
Cationic Antimicrobial Peptides Promote Microbial Mutagenesis and Pathoadaptation in Chronic Infections
Antimicrobial peptides (AMPs) are produced by the mammalian immune system to fight invading pathogens. The best understood function of AMPs is to interact with the membranes of microbes, thereby disrupting and killing cells. However, the amount of AMP available during chronic bacterial infections may not be sufficient to kill pathogens (sub-inhibitory). In this study, we found that at sub-inhibitory levels, AMPs promote mutations in bacterial DNA, a function not previously attributed to them. In particular, we found that in the bacteria Pseudomonas aeruginosa, one AMP called LL-37 can promote mutations, which enable the bacteria to overproduce a protective sugar coating, a process called mucoid conversion. P. aeruginosa mucoid conversion is a major risk factor for those suffering from cystic fibrosis (CF), the most common lethal, heritable disease in the US. We found that LL-37 is able to produce these mutations by penetrating the bacterial cell and binding to the bacterial DNA. DNA binding disrupts normal DNA replication and allows mutations to occur. Furthermore, we observed LL-37 induced mutagenesis in processes apart from mucoid conversion, in both P. aeruginosa and E. coli. This suggests that AMP-induced mutagenesis may be important for a broad range of chronic diseases and pathogens.
Published in the journal:
. PLoS Pathog 10(4): e32767. doi:10.1371/journal.ppat.1004083
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004083
Summary
Antimicrobial peptides (AMPs) are produced by the mammalian immune system to fight invading pathogens. The best understood function of AMPs is to interact with the membranes of microbes, thereby disrupting and killing cells. However, the amount of AMP available during chronic bacterial infections may not be sufficient to kill pathogens (sub-inhibitory). In this study, we found that at sub-inhibitory levels, AMPs promote mutations in bacterial DNA, a function not previously attributed to them. In particular, we found that in the bacteria Pseudomonas aeruginosa, one AMP called LL-37 can promote mutations, which enable the bacteria to overproduce a protective sugar coating, a process called mucoid conversion. P. aeruginosa mucoid conversion is a major risk factor for those suffering from cystic fibrosis (CF), the most common lethal, heritable disease in the US. We found that LL-37 is able to produce these mutations by penetrating the bacterial cell and binding to the bacterial DNA. DNA binding disrupts normal DNA replication and allows mutations to occur. Furthermore, we observed LL-37 induced mutagenesis in processes apart from mucoid conversion, in both P. aeruginosa and E. coli. This suggests that AMP-induced mutagenesis may be important for a broad range of chronic diseases and pathogens.
Introduction
Cystic fibrosis (CF) is the most common lethal, heritable disease in the US and results from mutations in the gene encoding the CF transmembrane conductance regulator. One of the most concerning effects of this mutation is altered anion transport of the airway epithelial cells resulting in increased susceptibility to infections and enhanced innate immune responses (reviewed in ref. [1]). The combinatorial effect of defects in the CF airway and chronic bacterial infections results in a hyper inflammatory environment dominated by an influx of polymorphonucleocytes (PMNs). Chronic pulmonary infections with Pseudomonas aeruginosa are a leading cause of death in CF patients [2], [3]. During the course of infection, P. aeruginosa often undergoes a phenotypic change from a non-mucoid to a mucoid appearance, which directly correlates with a worsening clinical prognosis [2], [4]. Mucoid conversion is characterized by the overproduction of the polysaccharide alginate, which confers a selective advantage for P. aeruginosa in the CF lung by providing recalcitrance to currently available therapeutics and host antimicrobials (reviewed in refs. [5], [6]).
Mucoid conversion occurs when the negative regulator of alginate synthesis, MucA, is genetically or physiologically disrupted [7], [8]. Up to 84% of mucoid isolates from patients possess mutations within mucA resulting in constitutive overexpression of alginate [9]–[12]. Hyper inflammation in the CF lung environment generates an abundance of mutagenic factors, which may be responsible for directly inducing mucA mutations. For example, PMNs and hydrogen peroxide (H2O2) elevate mucoid conversion in vitro by promoting mutagenesis [13]–[15]. However, the robust adaptive nature of the P. aeruginosa genome in chronic CF infections is evident and perpetuated by the appearance of mutator strains, which likely contribute to selection of mucoid variants (reviewed in [16]–[19]). Therefore, it will be of therapeutic utility to determine if specific host factors in CF promote mucA mutagenesis and investigate if intervention at this initial stage would prove effective.
This study aimed to examine the specific role of host inflammatory factors in promoting mucA mutations leading to mucoid conversion. PMNs derived from both healthy and CF individuals stimulated mucoid conversion independent of selection. Surprisingly, while reactive oxygen species (ROS) have the capacity to induce mucoid conversion in vitro, PMNs still efficiently promote mucoid conversion in the absence of an oxidative burst response. This led to the discovery that non-oxidative PMN components, including the antimicrobial peptide LL-37, are important for promoting mucoid conversion in CF. Importantly, LL-37 also elevated spontaneous rifampin resistance in P. aeruginosa and E. coli, indicating a new role for LL-37 as a bacterial mutagen. LL-37-induced mutagenesis required the translesion DNA polymerase, DinB (Pol IV); whose error-prone replication is responsible for generating mucA mutations. Using several independent methods, it was determined that, at sub-inhibitory concentrations, LL-37 enters P. aeruginosa cells, interacts with DNA, and promotes mucA mutations. Finally, conversion of P. aeruginosa to the mucoid phenotype then protects the bacterial cells from killing by lethal concentrations of LL-37. These data reveal a novel mechanism to describe how antimicrobial peptides interact with bacterial cells and demonstrate that LL-37 may promote mutations leading to persistence and chronic infection.
Results and Discussion
Development of a genetic scheme to measure the frequency of mucoid conversion and select for stable P. aeruginosa mucoid variants
The study of mucoid conversion in the laboratory has been hampered by the difficulty in isolating rare mucoid variants that arise in a population (∼1×10−9). To circumvent this problem, a system for selecting mucoid colonies was developed (Figure S1 and Materials and Methods). A promoterless gene encoding chloramphenicol-resistance (cat) was placed under control of the promoter of the alginate biosynthetic operon (algD) [20], [21] and integrated into the chromosome of non-mucoid, chloramphenicol sensitive P. aeruginosa strain PAO1 to generate PAO1algD-cat (WFPA934). Under this system, mucoid bacteria that are producing alginate and therefore transcribing algD will be chloramphenicol-resistant, allowing for selection of mucoid variants by growth on chloramphenicol-containing media (Figure S1A). By comparing the numbers of colonies on non-selective versus chloramphenicol media the relative frequency of mucoid conversion was determined.
To investigate the utility of this selection strategy, non-mucoid P. aeruginosa were exposed to a sub-inhibitory concentration (1/10 the minimum inhibitory concentration (MIC; 0.1 µM)) of H2O2 for one hour, followed by overnight recovery in media alone. Cultures are then serially diluted and plated on non-selective media to determine the total number of bacteria (∼1×1010) and whole culture plated on chloramphenicol containing media to determine the number of mucoid colonies (0–300 depending on treatment). The mucoid conversion frequency is then determined by dividing the number of mucoid colonies by the total. There was no significant difference among treatments in the total number of bacteria after the one-hour treatment or after the overnight recovery period, therefore changes in mucoid conversion frequencies are a direct result of induction of mucoid variants. In agreement with previous studies [13], [14], [22], treatment with H2O2 significantly increased the frequency of mucoid conversion (Figure S1B). While non-mucoid chloramphenicol isolates were observed, the frequency was very low (∼1.9×10−10) and was not altered by the addition of H2O2 (or any of the other mutagens used in this study, see below). Since the translesion DNA polymerase DinB (Pol IV) and defects in the mismatch repair protein MutS contribute to mucoid conversion [14], [15], [22], the role of these proteins was also examined. As predicted, P. aeruginosa isolates lacking mutS exhibited increased mucoid conversion frequencies and dinB– deficient isolates had severely impaired mucoid conversion (Figure S1B). Importantly, no reduction in growth was observed during the short treatment periods utilized and resulting mucoid variants were stable upon multiple passages, where approximately 80% possessed mutations in mucA (Table S1). Therefore, we were able to identify mucoid variants, which acquire mutations by direct induction versus selection based on resistance. This selection strategy now provides a unique opportunity to interrogate the CF factors that directly promote mutations leading to mucoid conversion.
PMNs promote mucoid conversion independent of phagocytosis and the oxidative burst response
To examine the role of PMNs in mucoid conversion, opsonized PAO1algD-cat was incubated in the presence of PMNs derived from three sources: peripheral PMNs from healthy or CF human donors, or an inducible promyelocytic cell line (HL-60) as described in Supporting Material and Methods (Text S1). Treatment with each cell type significantly increased the frequency of mucoid conversion (Figure 1A). A trend was also observed for increased mucoid conversion in CF PMNs compared to healthy PMNs from multiple donors; however, this was not a statistically significant difference. Moreover, mucA from mucoid isolates treated with healthy human PMNs harbored mutations (Table S1), demonstrating that human PMNs can induce mutations promoting mucoid conversion.

To investigate the mechanism(s) by which PMNs induce mucoid conversion, we first sought to determine if bacterial uptake is necessary. Phagocytosis was blocked by the addition of cytochalasin D (Figure 1B; confirmed by microscopy (Figure S2A)), or by separating PMNs from the bacteria with transwells (Figure 1B). Efficient mucoid conversion was maintained when phagocytosis was inhibited by either method, demonstrating the factors promoting mucoid conversion are released from the PMN. Since we hypothesize ROS generation is responsible for PMN-induced mucoid conversion, we sought to determine if inhibition of bacterial uptake affected the oxidative burst response. Surprisingly, both cytochalasin D and separation by transwells blocked the PMN oxidative burst (Figure S2B, C respectively), suggesting PMNs can promote mucoid conversion in the absence of ROS. To confirm this, PMNs were specifically inhibited by pretreatment with the NADPH oxidase inhibitor diphenyleneiodonium (DPI). While treatment of PMNs with DPI significantly inhibited ROS production (Figure 1C), this did not lead to inhibition of mucoid conversion (Figure 1D). Inhibition of oxidative burst was further confirmed with lucigenin and scopoletin, which measure O2• generation, and H2O2, respectively (data not shown). Collectively, these data demonstrate that PMNs deficient in generating ROS can still efficiently induce mucoid conversion.
Non-oxidative PMN pathways promote mucoid conversion
The above data reveal that ROS-independent PMN factors may be important determinants of mucoid conversion. Moreover, while ROS clearly promote mucoid conversion in vitro and likely contribute to pathoadaptation in vivo, oxygen independent mechanisms may predominate in CF due to the hypoxic nature of the mucopurulent masses in the lumen of patient airways, where P. aeruginosa microcolonies reside [23], [24]. To test whether non-oxidative components are involved in mucoid conversion, PMN lysates that retained granular components but lack ROS were prepared (see Text S1. Supporting Materials and Methods). Treatment of PAO1algD-cat with PMN lysates or soluble components specifically isolated from granules increased the frequency of mucoid conversion compared to vehicle treated P. aeruginosa (Figure 2A). Moreover, PMN lysates and granule fractions promoted mutations within mucA, indicating non-oxidative PMN components possess mutagenic capacity (Table S1). To investigate which granule component(s) are responsible for mucoid conversion, PAO1algD-cat was incubated with sub-inhibitory concentrations (0.25 µM) of purified AMP including the human cathelicidin, LL-37, beta defensins (hBD) 1 and 2, and human neutrophil peptide 1 (HNP1). LL-37 and hBD2 treatment increased the frequency of mucoid conversion approximately four-fold and two-fold respectively compared to buffer controls, while hBD1 and HNP1 did not exhibit any effect (Figure 2B). Together these data reveal a novel property for non-oxidative PMN granular components, including specific antimicrobial peptides, in promoting P. aeruginosa mucoid conversion.

LL-37 is a human cationic host defense molecule elevated in CF sputum and found in the granules of PMNs and at mucosal surfaces [25]. In addition to possessing a broad range of antimicrobial action against bacteria, viruses, and fungi, LL-37 possess immunomodulatory and anti-biofilm activities at sub-inhibitory concentrations [26]–[28]. In PMNs, the propeptide form of LL-37 is stored within the specific granules and upon stimulation is released to the extracellular environment and cleaved to the mature form by serine proteases [29]. Since the PMN factors primarily responsible for mucoid conversion are released from the PMN into the extracellular environment (Figure 1B) and we were able to confirm the presence of mature LL-37 in PMN lysate preparations by immunoblot analysis (Figure S3), we hypothesized that LL-37 may promote mucoid conversion in CF and focused current studies on LL-37.
LL-37 contributes to P. aeruginosa mucoid conversion in CF
Despite the presence of elevated LL-37 in CF sputum, it has been argued that in the CF pulmonary environment some AMPs may not function optimally due to high salt concentrations [30], [31] or may be sequestered by extracellular DNA and F-actin bundles [32]. Therefore, to determine if LL-37 mediated mucoid conversion is a process capable of functioning in the CF pulmonary environment, the impact of sputum isolated from CF patients on mucoid conversion was investigated. Sputum isolated from four different CF individuals induced an average mucoid conversion frequency of 3×10−9 (Figure 2C). To determine if LL-37 contributes to sputum induced mucoid conversion, LL-37 was immune-depleted from each sputum sample. The mucoid conversion frequency decreased significantly upon depletion with antibodies directed toward LL-37, compared to an isotype control antibody (Figure 2C), demonstrating LL-37 contributes to mucoid conversion in CF sputum. However, immune-depletion of LL-37 did not completely abolish mucoid conversion. This is likely a combination of incomplete elimination of LL-37 from CF sputum, as well as potential contribution of other inflammatory factors in sputum to mucoid conversion (oxidative and nitrosative stress, other AMP, etc). Moreover, after incubation of P. aeruginosa with CF sputum, no change in the overall viability of the bacteria was observed (data not shown). These data suggest that antimicrobial factors present in CF sputum may not be present at bioavailable levels sufficient to control P. aeruginosa infections, but are sufficient to promote conversion to the mucoid phenotype.
LL-37 induces bacterial mutagenesis
To investigate how LL-37 promotes mucoid conversion, we first interrogated the primary mechanism of mucoid conversion observed in CF P. aeruginosa isolates, mutagenesis of the anti-sigma factor encoding gene mucA. We subjected ten LL-37-derived mucoid isolates to mucA sequence analysis (Table S2). All isolates possessed mutations within mucA and 70% of these were frameshifts predicted to eliminate mucA function. Mucoid clinical isolates from CF patients possess a range of mucA alterations; however, frameshift mutations and C to T transitions are the most common [33]–[36], a pattern represented here in LL-37 treated mucoid isolates (Table S2). To confirm the alterations in mucA were responsible for the mucoid phenotype, mucA was expressed on an arabinose inducible plasmid (pHERD20TmucA) in representative isolates with unique mucA alleles (WFPA934 LL-37 1.1 (ΔC at 184), 1.2 (C→A at 531), and (ΔT at 470)). The phenotype of each isolate harboring pHERD20TmucA was complemented to the non-mucoid phenotype upon growth on arabinose (Figure 3A), indicating these mutations are directly responsible for LL-37-dependent mucoid conversion.

To determine if LL-37 promotes global bacterial mutagenesis, the frequency of LL-37-induced rifampin resistance (RifR) was examined. Upon treatment of non-mucoid PAO1 with sub-inhibitory concentrations of LL-37 a moderate, but significant increase in RifR was observed compared to cells treated with buffer. The frequency of acquisition of RifR was similar to control treatments with H2O2 (Figure 3B), which is known to promote global mutagenesis in a range of microorganisms [37]. Furthermore, LL-37 also elevated the acquisition of RifR in E. coli, demonstrating this mechanism of mutagenesis is not specific to P. aeruginosa (Figure 3B). Collectively, these data demonstrate for the first time that LL-37 can function as bacterial mutagen and may contribute to the generation of mutations during chronic infection.
The translesion DNA polymerase DinB is essential for LL-37 induced mucoid conversion
To uncover the mechanism by which LL-37 induces mutations, we investigated the DNA repair enzymes MutS and DinB, due to their previously identified role in both P. aeruginosa and E. coli mutagenesis [14], [15], [22], [38]. The mucoid conversion frequency of ΔmutS, ΔdinB and double ΔmutSΔdinB mutants upon treatment with LL-37 was determined (Figure 3C). In the mutS mutant the spontaneous mutation frequency was increased nearly 1000-fold and this was further elevated with LL-37 treatment. Of notable significance, mucoid conversion was almost eliminated in a dinB-deficient strain, independent of the mutS status. These results illustrate an important role for DNA repair proteins in LL-37-induced mucoid conversion.
Cell envelope stress has been linked to production of intracellular ROS, which induce mutagenesis and bacterial SOS response genes [39]. Since DinB is a member of the SOS regulon [15]; we initially hypothesized that LL-37 interactions with the P. aeruginosa cell envelope would result in membrane stress, leading to induction of SOS response genes, including dinB. Therefore, the expression of P. aeruginosa regulators of membrane and SOS responses, algT/U (σE/22), and lexA, respectively, were evaluated by quantitative real time PCR (qRT-PCR) following LL-37 exposure. However, treatment of non-mucoid PAO1 with sub-inhibitory concentrations of LL-37 did not significantly alter the expression of lexA or algT/U, whereas positive control treatments did (mitomycin C and D-cycloserine, respectively) (Table S3). Surprisingly, expression of dinB was also not altered by LL-37 treatment (Table S3). Combined with the observation that DinB is required for spontaneous generation of mucA mutations (buffer treated, Figure 3C), these results suggest that basal levels of DinB present during normal replication are sufficient to promote mutagenesis leading to mucoid conversion. We propose that LL-37 elevates mutagenesis in P. aeruginosa by a mechanism independent of membrane or SOS stress responses.
Sub-inhibitory concentrations of LL-37 enter the P. aeruginosa cytosol and interact with DNA
Bacterial DNA has been proposed to be an alternative target for a subset of AMPs, whereby peptides gain entry to the cytosol and bind bacterial DNA, resulting in subsequent disruption of DNA or protein synthesis [40]–[45]. Since membrane stress and SOS response pathways were not induced by LL-37, we explored an alternative hypothesis that LL-37 may induce translesion DNA synthesis and mutagenesis by interacting directly with genomic DNA. The structure of LL-37 resembles classical cell-penetrating peptides and it has been postulated that LL-37 may enter both prokaryotic and eukaryotic cells. Lande et al demonstrated that LL-37 is capable of trafficking into dendritic cells, providing evidence that LL-37 can penetrate eukaryotic cells [46]. While multiple reviews have generalized this observation to include all cell types, it has yet to be formally determined if LL-37 can enter the cytosol of bacterial cells [27], [45]. Therefore, the capacity of sub-inhibitory concentrations of LL-37 to enter the P. aeruginosa cytosol was examined utilizing fluorescent confocal - and transmission-electron microscopy (TEM). Non-mucoid PAO1 was treated with LL-37, fixed, and labeled with anti-LL37. For confocal microscopy, the localization of anti-LL37 labeling of permeabilized PAO1 (to allow antibody access to the cytosol) was compared to non-permeabilized cells. Non-permeabilized cells showed a weak, diffuse signal on the surface, compared to permeabilized cells, which demonstrated a stronger, punctate signal in the center of the cell (indicated by white arrows, Figure 4A). This was clearly visible only in the cytosol, when sequential images where taken along the extent of the Z plane of the cells (shown in Movie S1). With transmission electron microscopy (TEM), LL-37 labeling was visible in the cytosol of approximately 28% (white arrows) of cells and in the membrane of 11% (black arrows)(Figure 4B, center).
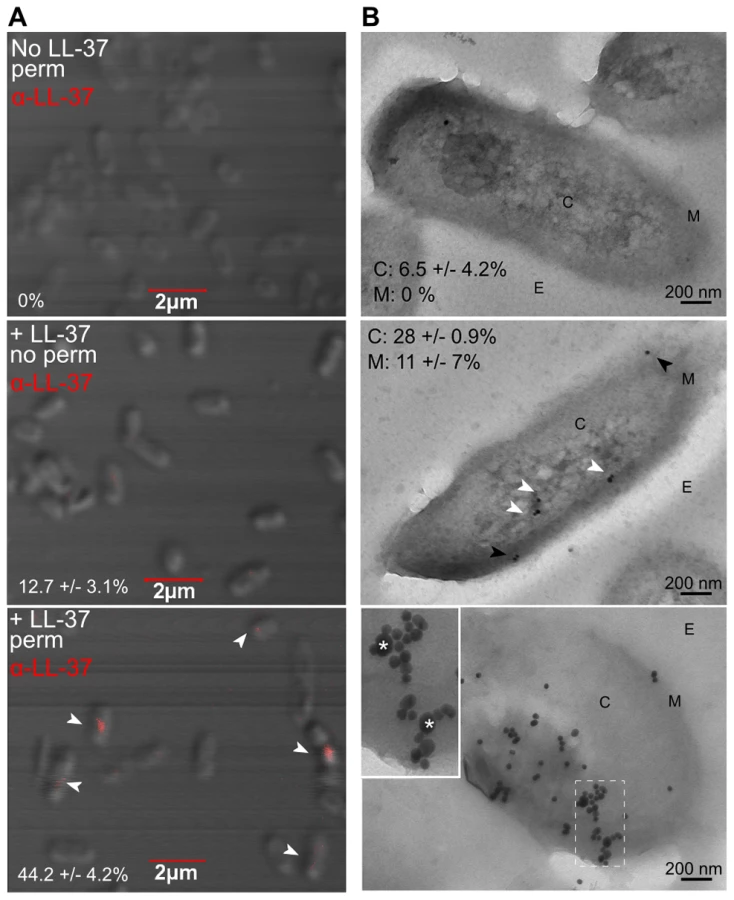
These data reveal for the first time that LL-37 can gain entry to the cytosol of P. aeruginosa. However, intracellular LL-37 was visualized in only a subset of cells. Of note, 43% of cells with cytosolic LL-37 labeling, as visualized by confocal microscopy, appeared to be actively dividing (Figure 4A). At lethal concentrations, LL-37 readily interacts with the septum of diving cells [47]; therefore active replication may also play a role in LL-37 gaining access to the P. aeruginosa cytosol at sub-inhibitory concentrations. Several models have been proposed to explain how AMPs interact with bacterial membranes, but accumulating evidence for LL-37 supports the Shai-Matsuzaki-Huang model [28], [48]–[51]. In this model, peptides carpet the outer leaflet of the bacterium and integrate into the membrane. This is followed by a transient pore forming stage, where lipids and peptides are transported to the inner leaflet, resulting in collapse of membrane fragments and disruption of the membrane. In some cases, transient pore formation results in diffusion of peptides into the cytosol, where peptides can then interact with intracellular targets [48]. At sub-inhibitory concentrations, transient pore formation may occur without significant disruption of the membrane or loss of cell viability.
To determine if bacterial DNA might be an intracellular target of LL-37, TEM experiments were performed as described previously, with a second label added for double-stranded DNA. 76% of intracellular LL-37 was localized with P. aeruginosa DNA (Figure 4B, bottom panel). Electrophoretic mobility shift assays (EMSA) were also performed, where LL-37 treatment resulted in a shift of all DNA tested with similar affinity (apparent KD = 12.8 µM, Figure S4), further demonstrating that LL-37 binds non-specifically to P. aeruginosa DNA. It was observed that upon the addition of increasing amounts of LL-37 a threshold concentration was reached where no migration of DNA was observed, instead of a step-wise migration, a phenotype that has been observed for other short DNA binding peptides [41], [52].
To investigate how LL-37 may interact with DNA and whether DNA binding is necessary for mucoid conversion, a structure-based search of LL-37 revealed homology to eukaryotic transcription factors containing basic region leucine zipper (bZIP) motifs. A three-dimensional model of LL-37 bound to DNA suggested that the amino-terminal basic residues of LL-37 occupy positions suitable for interactions with the negatively charged phosphate groups of the DNA backbone (Figure 5A). To investigate LL-37/DNA interactions, two synthetic peptides were generated: an LL-37 variant with the amino acid sequence randomly scrambled and a variant with the basic residues in the predicted DNA binding region replaced with glutamate residues (K/R7-19E, Figure 5B). Scrambled LL-37 had a moderate loss of DNA binding, as well as a decrease in the ability to induce mucoid conversion (Figure 5C and D). When basic residues within the putative DNA binding region were modified, a complete loss of both DNA binding and mucoid conversion was observed (Figure 5C and D). These data suggest that LL-37/DNA interactions are required for LL-37 to promote mucoid conversion. Studies are currently underway to further define LL-37/DNA interactions, their impact upon DinB and/or the DNA replisome, and the precise mechanism of mutagenesis.

LL-37 contributes to pathoadaption of P. aeruginosa
Herein, we show that LL-37 induces mucoidy but it is unknown if this conversion provides P. aeruginosa adaptive protection from lethal concentrations of LL-37. We observed a 10-fold increase in survival of two mucoid isolates derived from LL-37 treatment when compared with parental non-mucoid PAO1 (Figure 6A). To determine if protection was dependent upon prior treatment with LL-37, a H2O2-derived mucoid isolate, and a genetically engineered mucoid strain, PDO300 (PAO1ΔmucA22) were examined and both showed enhanced resistance to LL-37 mediated killing (Figure 6A). These data demonstrate that conversion to the mucoid phenotype provides P. aeruginosa protection from lethal concentrations of LL-37, independent of prior exposure to LL-37. Since mucA inactivation and subsequent algT/U induction controls an entire regulon, of which only a subset are dedicated to alginate production, it was necessary to determine if alginate overproduction is specifically responsible for LL-37 protection. FRDmucA22ΔalgD (deficient in alginate production) was 10-fold more susceptible to LL-37 compared to the isogenic mucoid parental strain FRD1 (mucA22, Figure 6B). These data demonstrate that alginate overproduction contributes to protection of P. aeruginosa from LL-37 killing and this might promote P. aeruginosa pathoadaptation in the CF pulmonary environment.

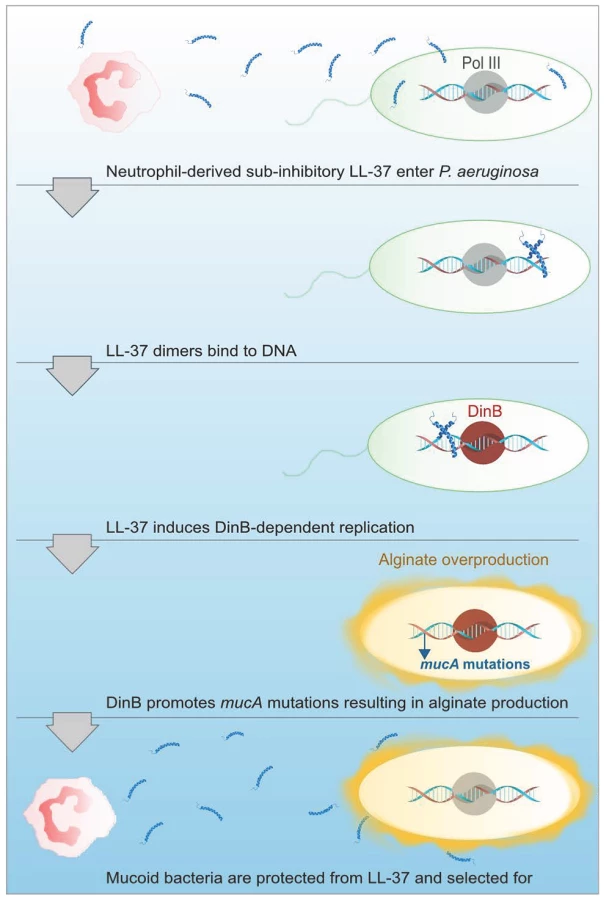
Collectively, these data provide evidence for an additional role for LL-37 beyond the antimicrobial, anti-biofilm, and immunomodulatory functions previously described. We demonstrate that at sub-inhibitory levels, LL-37 promotes bacterial mutagenesis, which may contribute to evolution and pathoadaptation during chronic infections. Importantly, LL-37 induced mutations within mucA mimic what is observed in mucoid P. aeruginosa strains isolated from the CF airway and the sub-inhibitory conditions utilized may be representative of the level of bioavailable peptide present in the CF pulmonary environment. Furthermore, LL-37 induced mutagenesis of mucA leading to mucoid conversion was modulated exclusively by the error-prone polymerase DinB. We were intrigued by the stringent dependence upon DinB in this process, particularly since previous studies demonstrate DinB is not required for spontaneous or UV-induced RifR [15]. We observed that LL-37 can promote RifR in both P. aeruginosa and E. coli, suggesting perhaps the mechanism for acquisition of LL-37-induced mucA mutations and RifR occur via different pathways. Moreover, LL-37 did not increase the expression of DinB under these conditions demonstrating basal levels of DinB are sufficient to promote mutagenesis. Typical translesion DNA synthesis occurs when the replisome stalls upon encountering damaged DNA or a challenging template and low-fidelity polymerases like DinB will displace Pol III in order to perpetuate replication [53]. Since LL-37 interactions with DNA are required for LL-37-incuded mutagenesis we postulate that LL-37 presents a physical barrier that stalls Pol III, inducing a switch to DinB, whose error-prone replication promotes mutagenesis (see Figure 7 for model). EMSAs suggest that LL-37 non-specifically interacts with DNA; however, peptides have been identified which specifically interact with DNA repair intermediates, such as Holliday junctions [54], [55]. Therefore, an alternative hypothesis could be that LL-37 perturbs effective DNA repair by binding to repair intermediates. In this regard, Overhage et al performed microarray experiments with sub-inhibitory concentrations of LL-37 and identified changes in expression of genes involved in alginate regulation and DNA repair [26]. However, differences in the conditions utilized in these experiments and a lack of convergence upon a single pathway do not support any one hypothesis. Additionally, sub-inhibitory levels of LL-37 have been found to stimulate expression of the capsule synthesis operon in Group A Streptococci [56]. Together these studies suggest additional functions for sub-inhibitory levels of LL-37 impacting the expression of virulence genes. While the current study identified stable variants generated by mutagenesis, it is interesting to speculate how transient gene expression may impact their generation and selection in the host environment and further studies are clearly warranted.
While data presented here demonstrate PMNs can promote mucoid conversion in the absence of an oxidative burst response, it is likely that mucoid conversion in the CF lung results from a combination of non-oxidative, oxidative, and nitrosative stresses. Moreover, other chronic pneumonias, such as COPD, are also characterized by elevated levels of PMNs, generally on the order of two to three-fold higher than healthy controls [57], [58]. Mucoid P. aeruginosa have been isolated from patients with COPD, therefore, the mechanisms elucidated in this study may be important in other infections where chronic inflammation ensues [59]. Bronchial alveolar lavage fluid recovered from CF patients can have up to a 380-fold increase in PMNs recovered compared to healthy patients, even in patients without symptoms of an active infection [60]. Moreover, elevated levels of PMNs are detectable in CF newborns, which persist and undergo cycles of exacerbation throughout the life-time of the patient [61]. We therefore hypothesize that persistent exposure of P. aeruginosa to chronic inflammation for decades significantly contributes to microbial pathoadaptation in CF patients. Future investigation of how these inflammatory factors function in combination to promote P. aeruginosa pathoadaptation will be important for comprehensive understanding of these processes and the development of rational therapeutics for chronic infections.
Aggressive antibiotic and anti-inflammatory use over the past decade has drastically improved the life expectancy and disease outcome for CF patients. However, increased acquisition of antibiotic resistance mechanisms is presenting a significant challenge for future treatment options [62]. Many are turning towards cationic antimicrobial peptides as a promising alternative for developing antimicrobials, as they are thought to be relatively insusceptible to the development of resistance mechanisms by mutations [63]. This study demonstrates that some antimicrobial peptides may instead act to promote mutagenesis and the acquisition of resistance at sub-inhibitory levels. These data reinforce how important it is to consider the impact of current and novel treatments and the host immune response on evolution of microbial communities during chronic infections.
Materials and Methods
Ethics statement
Human PMNs and serum were obtained from healthy adult human donors according to the protocol approved by The Ohio State University Biomedical Sciences Institutional Review Board (2009H0314), where informed consent was obtained from all donors. Sputum, PMNs and serum were obtained from CF patients (adults and minors) according to the protocol approved by The Nationwide Children's Hospital Institutional Review Board (IRB12-00405) with informed consent obtained from all adult donors and from the parents/guardians of minors who participated. For children between the age of 9 and 18 an assent form was also obtained.
Mucoid conversion studies
Overnight cultures of WFPA934 (PAO1algD-cat, Table S4) were diluted into fresh M63 media and grown to mid-log phase. 2×108 colony forming units (CFU)/ml were resuspended in Hank's buffered saline (HBSS) or 10 mM sodium phosphate buffer (pH 6.2, SPB), as indicated and treated with sub-inhibitory concentrations of antimicrobials for one hour. Reactive oxygen species were used at 1/10 the minimum inhibitory concentration (MIC): hydrogen peroxide, 0.1 µM, hypochlorous acid, 0.1 µM, and paraquat to generate superoxide, 100 mM, antimicrobial peptides (LL-37, human beta-defensin 1, human beta-defensin 2 and human neutrophil peptide 1) were used at 0.25 µM (PeproTech, Rocky Hill, NY). Human PMNs and serum were isolated according to previously described protocols [64]. Mid-log phase P. aeruginosa suspensions were opsonized with 10% fresh human serum at 37°C for 30 min, added (MOI of 50) to the wells and centrifuged at 100 g for 2 min at 4°C to synchronize the infection. PMN treatments were performed according to previously described protocols and are described in detail in Supporting Materials and Methods (Text S1). P. aeruginosa cells were then washed twice and resuspended in M63 media for an overnight growth recovery period. Cultures were then serially diluted for plating on non-selective Pseudomonas isolation agar (PIA) and incubated at 37°C overnight to determine the total CFU and plated straight onto PIA containing chloramphenicol (250 µg/ml) and incubated at 37°C for 48 hours to determine the number of mucoid CFU. The mucoid conversion frequency was then determined by dividing the number of mucoid variants by the total number of CFU.
CF sputum samples
CF sputum samples were collected by spontaneous expectoration from patients attending Nationwide Children's Hospital in Columbus, OH (IRB12-00405). The samples were diluted 1∶1 in buffer containing 140 mM NaCl, 10 mM Tris, 0.2 mM CaCl2 (pH 7.4) and physically disrupted by pipetting up and down with decreasing sized serological pipettes, followed by pushing through decreasing sized needles until sample is easily pushed through a 27 gauge needle. Samples were then centrifuged (10 min; 15,500 g) to pellet the remnant cells and bacteria. For immune-depletion of LL-37, sputum was incubated with 1.6 µg/ml monoclonal anti-LL37 antibody (Santa Cruz Biotechnology, Dallas, TX) or mouse IgG1 isotype control antibody (R & D Systems, Minneapolis, MN) overnight at 4°C. The antibody/antigen complex was then pulled-down with Protein A/G Agarose Beads (Thermo Scientific, Rockford, IL) according to the manufacturer's instructions and confirmed by immunoblot analysis (See Text S1. Supporting Materials and Methods).
PCR and sequencing
Genomic DNA was harvested from the mucoid variants isolated in the mucoid conversion assay using the Wizard genomic purification kit (Promega). PCR amplification was performed using mucA-specific primers mucAupF and mucAdnR (Table S5). After verification of PCR product by agarose gel electrophoresis, The Ohio State University Medical Center Nucleic Acid Core sequenced the PCR products using Sanger sequencing techniques with mucAupF, mucAdnR, and mucA1F21. The sequence data produced for mucoid variants were then aligned with mucA gene sequence of the non-mucoid PAO1algD-cat parental to determine if and/or where mutations occurred in the mucA gene of the variants.
Complementation analyses and alginate quantification
The mucA allele was cloned into the shuttle vector pHERD20T [65] for complementation with gene expression driven by the pBAD arabinose-inducible promoter. P. aeruginosa strains were grown at 37°C on PIA plates supplemented with carbenicillin and 0.1% (wt/vol) arabinose for 24 h. Bacterial growth was removed from plates with phosphate-buffered saline (PBS) and the optical density at 600 nm (OD600) of the bacterial suspension in PBS was measured. Alginate was isolated and measured by a standard carbazole assay as previously described [66], [67].
Rifampin assays
Overnight cultures were diluted into fresh M63 media for P. aeruginosa or LB for E. coli and grown to mid-log phase. 2×108 CFU/ml were resuspended in 10 mM sodium phosphate buffer (pH 6.2)(SPB), and treated with sub-inhibitory concentrations of LL-37 or H2O2 (1.25 µM or for P. aeruginosa and 6.25 µM for E. coli.). Cells were then washed twice and resuspended in media for an overnight growth recovery period. Cultures were then serially diluted for plating on non-selective PIA or LB and plated straight onto media containing 100 µg/ml rifampin (Rif) and incubated at 37°C for 24 hours. The Rif resistance frequency was then determined by dividing the number of Rif resistant variants by the total number of CFU.
Microscopy studies
For determination of localization of sub-inhibitory concentration of LL-37 by confocal microcopy, PAO1algD-cat was grown to mid-log phase and 1×106 cells were incubated with 0.25 µM LL-37 or SPB for one hour. Cells were washed, fixed in 4% paraformaldehyde for 10 min, permeabilized with 0.2% Triton-X for 1 min, blocked with 2% bovine serum albumin (BSA) and stained with anti-LL-37 antibody (Santa Cruz) (1∶100) conjugated directly to Alexa Fluor 647 (Invitrogen). Bacteria were wet-mounted onto coverslips and visualized by confocal microscopy (Olympus FV 1000 Spectral) using a 100× oil objective. For quantification, 100 cells were chosen at random and intracellular and membrane-associated peptides counted and averaged by two readers blinded to the treatment conditions. For transmission electron microscopy, PAO1 was grown to mid-log phase and 1×106 cells were incubated with 0.25 µM LL-37 or SPB for one hour. Cells were washed and fixed in 4% paraformaldehyde for 12 h. Free aldehyde was quenched by the addition of 0.1 M glycine for 20 min and cells were resuspended in 0.2 M sucrose and incubated at 4°C overnight. Samples were embedded in LR white, cut into ultrathin sections (Leica EMU 6 ultramicrotome) (60–90 nm) and collected into formvar-coated nickel grids. Sections were stained by anti-LL37 (Santa Cruz) antibody conjugated to Protein-G colloidal gold (20 nm)(1∶500)(EY Laboratories) and/or anti-dsDNA (Abcam) conjugated to Protein-G colloidal gold (10 nm, 1∶100, EY Laboratories). Sections were viewed by transmission electron microscopy (FEI Tecnai G2 Spirit) operating at 80 kV. Twenty images were chosen at random and intracellular and membrane-associated peptides counted and averaged by two readers blinded to the treatment conditions.
Structure analysis and molecular modeling
A structural based homology search was performed using the DALI server. Homology modeling of LL-37 bound to B-DNA was performed manually on the basis of the backbone atomic coordinates of the homologous protein, sterol regulatory element binding protein, bound to DNA, whose crystal structure is known (pdbid: 1am9) [68].
LL-37 susceptibility assay
2×108 CFU of mid-log phase P. aeruginosa strains were incubated with lethal doses of LL-37 (2.5 µM) of LL-37 for 1 hour at 37°C washed, serially diluted and plated on PIA to determine the total CFU.
Statistical analysis
Results of mutagenesis studies presented significant variation (including spontaneous untreated controls) in accordance with previous observations [69]. Therefore all mutagenesis studies (including rifampin resistance) were performed in triplicate on at least four independent occasions. For all statistical analyses, data were tested for normality using Prism Version 5.0b. Data determined to be normally distributed were analyzed for statistical significance using parametric unpaired two-tailed students t-test in Prism. Data not normally distributed were analyzed using non-parametric unpaired two-tailed Mann-Whitney test using Stata Version 10.1. For experiments with multiple treatments (Figure 3BC, 5D, and S1B) ANOVA or Kruskal-Wallis (for data not normally distributed) with a Dunn's post-test yielded similar results as the indicated t-tests.
Accession numbers
Proteins discussed in this manuscript are listed followed by their corresponding UniProtKB (Universal Protein Knowledgebase) number: MucA (MUCA_PSEAE), DinB (DPO4_PSEAE), AlgT/U (RPSH_PSEAE), and LexA (LEXA_PSEAE).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. BoucherR (2004) New concepts of the pathogenesis of cystic fibrosis lung disease. European Respiratory Journal 23 : 146.
2. DoullIJ (2001) Recent advances in cystic fibrosis. Archives of Disease in Childhood 85 : 62–66.
3. GasparMC, CouetW, OlivierJC, PaisAACC, SousaJJS (2013) Pseudomonas aeruginosa infection in cystic fibrosis lung disease and new perspectives of treatment: a review. European Journal of Clinical Microbiology & Infectious Diseases 32 : 1–22 doi:10.1007/s10096-013-1876-y
4. FickR, SonodaF, HornickD (1992) Emergence and persistence of Pseudomonas aeruginosa in the cystic fibrosis airway. Sem Resp Infec 7 : 168–178.
5. GovanJR, DereticV (1996) Microbial pathogenesis in cystic fibrosis: mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol Rev 60 : 539–574.
6. RamseyD, WozniakD (2005) Understanding the control of Pseudomonas aeruginosa alginate synthesis and the prospects for management of chronic infections in cystic fibrosis. Mol Microbiol 56 : 309–332.
7. MartinDW, SchurrMJ, MuddMH, GovanJR, HollowayBW, et al. (1993) Mechanism of conversion to mucoidy in Pseudomonas aeruginosa infecting cystic fibrosis patients. Proc Natl Acad Sci USA 90 : 8377–8381.
8. DamronFH, GoldbergJB (2012) Proteolytic regulation of alginate overproduction in Pseudomonas aeruginosa. Mol Microbiol 84 : 595–607 doi:10.1111/j.1365-2958.2012.08049.x
9. FyfeJ, GovanJ (1981) A revised chromosomal location for muc: a locus involved in the control of alginate production by mucoid Pseudomonas aeruginosa. Soc Gen Microbiol Q 8 : 250–251.
10. MacGeorgeJ, KorolikV, MorganA, AsheV, HollowayB (1986) Transfer of a chromosomal locus responsible for mucoid colony morphology in Pseudomonas aeruginosa isolated from cystic fibrosis patients to P. aeruginosa PAO. J Med Microbiol 21 : 331–336.
11. FlynnJ, OhmanD (1988) Cloning of genes from mucoid Pseudomonas aeruginosa which control spontaneous conversion to the alginate production phenotype. J Bacteriol 170 : 1452–1460.
12. FyfeJA, GovanJR (1980) Alginate synthesis in mucoid Pseudomonas aeruginosa: a chromosomal locus involved in control. J Gen Microbiol 119 : 443–450.
13. MatheeK, CiofuO, SternbergC, LindumPW, CampbellJI, et al. (1999) Mucoid conversion of Pseudomonas aeruginosa by hydrogen peroxide: a mechanism for virulence activation in the cystic fibrosis lung. Microbiology 145 : 1349–1357.
14. MoyanoAJ, LujánAM, ArgarañaCE, SmaniaAM (2007) MutS deficiency and activity of the error-prone DNA polymerase IV are crucial for determining mucA as the main target for mucoid conversion in Pseudomonas aeruginosa. Mol Microbiol 64 : 547–559 doi:10.1111/j.1365-2958.2007.05675.x
15. SandersLH, RockelA, LuH, WozniakDJ, SuttonMD (2006) Role of Pseudomonas aeruginosa dinB-encoded DNA polymerase IV in mutagenesis. J Bacteriol 188 : 8573–8585 doi:10.1128/JB.01481-06
16. CiofuO, MandsbergLF, BjarnsholtT, WassermannT, HoibyN (2010) Genetic adaptation of Pseudomonas aeruginosa during chronic lung infection of patients with cystic fibrosis: strong and weak mutators with heterogeneous genetic backgrounds emerge in mucA and/or lasR mutants. Microbiology 156 : 1108–1119 doi:10.1099/mic.0.033993-0
17. HogardtM, HeesemannJ (2010) Adaptation of Pseudomonas aeruginosa during persistence in the cystic fibrosis lung. International Journal of Medical Microbiology 300 : 557–562.
18. HogardtM, HobothC, SchmoldtS, HenkeC, BaderL, et al. (2007) Stage-specific adaptation of hypermutable Pseudomonas aeruginosa isolates during chronic pulmonary infection in patients with cystic fibrosis. J Infect Dis 195 : 70–80 doi:10.1086/509821
19. MatheeK, NarasimhanG, ValdesC, QiuX, MatewishJM, et al. (2008) Dynamics of Pseudomonas aeruginosa genome evolution. Proc Natl Acad Sci U S A 105 : 3100–3105 doi:10.1073/pnas.0711982105
20. WoodLF, LeechAJ, OhmanDE (2006) Cell wall-inhibitory antibiotics activate the alginate biosynthesis operon in Pseudomonas aeruginosa: Roles of sigma (AlgT) and the AlgW and Prc proteases. Mol Microbiol 62 : 412–426 doi:10.1111/j.1365-2958.2006.05390.x
21. WozniakD, OhmanD (1991) Pseudomonas aeruginosa AlgB, a two-component response regulator of the NtrC-family, is required for algD transcription. J Bacteriol 173 : 1406–1413.
22. SandersLH, DevadossB, RajaGV, O'ConnorJ, ShengchangS, et al. (2011) Epistatic Roles for Pseudomonas aeruginosa MutS and DinB (DNA Pol IV) in Coping with Reactive Oxygen Species-Induced DNA Damage. PLoS ONE 6: e18824 doi:10.1371/journal.pone.0018824
23. WorlitzschD, TarranR, UlrichM, SchwabU, CekiciA, et al. (2002) Effects of reduced mucus oxygen concentration in airway Pseudomonas infections of cystic fibrosis patients. J Clin Inves 109 : 317–325.
24. SchobertM, JahnD (2010) Anaerobic physiology of Pseudomonas aeruginosa in the cystic fibrosis lung. Int J Med Microbiol 300 : 549–556 doi:10.1016/j.ijmm.2010.08.007
25. XiaoW (2005) Sputum Cathelicidin, Urokinase Plasminogen Activation System Components, and Cytokines Discriminate Cystic Fibrosis, COPD, and Asthma Inflammation. Chest 128 : 2316–2326 doi:10.1378/chest.128.4.2316
26. OverhageJ, CampisanoA, BainsM, TorfsECW, RehmBHA, et al. (2008) Human host defense peptide LL-37 prevents bacterial biofilm formation. Infect Immun 76 : 4176–4182 doi:10.1128/IAI.00318-08
27. FjellCD, HissJA, HancockREW, SchneiderG (2011) Designing antimicrobial peptides: form follows function. Nat Rev Drug Discov 11 : 37–51 doi:10.1038/nrd3591
28. VandammeD, LanduytB, LuytenW, SchoofsL (2012) A comprehensive summary of LL-37, the factotum human cathelicidin peptide. Cellular Immunology 280 : 22–35 doi:10.1016/j.cellimm.2012.11.009
29. SørensenOE, FollinP, JohnsenAH, CalafatJ, TjabringaGS, et al. (2001) Human cathelicidin, hCAP-18, is processed to the antimicrobial peptide LL-37 by extracellular cleavage with proteinase 3. Blood 97 : 3951–3959.
30. BalsR, WeinerDJ, MeegallaRL, AccursoF, WilsonJM (2001) Salt-Independent Abnormality of Antimicrobial Activity in Cystic Fibrosis Airway Surface Fluid. Am J Respir Cell Mol Biol 25 : 21.
31. FelgentreffK, BeisswengerC, GrieseM, GulderT, BringmannG, et al. (2006) The antimicrobial peptide cathelicidin interacts with airway mucus. Peptides 27 : 3100–3106 doi:10.1016/j.peptides.2006.07.018
32. BuckiR, ByfieldFJ, JanmeyPA (2007) Release of the antimicrobial peptide LL-37 from DNA/F-actin bundles in cystic fibrosis sputum. Eur Respir J 29 : 624–632 doi:10.1183/09031936.00080806
33. SpencerDH, KasA, SmithEE, RaymondCK, SimsEH, et al. (2003) Whole-Genome Sequence Variation among Multiple Isolates of Pseudomonas aeruginosa. J Bacteriol 185 : 1316–1325 doi:10.1128/JB.185.4.1316-1325.2003
34. AnthonyM, RoseB, PeglerMB, ElkinsM, ServiceH, et al. (2002) Genetic Analysis of Pseudomonas aeruginosa Isolates from the Sputa of Australian Adult Cystic Fibrosis Patients. Journal of Clinical Microbiology 40 : 2772–2778 doi:10.1128/JCM.40.8.2772-2778.2002
35. BragonziA, WiehlmannL, KlockgetherJ, CramerN, WorlitzschD, et al. (2006) Sequence diversity of the mucABD locus in Pseudomonas aeruginosa isolates from patients with cystic fibrosis. Microbiology 152 : 3261–3269 doi:10.1099/mic.0.29175-0
36. BoucherJ, YuH, MuddM, DereticV (1997) Mucoid Pseudomonas aeruginosa in cystic fibrosis: characterization of muc mutations in clinical isolates and analysis of clearance in a mouse model of respiratory infection. Infection and Immunity 65 : 3838–3846.
37. ImlayJA (2008) Cellular defenses against superoxide and hydrogen peroxide. Annu Rev Biochem 77 : 755–776 doi:10.1146/annurev.biochem.77.061606.161055
38. WagnerJ, GruzP, KimS, YamadaM, MatsuiK, et al. (1999) The dinB gene encodes a novel E. coli DNA polymerase, DNA pol IV, involved in mutagenesis. Molecular Cell 4 : 281–286.
39. MillerC (2004) SOS Response Induction by Beta-Lactams and Bacterial Defense Against Antibiotic Lethality. Science 305 : 1629–1631 doi:10.1126/science.1101630
40. HeJ, FurmanskiP (1995) Sequence specificity and transcriptional activation in the binding of lactoferrin to DNA. Nature 373 : 721–724 doi:10.1038/373721a0
41. HsuCH (2005) Structural and DNA-binding studies on the bovine antimicrobial peptide, indolicidin: evidence for multiple conformations involved in binding to membranes and DNA. Nucleic Acids Research 33 : 4053–4064 doi:10.1093/nar/gki725
42. MarchandC, KrajewskiK, LeeHF, AntonyS, JohnsonAA, et al. (2006) Covalent binding of the natural antimicrobial peptide indolicidin to DNA abasic sites. Nucleic Acids Research 34 : 5157–5165 doi:10.1093/nar/gkl667
43. ParkCB, KimHS, KimSC (1998) Mechanism of action of the antimicrobial peptide buforin II: buforin II kills microorganisms by penetrating the cell membrane and inhibiting cellular functions. Biochem Biophys Res Commun 244 : 253–257 doi:10.1006/bbrc.1998.8159
44. SubbalakshmiC, SitaramN (1998) Mechanism of antimicrobial action of indolicidin. FEMS Microbiol Lett 160 : 91–96.
45. SeilM, NagantC, DehayeJP, VandenbrandenM, LensinkMF (2010) Spotlight on human LL-37, an immunomodulatory peptide with promising cell-penetrating properties. Pharmaceuticals 3 : 3435–3460.
46. LandeR, GregorioJ, FacchinettiV, ChatterjeeB, WangY-H, et al. (2007) Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature 449 : 564–569 doi:10.1038/nature06116
47. SochackiKA, BarnsKJ, BuckiR, WeisshaarJC (2011) Real-time attack on single Escherichia coli cells by the human antimicrobial peptide LL-37. Proc Natl Acad Sci U S A 108: E77–E81 doi:10.1073/pnas.1101130108
48. ZasloffM (2002) Antimicrobial peptides of multicellular organisms. Nature 415 : 389–395 doi:10.1038/415389a
49. ShaiY (1999) Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by α-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochimica et Biophysica Acta (BBA) - Biomembranes 1462 : 55–70 doi:10.1016/S0005-2736(99)00200-X
50. MatsuzakiK (1999) Why and how are peptide–lipid interactions utilized for self-defense? Magainins and tachyplesins as archetypes. Biochimica et Biophysica Acta (BBA) - Biomembranes 1462 : 1–10 doi:10.1016/S0005-2736(99)00197-2
51. YangL, WeissTM, LehrerRI, HuangHW (2000) Crystallization of antimicrobial pores in membranes: magainin and protegrin. Biophys J 79 : 2002–2009 doi:10.1016/S0006-3495(00)76448-4
52. LiL, ShiY, CheserekMJ, SuG, LeG (2013) Antibacterial activity and dual mechanisms of peptide analog derived from cell-penetrating peptide against Salmonella typhimurium and Streptococcus pyogenes. Appl Microbiol Biotechnol 97 : 1711–1723 doi:10.1007/s00253-012-4352-1
53. FriedbergE, LehmannA, FuchsR (1995) Trading Places: How Do DNA Polymerases Switch During Translesion DNA Synthesis? Molecular Cell 18 : 499–505 doi:10.1016/j.molcel.2005.03.032
54. KeppleKV, PatelN, SalamonP, SegallAM (2008) Interactions between branched DNAs and peptide inhibitors of DNA repair. Nucleic Acids Research 36 : 5319–5334 doi:10.1093/nar/gkn512
55. Su DLWAMSLeo Y (2010) An Antimicrobial Peptide That Targets DNA Repair Intermediates In Vitro Inhibits Salmonella Growth within Murine Macrophages. Antimicrobial Agents and Chemotherapy 54 : 1888 doi:10.1128/AAC.01610-09
56. Induction of group A Streptococcus virulence by a human antimicrobial peptide (2008) Induction of group A Streptococcus virulence by a human antimicrobial peptide. 105 : 16755–16760.
57. StockleyRA (2002) Neutrophils and the Pathogenesis of COPD. Chest 121 : 151S–155S doi:_10.1378/chest.121.5_suppl.151S
58. HoenderdosK, CondliffeA The Neutrophil in Chronic Obstructive Pulmonary Disease. Too Little, Too Late or Too Much, Too Soon? Am J Respir Cell Mol Biol 48 : 531–539 doi:0.1165/rcmb.2012-0492TR
59. MurphyTF, BrauerAL, EschbergerK, LobbinsP, GroveL, et al. (2008) Pseudomonas aeruginosa in Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med 177 : 853–860 doi:10.1164/rccm.200709-1413OC
60. KonstanMW, HilliardKA, NorvellTM, BergerM (1994) Bronchoalveolar lavage findings in cystic fibrosis patients with stable, clinically mild lung disease suggest ongoing infection and inflammation. Am J Respir Crit Care Med 150 : 448–454 doi:10.1164/ajrccm.150.2.8049828
61. DhoogheB, NoëlS, HuauxF, LealT (2013) Lung inflammation in Cystic Fibrosis: Pathogenesis and novel therapies. Clin Biochem pii S0009-9120(13)00605-X doi:10.1016/j.clinbiochem.2013.12.020
62. McCaugheyG, GilpinDF, ElbornJS, TunneyMM (2013) The future of antimicrobial therapy in the era of antibiotic resistance in cystic fibrosis pulmonary infection. Expert Review of Respiratory Medicine 7 : 385–396.
63. MercerDK, O'NeilDA (2013) Peptides as the next generation of anti-infectives. Future Medicinal Chemistry 5 : 315–337 doi:10.4155/fmc.12.213
64. MishraM, ByrdMS, SergeantS, AzadAK, ParsekMR, et al. (2012) Pseudomonas aeruginosa Psl polysaccharide reduces neutrophil phagocytosis and the oxidative response by limiting complement-mediated opsonization. Cell Microbiol 14 : 95–106 doi:10.1111/j.1462-5822.2011.01704.x
65. QiuD, DamronFH, MimaT, SchweizerHP, YuHD (2008) PBAD-Based Shuttle Vectors for Functional Analysis of Toxic and Highly Regulated Genes in Pseudomonas and Burkholderia spp. and Other Bacteria. Appl Environ Microbiol 74 : 7422–7426 doi:10.1128/AEM.01369-08
66. KnutsonC, JeanesA (1968) A new modification of the carbazole analysis: application to heteropolysaccharides. Anal Biochem 24 : 470–481.
67. MaS, SelvarajU, OhmanDE, QuarlessR, HassettDJ, et al. (1998) Phosphorylation-independent activity of the response regulators AlgB and AlgR in promoting alginate biosynthesis in mucoid Pseudomonas aeruginosa. J Bacteriol 180 : 956–968.
68. ParragaA, BellsolellL, Ferré-D'AmaréAR, BurleySK (1998) Co-crystal structure of sterol regulatory element binding protein 1a at 2.3 å resolution. Structure 6 : 661–672 doi:10.1016/S0969-2126(98)00067-7
69. LuriaSE, DelbrückM (1943) Mutations of Bacteria from Virus Sensitivity to Virus Resistance. Genetics 28 : 491–511.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 4
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- The 2010 Cholera Outbreak in Haiti: How Science Solved a Controversy
- , , , Genetic Variability: Cryptic Biological Species or Clonal Near-Clades?
- Efficient Parvovirus Replication Requires CRL4-Targeted Depletion of p21 to Prevent Its Inhibitory Interaction with PCNA
- An Overview of Respiratory Syncytial Virus
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy