
Mycolactone-Dependent Depletion of Endothelial Cell Thrombomodulin Is Strongly Associated with Fibrin Deposition in Buruli Ulcer Lesions
Buruli ulcer (BU) is a neglected tropical disease that is most common in West Africa and parts of Australia, but has been reported from over 30 countries worldwide. The symptoms are painless ulcers of the skin caused by a bacterial infection. The bacteria, Mycobacterium ulcerans, produce a macrolide toxin called mycolactone. In this manuscript, we have studied the effects of mycolactone on endothelial cells, specialised cells that line blood vessels and form capillaries. One of the most important functions of these cells is to prevent blood from clotting inside the vessels. We show that mycolactone reduces the ability of cultured endothelial cells to anticoagulate blood, by blocking the expression of a protein called thrombomodulin. We went on to examine samples of BU patient skin and found that thrombomodulin is also reduced here, and that in contrast to normal skin large amounts of fibrin (one of the main constituents of blood clots) were present. This means that it may be useful to consider whether anticoagulants might improve the response to antibiotics and thereby improve treatment outcomes for BU patients.
Published in the journal:
. PLoS Pathog 11(7): e32767. doi:10.1371/journal.ppat.1005011
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1005011
Summary
Buruli ulcer (BU) is a neglected tropical disease that is most common in West Africa and parts of Australia, but has been reported from over 30 countries worldwide. The symptoms are painless ulcers of the skin caused by a bacterial infection. The bacteria, Mycobacterium ulcerans, produce a macrolide toxin called mycolactone. In this manuscript, we have studied the effects of mycolactone on endothelial cells, specialised cells that line blood vessels and form capillaries. One of the most important functions of these cells is to prevent blood from clotting inside the vessels. We show that mycolactone reduces the ability of cultured endothelial cells to anticoagulate blood, by blocking the expression of a protein called thrombomodulin. We went on to examine samples of BU patient skin and found that thrombomodulin is also reduced here, and that in contrast to normal skin large amounts of fibrin (one of the main constituents of blood clots) were present. This means that it may be useful to consider whether anticoagulants might improve the response to antibiotics and thereby improve treatment outcomes for BU patients.
Introduction
The neglected tropical disease Buruli ulcer (BU), caused by subcutaneous infection with Mycobacterium ulcerans, is most common in West Africa, but has also reported from Australia and altogether more than 30 countries worldwide [1]. These indolent, painless, ulcers affect at least 5,000 patients/year and are thought to be heavily under-reported. Without treatment, ulcers can extend to 15% of the skin surface area and cause significant morbidity [2]. The route of transmission of the infection is still controversial with proposed routes spanning from biting water insects to passive entry from an environmental reservoir via physical damage to skin [3,4]. The disease may present in different forms including indurated subcutaneous nodules, plaques, ulcers and oedema. The WHO recommended standard treatment consists of 8 weeks combination antibiotic therapy with streptomycin and rifampicin [5,6]. Adjunct surgical debridement followed by skin grafting is often required for larger lesions [7–9]. The specific treatment of the M. ulcerans infection remains burdened by the long duration, route of application and potential toxicity of the current drug regimen. The adjunct surgery suffers from a lack of skilled personnel, material supplies and inadequate wound care. As such, a therapeutic approach reducing the lesion size could profoundly improve the status quo in BU care.
Much insight into the pathogenesis and treatment of BU has been gained by studying the histopathology of infected tissues [9–11]. Strikingly, lesions show large clusters of extracellular acid-fast bacilli, extensive necrosis, and a relative lack of infiltrating immune cells [12]. All of these features are attributed to a lipid-like virulence factor produced by M. ulcerans called mycolactone [13]. Mycolactone is detected at high levels within ulcers [14,15], but pathology associated with its production is also evident some distance away from the microbes. The toxin may even cause some systemic immunosuppression [14]. Mycolactone purified from the acetone-soluble lipids of M. ulcerans was first described in 1999 [13]. It was subsequently shown that it can recapitulate the effects of the microbe, since injection of 100μg mycolactone in guinea pig skin caused ulceration within 5 days [13] with pathological features similar to those caused by M. ulcerans infection. Mycolactone causes apoptosis within the skin of infected guinea pigs, as revealed by TUNEL staining, in primary human keratinocytes [16] and also in cultured L929 and J774 cells which undergo cytoskeletal rearrangement and round up [17]. By 48 hours most of the cells have completely detached from the tissue culture plate, although these cells are still viable up to several days after they detach [17]. Nevertheless, mycolactone causes cell cycle arrest in G0/G1 stage of the cell cycle [13], and mycolactone treated cells are known to display growth inhibition [13,18].
The extensive tissue necrosis and cell death seen in BU would normally be expected to serve as a trigger for an inflammatory response in addition to that induced by the invading bacteria itself. However, the reverse is the case as there is no pain and little inflammation even in the presence of high bacterial loads in the centre of the lesions [1]. Mycolactone has been demonstrated to have a suppressive effect on the cells of both the innate and adaptive immune system, including dendritic cells, monocytes and T lymphocytes [19–23], preventing an effective immune response against the infection. Successful antibiotic treatment has been shown to be associated with a strong local immune response, with large numbers of infiltrating mononuclear cells and granuloma formation at the site of infection [10]. A possible mechanism may be cessation of mycolactone production prior to sterilisation, allowing the body’s own defences to play a role in healing [15].
Despite this wealth of research described above, to date there has been no molecular explanation of the coagulative necrosis seen in the ulcers beyond the cytopathic activity of mycolactone or its immunosuppressive effects, nor any description of the effects of mycolactone on endothelial cells. We recently showed that mycolactone is an inhibitor of Sec61-dependent translocation, thereby preventing the production of new secreted and membrane proteins that transit through the ER [18], including in these cells. We were intrigued as to how coagulative necrosis, and reports of clots and fibrin deposition evident within BU-associated vessels [24,25] could be linked to this activity and postulated that mycolactone may reduce the ability of endothelial cells to inhibit clot formation.
Endothelial cells form a highly dynamic organ that, together, regulate many physiological functions, and crucial to these is the regulation of coagulation [26]. One of the ways this is achieved is by the expression of specific receptors that together activate the protein C anticoagulant pathway [27]. When thrombin binds to thrombomodulin (TM), it ceases to be a procoagulant and instead becomes a potent anticoagulant, activating protein C which can then go on to inactivate factors Va and VIIIa together with its cofactor protein S, thereby down-regulating thrombin production over the endothelial cell surface. A second receptor, the endothelial cell protein C receptor (EPCR), enhances this and mediates the cytoprotective effects of activated protein C (APC) [28]. The microvasculature of the skin expresses both TM and the EPCR [29].
In this manuscript we have investigated the effect of mycolactone on these anticoagulant endothelial receptors, the molecular basis of depletion and the ability of mycolactone-exposed endothelial cells to activate protein C. Since our findings show that TM expression is highly sensitive to mycolactone, we determined its abundance in punch biopsies from BU lesions, proving that our findings are relevant in vivo. Indeed, we also found that fibrin deposition is a common feature of untreated BU lesions.
Results
Mycolactone causes depletion of protein C receptors from the surface of primary human dermal microvascular endothelial cells
Since the coagulative necrosis is primarily seen in the subcutaneous layer of the skin that contains abundant pervasive microvasculature, we have used primary human dermal microvascular endothelial cells to investigate the effect of mycolactone on the anticoagulant functions of the capillaries. Using flow cytometry we quantified expression of TM and the EPCR on the surface of the cells after 24hrs of exposure to 0.98–125ng/ml synthetic mycolactone A/B (Fig 1), which did not alter the physical characteristics of the exposed cells (Fig 1A). In line with previous observations [30,31], the proinflammatory cytokine IL-1β reduced the expression of TM by ~40% whereas DMSO (the mycolactone solvent [18]) had no effect (Fig 1B and 1C). Mycolactone dose-dependently and profoundly depleted the expression of TM, resulting in fluorescence levels 80% lower than solvent exposed cells at doses as low as 3.9ng/ml (Fig 1B and 1C). This same dose of mycolactone A/B could not prevent cytokine production by RAW264.7 cells [18], reinforcing the view that different types of cells display profoundly different sensitivities to mycolactone. Expression of the EPCR was also inhibited by mycolactone exposure, but a higher dose (31.3ng/ml; Fig 1C) was required for a statistically significant effect and the maximum inhibition was ~30%.

Based on this titration of mycolactone, a dose of 7.8ng/ml mycolactone was chosen to investigate the time dependence of protein depletion. Loss of TM increased over time (T½ 11.7±1.4hrs) and reached statistical significance at 16hrs of mycolactone exposure (Fig 1D). We also observed a time-dependent decrease in EPCR abundance, and although the rate and extent of depletion was slower and smaller than TM (Fig 1D), the cells had still lost ~60% of their EPCR by 48hrs.
The mechanism of thrombomodulin depletion from endothelial cells is driven by the translocation-blocking activity of mycolactone
Loss of TM was not restricted to surface protein, rather total cellular abundance was reduced (Fig 2A). We also examined whether the Wiskott–Aldrich syndrome protein (WASP) inhibitor wiskostatin could restore TM expression [32], but could find no effect, suggesting that a change in actin dynamics does not contribute to the decreased abundance (Fig 2B). Furthermore, while elastase caused soluble TM (sTM) to be released from the cells as expected [33], no such increase could be observed following mycolactone treatment (Fig 2C). This was in contrast to IL-1β treatment, where at least part of the loss of cell surface TM could be attributed to shedding (Fig 2C). However, similarly to Cox-2 and TNF in macrophages [18], TM depletion could be reversed by inhibiting the proteasome (Fig 2D).
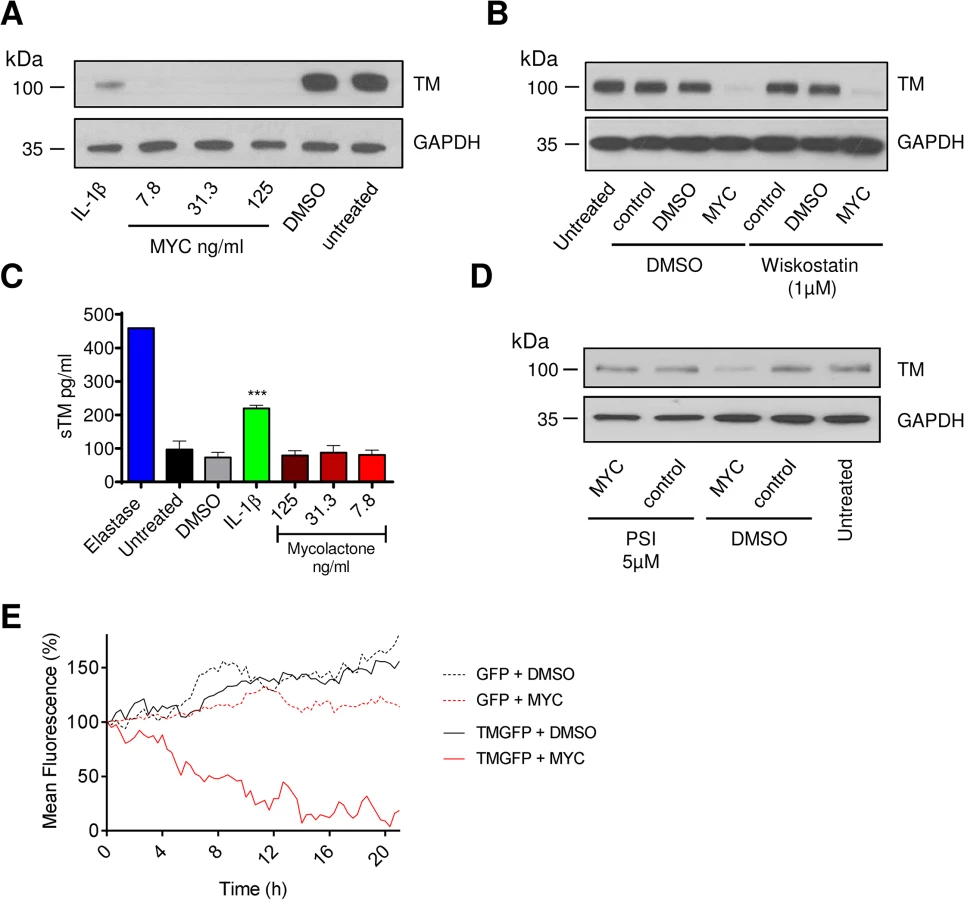
We also carried out time-lapse live fluorescence microscopy of HeLa cells (Fig 2E and S1–S4 Videos). The cells stably expressed recombinant TM as an in-frame fusion protein with carboxy-terminal GFP ("TM-GFP"). Production and membrane expression of this protein, like endogenous TM, was dependent on its co-translational translocation into the ER. As a control we used cells stably expressing recombinant GFP alone ("GFP") produced in the cytosol and thus not dependent on translocation. Exposure of cells to 125ng/ml mycolactone caused ER-transiting membrane TM-GFP (S2 Video and Fig 2E solid red line), but not cytosolic GFP (S4 Video and Fig 2E dotted red line), to undergo a gradual reduction in the cell-surface fluorescence of each cell (T½ 8.5±0.6hrs, Fig 2E). Exposure of cells expressing TM-GFP to DMSO did not cause the same effect (S1 Video and Fig 2E solid black line). Taken together our data strongly support a mechanism of TM loss involving Sec61-dependent translocation blockade and consequent degradation of mislocated TM in the cytosol [18].
It is also interesting to note that the mean whole-field fluorescence of cytosolic GFP-expressing cells exposed to mycolactone did not increase similarly to DMSO exposed cells (Fig 2E, compare GFP + DMSO [dotted black line] with GFP + MYC [dotted red line]). This is a clear demonstration of the growth inhibition caused by mycolactone [13,18] that prevents normal cell division and associated increase in whole-field fluorescence (S3 Video). The effect of mycolactone on the adhesion of these cells can also be clearly seen; over the 20hr experiment, the cells became increasingly rounded. Notably, mycolactone-exposed cells were apparently still able to form lamellipodia, but these structures were unable to result in functional attachment to the tissue culture well surface (see S4 Video).
Mycolactone has no direct effect on thrombin generation or platelet function, but causes a profound inhibition of endothelial cells’ ability to activate protein C
TM is an essential protein that converts thrombin from a pro - to an anti-coagulant role in the protein C anticoagulant pathway, so we went on to investigate any direct or indirect effects of mycolactone on coagulation. The coagulation cascade is dependent on Ca2+-dependent interactions of the Gla domains of γ-carboxylated proteins with negatively charged phospholipids and activated platelets, and given mycolactone’s hydrophobic lipid-like nature [13] it seemed possible that it might directly alter this process. We used calibrated automated thrombography (CAT) to investigate this but could find no changes in the rate of thrombin generation or lag phase at any dose of mycolactone tested (Fig 3A), or even at doses as high as 0.67μg/ml (S1A Fig). We also investigated whether mycolactone might alter the function of platelets. Mycolactone did not alter the aggregation of platelets in response to collagen (Fig 3B), or alone (S1B Fig). Similarly it did not alter fibrinogen binding or P-selectin exposure after activation by collagen-related peptide (CRP, Fig 3C), or alone (S1B Fig).

On the other hand, mycolactone did profoundly block the ability of HDMVEC to activate protein C. First we estimated the apparent Km for protein C activation on these cells according to a previously optimised method [34,35] (Fig 3D), which was 444nM, compatible with the literature. We then went on to compare the rate at which 0.5μM protein C was activated over cells treated with mycolactone. The specificity of this assay for TM-dependent activation was determined using the function-blocking antibody CTM1009, which prevents thrombin binding to TM [36]. This resulted in an ~80% reduction in the amount of activated protein C (APC) formed (Fig 3E). IL-1β treatment reduced APC generation, as expected and, strikingly, 7.8ng/ml mycolactone reduced APC generation almost to the same extent as CTM1009 (~75%). Even 1.9ng/ml mycolactone reduced APC generation ~20% in the 30 minute activation assay. If a similar depletion of TM could be demonstrated in endothelial cells of BU lesions, then the vessels they line would be severely impeded in their ability to anticoagulate the circulating blood.
Thrombomodulin is greatly reduced in the infected skin of patients with Buruli ulcer
Healthy skin is a highly heterogeneous tissue composed of diverse tissue types and, in BU, a wide variation in histopathological changes is seen in lesions of the same category. Therefore, in order to establish the in vivo relevance of these findings, the abundance of TM in 4mm punch biopsy samples taken from BU lesions was assessed, using the delberately cautious approach described in the materials and methods. The expression of TM was scored using a semi-quantitative method, reflecting both the intensity of staining and the proportion of skin vessels that were affected, similar to methods used for biomarkers of cancer [37]. Scores were relative to normal TM staining in healthy skin from an unaffected individual, where TM is found abundantly in both keratinocytes of the epidermis and the endothelium of subcutaneous tissue ([38], see Fig 4A). This pattern and intensity of staining gained a maximum score of 9.

Using the strategy described, we investigated TM expression in lesions of 40 untreated, laboratory confirmed, BU patients (31 ulcers and nine plaque lesions; Table 1). Twenty five of the patients had Category 2 ulcers, however the cohort included also patients with Category 1 (8 patients) and Category 3 (6 patients). None of the patients had oedematous disease. In order to minimise topological variability between patients the location of punch sampling was standardised as far as possible (see Materials and Methods), and for ulcers was performed 1cm inside the outer margin of the induration surrounding the ulcer. In 32/40 lesions, histopathology revealed the presence of necrotic tissue, consistent with coagulative necrosis, within the biopsy. For SMA (Table 2), we found that it usually stained vessels both in the dermis and the subcutis (Fig 4B6 and 4B7). However, in four patients, SMA staining could not be detected in the subcutis, and these patients were therefore not considered in the TM analysis (Table 2). In comparison, CD31/PECAM-1 staining was fainter although it was still able to mark capillaries and small vessels in the skin (Fig 4B4 and 4B5).


Our analysis of TM expression revealed that, for most (but not all) BU lesions, TM abundance was reduced within the biopsy (Fig 4B and 4D). A major effect on the abundance of TM (defined as a combined score of less than 4 out of maximum of 9) was found in 78% of patient’s subcutis and 63% of patient’s dermis (Table 2, Fig 4B2 and 4B3 and 4D). Of these, TM was considered absent (combined score of <1) in either the subcutis or dermis of 21 lesions (53%). A further 15 and 22% showed a moderate reduction, respectively (Table 2). Therefore, the abundance of TM appeared to be more greatly affected in the subcutis than the dermis. The range in scores observed for ulcer and plaque category lesions were similar, suggesting that reduction of TM is a common feature of BU disease (Fig 4D).
In addition to changes to endothelial TM, we also observed changes to keratinocyte TM staining in the epidermis in some BU patients although this was more variable. In 8/40 (20%) samples TM could no longer be detected, whereas 24/40 (60%) showing moderate and 8/40 (20%) samples showing strong expression of TM in the hyperplastic regions (Fig 4C). As in endothelium, a similar range in scores was observed for both ulcer and plaque category lesions (Fig 4E), suggesting that changes to keratinocyte TM is also a common feature of BU disease. It should be noted that the wider variation in keratinocyte TM could reflect the location at which the punch biopsy was taken within the lesion. Indeed, there is a strong positive correlation between the score for dermal TM and epidermis TM (r = 0.693, p<0.0001) in these patients, suggesting that the two are linked.
Abundant fibrin deposition in the infected skin of patients with Buruli ulcer
It is well established that loss of TM in the presence of otherwise normal coagulation factors can cause fibrin deposition. While a single report from 1966 exists in the literature of fibrin deposition in BU lesions using phosphotungstic acid-haematoxylin stain (PTAH) [24], it is not known how widely occurring this phenomenon is. Immunohistochemistry analyses for fibrin deposition were therefore performed using a well-established monoclonal antibody that binds specifically to fibrin and not fibrinogen [39] (Fig 5 and Table 2). Semi-quantitative scoring was performed as for TM.

As expected, no fibrin deposition was observed in skin from a healthy individual, (Fig 5A, scored 0 out of 3). In contrast, lesions of all 40 BU patients that have been tested had fibrin deposited in the subcutis of the skin (Fig 5B), with 50% of them showing substantial deposition (scoring 2.5–3 out of 3). Furthermore, 75% of patients also had staining in the dermis although the effect was more moderate (Table 2 and Fig 5C). We found a clear positive correlation between the presence and degree of tissue necrosis (Fig 5D) with the amount of fibrin staining. This was in both the dermis (p = 0.0002) and subcutis (p = 0.0000002) (Table 3). Notably, subcutaneous tissue, where more fibrin is deposited, is the first skin tissue area to become necrotic in BU patients [40].

Mycolactone’s effects on cellular thrombomodulin are not due to cell death
Although reductions in TM abundance were found in live cells in patients (Fig 4) (as evidenced by their expression of SMA and CD31/PECAM-1 in nucleated cells), the link between fibrin staining and necrosis observed in the patients led us to investigate whether loss of TM is synchronised with cell death. Importantly, after 24 and even 48hrs of exposure to 7.8ng/ml mycolactone did not directly cause any significant loss of viability of the endothelial cells using a Live/Dead (calcein/Ethidium Bromide [EtBr] homodimer) assay (Fig 6A). After 24hrs ~20% of cells were detached from the plate, but were still alive when retrieved from the supernatant (Fig 6A). Therefore, the loss of TM from these cells appears to precede (or coincide with) changes in morphology, detachment or death. Indeed, when time to cell death was assessed using the CellEvent system (which does not require cells to be washed or manipulated in any way prior to analysis, therefore capturing attached, loosely attached and detached cells), significant numbers of HDMVEC did not enter late apoptosis until day 3, and the largest increase was observed between days 3 and 4, after which ~80% of the cells had died (Fig 6B).

Furthermore, since endothelial PECAM-1/CD31 staining was reduced in some BU lesions we investigated whether loss of adhesion molecules occurred in vitro, and might be linked to cell death. The impact of mycolactone on the expression of PECAM-1/CD31 and VE-cadherin was more variable, probably due to changes in turnover rates as the cells become confluent [41]. However, we found that both VE-cadherin and PECAM-1/CD31 were ~50% reduced by mycolactone exposure at 24hrs (Fig 6C).
Discussion
The hallmark of BU disease is the presence of extensive subcutaneous necrosis, which is due to the cytotoxic activity of mycolactone [42]. However what is less clear is the earliest molecular event that triggers cellular death in M. ulcerans infected skin tissue. In the scientific literature to date there are many reports that have examined the cytopathic and cytotoxic effects of mycolactone in vivo and in vitro. For instance, TUNEL staining has been reported 2 days following injection of purified mycolactone in experimental animals as well as in BU patients [17,25]. More recently, conflicting data has been presented with more specific assays. While Bax and total caspase 3 and 9 have been detected in BU lesion [25], cleaved (i.e. activated, but possibly also unstable) caspase 3 was rarely detected in necrotic areas [43]. In this manuscript we provide compelling evidence that mycolactone-dependent depletion of TM occurs on HMDVEC and in diseased microvasculature, and that fibrin deposition is a common feature of BU lesions. Our findings suggest that the effect of mycolactone on tissue is probably both direct (due to cytotoxic/cytopathic effects on cells) and indirect, due to mycolactone-dependent loss of coagulation control and resultant tissue ischemia.
There are a number of potential mechanisms by which mycolactone might cause such a loss of TM. In the presence of thrombin, the thrombin-TM complex is internalised by endocytosis and unbound TM is returned to the cell surface [44]. Mycolactone is known to cause inappropriate actin polymerisation [32] that might credibly inhibit this process. However, whole cell lysate blots showed that TM did not accumulate in intracellular compartments, and depletion of TM was not restored by inhibition of WASP activity [32]. Instead, TM was found to deplete steadily from the cell surface due to an inability to replace TM during the normal protein turnover process, since newly synthesised, misolocalised, TM is degraded by the proteasome [18]. The rate of loss of TM was determined by flow cytometry (T1/2 11.7±1.4hrs) and confocal microscopy of a recombinant GFP-tagged protein (T1/2 8.5±0.6hrs). Both of these correspond well with the literature where human TM was shown to turn over at a rate of 5–10%/hour [45] (equating to a T1/2 of 5–10hrs). Notably other membrane proteins were less sensitive to mycolactone, presumably due to a lower turnover rate. For instance both the EPCR and PECAM-1 (CD-31) were only reduced by about 40–50% in 24hrs. Therefore TM's relatively higher turnover rate would appear to have rendered it more sensitive to mycolactone than other proteins.
Total deletion of TM is incompatible with life [46], whereas endothelial cell-specific deletion causes spontaneous thrombosis and a life-threatening coagulopathy including necrosis of the skin [47]. Conversely, a polymorphism in TM (THBD c.1418C>T) that increases surface TM due to decreased shedding, reduces the risk of deep vein thrombosis in carriers [48]. Furthermore, homozygous congenital deficiency of either protein S or protein C causes neonatal purpura fulminans; a combined disseminated intravascular coagulation and haemorrhagic disease that often presents with skin necrosis. Therefore, loss of the protein C anticoagulant pathway alone is sufficient to drive fibrin deposition and thrombosis. Indeed these pathways have been mechanistically linked in cerebral malaria [49] and sepsis [50]. There was not a complete correlation between fibrin deposition and TM abundance in the patients we examined in this study. Furthermore, in most patients, fibrin staining was observed predominantly in the tissue rather than the vessels, suggesting that other mechanisms also contribute to fibrin deposition such as reduced endothelial cell barrier function due to APC’s known cytoprotective effects [51,52]. However, it should be noted that fibrin deposited in vessels can be effectively cleared by the fibrinolytic system after a clotting event has taken place. A certain degree of caution is necessary due to the subjective nature of the immunohistochemistry scoring and the heterogeneity of BU skin lesions that presents a substantial challenge in quantification. However, qualitatively, strong fibrin and low thrombomodulin staining were frequently observed in BU lesions.
In this manuscript we have provided evidence that disruption of the protein C anticoagulant pathway due to mycolactone-dependent depletion of TM from endothelial cells is strongly associated with the fibrin deposition we observe in BU skin lesions. However, whether this is the direct cause of fibrin deposition is not easily answered in clinical samples from BU patients. This is because, at all clinical stages of disease, tissue necrosis is the defining feature. Indeed, there may be even vessel to vessel differences due to such factors as local mycolactone concentration gradients and local extent of necrosis. Thus it is not possible to determine the order in which TM depletion, fibrin deposition and death of skin tissue occurred in such samples.
There are alternative explanations for the fibrin deposition observed. Mycolactone reduced the expression of PECAM-1/CD31 and VE-Cadherin on the surface of endothelial cells. Loss of such adhesion molecules (including TM, itself a Ca2+-dependent adhesion molecule [53]) would increase the permeability of vessels. Indeed, endothelial cells are known to become ‘leaky’ in this way in many pathological states [54]. This might explain why fibrin deposition is frequently observed in BU lesions, since it would allow soluble proteins in the plasma to access this compartment even if erythrocytes or platelets cannot. Alternatively, the generation of high levels of Tissue Factor by dying cells or the exposure of collagen, von Willebrand factor (vWF) and other pro-coagulant surfaces should vessels be denuded would also promote fibrin deposition irrespective of TM. Depending on the concentration of mycolactone, and the duration of exposure (which will also vary throughout the evolution of the disease), different mechanisms may predominate. Since loss of TM precedes cell death by 2–3 days (at least in vitro), loss of coagulation control and resultant fibrin deposition/ischemia would be expected to precede cellular apoptosis caused directly by mycolactone. Therefore the promotion of a “procoagulant endothelial cell” is likely to be the one of the first triggers for disease at early (preclinical) stages of infection, as well as at the edges of the growing ulcer and encroaching mycolactone.
The expression of other ER-transiting components of the coagulation and anticoagulation systems may also be directly affected by mycolactone, and these are currently the subject of further ongoing investigations. The finding of fibrin deposition however, strongly suggests that the combined result of such effects is excessive thrombin generation. However, platelets do not seem to play a major role, since we found that mycolactone does not affect directly in vitro platelet function.
In addition to endothelial TM, keratinocyte TM [55] expression in the epidermis was also altered in punch biopsies from BU patients. Changes were variable and not as severe as in vessels. However loss or reduction of keratinocyte TM does appear to be a common feature of BU lesions and correlated with abundance of TM in the dermis. Heterogeneity may be related both to differences in the location from where tissue biopsies were taken and disease activity. Notably a keratinocyte-specific knockout of TM causes a substantial reduction in the ability to heal wounds [56] and is itself connected to the formation of cell-cell adhesion dependent on E-cadherin [57]. Any role for TM in wound healing would add to previously described mechanisms involving changes to actin dynamics, where mycolactone was shown to decrease migration of transformed epithelial cells (HeLa) in to a scratch wound in vitro [32].
In the literature there is a single anecdotal report of a patient with BU who was successfully treated by anticoagulant therapy (300mg heparin per day) alongside with rifampicin [58]. At the time, there was no supporting information to explain the success of this treatment and to date it has not lead to a change in treatment regimens, which depend primarily on rifampicin and streptomycin or clarithromycin [59]. However, our findings suggest that the use of anticoagulant support to go alongside antibiotic therapy might increase perfusion of the skin by decreasing fibrin deposition. This would improve the efficacy of antibiotic therapy, especially since mycolactone can persist in the skin (albeit at reduced levels) after sterilisation has been achieved [15].
In summary, in this manuscript we have shown that TM expression is highly sensitive to mycolactone exposure both in vitro and in vivo. We also demonstrated that fibrin deposition is a common feature of untreated BU lesions. The coagulation cascade is in a delicate balance, poised to prevent excessive blood loss due to injury whilst maintaining intravascular fluidity [50]. We therefore conclude that loss of coagulation control, driven in part by loss of TM, is strongly associated with the coagulative necrosis seen in BU.
Materials and Methods
Reagents
We used synthetic mycolactone A/B (kind gift of Prof. Yoshito Kishi, Harvard University) throughout these investigations [60]. Unless specified all other reagents are from Sigma-Aldrich.
Cell culture and treatment
Primary human dermal microvascular endothelial cells (HDMVEC; LONZA) from two donors were used and cultured according to manufacturer’s recommendation in EGM-2MV (LONZA). Reagents for controls included DMSO (equivalent to top mycolactone dose; solvent control), 5μM protease inhibitor I (PSI), 10ng/ml TNF-α/IL-1β (Peprotech), 10μg/ml cycloheximide (CHX) and 5μg/ml human leukocyte elastase.
Immunochemistry
Flow cytometry of live HDMVEC was carried out using standard techniques on a FACSCanto flow cytometer (BD Biosciences). Cells were detached with trypsin-EDTA (LONZA). Antibodies were from BD Pharmingen (EPCR; RCR-252, TM; 1A4, VE-Cadherin; 55-7H1, isotype controls) or Sigma (PECAM-1; WM-59). Western blot analysis was carried out using standard techniques, following lysis of cells with Gel sample buffer, separation by SDS-PAGE on 10% polyacrylamide gels and transfer to PVDF membranes. Antibodies were from Dako (TM; 1009) or Ambion (GAPDH). Soluble TM (sTM) was quantified in cell supernatants by ELISA (Stratech) according to the manufacturer’s protocol.
Time-lapse confocal microscopy
Vector TMG was generated previously [53] and contains the full-length TM coding region cDNA upstream of eGFP. A control plasmid (expressing only eGFP) was prepared by removing the TM cDNA using BamHI and EcoRI restriction sites by standard cloning techniques. Both plasmids were stably transfected into HeLa cell lines by standard methodology and selection with G418. These cell lines are referred to as TM-GFP (transfected with vector TMG) and GFP (transfected with the GFP only plasmid). For confocal microscopy, cells were seeded into 6 well plates, treated as necessary and placed in a heated/humidified/5% CO2 chamber for 20hrs. Images were captured using in three different fields for each condition at 20min intervals using a Nikon A1 confocal laser scanning unit attached to an Eclipse Ti microscope. For quantitation, whole field fluorescence was calculated using NIS elements software (Nikon) with per-field background subtraction at each time-point and expressed as a percentage of starting fluorescence.
Calibrated automated thrombography
The effect of mycolactone on thrombin generation was assessed by CAT according to a previously published method [61] using a Fluoroskan Ascent FL plate reader (Thermo Labsystem) in combination with Thrombinoscope software (Synapse BV). Thrombin generation was quantified in human pooled plasma containing various concentrations of mycolactone or 0.013% DMSO as a control. The reaction was initiated with 4pM tissue factor (Dade Innovin, Dade Behring), 4μM phospholipid vesicles (DOPS/DOPC/DOPE, 20 : 60 : 20) and 16.6mM CaCl2. Thrombin generation was monitored using 0.42mM of the fluorogenic substrate Z-GlyArg-AMC-HCl (Bachem).
Platelet preparation, aggregation and activation assay
Human platelet preparation, aggregation and activation assays were performed according to well-established methods [62,63]. Briefly, blood was obtained from aspirin-free, healthy human volunteers with written informed consent as approved by the University of Reading Research Ethics Committee. Platelets were prepared and resuspended in modified Tyrodes-HEPES buffer (134mM NaCl, 2.9mM KCl, 0.34mM Na2HPO4, 12mM NaHCO3, 20mM HEPES and 1mM MgCl2, pH 7.3) and used at a final density of 2x108cells/ml. Aggregation assays were performed as described, using collagen (Nycomed, Austria) in the presence or absence of either DMSO (final concentration 0.1%v/v) or mycolactone (at the indicated concentrations). Aggregation was quantified using an optical platelet aggregometer (Chrono-log, Havertown, PA) with continuous stirring (1200 rpm). Fibrinogen binding and P-selecting exposure were quantified by flow cytometry using FITC labelled rabbit anti-human fibrinogen antibody (Dako UK Ltd) and mouse anti-human CD62P antibody (BD Biosciences, UK), respectively. Washed platelets were incubated with mycolactone or DMSO as above before activation with CRP-XL (a selective agonist for platelet collagen receptor GPVI) at room temperature. The cells were fixed in 0.2% (v/v) formyl saline before analysis by flow cytometry. Data were analysed by calculating the median fluorescence intensity.
Protein C activation assay
Protein C activation by cell surface-bound TM was quantified according to the method of Stearns-Kurosawa et al [36], with minor modifications. Purified human plasma proteins were obtained from Enzyme Research Laboratories. HDMVEC were washed three times with HBSS containing 3mM CaCl2, 0.6mM MgCl2 and 1% BSA (‘Activation Buffer’). Human protein C in Activation Buffer was added at the indicated concentration and protein C activation was initiated by the addition of 13.5nM α-thrombin. The cells were incubated for 30mins at 37°C at which point the reaction was stopped with 5μg antithrombin III and 3U heparin. In some cases, untreated cells were exposed to 50μg/ml of CTM1009, an anti-TM antibody known to inhibit protein C activation by preventing thrombin binding [36] for 30mins prior to the assay in HBSS containing 0.1% BSA and 0.02% sodium azide prior to washing as above. Activated protein C (APC) was quantified by assessing cleavage of the chromogenic substrate S-2366 (Chromogenix). Aliquots were transferred to 96-well microplates and S-2366 added at a final concentration of 200μM. The rate of substrate conversion was quantified at 405nm using a FLUOstar Omega plate reader (BMG LABTECH), and the concentration of APC interpolated from a standard curve of purified APC using Graphpad Prism v.6.
Viability assays
For the viability assay using the LIVE/DEAD Viability/Cytotoxicity Kit (Molecular Probes Inc.), cells were washed twice with PBS incubated with 0.625μM CalceinAM and 10μM Ethidium homodimer-1 for 10min and then the plate was read on a FLUOstar Omega plate reader. During washing, the supernatants for retained and concentrated by gentle centrifugation in order to analyse the degree of cell detachment and the viability of those cells. Total live and dead cells were determined by the fluorescence of untreated or digitonin treated cells, respectively, considering both attached and detached portions. Live and dead cells were then expressed as a percentage of total cells.
In order to investigate apoptosis of cells, the CellEvent Caspase-3/7 green ready probes reagent was used. This fluorogenic indicator of activated caspase-3/7 can be used on live cells without washing, making it ideal for assessing mycolactone treated cells over longer time courses due to its cytopathic effects. After treatment, cells were stained with 0.3μg/ml PI (BD Biosciences, UK), 3μM DRAQ5 (Biostatus, UK) and 5μl CellEvent Caspase-3/7 green ready probes reagent (Invitrogen, Carlsbad, CA). Stained cells were observed by confocal microscopy using a Nikon A1 confocal laser scanning unit attached to an Eclipse Ti microscope. Active caspase-3/7 (green fluorescence), PI (red fluorescence) and nuclear (blue fluorescence) staining were analysed for 3 fields per treatment (minimum of 20 cells per field). Total cell numbers were determined by DRAQ5 staining, late apoptosis is expressed as a proportion of these cells which stained for both active caspase 3/7 and PI.
Ethics statement
Ethical approval for analysing BU patient specimens was obtained from the National Ethics Committee of Cameroon, the Ethics Committee of the Heidelberg University Hospital, Germany, the Ethikkommission beider Basel, Basel, Switzerland and the provisional national ethical review board of the Ministry of Health Benin. Written informed consent was obtained from adult patients, or the guardians of patients who were children.
Immunohistochemistry of punch biopsies
4mm BU punch biopsies were collected during other studies (ISRCTN72102977, IRB00006860 and EK.242/13) and were reanalysed for the current purpose. The clinical characteristics of the 40 untreated, laboratory reconfirmed, BU patients from whom biopsies were obtained are shown in (Table 1). In ulcerated BU lesions (31 patients) punch biopsies were taken 1cm inside the outer margin of the induration surrounding the ulcer. In contrast, from BU plaque lesions (9 patients) punch biopsies were collected from the non-ulcerated center of the lesion. After removal, tissue was fixed, transported, embedded into paraffin and cut into 5μm sections.
Immunohistochemistry was performed according to an established method [64]. Primary antibodies were from Dako (CD31; M0823, TM; M0617), Novocastra (SMA; NCL-SMA) Fibrin; 59D8 [39]). Biotin-conjugated secondary antibody was from Vectorlabs (BA2000). Staining was performed using the Vector ABC elite and the NovaRED system (Vectorlabs). Haematoxylin was used as a counter stain. Slides were analyzed with a DM2500 Microscope (Leica). Pictures were taken either with an Aperio scanner (Fig 4B1 and Fig 5) or with a Leica DFC 420 camera and the Leica application Suite V4 software. We have not been able to obtain 4mm punch biopsies from several healthy individuals, due to the invasive nature of this technique. Staining patterns and intensities for reference therefore included only one uninfected skin sample from a healthy individual. For further confirmation of TM and fibrin antibody staining performance in fixed tissues, we also examined additional samples from individuals with keloids and contact dermatitis.
Analysis of the immunohistochemistry results was not straightforward, due to the heterogeneity of skin and BU histopathology. This caused us to take a deliberately cautious approach to considering our findings. First, the integrity of vessels and endothelial cells within the biopsy tissue was assessed using the well-established markers smooth muscle alpha-actin (SMA) and CD31/PECAM-1, respectively. We scored each biomarker semi-quantitatively as follows; 0; no staining, 1; minimal staining, 2; medium staining and 3; strong staining. Scoring was performed separately for the epidermal, dermal and subcutaneous layer of the BU punch biopsies. Additionally, TM scoring was only carried out in regions of samples where vessels could be identified by SMA staining, in order to minimise the risk of scoring TM staining in vessels that had be destroyed by the infection. The semi-quantitative scoring for TM abundance incorporated both the intensity of staining and the extent of staining, according to the method of d’Adhemar [37], resulting in a maximum score of 9, which was equivalent to the staining observed in normal tissue. Fibrin staining and tissue necrosis had a maximum score of 3, but here normal tissue scored 0 for both biomarkers. Furthermore, in order to most accurately portray the trends and variations observed, the dermis and subcutis have been considered separately. We have not attempted statistical analysis of our results, due to the factors described above, but the scores are useful to indicate the range of findings we observed.
Statistical analysis
Data were analysed using Graphpad Prism v.6 software. In vitro data was assessed using a one way ANOVA and Dunnett’s post-hoc test. Scoring data was assessed for a Gaussian distribution using D'Agostino & Pearson omnibus normality test. Since many data sets were not normally distributed, correlations used the method of Spearman.
Accession numbers
β3 integrin: P05106, caspase 3: P42574, caspase 7: P55210, EPCR: Q9UNN8, fibrin (from fibrinogen α,β,γ): P02671, P02675, P02679, GAPDH: P16858, IL-1β: P01584, PECAM1/CD31: P16284, protein C: P04070, SMA: P62736, Thrombomodulin: P07204, TNF: P01375 ve-cadherin: P33151.
Supporting Information
{kind=link}
Zdroje
1. Chany AC, Tresse C, Casarotto V, Blanchard N (2013) History, biology and chemistry of Mycobacterium ulcerans infections (Buruli ulcer disease). Nat Prod Rep 30 : 1527–1567. doi: 10.1039/c3np70068b 24178858
2. Walsh DS, Portaels F, Meyers WM (2011) Buruli ulcer: Advances in understanding Mycobacterium ulcerans infection. Dermatol Clin 29 : 1–8. doi: 10.1016/j.det.2010.09.006 21095521
3. Merritt RW, Walker ED, Small PL, Wallace JR, Johnson PD, et al. (2010) Ecology and transmission of Buruli ulcer disease: a systematic review. PLoS Negl Trop Dis 4: e911. doi: 10.1371/journal.pntd.0000911 21179505
4. Roche B, Benbow ME, Merritt R, Kimbirauskas R, McIntosh M, et al. (2013) Identifying the Achilles' heel of multi-host pathogens: The concept of keystone "host" species illustrated by transmission. Environ Res Lett 8 : 045009. 24554969
5. Converse PJ, Nuermberger EL, Almeida DV, Grosset JH (2011) Treating Mycobacterium ulcerans disease (Buruli ulcer): from surgery to antibiotics, is the pill mightier than the knife? Future Microbiol 6 : 1185–1198. doi: 10.2217/fmb.11.101 22004037
6. Simpson C, O'Brien DP, McDonald A, Callan P (2013) Mycobacterium ulcerans infection: evolution in clinical management. ANZ J Surg 83 : 523–526. doi: 10.1111/j.1445-2197.2012.06230.x 22989109
7. Ruf MT, Chauty A, Adeye A, Ardant MF, Koussemou H, et al. (2011) Secondary Buruli ulcer skin lesions emerging several months after completion of chemotherapy: paradoxical reaction or evidence for immune protection? PLoS Negl Trop Dis 5: e1252. doi: 10.1371/journal.pntd.0001252 21829740
8. Nienhuis WA, Stienstra Y, Abass KM, Tuah W, Thompson WA, et al. (2012) Paradoxical responses after start of antimicrobial treatment in Mycobacterium ulcerans infection. Clin Infect Dis 54 : 519–526. doi: 10.1093/cid/cir856 22156855
9. Yeboah-Manu D, Kpeli GS, Ruf MT, Asan-Ampah K, Quenin-Fosu K, et al. (2013) Secondary bacterial infections of buruli ulcer lesions before and after chemotherapy with streptomycin and rifampicin. PLoS Negl Trop Dis 7: e2191. doi: 10.1371/journal.pntd.0002191 23658847
10. Schutte D, Pluschke G (2009) Immunosuppression and treatment-associated inflammatory response in patients with Mycobacterium ulcerans infection (Buruli ulcer). Expert Opin Biol Ther 9 : 187–200. doi: 10.1517/14712590802631854 19236249
11. Ruf MT, Schutte D, Chauffour A, Jarlier V, Ji B, et al. (2012) Chemotherapy-associated changes of histopathological features of Mycobacterium ulcerans lesions in a Buruli ulcer mouse model. Antimicrob Agents Chemother 56 : 687–696. doi: 10.1128/AAC.05543-11 22143518
12. Silva MT, Portaels F, Pedrosa J (2009) Pathogenetic mechanisms of the intracellular parasite Mycobacterium ulcerans leading to Buruli ulcer. Lancet Infect Dis 9 : 699–710. doi: 10.1016/S1473-3099(09)70234-8 19850228
13. George KM, Chatterjee D, Gunawardana G, Welty D, Hayman J, et al. (1999) Mycolactone: a polyketide toxin from Mycobacterium ulcerans required for virulence. Science 283 : 854–857. 9933171
14. Sarfo FS, Le Chevalier F, Aka N, Phillips RO, Amoako Y, et al. (2011) Mycolactone diffuses into the peripheral blood of Buruli ulcer patients—implications for diagnosis and disease monitoring. PLoS Negl Trop Dis 5: e1237. doi: 10.1371/journal.pntd.0001237 21811642
15. Sarfo FS, Phillips RO, Zhang J, Abass MK, Abotsi J, et al. (2014) Kinetics of mycolactone in human subcutaneous tissue during antibiotic therapy for Mycobacterium ulcerans disease. BMC Infect Dis 14 : 202. doi: 10.1186/1471-2334-14-202 24731247
16. Bozzo C, Tiberio R, Graziola F, Pertusi G, Valente G, et al. (2010) A Mycobacterium ulcerans toxin, mycolactone, induces apoptosis in primary human keratinocytes and in HaCaT cells. Microbes Infect 12 : 1258–1263. doi: 10.1016/j.micinf.2010.08.005 20800104
17. George KM, Pascopella L, Welty DM, Small PL (2000) A Mycobacterium ulcerans toxin, mycolactone, causes apoptosis in guinea pig ulcers and tissue culture cells. Infect Immun 68 : 877–883. 10639458
18. Hall BS, Hill K, McKenna M, Ogbechi J, High S, et al. (2014) The Pathogenic Mechanism of the Mycobacterium ulcerans Virulence Factor, Mycolactone, Depends on Blockade of Protein Translocation into the ER. PLoS Pathog 10: e1004061. doi: 10.1371/journal.ppat.1004061 24699819
19. Pahlevan AA, Wright DJ, Andrews C, George KM, Small PL, et al. (1999) The inhibitory action of Mycobacterium ulcerans soluble factor on monocyte/T cell cytokine production and NF-kappa B function. J Immunol 163 : 3928–3935. 10490994
20. Coutanceau E, Decalf J, Martino A, Babon A, Winter N, et al. (2007) Selective suppression of dendritic cell functions by Mycobacterium ulcerans toxin mycolactone. J Exp Med 204 : 1395–1403. 17517970
21. Coutanceau E, Marsollier L, Brosch R, Perret E, Goossens P, et al. (2005) Modulation of the host immune response by a transient intracellular stage of Mycobacterium ulcerans: the contribution of endogenous mycolactone toxin. Cell Microbiol 7 : 1187–1196. 16008585
22. Torrado E, Adusumilli S, Fraga AG, Small PL, Castro AG, et al. (2007) Mycolactone-mediated inhibition of tumor necrosis factor production by macrophages infected with Mycobacterium ulcerans has implications for the control of infection. Infect Immun 75 : 3979–3988. 17517872
23. Simmonds RE, Lali FV, Smallie T, Small PL, Foxwell BM (2009) Mycolactone inhibits monocyte cytokine production by a posttranscriptional mechanism. J Immunol 182 : 2194–2202. doi: 10.4049/jimmunol.0802294 19201873
24. Connor DH, M. WC, Fletcher Lunn H (1966) Buruli ulceration. A clinicopathologic study of 38 Ugandans with Mycobacterium ulcerans ulceration. Arch Path 81 : 183–189.
25. Zavattaro E, Boccafoschi F, Borgogna C, Conca A, Johnson RC, et al. (2012) Apoptosis in Buruli ulcer: a clinicopathological study of 45 cases. Histopathology 61 : 224–236. doi: 10.1111/j.1365-2559.2012.04206.x 22439755
26. van Hinsbergh VW (2012) Endothelium—role in regulation of coagulation and inflammation. Semin Immunopathol 34 : 93–106. doi: 10.1007/s00281-011-0285-5 21845431
27. Simmonds RE, Lane DA (1998) Regulation of Coagulation. In: Loscalzo J, Schafer AI, editors. Thrombosis and Haemorrhage. 2nd Ed ed. Baltimore: Williamsn & Wilkins. pp. 45–76.
28. Esmon CT (2012) Protein C anticoagulant system—anti-inflammatory effects. Semin Immunopathol 34 : 127–132. doi: 10.1007/s00281-011-0284-6 21822632
29. Ye X, Fukudome K, Tsuneyoshi N, Satoh T, Tokunaga O, et al. (1999) The endothelial cell protein C receptor (EPCR) functions as a primary receptor for protein C activation on endothelial cells in arteries, veins, and capillaries. Biochem Biophys Res Commun 259 : 671–677. 10364477
30. Archipoff G, Beretz A, Freyssinet JM, Klein-Soyer C, Brisson C, et al. (1991) Heterogeneous regulation of constitutive thrombomodulin or inducible tissue-factor activities on the surface of human saphenous-vein endothelial cells in culture following stimulation by interleukin-1, tumour necrosis factor, thrombin or phorbol ester. Biochem J 273 (Pt 3): 679–684.
31. Nawroth PP, Handley DA, Esmon CT, Stern DM (1986) Interleukin 1 induces endothelial cell procoagulant while suppressing cell-surface anticoagulant activity. Proc Natl Acad Sci U S A 83 : 3460–3464. 3486418
32. Guenin-Mace L, Veyron-Churlet R, Thoulouze MI, Romet-Lemonne G, Hong H, et al. (2013) Mycolactone activation of Wiskott-Aldrich syndrome proteins underpins Buruli ulcer formation. J Clin Invest 123 : 1501–1512. doi: 10.1172/JCI66576 23549080
33. Boehme MW, Galle P, Stremmel W (2002) Kinetics of thrombomodulin release and endothelial cell injury by neutrophil-derived proteases and oxygen radicals. Immunology 107 : 340–349. 12423310
34. Fukudome K, Ye X, Tsuneyoshi N, Tokunaga O, Sugawara K, et al. (1998) Activation mechanism of anticoagulant protein C in large blood vessels involving the endothelial cell protein C receptor. J Exp Med 187 : 1029–1035. 9529319
35. Kunz G, Ohlin AK, Adami A, Zoller B, Svensson P, et al. (2002) Naturally occurring mutations in the thrombomodulin gene leading to impaired expression and function. Blood 99 : 3646–3653. 11986219
36. Stearns-Kurosawa DJ, Kurosawa S, Mollica JS, Ferrell GL, Esmon CT (1996) The endothelial cell protein C receptor augments protein C activation by the thrombin-thrombomodulin complex. Proc Natl Acad Sci U S A 93 : 10212–10216. 8816778
37. d'Adhemar CJ, Spillane CD, Gallagher MF, Bates M, Costello KM, et al. (2014) The MyD88+ phenotype is an adverse prognostic factor in epithelial ovarian cancer. PLoS One 9: e100816. doi: 10.1371/journal.pone.0100816 24977712
38. Peterson JJ, Rayburn HB, Lager DJ, Raife TJ, Kealey GP, et al. (1999) Expression of thrombomodulin and consequences of thrombomodulin deficiency during healing of cutaneous wounds. Am J Pathol 155 : 1569–1575. 10550314
39. Hui KY, Haber E, Matsueda GR (1983) Monoclonal antibodies to a synthetic fibrin-like peptide bind to human fibrin but not fibrinogen. Science 222 : 1129–1132. 6648524
40. Ruf MT, Sopoh GE, Brun LV, Dossou AD, Barogui YT, et al. (2011) Histopathological changes and clinical responses of Buruli ulcer plaque lesions during chemotherapy: a role for surgical removal of necrotic tissue? PLoS Negl Trop Dis 5: e1334. doi: 10.1371/journal.pntd.0001334 21980547
41. Le TL, Yap AS, Stow JL (1999) Recycling of E-cadherin: a potential mechanism for regulating cadherin dynamics. J Cell Biol 146 : 219–232. 10402472
42. Adusumilli S, Mve-Obiang A, Sparer T, Meyers W, Hayman J, et al. (2005) Mycobacterium ulcerans toxic macrolide, mycolactone modulates the host immune response and cellular location of M. ulcerans in vitro and in vivo. Cell Microbiol 7 : 1295–1304. 16098217
43. Andreoli A, Ruf MT, Sopoh GE, Schmid P, Pluschke G (2014) Immunohistochemical monitoring of wound healing in antibiotic treated Buruli ulcer patients. PLoS Negl Trop Dis 8: e2809. doi: 10.1371/journal.pntd.0002809 24762629
44. Maruyama I, Majerus PW (1985) The turnover of thrombin-thrombomodulin complex in cultured human umbilical vein endothelial cells and A549 lung cancer cells. Endocytosis and degradation of thrombin. J Biol Chem 260 : 15432–15438. 2999116
45. Chu M, Bird CH, Teasdale M, Bird PI (1998) Turnover of thrombomodulin at the cell surface occurs at a similar rate to receptors that are not actively internalized. Thromb Haemost 80 : 119–127. 9684797
46. Healy AM, Rayburn HB, Rosenberg RD, Weiler H (1995) Absence of the blood-clotting regulator thrombomodulin causes embryonic lethality in mice before development of a functional cardiovascular system. Proc Natl Acad Sci U S A 92 : 850–854. 7846065
47. Isermann B, Hendrickson SB, Zogg M, Wing M, Cummiskey M, et al. (2001) Endothelium-specific loss of murine thrombomodulin disrupts the protein C anticoagulant pathway and causes juvenile-onset thrombosis. J Clin Invest 108 : 537–546. 11518727
48. Navarro S, Medina P, Bonet E, Corral J, Martinez-Sales V, et al. (2013) Association of the thrombomodulin gene c.1418C>T polymorphism with thrombomodulin levels and with venous thrombosis risk. Arterioscler Thromb Vasc Biol 33 : 1435–1440. doi: 10.1161/ATVBAHA.113.301360 23520161
49. Moxon CA, Wassmer SC, Milner DA Jr., Chisala NV, Taylor TE, et al. (2013) Loss of endothelial protein C receptors links coagulation and inflammation to parasite sequestration in cerebral malaria in African children. Blood 122 : 842–851. doi: 10.1182/blood-2013-03-490219 23741007
50. Esmon CT (2001) The normal role of Activated Protein C in maintaining homeostasis and its relevance to critical illness. Crit Care 5: S7–12. 11379986
51. Mosnier LO, Zlokovic BV, Griffin JH (2007) The cytoprotective protein C pathway. Blood 109 : 3161–3172. 17110453
52. Esmon CT (2006) Inflammation and the activated protein C anticoagulant pathway. Semin Thromb Hemost 32 Suppl 1 : 49–60. 16673266
53. Huang HC, Shi GY, Jiang SJ, Shi CS, Wu CM, et al. (2003) Thrombomodulin-mediated cell adhesion: involvement of its lectin-like domain. J Biol Chem 278 : 46750–46759. 12951323
54. Mehta D, Malik AB (2006) Signaling mechanisms regulating endothelial permeability. Physiol Rev 86 : 279–367. 16371600
55. Raife TJ, Lager DJ, Madison KC, Piette WW, Howard EJ, et al. (1994) Thrombomodulin expression by human keratinocytes. Induction of cofactor activity during epidermal differentiation. J Clin Invest 93 : 1846–1851. 8163684
56. Cheng TL, Wu YT, Lai CH, Kao YC, Kuo CH, et al. (2013) Thrombomodulin regulates keratinocyte differentiation and promotes wound healing. J Invest Dermatol 133 : 1638–1645. doi: 10.1038/jid.2013.8 23321922
57. Hsu YY, Shi GY, Kuo CH, Liu SL, Wu CM, et al. (2012) Thrombomodulin is an ezrin-interacting protein that controls epithelial morphology and promotes collective cell migration. FASEB J 26 : 3440–3452. doi: 10.1096/fj.12-204917 22593542
58. Kanga JM, Dion-Laine M, Kacou DE, Menan EI (2001) Contribution of heparin therapy in the medical treatment of Buruli ulcer apropos of 1 case. Bull Soc Pathol Exot 94 : 32–35. 11346980
59. Phillips RO, Sarfo FS, Abass MK, Abotsi J, Wilson T, et al. (2014) Clinical and bacteriological efficacy of rifampin-streptomycin combination for two weeks followed by rifampin and clarithromycin for six weeks for treatment of Mycobacterium ulcerans disease. Antimicrob Agents Chemother 58 : 1161–1166. doi: 10.1128/AAC.02165-13 24323473
60. Song F, Fidanze S, Benowitz AB, Kishi Y (2002) Total synthesis of the mycolactones. Org Lett 4 : 647–650. 11843613
61. Ahnstrom J, Andersson HM, Canis K, Norstrom E, Yu Y, et al. (2011) Activated protein C cofactor function of protein S: a novel role for a gamma-carboxyglutamic acid residue. Blood 117 : 6685–6693. doi: 10.1182/blood-2010-11-317099 21508412
62. Vaiyapuri S, Hutchinson EG, Ali MS, Dannoura A, Stanley RG, et al. (2012) Rhinocetin, a venom-derived integrin-specific antagonist inhibits collagen-induced platelet and endothelial cell functions. J Biol Chem 287 : 26235–26244. doi: 10.1074/jbc.M112.381483 22689571
63. Vaiyapuri S, Jones CI, Sasikumar P, Moraes LA, Munger SJ, et al. (2012) Gap junctions and connexin hemichannels underpin hemostasis and thrombosis. Circulation 125 : 2479–2491. doi: 10.1161/CIRCULATIONAHA.112.101246 22528526
64. Ruf MT, Pluschke G (2014) Histopathological Staining Techniques. In: Portaels F, editor. Laboratory diagnosis of Buruli ulcer: A manual for healthcare providers. Geneva: World Health Organization. pp. 76–93.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 7
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- RNA Virus Reassortment: An Evolutionary Mechanism for Host Jumps and Immune Evasion
- Activation of TLR2 and TLR6 by Dengue NS1 Protein and Its Implications in the Immunopathogenesis of Dengue Virus Infection
- N-acetylglucosamine Regulates Virulence Properties in Microbial Pathogens
- Characterization of a Prefusion-Specific Antibody That Recognizes a Quaternary, Cleavage-Dependent Epitope on the RSV Fusion Glycoprotein
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy