport of the viral genome into/from the host cell nucleus by forming the viral ribonucleoprotein complex (vRNP). Because NP regulates replication and transcription of the viral genome in addition to its role in nuclear export (all of which are essential for the production of viral progeny), it is a promising drug target. Here, we used the antiviral compound RK424 to identify a novel pocket structure within NP. This structure encompassed three different functional domains that are involved in the above-mentioned replication steps. RK424 inhibits viral genome replication/transcription and nuclear export of NP by destabilizing the NP oligomer and inhibiting the binding of chromosome region maintenance 1 (CRM1) to NP via nuclear export signal (NES) 3, which is located in close proximity to the NP pocket. Taken together, these findings suggest that this small NP pocket is a novel antiviral target.
Introduction
Influenza A virus (IAV) causes periodic and widespread epidemics or pandemics, which take the form of respiratory diseases with cold-like symptoms; however, the virus can sometimes cause serious disease with high mortality rates [1]. One of the few options for treating influenza infection is antivirals. Currently, the only approved classes of anti-influenza virus drugs are viral M2 ion channel inhibitors and neuraminidase (NA) inhibitors [2,3]. Although these drugs can be effective for treating influenza infection, the emergence of drug-resistant viral strains is a serious problem [4,5,6]. It is for this reason that M2 ion channel blockers are no longer used clinically to treat circulating IAV strains. Moreover, NA inhibitor-resistant viruses are beginning to emerge. In addition to these conventional antivirals, a novel antiviral drug, T705 (favipiravir), was approved by the Japanese Ministry of Health. T705 is administered as a prodrug, which is metabolized to its active form, T-705-4-ribofuranosyl-5-triphosphate (T-705RTP), inside the cell. T-705RTP is a purine analog that selectively inhibits the influenza virus RNA-dependent RNA polymerase (RdRp) [7]. However, because T-705 is a ribonucleic analog, adverse effects may occur (indeed, such effects are observed after ribavirin treatment) [8]. Indeed, the use of this drug is limited to the treatment of newly-emergent influenza viruses that are resistant to current antivirals; therefore, NA inhibitors are the only drugs currently used to treat the majority of influenza virus infections. Thus, there is an urgent need for new antiviral drugs with novel mechanisms of action.
Recently, the viral nucleoprotein (NP) has attracted interest as a target for new antiviral drugs. NP is the major component of the viral ribonucleoprotein complex (vRNP), which comprises three polymerase subunit proteins (PB2, PB1, and PA) and viral RNA (vRNA) [9]. The vRNP is maintained by a double-helical structure derived from a NP homo-oligomer coupled to the vRNA [10]. This double-helical structure is associated with vRNP functions such as viral replication, nuclear export, and genome packaging. The NPs within the vRNP complex oligomerize via two interaction forces. One comprises an intra-strand interaction through which the tail loop (amino acid (aa) residues 402–428) is inserted into the neighboring NP tail loop pocket [11]. The other comprises an inter-strand interaction between two NP strands with opposite-polarity. The mechanism underlying the interaction between the two NP strands is still not clear; however, two dimer interfaces, the helix-turn-helix motif (aa 149–167) and the C-terminal region (aa 482–498), of NP are involved in this process [12]. After the influenza virus is adsorbed and internalized into the host cell, the vRNP complex is released from the endosome into the cytoplasm from where it is imported into the nucleus. Following virus replication in the nucleus, the vRNP is exported back into the cytoplasm and moves to the cell membrane. During this step, the C - and N-terminal domains of nonstructural protein 2/nuclear export protein (NS2/NEP) mediate the complexation of vRNP-matrix protein 1 (M1) and chromosome region maintenance 1 (CRM1), respectively, to export the vRNP from the nucleus to the cytoplasm [13,14,15,16,17]. In addition, NP is exported from the nucleus via the NES. NP contains functional NESs at aa 24–49 (NES1), 183–197 (NES2), and 248–274 (NES3) [18,19]. Although nuclear export via NES1 and NES2 is CRM-independent, export via NES3 is CRM1-dependent [18,19]. Binding assays show that NP binds directly to CRM1 and that the cytoplasmic localization of NP is inhibited by leptomycin B (LMB), which inhibits the CRM1-mediated nuclear export of cargo proteins [19, 20]. Thus, the direct interaction of NP with CRM1 may be associated with the nuclear export of the vRNP, because NP is a structural component of the vRNP.
The multiple functions of NP mean that it has attracted interest as a novel target for antiviral drugs [21]. A recent study shows that viral replication is inhibited by small-molecule compounds that target NP, including nuclear localization signal (NLS)-binding compounds (i.e., mycalamide analogs) [22] and NP salt bridge inhibitors (i.e., compound 3) [23]. Mycalamide analogs, which were identified by a chemical array screening method, inhibit influenza virus replication by binding to unconventional NLS within the NP [22]. Furthermore, compound 3 inhibits formation of the NP trimer by disrupting the salt bridge between E339 and R416 within the NP tail loop binding pocket, thereby inhibiting the functional oligomerization of NP [23]. Notably, nucleozin inhibits the formation of higher-order NP oligomers by cross-linking two composition of NP, showing a potent antiviral effect both in vitro and in vivo [24,25]. These results clearly show that influenza virus NP is a valid target for new antiviral compounds.
We previously screened a chemical library containing 50,000 compounds with diverse structures and identified RK424, a compound with antiviral effects. Here, we showed that RK424 inhibits IVA replication via a mechanism that is different from that of existing antiviral drugs. RK424 bound to a small pocket structure within NP, which is surrounded by three different domains (the RNA binging groove, the NP dimer interface, and NES3), thereby inhibiting the function of all three. Furthermore, the amino acid residues that comprise the NP pocket are well-conserved, and mutations at these residues are functionally restricted. Taken together, the results of the present study show that the newly-identified NP pocket is a promising target for antiviral drugs that inhibit the multiple functions of NP by simultaneously disrupting the function of three different functional domains.
Results
RK424 shows antiviral activity both in vitro and in vivo
We previously identified four hit compounds with antiviral activity higher than a 50% inhibitory concentration of 3 μM (inhibitory concentration (IC50) ≦3 μM) by screening 50,000 compounds in a Madin-Darby canine kidney (MDCK) cell-based influenza A infection assay [26]. Among these initial hit compounds, RK424 showed the greatest potency (IC50 = 0.48±0.19 μM and IC90 = 1.19±0.36 μM) against the A/WSN/1933 (H1N1) virus in a plaque assay without inhibiting the growth of MDCK cells (50% cytotoxicity concentration (CC50) ≧50 μM) (Fig 1A and 1B). RK424 showed a similar level of antiviral activity against many different subtypes of IAVs including currently circulating A/California/7/2009 virus (H1N1) (IC50 = 0.40±0.07 μM and IC90 = 0.88±0.02 μM). Interestingly, RK424 inhibited the replication of the highly pathogenic influenza viruses, A/CX/yamaguchi/7/2004 (H5N1) and A/Anhui/1/2013(H7N9), with IC50 values of 0.57±0.04 μM and 0.60±0.12 μM and IC90 values of 0.93±0.02 μM and 1.14±0.26 μM, respectively. These results clearly show that RK424 inhibits the replication of IAVs.

We also examined the efficacy of RK424 in vivo. Intravenous administration of 1mg/kg RK424 to mice resulted in a free drug maximum concentration (Cfree max) of RK424 (11.4 ng/mL) that was considerably lower than the IC50 of RK424 (162.7 ng/mL) in plasma (S1 Table). The dose could not be increased further because levels above 1mg/kg resulted in the precipitation of RK424 from the aqueous vehicle. Besides, the bioavailability of RK424 via the oral route relative to that via the intraperitoneal route was 50% lower and the Cfree max of RK424 (46.4 ng/mL) did not exceed the IC50 of RK424 (162.7 ng/mL) when RK424 was administered at 10 mg/kg via the oral route (S1 Fig and S2 Table). Therefore, mice were intraperitoneally injected with RK424 and then inoculated with ten 50% lethal doses (LD50) of A/WSN/1933 (H1N1). Morbidity and mortality were monitored daily by measuring body weight (Fig 1C and 1D). Phosphate buffered saline (PBS)-treated control mice showed severe morbidity and 100% mortality at 6 days post-infection. By contrast, mice treated with RK424 (10 mg/kg) lost less weight and 25% survived until the end of the observation period. RK424 provided high levels of protection against lethal virus infections at early time points (up to 6 days post-infection); however, in vivo data showed that RK424 was less effective than oseltamivir phosphate. To confirm the antiviral effects of RK424 in vivo, mice were euthanized at 6 days post-infection (the point at which RK424 showed the greatest antiviral effect) and the lungs were harvested for use in a virus titration assay. Lungs from the negative control group showed efficient virus replication whereas those from the RK424-treated group showed a ~2 log reduction in the virus titer (2.9×103 PFU/mL) compared with the titers of the controls (1.6×105 PFU/mL) (Fig 1E and 1F). These results show that RK424 is also effective for the inhibition of viral replication in vivo. Moreover, we investigated for synergy in the antiviral effect of RK424 treated with oseltamivir phosphate. First, we determined the most appropriate dose of oseltamivir phosphate (0.02mg/kg Os) for co-administration with RK424 by evaluating the antiviral effect of serial dilutions of oseltamivir phosphate in vivo (S2 Fig). Monotherapy groups (10mg/kg RK424 or 0.02 mg/kg Os) showed almost the same body weight changes and survival rates, and 29% of the mice in these groups survived until the end of the observation period. On the other hand, co-administration of RK424 (5 mg/kg or 10 mg/kg) with oseltamivir phosphate (0.02 mg/kg) resulted in less weight loss and a higher survival rate (up to 57%) than the administration of RK424 or oseltamivir phosphate alone. This result further supports the notion that RK424 and oseltamivir phosphate act via different mechanisms and that they elicit a synergistic effect when combined in vivo.
However, in vivo efficacy with respect to morbidity and mortality was modest compared with the in vitro antiviral effect. This discrepancy is perhaps explained by the high plasma protein binding (99.9%) of RK424 (S1 Table), which means that only 0.4% of drug is free from plasma protein. This may have caused a decrease in drug efficacy in vivo. Moreover, the volume of distribution (Vd) of RK424 (583 mL/kg) was lower than that of mouse total body water (725 mL/kg) (S2 Table) [27]. These PK parameters may explain its weak pharmacologic efficacy and low tissue absorption in vivo. Thus, the plasma protein binding property of RK424 should be optimized to further improve its efficacy in vivo.
Taken together, these data suggest that RK424 has antiviral activity both in vitro and in vivo.
RK424 inhibits vRNP activity
In contrast to treatment with oseltamivir phosphate (which inhibits the budding step of the viral life cycle), treatment with RK424 led to a reduction in the number, but not the size, of viral plaques (S3 Fig) and it was effective against the oseltamivir resistant virus, A/Tex/131/E119V (H3N2) (Fig 1B) [28]. Furthermore, RK424 did not interfere with viral replication when added to MDCK cells prior to virus inoculation (S4 Fig) and exhibited a prominent antiviral effect at an early stage in the virus life cycle (S5 Fig). These results indicate that the RK424 may target the post-entry and early replication step of the viral life cycle. To examine this further, we first explored the possibility that RK424 affects viral genome replication and/or transcription, both of which occur during the early stages in viral life cycle. To examine the effect of RK424 on vRNP activity (vRNP regulates viral replication and transcription), we performed a mini-genome assay using five expression plasmids harboring influenza A/WSN/1933 (H1N1): PB2/pCAGGS, PB1/pCAGGS, PA/pCAGGS and NP/pCAGGS, which control viral genome replication and transcription, and the luciferase-containing plasmid vNP-luc/pHH21, which encodes a viral-like genome in the absence or presence of RK424. We found that RK424 caused a notable and dose-dependent reduction in luciferase activity (Fig 2A) compared with control (dimethyl sulfoxide (DMSO)) treatment. By contrast, treatment with 10 μM of oseltamivir phosphate did not significantly inhibit luciferase activity.

To confirm the inhibitory effect of RK424 on viral genome replication in infected cells, we next performed single-molecule fluorescence in situ hybridization (FISH) analysis with 48 single Quasar 670-labeled DNA oligos against vRNA encoding the PB2 subunit [26, 29] (S3 Table). Large numbers of vRNAs were observed in the cytoplasm of DMSO - and oseltamivir phosphate-treated MDCK cells at 6 h post-infection. By contrast, RK424 treatment resulted in a significant reduction in vRNA expression, and vRNAs mainly localized in the nucleus (Fig 2B).
Furthermore, a reduction in viral protein expression was also detected by western blotting (Fig 2C). Taken together, these results suggest that RK424 affects the function of the vRNP complex (PB2, PB1, PA, and NP), which is associated with viral polymerase activity.
RK424 inhibits the cytoplasmic localization of NP
NP is the major component of the vRNP complex [30]. Therefore, to further investigate the effects of RK424 on the nuclear import and export of the vRNP complex in virus-infected cells, we examined NP localization by fluorescence microscopy. We found that NP localized in both the nucleus and cytoplasm at 6 h post-infection in HeLa cells treated with either DMSO (control) or 10 μM of oseltamivir phosphate. By contrast, treatment with RK424 (0.5 or 2 μM) led to nuclear rather than cytoplasmic localization of NP, indicating that the vRNP complex could enter the nucleus but could not be exported to the cytoplasm (Fig 3A). Importantly, the effects of RK424 in virus-infected HeLa cells were also observed in HeLa cells transiently expressing NP (Fig 3B). The cytoplasmic distribution of NP was dependent upon the dose of RK424. As shown in Fig 3C, the cytoplasmic distribution of NP in HeLa cells transiently expressing NP in the absence and presence of 0.5, 2, and 10 μM of RK424 was 75%, 77%, 57%, and 30%, respectively. This shows that RK424 interferes with the nuclear export of NP in the absence of other viral components. Taken together, these results suggest that RK 424 may exert its antiviral effects by inhibiting the function of NP.

RK424 binds to a small pocket structure within NP that is involved in oligomerization and nuclear export
To further examine how RK424 inhibits NP function, we used in silico docking analysis to create a potential model for the binding of RK424 to NP. To establish unbiased predictive virtual docking models, we obtained the crystal structure of monomeric influenza A/WSN/1933 (H1N1) NP from the Protein Data Bank (PDB) and performed docking studies using AutoDock molecular modeling simulation software [31]. Three potential binding sites were identified; however, the potential binding models showed that the interaction with the highest binding free energy (ΔG) occupied binding site 1 (S6 Fig). Moreover, binding site 1 was surrounded by three functionally important domains: the RNA binding groove (orange) [11], the NP dimer interface (purple), [12] and NES3 (yellow) [18] (Fig 4A and S6 Fig, front side). Therefore, we focused on the binding model based on binding site 1. The interaction map predicted six different configurations for binding site 1, revealing that four amino acid residues (R162, S165, L264, and Y487) were predominantly involved in the interaction with RK424 (S7 Fig); the amino acid residues within binding sites 2 and 3 that were predicted to interact with RK424 did not correlate with any known NP functions (S8 Fig). RK424 occupied a small pocket on NP; configuration 0 had the best fitting score (ΔG of −8.03 kcal/mol) (Fig 4B and S7 Fig).

To confirm the accuracy of this binding model, we first prepared RK424-cross-linked affinity beads (RK424 beads) using a photo-cross-linking method [32] and examined whether RK424 binds to wild-type and mutant NP proteins harboring alanine substitutions at all four potential binding sites (R162A, S165A, L264A, and Y487A). Recombinant wild-type NP (NP-Flag) and Flag-tagged mutant NP proteins (mutant NP-Flag) were purified by FLAG affinity agarose beads. The purity of the purified proteins was checked by running samples in 10% SDS-PAGE gels followed by Coomassie Brilliant Blue (CBB) staining (Fig 4C, upper panel). The purified proteins were then incubated with control and RK424 beads and binding analyzed by western blotting. Wild-type NP-Flag protein bound to RK424 beads but not to control beads; however, the amount of mutant NP-Flag protein bound to RK424 beads was significantly lower than that of wild-type NP-Flag protein (Fig 4C, lower panel). These results suggest that RK424 binds to NP through the amino acid residues predicted by the interaction model.
Therefore, we next explored the structure-activity relationship (SAR) of RK424 using RK424 analogs. Substituting the carboxyl group with an amide group (Analog #1) led to a partial reduction in the antiviral effect, whereas the loss of the biphenyl ring (Analog #2) and quinolone backbone (Analog #3) led to a complete loss of antiviral activity. By contrast, replacing the biphenyl ring with a diphenyl ether moiety (Analog #4) resulted in an antiviral effect comparable with that of RK424 (Fig 4D). Analog #4 showed almost the same antiviral effect (IC50 = 0.43±0.15 μM) as RK424 (IC50 = 0.48±0.19 μM), but we could not obtain sufficient Analog #4 for further analyses. Analog #4 is synthesized by Wakunaga Pharmaceuticals and is not commercially provided by any supplier. Therefore, we used RK424 for further analysis. Taken together, these results agree with those obtained from the docking model (S7 Fig), i.e., that the carboxyl group, quinoline backbone, and biphenyl ring of RK424 may be required for the inhibitory effects of RK424.
RK424 disrupts NP-RNA and NP-NP interactions, thereby inhibiting NP oligomerization
The four amino acids residues that play a potential role in RK424 binding to NP reside within different functional domains. These domains are involved in (i) oligomerization of NP via NP-RNA and NP-NP interactions, and (ii) nuclear export of NP. We first used surface plasmon resonance (SPR; Biacore) to examine the effect of inhibiting the NP-RNA interaction. A 24-mer RNA oligomer was biotinylated at the 5’-end (to facilitate immobilization) and methylated at the 2’-OH of ribose (to provide nuclease resistance) was immobilized on an SA sensor chip. We chose a 24-mer because studies show that this length approximates the binding distance between NP and vRNA [33,34,35,36]. Samples containing RK424 (10 μM) showed a reduced binding response to RNA. This reduction was dependent upon the dose of RK424. The RNA binding observed for treated samples was almost 50% less than that observed for control samples (Fig 5A and 5B). Notably, RK424 affected the rate of association without affecting the rate of disassociation. This suggests that RK424 interferes with the binding of NP to RNA.

We next used a pull-down assay incorporating NP proteins fused to different tags to examine whether RK424 caused the NP-NP homo-oligomer to dissociate. In the absence of RK424, a purified recombinant wild-type NP protein fused to a HA tag (NP-HA) bound to FLAG agarose beads coupled to purified recombinant wild-type NP protein fused to mRFP-Flag-tag (NP-mRFP-Flag beads); however, NP-HA did not bind to control FLAG agarose beads coupled to mRFP-Flag protein (mRFP-Flag beads). RK424 inhibited the binding of NP-HA to NP-mRFP-Flag beads in a dose-dependent manner (Fig 5C and 5D). These results suggest that RK424 inhibits the NP-NP interaction.
NP shapes the main backbone and regulates the conformation of vRNP by interacting with RNA and with itself (NP-NP homodimers) [11,34,37]. Therefore, to examine whether RK424 prevents the formation of large NP oligomer/vRNA complexes, we incubated purified NP proteins (S9A Fig) with vRNA encoding the M segment (synthesized by in vitro transcription; S9B Fig) in the absence or presence of RK424. Next, the incubated samples were subjected to size exclusion chromatography and the particle sizes monitored using an ÄKTA purifier and UNICORN software. In the absence of RNA, purified NP proteins exist in equilibrium as monomers and trimers/tetramers (S9C Fig upper panel); this equilibrium is regulated by the salt concentration [33]. Salt concentrations higher than those encountered under physiological conditions shift the equilibrium toward trimers/tetramers, whereas lower salt concentrations shift it toward monomers. In solution, the NP trimers/tetramers gradually dissociate to form monomers [12]. Thus, we maintained the purified NP proteins in a 100 mM NaCl solution at 4°C for 48 h. This shifted the equilibrium toward monomer formation, a situation confirmed by size exclusion chromatography (S9C Fig lower panel). The elution peak for NP proteins incubated in the absence of vRNA occurred at around 14.6 mL, and was attributed to NP monomers; however, molecular mass of the NP protein eluted in this peak was estimated to be around 40 kDa, which is smaller than the molecular mass measured by 10% SDS-PAGE (56 kDa) (Fig 5E and S9A Fig) [11]. This suggests that the NP molecule adopts a more globular and compact shape than the proteins used for calibration. On the other hand, NP proteins incubated in the presence of vRNA but in the absence of RK424 eluted at around 8.5 mL; this peak was attributed to the presence of large NP oligomers (as calculated from the standard curve) (Fig 5E). These results show that the NP protein was able to form large oligomers by interacting with vRNA. Next, we examined the inhibitory effect of RK424 on NP oligomer formation. In the presence of vRNA and RK424, the peak height of the large NP oligomer was reduced; instead, a broad peak was observed and the elution volume increased to around 10 mL (Fig 5E). This suggests that RK424 inhibited NP-NP and NP-RNA interactions, thereby interfering with oligomerization. Because the NP peak in the presence of vRNA occurred between 8 mL and 9 mL in the absence of RK424, we quantified the rate of NP oligomerization by evaluating the peak integration between these elution volumes. There was a dose-dependent reduction in the rate of oligomerization in the presence of RK424 (Fig 5F). Taken together, these results demonstrate that RK424 inhibits NP oligomerization by disrupting NP-RNA and NP-NP interactions.
RK424 inhibits the nuclear export of NP and NP-CRM
In addition to the NP amino acids involved in NP-RNA and NP-NP interactions, the docking model showed that RK424 partially bound to amino acids that form the NES3 domain (Fig 4B). To investigate the inhibitory effect of RK424 on the nuclear export of NP, we performed an in vitro nuclear export assay using digitonin-permeabilized, semi-intact HeLa cells (Fig 6A and 6B). The nuclear export activity of NP-mRFP-Flag protein was examined by observation and quantification of mRFP fluorescence under a confocal laser scanning microscope. In the absence of cell lysate, many cells showed mRFP fluorescence (Fig 6A; Buffer). By contrast, the number of cells expressing NP-mRFP-Flag was clearly lower in presence of cell lysate (Fig 6A; +Lys). The fluorescence intensity of these cells was reduced to 42% (of that in untreated cells) in the presence of cell lysate (Fig 6B). In the presence of leptomycin B (LMB), many cells showed levels of mRFP fluorescence comparable with those in buffer-treated samples and 97% of the fluorescence intensity was retained in the nucleus compared with that in buffer-treated cells (Fig 6A; LMB, and Fig 6B). The fluorescence intensity in the presence of 2 μM and 10 μM RK424 was 76% and 134% of that in buffer-treated cells, respectively (Fig 6A; RK424, and Fig 6B), indicating that RK424 inhibited the nuclear export activity of NP.

Next, to investigate whether RK424 inhibits nuclear export by preventing NP from binding to CRM1, we examined the binding of endogenous CRM1 to NP in the presence or absence of RK424. CRM1 bound to FLAG agarose beads coupled to the purified NP-Flag protein (NP-Flag beads); however, binding was inhibited in the presence of RK424 (5 μM or 10 μM) and LMB (positive control) (Fig 6C). RK424 inhibited CRM1 binding to NP-Flag beads in a dose-dependent manner.
The small pocket within NP has a unique surface structure
When developing new drugs, identifying potential off-target effects is very important. With this in mind, we superimposed the NP pocket on to other known binding pocket surfaces to identify structural similarities. To do this, we searched two comprehensive databases: Pocket Similarity Search using Multiple-sketches (PoSSuM) [38] and ProBiS [39]. PoSSuM covers 1.8 million known and potential binding sites in the PDB, and ProBiS covers over 29,000 non-redundant proteins in the PDB. These databases allow researchers to compare the protein structure of the test molecule with those in the data base. In addition to the NP Pocket Similarity Search, we also examined the influenza A PA pocket, which is positioned at the active site of the endonuclease domain because this structure is a potential novel antiviral target [40,41,42] (S10A Fig). The ProBiS search identified 20 pocket structures that were similar to the PA structure, some of which are involved in host cell function (S10B and S10C Fig). By contrast, no proteins with a structure similar to the NP pocket were identified, suggesting that the NP pocket has a unique surface structure. Thus, targeting the NP structure rather than the PA structure may be much less likely to cause off-target effects.
The amino acid residues that form the predicted RK424 binding pocket within NP are highly conserved and play essential roles in viral replication
Finally, to demonstrate the accuracy of our docking model and confirm that RK424 binds to the binding pocket within NP, we selected escape mutant viruses by passaging an IAV in the presence of RK424. In this experiment, we selected nucleozin as the positive control, because nucleozin exerts a potent antiviral effect via its ability to cross-link two NP molecules [25]. The structures of the NP inhibitors are listed in S11 Fig. The selection was carried out for a total of four passages with serially increasing concentrations of RK424 and nucleozin. In the cells passaged with RK424, CPE was observed until 3rd passage, but no CPE occurred at the 4th passage. On the other hand, as the number of passages increased, the concentration of nuclozin at which CPE occurred was up to 10 μM by the 4th passage (Fig 7A). Furthermore, viral replication imposed by nucleozin treatment increased as the number of passages increased but viral replication imposed by RK424 treatment decreased (Fig 7B).These results suggest that RK424 makes viral infected cells produce more propagation-deficient virions than fully infectious virions. To confirm this notion, we evaluated the infectivity of RK424 3rd passage virus by comparing the viral titer of this virus normalized to absolute virion numbers (estimated by hemagglutination units; HAU) with that of the initial WT viral titer used in the selection of the escape mutant. The titer of the RK424 3rd passage virus was 1.41×103 PFU/mL per HAU, which was lower than that of the WT (2.19×105 PFU/mL per HAU) (Fig 7C). These results indicate that the number of propagation-deficient virions produced by RK424 3rd passage virus had increased. After four passages, we confirmed sensitivity of RK424 - and nucleozin-selected viruses to 10 μM RK424 and nucleozin (Fig 7D). Nucloezin 4th passage virus exhibited resistance to 10 μM nucleozin but not RK424. However, RK424 3rd passage virus was susceptible to both 10 μM RK424 and nucleozin. These results showed that nucleozin-resistant viruses appeared after four passages with nucleozin but RK424 3rd passage viruses were unable to acquire resistance to RK424. Sequence analysis revealed that nucleozin 4th passage virus had three amino acid mutations (Y52H, Y289H and Y313S) which were previously reported to be result in resistance to nucleozin [25], but no amino acid mutation was detected in RK424 3rd passage virus, which also support the result of sensitivity of RK424 - and nucleozin-selected viruses to RK424 and nucleozin.
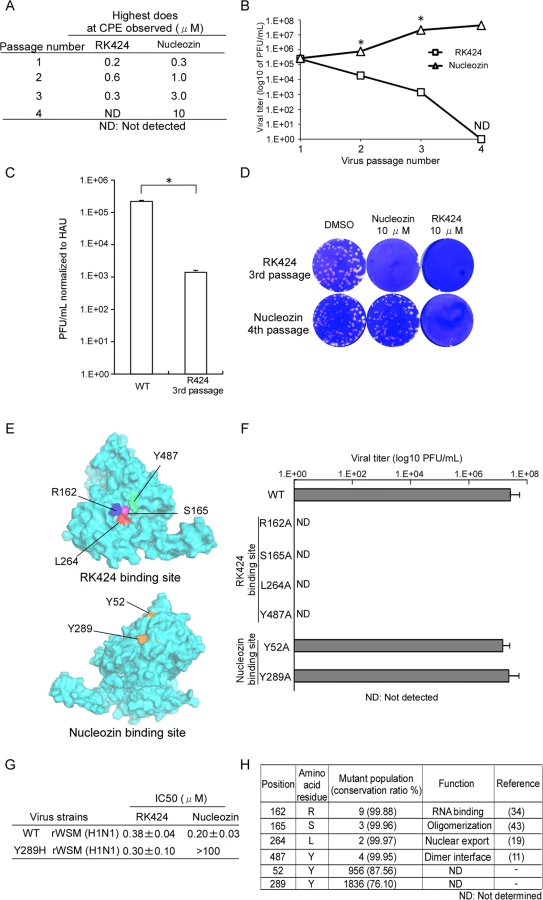
Therefore, we next used a reverse genetics approach to generate mutant viruses harboring an alanine substitution at each potential RK424 binding site within NP (Fig 7E). We successfully generated recombinant wild-type WSN (rWSN) and mutant rWSN viruses harboring Y52H and Y289H mutations within the NP, which have previously been shown to be major mutations resulting in resistance to nucleozin [24,25]. Although these viruses grew well, none of the mutant rWSN viruses harboring alanine substitutions at potential RK424 binding sites were rescued at 72 h after transfection (Fig 7F). Interestingly, we found that although nucleozin did not inhibit the replication of Y289H rWSN, RK424 did (Fig 7G). These results suggest that the binding of RK424 to the amino acids in the NP pocket (or to a site different from that recognized by nucleozin) may inhibit a process that is essential for the viral life cycle. In addition, we investigated the conservation of amino acid residues within the potential RK424 binding site (R162, S165, L264, and Y487), and within the nucleozin binding site (Y52 and Y289), among IAVs of human, avian, and swine origin (Fig 7H). To do this, we used perl script to analyze NP sequences obtained from the NCBI Influenza Virus Resource. The predicted amino acids responsible for RK424 binding showed > 99% conservation among avian, human, and swine IAVs; however, the tyrosine (Y) residues at positions 52 and 289 (which are crucial for nucleozin binding) were poorly conserved. These data indicate that mutations at potential RK424 binding residues are functionally restricted. In actuality, these amino acid residues are reported to regulate various NP functions during the viral life cycle [11, 19, 34, 43]. These findings support the hypothesis that the small pocket within NP may be a promising target for antiviral drugs, particularly a multifunctional NP inhibitor.
Discussion
Here, we identified a novel small pocket structure within NP as a potential target for antiviral drugs. Moreover, the in vivo co-administration (RK424 and oseltamivir phosphate) data and the effectiveness of RK424 against the oseltamivir resistant virus indicated that RK424 inhibited viral replication via a pathway different that of existing antiviral drugs (neuraminidase inhibitors). Further analyses showed that RK424 bound to the small pocket of NP and interfered with three core NP functions: i) the NP-RNA interaction, ii) the NP-NP interaction, and iii) the nuclear export of NP. In addition, we demonstrated that the NP pocket structure comprised three functional domains: the RNA binding groove, the dimer interface, and NES3. Previous reports [11, 12, 34, 43] show that several amino acid residues within the RNA binding groove and dimer interface are essential for NP-RNA and NP-NP interactions. Moreover, Chutiwitoonchai et al. showed that NP-NES3 is the most important NP-NES sequence for nuclear export of NP and viral replication [19]. Taken together, these reports strongly suggest that these three domains regulate the oligomerization and nuclear export of NP, thereby maintaining the structure and function of the vRNP, which plays a key role in viral replication. Moreover, we showed that the identified NP pocket has a surface structure different from that of host cell molecules, suggesting that it may be an ideal drug target. Finally, we generated predicted configurations for a potential RK424 binding site (binding site 1) and showed that RK424 partially overlapped all three functional domains and interacted predominantly with four amino acids residues, R162, S165, L264, and Y487, which play important roles within the three functional domains [11,18,34,43]. According to these binding models, RK424 inhibits the oligomerization and nuclear export of NP, suggesting that the NP pocket is involved in multiple NP functions.
In silico docking studies incorporating RK424 and monomeric NP identified three potential binding sites for RK424 (S6 Fig). Binding site 1 is surrounded by the three different functional domains (S7 Fig), whereas potential binding sites 2 and 3 are not (S8 Fig). The docking model for binding site 2 revealed that RK424 interacts with four amino acids residues: R422, S450, R452, and D455 (S8 Fig). However, these amino acid residues are not well-conserved among influenza virus strains and no relationship between these residues and viral protein function has been reported. The model for potential binding site 3 revealed that RK424 binds to only three residues, R461, G462, and P474; this binding model had the lowest binding energy of all models tested (S8 Fig). Although these three amino acids are well-conserved between species, their functional importance for viral replication is unclear. However, a previous report used mutational analysis and plasmid-driven reverse genetics to show that P474 has no significant role in viral replication [44]. Taken together, these data suggest RK424 is unlikely to bind to sites 2 and 3. By contrast, the predicted NP pocket at potential binding site 1 comprises functionally well-established domains, and the configuration of RK424 at this pocket has high binding energy. The experiments involving RK424 cross-linked beads support the data from the docking model. Moreover, amino acid residues R162, S165, L264, and Y487 are well-conserved and play roles in essential viral protein function. The data derived from these docking models may explain why RK424 shows a broad spectrum of activity against IAV.
The results of the SAR experiments showed that the carboxyl group, the quinoline backbone, and the biphenyl ring of RK424 are essential for its antiviral effects. Fitting these results to configuration 0 (the RK424 binding model) revealed that the NP pocket had the best fitting score. The carboxyl group of RK424 forms a hydrogen bond with R162 within the NP pocket and the phenyl ring derived from the quinolone backbone of RK424 interacts with the L264 side chain via Van der Waals forces. Furthermore, the biphenyl ring of RK424 forms a CH-π hydrogen bond with the Y487 side chain. The S165 side chain does not bind directly to RK424 in this model; however, other configuration models (configuration 1, 2, 3, and 5) suggested a direct interaction between residue S165 and the carboxyl group or the phenyl ring derived from the quinolone backbone of RK424. Moreover, other amino acid sequences, such as F488 and F489, were also predicted to interact with RK424 (S7 Fig). Thus, it is difficult to predict the exact mechanism via which RK424 binds to the NP pocket from this study. Although preliminary data from the potential docking models allow us to speculate as to how RK424 binds to the NP pocket, further studies (e.g., co-crystallization of the NP protein/RK424 complex) are needed to definitively identify the precise interaction model.
The vRNP, comprising the viral genome, polymerase subunits PB2, PB1, and PA, and NP, regulates viral transcription and replication in the host cell nucleus, and is maintained via NP-NP and NP-RNA interactions [11,34,37]. After viral transcription and replication, the vRNP components must be exported from the nucleus to the cytoplasm, where they assemble at the cell membrane prior to budding. This process is crucial for the viral life cycle; therefore, it is a potential target for novel antiviral agents. RK424 inhibited several important processes; namely, the RNA-induced oligomerization and nuclear export of NP. Three of the amino acid residues predicted to be involved in RK424 binding (R162, S165, and Y487) are located within domains involved in NP oligomerization. Residues R162 and S165 are located in the RNA binding groove of NP; however, they play different functional roles. Many positively-charged residues, e.g., arginine (R) and lysine (K), are present in the RNA binding groove, where they interact with negatively-charged RNA molecules. In addition, the electropositive groove between the head and the body domains of NP (NP-G2) contains four arginine residues, R150, R152, R156, and R162 [34]. Mutating these arginine residues to alanine leads to a significant reduction in the affinity of NP for RNA, suggesting that R162 is involved in RNA binding [34]. On the other hand, a previous report showed that the phosphorylation status of residue S165 within NP is closely associated with NP oligomerization. An S165D mutant (which mimics S165 phosphorylation) attenuates the NP-NP and NP-RNA interactions by stabilizing the monomeric structure [43]. In addition, the S165A mutation attenuates vRNA transcription, thereby preventing the recovery of recombinant virus [45]. Moreover, the crystal structure of an ∆402–429 NP mutant suggests a new NP-NP interaction model that does not involve the tail loop binding pocket [12]. This interaction comprised amino acid residues 149–167 and 482–498 of NP. Notably, mutating Y487 attenuates the transcription and replication of vRNA by disrupting the NP-NP interaction [12]. RK424 inhibited NP oligomerization by directly inhibiting the NP-RNA and NP-NP interactions, thereby causing a significant reduction in viral RNA synthesis. This suggests that RK424 may disrupt NP functions that are dependent on residues R162, S165, and Y487. After NP oligomerization, vRNP is exported from the nucleus to cytoplasm via the NS2/NEP and M1 complex pathways [13,14,15,16,17]. Recent studies suggest that nuclear export of NP is closely associated with vRNP export because NP, which has an NES, is a major component of the vRNP [18,20]. NP contains three functional NESs: amino acids 24–49 (NES1), 183–197 (NES2), and 248–274 (NES3) [18]. Export via NES3 is CRM1-dependent whereas export through NES1 and NES2 is CRM-independent [18]. Our previous study indicated that mutating NP-NES1 and NP-NES2 did not alter their cytoplasmic localization, and had only a partial effect on replication kinetics; however, nuclear export of an NES3 mutant was attenuated and mutant virus could not be rescued by reverse genetics [19]. Two hydrophobic residues in particular, L264 and L266, play an essential role in nuclear export, CRM-1 binding, and viral production [19]. The present study showed that LMB (a specific inhibitor of CRM1) inhibited the cytoplasmic localization and nuclear export of NP. These results also suggest that nuclear export of NP is mostly CRM1-dependent. A nuclear export assay using digitonin-permeabilized cells showed that NP was able to move from the nucleus to the cytoplasm by interacting with CRM1, and that RK424 inhibited this process. Interestingly, the present study shows that RK424 directly binds to NP and inhibits its binding to CRM1, whereas LMB covalently binds to S528 within CRM1, thereby inhibiting NP nuclear export by disrupting CRM1 binding to the NES [46]. The docking model described herein showed that RK424 partially obstructs NP NES3 by binding to L264, which is critical for nuclear export. Based on this model, binding of RK424 to the NP pocket may inhibit its nuclear export by interfering with CRM1 binding. Furthermore, nuclear export of NP is closely associated with NP oligomerization [14]. The NES, which comprises many hydrophobic amino acid residues, is presented at the surface of the oligomeric NP [18], and the hydrophobicity of NP increases upon oligomerization; therefore, surface exposure of the NES may be associated with NP oligomerization [47]. Indeed, the alanine substitution mutants, E339A and R416A, completely abrogate NP oligomerization, thereby preventing it from localizing to the cytoplasm [11,18,48]. Thus, by inhibiting NP oligomerization, RK424 may also inhibit the nuclear export of NP.
Predicting possible side effects is an important issue when designing new drugs. The potential utility of a molecule as a targetable drug can be examined by comparing the structure of its target binding pocket with the structures of host proteins [49]. Molecular targets that show high similarity to host molecules may cause severe medical problems; such molecules should not be pursued. The NP pocket structure identified herein showed no similarity to any other molecules in the database. By contrast, the pocket in influenza A PA which regulates endonuclease activity and is a promising target for the development of new anti-influenza drugs [40,41,42], showed high similarity to several host molecules. This suggests that the structure of the NP pocket is unique, making it a more promising target for antiviral target drugs. Drugs that target this binding pocket would be expected to have fewer side effects.
Recently, several compounds targeting NP have been identified as novel antiviral drug candidates, including NLS-binding compounds (e.g., mycalamide analogs), NP salt bridge inhibitors (e.g., compound 3), and NP-aggregation inducers (e.g., nucleozin) [22,23,24,25]. NLS-binding compounds and NP salt bridge inhibitors have weak antiviral effects (sub-micromolar order) because they disrupt only one of the NP functions. On the other hand, nucleozin inhibits multiple NP functions and has a potent antiviral effect (nanomolar order). These findings suggest that developing a drug that inhibits multiple NP functions will be extremely effective because NP is the most abundant multifunctional viral protein in infected cells [50]. However, viruses rapidly develop resistance to nucleozin because its target amino acids (Y52 and Y289) do not play an essential role in the viral lifecycle [24,25]. Furthermore, these residues are not highly conserved among IAVs. In particular, swine influenza viruses harbor a natural Y289H mutation, making them resistant to nucleozin [24]. This is the reason for the rapid appearance of resistant viruses and the limited antiviral spectrum of nucleozin. RK424 inhibited several NP functions and showed potent antiviral effects in vitro against many different subtypes of IAVs including currently circulating A/California/7/2009 (H1N1) virus and the highly pathogenic avian influenza virus strains, A/Ck/Yamaguchi/7/2004 (H5N1) and A/Anhui/1/2013 (H7N9). These results support the findings that RK424 targets conserved amino acids in different virus strains. Indeed, although we were able to generate nucleozin-resistant virus, we were unable to generate the RK424-resistant virus, even after four passages (Fig 7A–7D). The inability to generate resistant virus, coupled with the inability to rescue viruses harboring R162A, S165A, L264A, and Y487A mutations indicates that RK424 inhibits processes that are essential for the viral life cycle. For example, RK424 inhibits NP-RNA binding and the nuclear export of NP. These processes are important for viral genome packaging into virions. Thus, RK424 might prevent viral genome packaging, resulting in the production of propagation-deficient virions lacking viral genome RNA. These observations may also provide reasons why a RK424 resistant virus was not generated. However, further experiments are needed to fully characterize the underlying mechanism(s).
In addition to demonstrating antiviral activity in vitro, we also showed that RK424 exhibited antiviral effects in vivo. Remarkably, significant (~2 log) reductions in viral titers in the lung were observed after day 6 post-infection. However, mice body weight was lower after the administration of 10 mg/kg RK424 than after the administration of 10 mg/kg oseltamivir phosphate, which resulted in no weight loss, and recovery from morbidity was modest in the 10 mg/kg RK424-treated group. Therefore, viral titers may rebound after day 6 post-infection and thus mice may continue to succumb to viral infection. Another group has reported that administration of 50 mg/kg T-705 for 5 days, beginning 1 h post-infection, results in significantly lower(~2 log) viral titers (3.50 log10 PFU/mL) in the lung than in negative control group (5.47 log10 PFU/mL). This was observed after day 6 post-infection, but recovery from morbidity was only 20% of that of the negative control group [51]. These results also suggest that ~2 log reductions in viral titers in the lung after cessation of antiviral compound administration are not enough to prevent viral replication and a rebound in viral titers, which can cause severe morbidity in vivo. The reason for this limited antiviral effect of RK424 in vivo may be because RK424 is highly lipophilic, a property that affects important parameters such as its solubility, absorbency, and stability, all of which affect PK [52]. Thus, the free plasma concentration of RK424 is not sufficient to exert a potent antiviral efficacy in vivo. RK424 has a high plasma protein binding rate because of its high lipophilicity. The efficacy of a drug is affected by the degree to which it exists in a form free from plasma proteins (free drug) within blood plasma. Unbound drugs can traverse cell membranes and become absorbed by target tissues, where they can interact with the drug target and exhibit pharmacologic effects. The plasma protein binding of RK424 is 99.6%, which means only 0.4% of the drug exists in free form in blood plasma. The Cmax of RK424 (39200 ng/mL) is more than 200 times higher than the IC50 concentration used in vitro (0.5 μM = 162.7 ng/mL), but the free drug maximum concentration (Cfree max) was 156.8 ng/mL because almost all of the drug was bound to plasma proteins. Taking into account this property, the free plasma exposure of RK424 was adjusted to obtain the in vitro IC50 (0.5 μM = 162.7 ng/mL) concentration by the intraperitoneal administration of 10 mg/kg RK424 twice per day, but the antiviral effect was limited. This may mean that it will be necessary to increase the plasma free drug concentration to arrive at the in vitro IC90 concentration (1.2 μM = 390.5 ng/mL). However, the intraperitoneal administration of RK424 at 10 mg/kg did not result in the in vitro IC90 concentration. We could not increase the dose of RK424 further, because RK424 precipitated from the viecle solution. The oral administration route provides a way to administer a drug with low solubility to animals. However, to achieve and maintain the in vitro IC90 concentration (1.2 μM = 390.5 ng/mL) in plasma via oral administration, multiple does of around 100 mg/kg RK424 would be needed because the bioavailability and t1/2 of oral administration are lower than those obtainable by intraperitoneal administration. Indeed, to design a multiple dosing regimen precisely, the PK study has to be done as a multiple dose of RK424. Moreover, the Vd (583mL/kg) parameter, which is lower than mouse total body water (725 mL/kg), suggests that RK424 may not be efficiently absorbed into target tissues. In conclusion, the PK properties of RK424 are insufficient to exert an antiviral effect in vivo. To acquire antiviral activity in vivo, the pharmacologic parameters of RK424 will need to be optimized by introducing changes in its chemical structure.
To summarize, the results presented herein highlight a novel strategy for developing new drugs that target the NP pocket; such drugs may have multifunctional effects and reduce the emergence of drug-resistant viruses. One possible approach to developing a potent antiviral drug targeting the NP pocket is to optimize the structure of RK424. This could be achieved by replacing its aromatic rings, the cause of its high lipophilicity, with aromatic heterocyles or by introducing side chains modified with hydrophilic groups, as long as these changes do not affect its antiviral activity. In addition to this approach, target-oriented drug screening may be a more desirable approach because the NP pocket has a very unique surface structure and the amino acid residues within this pocket are highly conserved. These characteristics make the NP pocket an attractive target for drug development. Thus, another promising strategy is computational virtual screening, which is a useful way of identifying small molecular compounds with backbone structures that differ from those of RK424.
Materials and Methods
Compounds
RK424 was purchased from Enamine Ltd (Kiev, Ukraine). RK424 analogs were synthesized by Wakunaga Pharmaceutical Co. Ltd. (Akitakata, Japan) and a stock solution (10 mM) was prepared in dimethyl sulfoxide (DMSO). Oseltamivir phosphate was purchased from Sundia MediTech Company, Ltd (Shanghai, China) and a stock solution was prepared in DMSO. Leptomycin B (LMB) was purchased from Sigma (St. Louis, MO). LMB was dissolved in ethanol (EtOH) and then diluted in DMSO.
Cells, transfection, and viruses
Madin-Darby canine kidney (MDCK) cells, HeLa cells, and human embryonic kidney 293T (HEK293T) cells from the repository of our laboratory, were grown in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen, Carlsbad, CA) containing Pen Strep Glutamine (PSG, GIBCO Industries Inc., Los Angeles, CA) and 10% fetal bovine serum (Sigma). Cells were cultured at 37°C in 5% CO2. Transfections were performed using FuGENE HD (Roche Diagnostics, Basel, Switzerland) according to the manufacturer's instructions. The influenza A viruses were propagated in MDCK cells at 37°C for 48 h in 5% CO2.
Plasmid construction
The mammalian expression vector, pCAGGS, encoding monomeric red fluorescent protein (mRFP) fused to a Flag tag (mRFP-Flag), wild-type NP fused to mRFP-Flag tag (NP-mRFP-Flag), and wild-type NP fused to a Flag tag (NP-Flag) have been described previously [19,22]. The viral genome-expressing plasmids PB2/pHH21, PB1/pHH21, PA/pHH21, HA/pHH21, NP/pHH21, NA/pHH21, M/pHH21, NS/pHH21, and empty-pHH21, the expression plasmids PB1/pCAGGS, PB2/pCAGGS, PA/pCAGGS and NP/pCAGGS, and the vNP-luc/pHH21 plasmid (derived from the A/WSN/1933 (H1N1) virus) were kind gifts from Dr. Y Kawaoka (University of Tokyo). The plasmid encoding wild-type NP fused to a HA tag (NP-HA) was generated by polymerase chain reaction (PCR) using primers XhoI-NP (F), 5’-AAACTCGAGATGGCGACCAAAGGCACCAA-3’ and NotI-HA-NP(R), 5’-TAGCGGCCGCTTAAGCGTAATCTGGAACATCGTATGGGTAATTGTCGTACTCCTCTGCAT-3’ (restriction enzyme sites underlined) and NP/pCAGGS as a template. The PCR product was cloned into the XhoI and NotI sites within pCAGGS. The plasmid encoding wild-type NP fused to glutathione S-transferase (GST) (NP-GST) was generated by PCR using primers EcoRI-NP (F), 5’-GGGAATTCCATGGCGACCAAA-3’ and Xho1-GST-NP (R), 5’-AACTCGAGTTAATTGTCGTACTCCTCTGC-3’ (restriction enzyme sites underlined) and NP/pCAGGS as the template. The PCR product was cloned into the EcoRI and XhoI sites within pGEX6p-3 (GE Healthcare, Buckinghamshire, UK). Plasmids NP/pHH21 and NP/pCAGGS harboring site specific mutations (Y52H, R162A, S165A, L264A, Y289H, and Y487A) were created by Prime STAR Max DNA Polymerase (Takara Bio, Otsu, Japan) using the following primers Y52H-(F), 5’-AGTGATCATGAGGGACGGCTGATTCAG-3’ and Y52H-(R), 5’-TCCCTCATGATCACTGAGTTTAAGTTC-3’; R162A-(F), 5’-GATCCCGCGATGTGCTCACTGATGCAG-3’ and R162A-(R), 5’-GCACATCGCGGGATCCATTCCTGTGCG-3’; S165A-(F), 5’-ATGTGCGCACTGATGCAGGGCTCAACC-3’ and S165A-(R), 5’-CATCAGTGCGCACATCCTGGGATCCAT-3’; L264A-(F), 5’-TCTGCAGCCATATTGAGAGGGTCAGTT-3’ and L264A-(R), 5’-CAATATGGCTGCAGACCGTGCTAAAAA-3’; Y289H-(F), 5’-AGTGGACACGACTTTGAAAGAGAGGGA-3’ and Y289H-(R), 5’-AAAGTCGTGTCCACTGGCTACGGCAGA-3’; and Y487A-(F), 5’-GGATCTGCTTTCTTCGGAGACAATGCA-3’ and Y487A-(R), 5’-GAAGAAAGCAGATCCTTCATTACTCAT-3’ (point mutated sites underlined).
A pCAGGS plasmid encoding a Flag-tagged mutant NP (mutant NP-Flag) harboring all R162A, S165A, L264A, and Y487A mutations was generated using the method described above. All mutations were confirmed by DNA sequencing.
In vitro antiviral activity assay
The antiviral activity of the different compounds was measured in a plaque assay as described previously [26]. Briefly, MDCK cells were inoculated with IAV at a multiplicity of infection (MOI) of 0.008. After washing, the cells were incubated in 2 mL of minimum essential medium (MEM) (2 ×) containing 1% agarose, 1 μg/mL trypsin (Worthington Biochemical Corporation), and each of the test compounds. Cells were then incubated at 37°C for 48 h in a 5% CO2 incubator. The number of plaques was counted and the IC50 calculated.
Cell toxicity assay
Cell toxicity was measured in a water soluble tetrazolium salt-1 (WST-1) (Takara Bio) assay as previously described [26]. The 50% cytotoxic concentration (CC50) was calculated by comparing the viability of RK424-treated cells with that of DMSO-treated cells.
Ethics statement
All animal experiments were approved by RIKEN Institutional Animal Use and Care Administrative Advisory Committees and were performed in accordance with RIKEN institutional guidelines and regulations (Certificate number: H23-2-101). All procedures were performed under pentobarbital treatment (30–50 mg/kg), and all efforts were made to minimize suffering. Mice were euthanized at the endpoint (14 days after virus inoculation) by intraperitoneal injection of pentobarbital (more than three times the dosage of pentobarbital for anesthesia). Mice were monitored and their body weight were measured daily during the study. Mice were euthanized if rapid and lasting weight loss (loss of more than 20% of body weight in a few days) was observed (a humane endpoint).
In vivo antiviral activity assay
RK424 was dissolved in polyethylene glycol 400 (PEG400; Sigma) and then diluted with PBS to a final concentration of 1 mg/mL or 2 mg/mL. Six-week-old Balb/c mice (CREA Japan Inc, Tokyo, Japan) were infected intranasally with ten 50% lethal doses (LD50) of A/WSN/1933 (H1N1). Mice (eight per each RK424 treated-group) were intraperitoneally injected with RK424 2 h before virus exposure and then twice per day (2 × 5 mg/kg and 2 × 10 mg/kg) for 5 days beginning on the day of infection. Seven mice per positive control group also received 2 × 10 mg/kg of oseltamivir phosphate per day. Seven mice per negative control group received PBS. Bodyweight was measured daily and survival evaluated. Three mice from the negative control group and the 10 mg/kg RK424 group were euthanized on day 6 post-infection and virus titers in the lungs were measured in a plaque assay. Serial diluted oseltamivir phosphate was administered to three mice per each group (2 × 0.01, 2 × 0.03, 2 × 0.1, 2 × 0.3 and 2 × 1 mg/kg oseltamivir phosphate) twice per day according to the same protocol mentioned above and appropriate doses of oseltamivir phosphate were estimated for co-administration with RK424. In the co-administration study, 2 × 0.02 mg/kg oseltamivir phosphate together with 2 × 5 mg/kg or 2 × 10 mg/kg of RK424 were also administered twice per day in the same way and synergetic antiviral effect was evaluated with respect to mono therapy (0.02 mg/kg of oseltamivir phosphate or 5 or 10 mg/kg RK424) and the negative control group (PBS). Seven mice per each group were tested in the co-administration study.
Plasma concentration and pharmacokinetic parameters of RK424
These studies were performed at Nemoto Science Co., Ltd. (Tsukuba, Japan). RK424 sufficient to achieve 1 mg/kg was dissolved in N,N - Dimethylacetamide (Wako Pure Chemical Industries, Ltd., Osaka, Japan) and isotonic sodium chloride solution (Otsuka Pharmaceutical Factory, Inc., Naruto, Japan) and was intravenously administrated; RK424 sufficient to achieve 10 mg/kg was dissolved in PEG 400 (Sigma) and was intraperitoneally administrated; and RK424 sufficient to achieve 10 mg/kg was dissolved in 0.5% methyl cellulose (Wako Pure Chemical Industries, Ltd.) solution and was orally administrated to eight-week-old Balb/c mice (CHARLES RIVER LABORATORIES JAPAN, INC., Yokohama, Japan). Three mice were assigned to each experimental group. Venous blood samples were collected at 0, 0.25, 0.5, 1, 2, 4, 6 and 24 h after intraperitoneal administration and collected at 0, 0.25, 0.5, 1, 2, 6 and 24 h after intravenous and oral administration. The blood samples were deproteinized and subjected to LC/MS/MS (ACQUITY UPLC system and Xevo TQ MS; Nihon Waters K.K., Tokyo, Japan) analyses. Pharmacokinetic parameters (Cmax, Tmax, AUC0–24 h, t1/2, Vd, and relative bioavailability) were calculated using Winnolin Ver.6.1 (Pharsight Corporation, CA).
Plasma protein binding of RK424
These studies were performed at Wakunaga pharmaceutical Co., Ltd. (Akitakata, Japan).
Mouse plasma was prepared from seven-week-old mouse blood samples (CRJ, Yokohama, Japan). RK424 (50 μM) solution was prepared by mixing 5 mM RK424 in DMSO with mouse plasma and incubating the mixture at 37°C for 30 min. RK424 free from plasma protein was separated by centrifugal filtration using 10 kDa size cut filter (MultiScreen Ultracel-10; Merck Millipore, Darmstadt, Germany). The filtrate sample was measured by ACQUITY Ultra Performance Liquid chromatography (UPLC) system (Waters, Milford, MA) and the plasma protein binding rate was calculated.
Mini-genome assay
HEK293T cells were cultured in 24 well plates at 37°C for 24 h. Cells were transfected with plasmid (PB2/pCAGGS, PB1/pCAGGS, PA/pCAGGS, or NP/pCAGGS) DNA (0.5 μg) and a luciferase RNA expression (vNP-luc/pHH21) vector. Cells were treated (or not) with the test compounds and luciferase activity was measured according to manufacturer’s instructions after incubation at 37°C for 48 h.
Generation of recombinant viruses
Recombinant viruses were generated by DNA transfection as described previously [19]. Briefly, a co-culture of MDCK (4 × 105) and HEK-293T (6 × 105) cells was transfected with the eight viral genome-expressing plasmids or with the four viral protein-expressing plasmids. At 72 h post-transfection, recombinant viruses were harvested and the virus titer in the cell supernatants was determined in a plaque assay.
Indirect immunofluorescence
HeLa cells were seeded on coverslips and grown to 60–70% confluence. Cells were then transfected with 0.5 μg of NP/pCAGGS or infected with A/WSN/1933 (H1N1) virus at an MOI of 10 in the presence or absence of the test compounds. After 48 h of transfection or 6 h of infection, cells were fixed with 4% paraformaldehyde for 20 min at room temperature (RT) and then further fixed with cold methanol for 10 min at −20°C. Cells were washed three times with PBS and then incubated with a primary anti-NP monoclonal antibody (MAb) (Santa Cruz Biotechnology Inc., Santa Cruz, CA; diluted 1 : 50 in PBS) for 1 h at RT. The cells were then washed with PBS and incubated with an Alexa Fluor 488-conjugated goat secondary antibody against rabbit IgG (H+L, Invitrogen: 1 : 500 in PBS) for 1 h at RT. The cells were then incubated with PBS containing Hoechst 33342 for 5 min and then washed three times with PBS. The excitation and emission wavelengths were set at 488 nm and 519 nm, respectively. Prepared samples were observed under a confocal laser scanning microscope (FV 1000; Olympus, Tokyo, Japan).
Nuclear export assay
HeLa cells were attached to glass cover slips at 37°C for 24 h and then transiently transfected with NP-mRFP-Flag/pCAGGS. After 48 h, cells were washed twice with cold transport buffer (20 mM HEPES (pH 7.4), 110 mM potassium acetate, 2 mM magnesium acetate, 1 mM EGTA (pH 7.4), 2 mM DTT, 1 μg/mL aprotinin, 1 μg/mL leupeptin, and 1 μg/mL pepstatin) and permeabilized with digitonin (50 μg/mL; Sigma) in transport buffer for 5 min on ice. The cells were then washed with cold transport buffer and incubated with apyrase (10 U/mL; Sigma) for 5 min on ice. After washing twice with cold transport buffer, the cells were incubated in cold transport buffer for 10 min on ice before addition of a the reaction mixture (40 μg HeLa cell lysate, 10 mg/mL bovine serum albumin (BSA), 10 mM ATP, 5 mM creatine phosphate, 0.64 U creatine kinase) containing (or not) the test compounds. The mixture was the incubated at 30°C for 1 h. After washing three times with cold transport buffer, mRFP fluorescence was observed under a confocal laser scanning microscope (FV 1000; Olympus). The fluorescence intensity in the nuclei was analyzed with MetaMorph software (Molecular Devices Inc., Downingtown, PA). The fluorescence intensity of NP-mRFP-Flag was quantified in >100 nuclei in each of three independent experiments.
Purification of NP proteins fused to different tags
HEK293T cells were transfected with 10 μg of NP-Flag/pCAGGS, NP-HA/pCAGGS, NP-mRFP-Flag/pCAGGS, or mRFP-Flag/pCAGGS for 48 h. The cells were then washed with PBS and lysed in 500 μL of lysis buffer (10 mM Tris-HCl (pH 7.8), 150 mM NaCl, 1 mM EDTA and 1% NP-40) at 4°C for 1 h. The lysates were centrifuged at 20,000 × g for 10 min and the supernatants collected and mixed with ANTI-FLAG M2 agarose beads (Sigma) or ANTI-HA agarose beads (Sigma). After incubating at 4°C for 24 h, the beads were washed three times with wash buffer (10 mM Tris-HCl (pH 7.4), 150 mM NaCl, and 0.05% NP-40). The proteins were purified by adding 3 × Flag peptide (Sigma) or HA peptide (Sigma). The purified NP-Flag and NP-HA proteins were then subjected to 10% SDS-PAGE and analyzed by western blotting. FLAG agarose beads coupled to mRFP-Flag, NP-mRFP-Flag, and NP-Flag-bound were used to analyze the interactions between NP and CRM1, or between NP and NP, before elution with the 3 × Flag peptide.
Photo-cross-linked small-molecule RK424 affinity beads
RK424 was cross-linked to Sepharose beads as previously described [32]. Briefly, N-hydroxysuccinimide-activated beads were washed three times with 1 mM aqueous HCl followed by coupling solution (0.1 M NaHCO3 and 50% dioxane mixture). A solution of photoaffinity linker in coupling solution was then added to the beads, which were then incubated at 37°C for 2 h on a rotor. After washing five times with coupling solution, the beads were blocked with 1 M ethanolamine in 0.1 M Tris–HCl (pH 8.0) buffer at 37°C for 1 h on a rotor. The beads were then placed in a spin column and washed three times with Milli-Q water and methanol before being transferred to a glass sample vial. A methanol solution containing RK424 was then added to the beads and the mixture concentrated and dried in vacuo. The beads were then irradiated in a UV cross-linker at 365 nm (4 J/cm2) and washed with methanol to yield RK424-cross-linked affinity beads. Purified NP-Flag proteins were added to 20 μL of RK424-cross-linked affinity beads or uncross-linked beads. After incubating at 4°C for 24 h, the beads were washed five times with wash buffer and bound proteins eluted in 10% SDS-PAGE sample buffer at 100°C for 5 min. The proteins that bound to RK424 were then separated in 10% SDS–PAGE gels and detected by western blotting with an anti-Flag M2 MAb (Sigma).
Binding of CRM1 to FLAG agarose beads coupled to NP-Flag protein (NP-Flag beads)
NP-Flag beads were incubated with HeLa cell lysate and different concentrations of RK424. After incubation at 4°C for 24 h, the NP-Flag beads were washed five times with wash buffer and the bound proteins separated in 10% SDS-PAGE gels. Binding of CRM1 to NP-Flag beads was detected by western blotting with an anti-CRM1 MAb (BD Biosciences, Franklin Lakes, NJ). LMB was used as positive control (LMB inhibits the binding of NP to CRM1). EtOH and DMSO were used as vehicle controls for LMB and RK424, respectively.
Surface plasmon resonance (SPR) analysis of the NP-RNA interaction
A biotinylated 2-O-methylated RNA oligonucleotide (5-UUU GUU ACA CAC ACA CAC GCU GUG-3) was purchased from Hokkaido System Science CO., Ltd. (Sapporo, Japan) and immobilized on an streptavidin-coated sensor chip (GE Healthcare) at a surface density of 100 resonance units (RU), according to manufacturer’s instructions The inhibitory effect of RK424 to NP-RNA binding was examined by incubating 10 ng/μL NP-FLAG proteins with RK424 at RT for 1 h. The RK424-treated samples were then injected onto the sensor chip surface and the SPR signal measured in a Biacore T-100 (GE Healthcare). Samples were injected in running buffer (200 mM NaCl, 20 mM Tris-HCl (pH 7.4), and 0.05% Tween 20) at 25°C.
Analysis of the NP-NP interaction
FLAG agarose beads coupled to mRFP-Flag (mRFP-Flag beads), NP-mRFP-Flag (NP-mRFP-Flag beads), and purified NP-HA proteins were treated with RNase A (final concentration of 200 μg/ml) at RT for 10 min followed by a further incubation at 4°C overnight. The purified NP-HA proteins were then incubated with RK424 at 4°C for 1 h. Purified NP-HA proteins were then added to NP-mRFP-Flag beads or mRFP-Flag beads and incubated overnight at 4°C. The incubated beads were washed five times with wash buffer and then subjected to 10% SDS-PAGE. Binding of NP-HA to mRFP-Flag or NP-mRFP-Flag was detected by western blotting with an anti-HA Mab (MBL, Nagoya, Japan). An anti-Flag M2 Mab (Sigma) was used as a loading control.
In vitro transcription and vRNA synthesis
vRNA encoding the M gene was generated using a RiboMAX Large Scale RNA Production System-T7 (Promega, Medison, WI). Briefly, approximately 1027 bp of the A/WSN/1933 (H1N1) virus negative-sense segment 7 genome was amplified from an M/pHH21 plasmid by PCR and the products purified using a QIAquick PCR purification kit (Qiagen, Valencia, CA). Five μg of the purified PCR product were added to a reaction mixture containing transcription buffer, rNTP mix (25 mM ATP, CTP, GTP and UTP), and T7 enzyme mix (final volume 100 μL) and incubated at 37°C for 4 h. Synthesized vRNA was treated with 5 units of RQ1 RNAse-Free DNase I (Promega) at 37°C for 15 min. The following primers were used to amplify the negative-sense segment 7 genome: T7_WSN_seg7-(F), 5’-GACTCAGTTAATACGACTCACTATATAGTTTTTTACTCCAGCTCTATGTTG-3’ and T7_WSNseg7-(R), 5’-AGCAAAAGCAGGTAGATATTG-3’ (T7 promoter sequence underlined).
Size exclusion chromatography to assess oligomerization
E. coli strain BL21 CodonPlus (DE3)-RIL (Stratagene, La Jolla, CA) harboring the GST-NP/pGEX6p-3 plasmid was cultured in the presence of 1 mM isopropyl-b-D-1 thiogalactopyranoside (IPTG) for 10 h at 22°C and then collected by centrifugation. The cell pellets were resuspended and sonicated in lysis buffer (50 mM Tris-HCl (pH 7.4), 200 mM NaCl, 1% Triton X-100, 1 mg/mL lysozyme, and 0.15 mg/mL RNAse A). Glutathione Sepharose 4 Fast Flow beads (GSH beads; GE Healthcare) were then added to the cell lysate and rotated overnight at 4°C. After washing the beads, bound proteins were released from the beads using PreScission Protease (GE Healthcare). The GST-cleaved NP proteins were then incubated with Heparin Sepharose 6 Fast Flow beads (GE Healthcare) to dissociate RNA from NP. The bound NP proteins were then eluted with elution buffer (1.5 M NaCl and 50 mM Tris-HCl (pH 7.4)). The eluted samples were concentrated to 1 mg/mL and dialyzed into lower salt buffer (100 mM NaCl and 50 mM Tris-HCl (pH 7.4)) using an Amicon Ultra 50K device (Merck Millipore). The buffer exchanged proteins were maintained at 4°C to allow the equilibrium to shift from oligomers to monomers. The prepared NP proteins were then rotated with RK424 and vRNA at RT for 60 min. Samples were then loaded onto a Superdex 200 Increase 10/300 GL Column (GE Healthcare) attached to an ÄKTA purifier chromatography system (GE Healthcare). The size distribution data and integrated peak areas were calculated using the instruments software (UNICORN ver. 5.1; GE Healthcare).
RNA FISH analysis
RNA FISH was performed according as previously described [26,29]. The probes targeting the PB2 segment were designed by STELLARIS RNA FISH PROBE DESIGNER and purchased from Biosearch Technologies, Inc. (Novato, CA). One set of probes contained a blend of 48 oligos, which comprised 20 mer single-stranded DNAs, each labeled with Quasar 670. The primer sequences are listed in S3 Table. MDCK cells (2 × 105 cells/well) were seeded onto coverslips in 12-well plates and incubated overnight at 37°C. At 6 h post-infection with A/WSN/1933 (H1N1) at an MOI of 5 in the absence or presence of RK424, the cells were washed once with PBS and then fixed with 4% paraformaldehyde in PBS for 10 min at RT. After a brief wash with PBS, the cells were permeabilized in 0.5% Triton X-100 in PBS for 1 min at RT. Prior to hybridization, the cells were washed with PBS and incubated for 5 min in 2 × SSC (300 mM sodium chloride, 30 mM sodium citrate) containing 10% formamide. To detect viral RNAs, 2 μM of labeled probes in 15 μl of hybridization buffer (10% dextran sulfate, 2 mM vanadyl ribonucleoside complexes (VRC; New England BioLabs), 0.02% BSA, 50 mg E. coli tRNA, 2 × SSC, and 10% formamide) was added to each sample. Hybridization was performed in a humidified chamber maintained at 37°C for 16 h. The samples were then washed twice for 30 min at 37°C with 10% formamide containing 2 × SSC supplemented with 2 mM VRC. Nuclei were stained with Hoechst 33342 and the cells were washed with PBS. Prepared samples were observed under a confocal laser scanning microscope (FV 1000; Olympus).
Generation of escape viruses by in vitro passaging an IAV in the presence of antiviral compound
MDCK cells were cultured in 12 well plate at 37°C for 24 h. MDCK cells were infected with A/WSN/1933 (H1N1) strain at 100 PFU/well and cultured in the presence of serial dilutions of RK424 or nucleozin. Viral replication was monitored for one week to observe any cytopathic effect (CPE) in the culture plates. The supernatants were collected from wells treated with the highest concentration of compound in wells in which CPE was observed and then stored at -80°C for subsequent analysis. Virus was serially passaged by using 5 μL of viral stock from the preceding passage to infect fresh MDCK cells in the presence of increasing concentrations of compound. The compound concentrations used in the selection varied, depending on the highest concentration of compound at which CPE was observed in the preceding passage. These passages led to the generation of mutant viruses resistant to RK424 or nucleozin when mutations occurred in the viral protein targeted by the antiviral compound. The selection was carried out for a total of four passages with RK424 or nucleozin. RK424 concentrations ranged from 0.1–2 μM at 1st passage, 0.2–3 μM at 2nd passage, 0.3–3μM at 3rd passage and 0.4–3 μM at 4th passage. Nucleozin concentrations ranged from 0.1–2 μM at 1st passage, 0.3–3 μM at 2nd passage, 1–5 μM at 3rd passage and 3–10 μM at 4th passage. After the 4th passage, drug sensitivity was confirmed by using the plaque reduction assay with 10 μM RK424 or nucleozin as mentioned above.
Sequence analysis of the NP coding regions in the escape mutant
Viral RNA was isolated from the plaque-purified viruses at 3rd passage with RK424 and 4th passage with nucleozin using Trizol LS reagent (Invitrogen) according to the manufacturer’s protocol. Reverse transcription (RT)-PCR was carried out with the SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen) according to the manufacturer’s protocol using a minus-strand-specific universal primer; Uni12 (F), 5’-agcaaaagcagg-3’. The coding region of the NP sequence in the RT product was amplified with the above described EcoRI-NP (F) and Xho1-GST-NP (R) primers by using KOD-Plus-(Toyobo, Osaka, Japan) according to the manufacturer’s protocol. The PCR-amplified product was directly sequenced on an ABI3730xl DNA Analyzer using an ABI PRISM BigDye Terminator v 3.1 Ready Reaction Cycle Sequencing Kit (Applied Biosystems, Foster City, CA).
Calculation of the conservation ratio
Complete sequences for IAV NP were downloaded using the advanced database search at the NCBI’s Influenza Virus Resource [53]. Avian, human, and swine sequences were obtained using the following parameters: type A, full length only, and whole collection date. The MAFFT (version 6) program was used to align the sequences from both groups using a FFT-NS-2 strategy [54]. The 3,759 human-, 3,273 avian-, and 651 swine-origin NP sequences were then used to calculate the amino acid conservation ratio.
Statistical analysis
The two-tailed t-test and the log-rank test were used for statistical analysis. P-values <0.05 were considered statistically significant.
Docking analysis of RK424 and NP
In silico analysis of RK424 and NP binding was performed using AutoDock (version 4.2) [31]. The steric structure of monomeric influenza A/WSN/1933 (H1N1) NP was derived from a NP multimer structure deposited in the PDB (PDB ID: 2IQH). The NP structure was pre-processed, i.e., hydrogen atoms were added prior to docking simulation. During docking simulation, the optimal conformations for each ligand-receptor binding state were retreated in spacious searching space. Subsequent analysis of the interaction between RK424 and NP was conducted using MOE version 2013.0801 [55].
Search for molecules with binding pockets structurally similar to the NP pocket
The pocket structures of NP and PA were extracted from complete NP (PDB ID: 2IQH) and PA (PDB ID: 4E5E) structures by removing residues within 10 Å of the small pocket (R162, S165, L264, and Y487) and the endonuclease motif (P107, D108, E119, and K134), respectively. Protein structure libraries (PoSSuM [38] or ProBiS [39]) were then searched for structures similar to the NP and PA pockets.
Accession numbers
The accession numbers for genes and proteins mentioned in this study are shown below. PB2 (GenBank Accession No. J0217), PB1 (GenBank Accession No.J02178), PA (GenBank Accession No. X17336), HA (GenBank Accession No. J02176), NP (GenBank Accession No. CY010791), NA (GenBank Accession No. L25817), M (GenBank Accession No. L25818), NS (GenBank Accession No. M12597), firefly luciferase (GenBank Accession No. AAA89082.1), mRFP (GenBank Accession No. AF506027), and GST (GenBank Accession No. AAB37352).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Taubenberger JK, Kash JC (2010) Influenza virus evolution, host adaptation, and pandemic formation. Cell Host Microbe 7 : 440–451. doi: 10.1016/j.chom.2010.05.009 20542248
2. Hay AJ, Wolstenholme AJ, Skehel JJ, Smith MH (1985) The molecular basis of the specific anti-influenza action of Amantadine. EMBO J 4 : 3021–3024. 4065098
3. Moscona A (2008) Medical management of influenza infection. Annu Rev Med 59 : 397–413. 17939760
4. Bright RA, Shay DK, Shu B, Cox NJ, Klimov AI (2006) Adamantane resistance among influenza A viruses isolated early during the 2005–2006 influenza season in the United States. JAMA 295 : 891–894. 16456087
5. Hurt AC, Ernest J, Deng YM, Iannello P, Besselaar TG, et al. (2009) Emergence and spread of oseltamivir-resistant A(H1N1) influenza viruses in Oceania, South East Asia and South Africa. Antiviral Res 83 : 90–93. doi: 10.1016/j.antiviral.2009.03.003 19501261
6. Dharan NJ, Gubareva LV, Meyer JJ, Okomo-Adhiambo M, McClinton RC, et al. (2009) Infections with oseltamivir resistant influenza A(H1N1) virus in the United States. JAMA 301 : 1034–1041. doi: 10.1001/jama.2009.294 19255110
7. Furuta Y, Takahashi K, Kuno-Maekawa M, Sangawa H, Uehara S, et al. (2005) Mechanism of action of T-705 against influenza virus. Antimicrob Agents Chemother 49 : 981–986. 15728892
8. Manns MP, Wedemeyer H, Cornberg M (2006) Treating viral hepatitis C: efficacy, side effects, and complications. Gut 55 : 1350–1359. 16905701
9. Klumpp K, Ruigrok RW, Baudin F (1997) Roles of the influenza virus polymerase and nucleoprotein in forming a functional RNP structure. EMBO J 16 : 1248–1257. 9135141
10. Arranz R, Coloma R, Chichon FJ, Conesa JJ, Carrascosa JL, et al. (2012) The structure of native influenza virion ribonucleoproteins. Science 338 : 1634–1637. doi: 10.1126/science.1228172 23180776
11. Ye Q, Krug RM, Tao YJ (2006) The mechanism by which influenza A virus nucleoprotein forms oligomers and binds RNA. Nature 444 : 1078–1082. 17151603
12. Ye Q, Guu TS, Mata DA, Kuo RL, Smith B, et al. (2012) Biochemical and structural evidence in support of a coherent model for the formation of the double-helical influenza A virus ribonucleoprotein. MBio 4: e00467–00412. doi: 10.1128/mBio.00467-12 23269829
13. Yasuda J, Nakada S, Kato A, Toyoda T, Ishihama A (1993) Molecular assembly of influenza virus: association of the NS2 protein with virion matrix. Virology 196 : 249–255. 8356796
14. Akarsu H, Burmeister WP, Petosa C, Petit I, Müller CW, et al. (2003) Crystal structure of the M1 protein-binding domain of the influenza A virus nuclear export protein (NEP/NS2). EMBO J 22 : 4646–4655. 12970177
15. Shimizu T, Takizawa N, Watanabe K, Nagata K, Kobayashi N (2011) Crucial role of the influenza virus NS2 (NEP) C-terminal domain in M1 binding and nuclear export of vRNP. FEBS Lett 585 : 41–46. doi: 10.1016/j.febslet.2010.11.017 21081124
16. Ye Z, Liu T, Offringa DP, McInnis J, Levandowski RA (1999) Association of influenza virus matrix protein with ribonucleoproteins. J Virol 73 : 7467–7473. 10438836
17. Noton SL, Medcalf E, Fisher D, Mullin AE, Elton D, et al. (2007) Identification of the domains of the influenza A virus M1 matrix protein required for NP binding, oligomerization and incorporation into virions. J Gen Virol 88 : 2280–2290. 17622633
18. Yu M, Liu X, Cao S, Zhao Z, Zhang K, et al. (2012) Identification and characterization of three novel nuclear export signals in the influenza A virus nucleoprotein. J Virol 86 : 4970–4980. doi: 10.1128/JVI.06159-11 22345439
19. Chutiwitoonchai N, Kakisaka M, Yamada K, Aida Y (2014) Comparative Analysis of Seven Viral Nuclear Export Signals (NESs) Reveals the Crucial Role of Nuclear Export Mediated by the Third NES Consensus Sequence of Nucleoprotein (NP) in Influenza A Virus Replication. PLoS One 9: e105081. doi: 10.1371/journal.pone.0105081 25119991
20. Elton D, Simpson-Holley M, Archer K, Medcalf L, Hallam R, et al. (2001) Interaction of the influenza virus nucleoprotein with the cellular CRM1-mediated nuclear export pathway. J Virol 75 : 408–419. 11119609
21. Aida Y, Sasaki Y, Hagiwara K (2012) Discovery of Novel Antiviral Agents Directed Against the Influenza A Virus Nucleoprotein. Antiviral Drugs—Aspects of Clinical Use and Recent Advances; 99–120.
22. Hagiwara K, Kondoh Y, Ueda A, Yamada K, Goto H, et al. (2010) Discovery of novel antiviral agents directed against the influenza A virus nucleoprotein using photo-cross-linked chemical arrays. Biochem Biophys Res Commun 394 : 721–727. doi: 10.1016/j.bbrc.2010.03.058 20230782
23. Shen YF, Chen YH, Chu SY, Lin MI, Hsu HT, et al. (2011) E339…R416 salt bridge of nucleoprotein as a feasible target for influenza virus inhibitors. Proc Natl Acad Sci U S A 108 : 16515–16520. doi: 10.1073/pnas.1113107108 21930946
24. Kao RY, Yang D, Lau LS, Tsui WH, Hu L, et al. (2010) Identification of influenza A nucleoprotein as an antiviral target. Nat Biotechnol 28 : 600–605. doi: 10.1038/nbt.1638 20512121
25. Gerritz SW, Cianci C, Kim S, Pearce BC, Deminie C, et al. (2011) Inhibition of influenza virus replication via small molecules that induce the formation of higher-order nucleoprotein oligomers. Proc Natl Acad Sci U S A 108 : 15366–15371. doi: 10.1073/pnas.1107906108 21896751
26. Sasaki Y, Kakisaka M, Chutiwitoonchai N, Tajima S, Hikono H, et al. (2014) Identification of a novel multiple kinase inhibitor with potent antiviral activity against influenza virus by reducing viral polymerase activity. Biochem Biophys Res Commun 450 : 49–54. doi: 10.1016/j.bbrc.2014.05.058 24858693
27. Davies B, Morris T (1993) Physiological parameters in laboratory animals and humans. Pharm Res 10 : 1093–1095. 8378254
28. Okomo-Adhiambo M, Demmler-Harrison GJ, Deyde VM, Sheu TG, Xu X, et al. (2010) Detection of E119V and E119I mutations in influenza A (H3N2) viruses isolated from an immunocompromised patient: challenges in diagnosis of oseltamivir resistance. Antimicrob Agents Chemother 54 : 1834–1841. doi: 10.1128/AAC.01608-09 20194700
29. Chou YY, Heaton NS, Gao Q, Palese P, Singer RH, et al. (2013) Colocalization of different influenza viral RNA segments in the cytoplasm before viral budding as shown by single-molecule sensitivity FISH analysis. PLoS Pathog 9: e1003358. doi: 10.1371/journal.ppat.1003358 23671419
30. Chase GP, Rameix-Welti MA, Zvirbliene A, Zvirblis G, Gotz V, et al. (2011) Influenza virus ribonucleoprotein complexes gain preferential access to cellular export machinery through chromatin targeting. PLoS Pathog 7: e1002187. doi: 10.1371/journal.ppat.1002187 21909257
31. Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, et al. (2009) AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem 30 : 2785–2791. doi: 10.1002/jcc.21256 19399780
32. Kanoh N, Honda K, Simizu S, Muroi M, Osada H (2005) Photo-cross-linked small-molecule affinity matrix for facilitating forward and reverse chemical genetics. 17. Ketha KM, Atreya CD (2008) Application of bioinformatics-coupled experimental analysis reveals a new transport-competent nuclear localization signal in the nucleoprotein of influenza A virus strain. BMC Cell Biol 9 : 22. doi: 10.1186/1471-2121-9-22 18442378
33. Tarus B, Bakowiez O, Chenavas S, Duchemin L, Estrozi LF, et al. (2012) Oligomerization paths of the nucleoprotein of influenza A virus. Biochimie 94 : 776–785. doi: 10.1016/j.biochi.2011.11.009 22155087
34. Ng AK, Zhang H, Tan K, Li Z, Liu JH, et al. (2008) Structure of the influenza virus A H5N1 nucleoprotein: implications for RNA binding, oligomerization, and vaccine design. FASEB J 22 : 3638–3647. doi: 10.1096/fj.08-112110 18614582
35. Compans RW, Content J, Duesberg PH (1972) Structure of the ribonucleoprotein of influenza virus. J Virol 10 : 795–800. 4117350
36. Ortega J, Martin-Benito J, Zurcher T, Valpuesta JM, Carrascosa JL, et al. (2000) Ultrastructural and functional analyses of recombinant influenza virus ribonucleoproteins suggest dimerization of nucleoprotein during virus amplification. J Virol 74 : 156–163. 10590102
37. Chan WH, Ng AK, Robb NC, Lam MK, Chan PK, et al. (2010) Functional analysis of the influenza virus H5N1 nucleoprotein tail loop reveals amino acids that are crucial for oligomerization and ribonucleoprotein activities. J Virol 84 : 7337–7345. doi: 10.1128/JVI.02474-09 20463064
38. Ito J, Tabei Y, Shimizu K, Tsuda K, Tomii K (2012) PoSSuM: a database of similar protein-ligand binding and putative pockets. Nucleic Acids Res 40: D541–548. doi: 10.1093/nar/gkr1130 22135290
39. Konc J, Janezic D (2012) ProBiS-2012: web server and web services for detection of structurally similar binding sites in proteins. Nucleic Acids Res 40: W214–221. doi: 10.1093/nar/gks435 22600737
40. Dias A, Bouvier D, Crepin T, McCarthy AA, Hart DJ, et al. (2009) The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature 458 : 914–918. doi: 10.1038/nature07745 19194459
41. Yuan P, Bartlam M, Lou Z, Chen S, Zhou J, et al. (2009) Crystal structure of an avian influenza polymerase PA(N) reveals an endonuclease active site. Nature 458 : 909–913. doi: 10.1038/nature07720 19194458
42. DuBois RM, Slavish PJ, Baughman BM, Yun MK, Bao J, et al. (2012) Structural and biochemical basis for development of influenza virus inhibitors targeting the PA endonuclease. PLoS Pathog 8: e1002830. doi: 10.1371/journal.ppat.1002830 22876176
43. Chenavas S, Estrozi LF, Slama-Schwok A, Delmas B, Di Primo C, et al. (2013) Monomeric nucleoprotein of influenza A virus. PLoS Pathog 9: e1003275. doi: 10.1371/journal.ppat.1003275 23555270
44. Li Z, Watanabe T, Hatta M, Watanabe S, Nanbo A, et al. (2009) Mutational analysis of conserved amino acids in the influenza A virus nucleoprotein. J Virol 83 : 4153–4162. doi: 10.1128/JVI.02642-08 19225007
45. Hutchinson EC, Denham EM, Thomas B, Trudgian DC, Hester SS, et al. (2012) Mapping the phosphoproteome of influenza A and B viruses by mass spectrometry. PLoS Pathog 8: e1002993. doi: 10.1371/journal.ppat.1002993 23144613
46. Sun Q, Carrasco YP, Hu Y, Guo X, Mirzaei H, et al. (2013) Nuclear export inhibition through covalent conjugation and hydrolysis of Leptomycin B by CRM1. Proc Natl Acad Sci U S A 110 : 1303–1308. doi: 10.1073/pnas.1217203110 23297231
47. Prokudina-Kantorovich EN, Semenova NP (1996) Intracellular oligomerization of influenza virus nucleoprotein. Virology 223 : 51–56. 8806539
48. Elton D, Medcalf E, Bishop K, Digard P (1999) Oligomerization of the influenza virus nucleoprotein: identification of positive and negative sequence elements. Virology 260 : 190–200. 10405371
49. Schumann M, Armen RS (2013) Identification of distant drug off-targets by direct superposition of binding pocket surfaces. PLoS One 8: e83533. doi: 10.1371/journal.pone.0083533 24391782
50. Webster RG, Bean WJ, Gorman OT, Chambers TM, Kawaoka Y (1992) Evolution and ecology of influenza A viruses. Microbiol Rev 56 : 152–179. 1579108
51. Furuta Y, Takahashi K, Fukuda Y, Kuno M, Kamiyama T, et al. (2002) In vitro and in vivo activities of anti-influenza virus compound T-705. Antimicrob Agents Chemother 46 : 977–981. 11897578
52. Arnott JA, Planey SL (2012) The influence of lipophilicity in drug discovery and design. Expert Opin Drug Discov 7 : 863–875. 22992175
53. Bao Y, Bolotov P, Dernovoy D, Kiryutin B, Zaslavsky L, et al. (2008) The influenza virus resource at the National Center for Biotechnology Information. J Virol 82 : 596–601. 17942553
54. Katoh K, Misawa K, Kuma K, Miyata T (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30 : 3059–3066. 12136088
55. Molecular Operating Environment (MOE), 2013.08; Chemical Computing Group Inc., 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2014.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 7
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- RNA Virus Reassortment: An Evolutionary Mechanism for Host Jumps and Immune Evasion
- Activation of TLR2 and TLR6 by Dengue NS1 Protein and Its Implications in the Immunopathogenesis of Dengue Virus Infection
- N-acetylglucosamine Regulates Virulence Properties in Microbial Pathogens
- Characterization of a Prefusion-Specific Antibody That Recognizes a Quaternary, Cleavage-Dependent Epitope on the RSV Fusion Glycoprotein
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy