
Abortive T Follicular Helper Development Is Associated with a Defective Humoral Response in -Infected Macaques
We introduced a non-human primate model for visceral leishmaniasis by intravenous injection of L. infantum promastigotes in rhesus macaques and followed the animals for a period of eight months. In this model, parasites dock to the liver and spleen shortly after inoculation and remain in these visceral compartments during all the acute phase of infection. However, at the chronic phase, additional body locations appeared colonized (lymph nodes, bone marrow). During the acute phase, a Th1-polarized CD4 T cell response develops in the spleen, but, and concomitant with parasite growth, it waned at the chronic phase. Furthermore, we observed the acute expansion of a splenic T follicular helper (Tfh) cell population, a CD4+ T cell subset specialized to assist B cells in the production of antigen-specific antibody. These cells were localized in close association with B cell follicles but, interestingly, the Tfh population is lost at the chronic phase. Nevertheless, there was a close association between the development of Tfh cells and the differentiation of B cells that produce L. infantum-specific IgG. Thus, our results suggest that Tfh cells are important in instructing B cells to produce parasite-specific antibodies during VL, but their abortive differentiation precludes the continuous production of specific-IgG.
Published in the journal:
. PLoS Pathog 10(4): e32767. doi:10.1371/journal.ppat.1004096
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004096
Summary
We introduced a non-human primate model for visceral leishmaniasis by intravenous injection of L. infantum promastigotes in rhesus macaques and followed the animals for a period of eight months. In this model, parasites dock to the liver and spleen shortly after inoculation and remain in these visceral compartments during all the acute phase of infection. However, at the chronic phase, additional body locations appeared colonized (lymph nodes, bone marrow). During the acute phase, a Th1-polarized CD4 T cell response develops in the spleen, but, and concomitant with parasite growth, it waned at the chronic phase. Furthermore, we observed the acute expansion of a splenic T follicular helper (Tfh) cell population, a CD4+ T cell subset specialized to assist B cells in the production of antigen-specific antibody. These cells were localized in close association with B cell follicles but, interestingly, the Tfh population is lost at the chronic phase. Nevertheless, there was a close association between the development of Tfh cells and the differentiation of B cells that produce L. infantum-specific IgG. Thus, our results suggest that Tfh cells are important in instructing B cells to produce parasite-specific antibodies during VL, but their abortive differentiation precludes the continuous production of specific-IgG.
Introduction
Visceral leishmaniasis (VL) is a chronic and potentially fatal infectious disease caused by the protozoan parasite Leishmania infantum/chagasi or L. donovani. The generalized spread of the parasite to the reticuloendothelial system (spleen, liver and bone marrow) results in a clinical picture including weight loss, cyclic fever, hepatosplenomegaly, anemia and hypergammaglobulinemia [1]. Studies in murine models revealed that recovery from VL is crucially dependent on the development of a robust cellular-mediated immunity, with the production of cytokines such as IFN-γ and TNF as central components of the protective response [2]. In contrast, parasite persistence and chronicity are associated with the induction of immune suppressive mediators, such as IL-10 and TGF-β [3], [4].
Inbred mice strains invariably control infections with viscerotropic Leishmania species and develop a life-long latent infection [5], contrasting with the potentially fatal human VL in which progressive illness develops, even in the presence of detectable levels of IFN-γ and TNF in lesional tissue [3], [6]–[8]. Therefore, despite the notable usefulness of murine models, new insights into the immunopathogenesis of VL would potentially benefit from a more frequent employment of alternative animal models [9]. Non-human primates (NHP) constitute powerful experimental models for understanding host-pathogen interactions that are not directly observable in human patients, particularly the early events after infection which are usually poorly characterized in humans [10]–[12]. Concerning leishmaniasis, the Asian rhesus macaque has already been shown to mimic human VL [13], and NHP models are routinely used for pre-clinical evaluation of novel drug and vaccine candidates [14].
The role played by antibodies and B cells during leishmaniasis has always been contentious. High titers of both Leishmania-specific and non-specific antibodies are a recurrent finding in patients [15], implying the development of strong humoral response during infection. While several reports in mice models have revealed an increased resistance to infection upon B cell depletion [16]–[18], others have proposed protective roles for B cells and/or antibodies [19], [20].
Upon binding and internalization of specific antigen, B cells generally depend on cognate interactions with CD4 T cells to differentiate into antibody-producing plasma cells (PCs) [21]. The activated B cell can either follow the follicular pathway and form a germinal center (GC), or differentiate into an extra-follicular focus of immunoglobulin (Ig)-secreting PCs. While the GC pathway generates long-lived memory B cells as well as PCs that produce antibodies with high affinity for the antigen, the extra-follicular pathway is generally associated with short-lived plasmablasts and PCs that secrete antibodies of modest affinity, but nevertheless provides an early source of antibody that might be critical during infection [21], [22]. Recent studies have greatly increased our knowledge on the biology of GC-associated CD4 T cells, also known as T follicular helper cells (Tfh). Tfh cells are phenotypically characterized by expression of the follicular-homing chemokine receptor CXCR5, the transcriptional repressor Bcl-6 and an array of surface molecules that include ICOS, CD40L and PD-1 [23], [24]. These cells produce high levels of IL-21, a crucial mediator in the development of affinity-matured and class-switched B cells, as well as in the differentiation of long-lived plasma cells [21], [25]. In contrast, much less is known about the functional and phenotypic characteristics of the CD4 T cell helpers associated with extra-follicular antibody responses. Furthermore, infections with several types of pathogens, including Leishmania spp., induce strong polyclonal B cell activation, in a process independent of T cell help, that results in copious secretion of non-specific and potentially auto-reactive antibody [26]. Despite the strong humoral response that is usually associated with VL, the mechanisms underlying antibody production remain poorly explored.
To address these questions we performed a detailed immunological analysis in rhesus macaques infected with L. infantum. Tracking the CD4 T cell responses overtime revealed that parasite containment in visceral compartments during the acute phase was associated with the differentiation of splenic CD4 T cells and their increased expression of Th1-related transcripts. The acute expansion of a splenic CD4 T cell population expressing CXCR5 and Bcl-6, but not PD-1, was associated with the differentiation of activated memory B cells and production of parasite-specific IgG. These cells were localized in B cell areas and closely paralleled the development of germinal centers. In the chronic phase, parasite dissemination and growth were concomitant with IL10 mRNA accumulation in lymphoid tissues. Furthermore, the splenic CXCR5+Bcl-6+ CD4 T cell population contracted, which was paralleled by loss of the activated memory B cells, impacting the production of parasite-specific antibodies, despite the chronic persistence of hypergammaglobulinemia.
Results
Parasite load dynamics and pathology in L. infantum-infected rhesus macaques
First, we monitored over time the progression of a variety of parameters in our model of rhesus macaques intravenously infected with a high dose of L. infantum promastigotes. Parasite load was assessed during the course of infection employing a quantitative PCR (qPCR) assay [27]. Parasite clearance was evident in the blood during the first weeks of infection, with a steady decrease in parasitemia from about 400 parasites per million of host cells at day 7 post-infection (pi), to less than 20 at day 28 (Fig. 1A). Yet, blood parasite numbers rebounded as the infection progressed towards late stages, being, by day 250 pi, at a level comparable to that of day 7 and significantly higher than at day 28 pi (Fig. 1A, P<0.05).

Parasites were hardly detectable in lymph nodes (LNs) during the first weeks of infection (Fig. 1B). However, a significant increase in the parasite burden occurred during the chronic phase (P<0.001), revealing a pattern of parasite growth and/or infected cell migration to the LNs during chronic infection (Fig. 1B). In the bone marrow (BM), the parasite load kinetics was reminiscent of that found during the early weeks in peripheral blood, with an apparent early clearance phase resulting in a scarcity of parasites by day 28 pi. Similarly to the situation in LNs, a significant increase in parasite burden was found as the infection advanced towards the chronic phase (P<0.001; Fig. 1C).
In the spleen and liver the parasite burden remained relatively constant during the acute phase (Fig. 1D–E). Yet, during chronic infection we found an increase in parasite numbers in these organs, albeit not statistically significant (Fig. 1D–E).
Weight loss and intermittent fever were not observed in infected animals during the time-course of our experiments (not shown). Yet, the animals developed a transient state of anemia during the first weeks after inoculation, with a reduction in erythrocyte number (Fig. 2A), hematocrit values and blood hemoglobin (Fig. S1A–B). Additionally, an early and transient neutrophilia was detected (Fig. 2B), accompanied by increased levels of serum markers of acute phase response such as C-reactive protein (CRP; Fig. 2C) and the complement factors C3 and C4 (Fig. 2D–E). Hepatocellular damage was detected at late stages of infection, as revealed by elevated serum levels of alanine transaminase (ALT; Fig. 2F), albeit without any signs of biliary tract disease, as indicated by normal levels of γ-glutamil-transaminase (Gamma-GT) and total bilirubin (TBil, Fig. S1C–D) or the absence of hepatic synthetic function abnormalities (normal levels of serum albumin and total serum protein; (Fig. S1E–F).
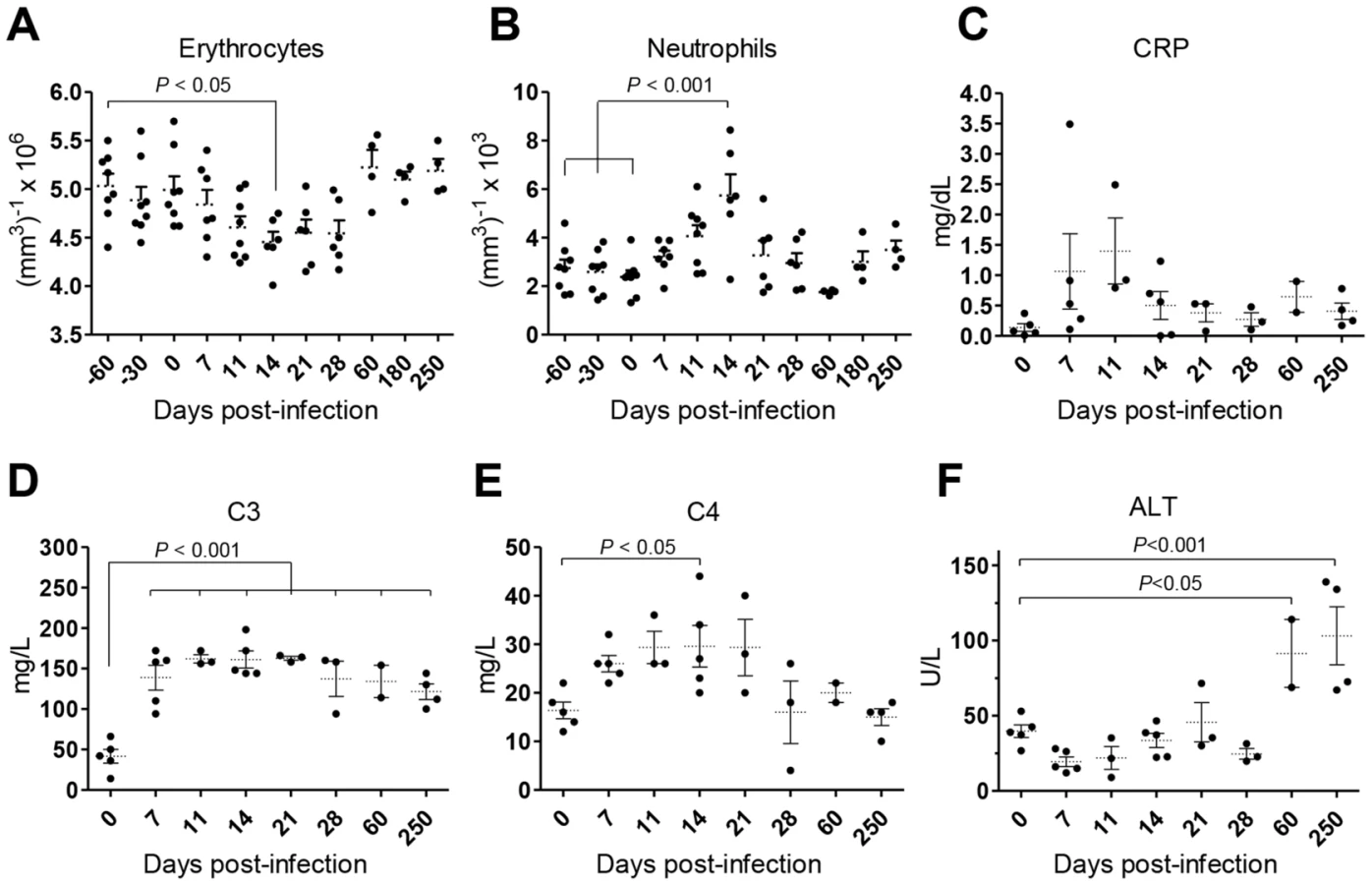
Collectively, our data point to a time-dependent and organ-specific establishment of L. infantum in rhesus macaques, with early parasite colonization of visceral compartments and posterior migration and/or growth in LNs.
L. infantum infection of rhesus macaques drives the expansion and differentiation of splenic CD4 T cells
CD4 T cells are crucial mediators of both protective and pathological immune responses during VL [2]. In rhesus macaques infected with L. infantum, the percentage of CD4 T cells in the spleen was significantly increased 11 days after infection (Fig. 3A, P<0.05), which resulted in a 3-fold increase in their total numbers that were thereafter maintained at constant levels (Fig. 3A). Despite the drastic increase in splenic CD4 T cells, the occurrence of splenomegaly was not evident in infected animals during the course of the experiments (not shown). In contrast, no significant variations occurred in the total numbers (not shown) or frequencies of CD4 T cells in the blood (Fig. 3B) and LNs (Fig. 3C).

By assessing the differentiation phenotype of CD4 T cells we observed that, in the spleen, L. infantum infection induced a significant decrease in the percentage of naïve (CD62L+ CD45RA+) CD4 T cells, at days 28 and 250 pi (Fig. 3D–E upper panel, P<0.05). This was paralleled by an increase in the percentage of CD4 T cells with an effector memory phenotype (CD62L− CD45RA−, P<0.05), while the frequencies of terminal effector (CD62L− CD45RA+) and central memory (CD62L+ CD45RA−) CD4 T cells remained roughly constant throughout infection (Fig. 3D–E, upper panel). In contrast, no significant alterations were detected in the differentiation phenotype of CD4 T cells in the blood or LNs during both acute and chronic stages of infection (Fig. 3E middle and lower panels).
T cell differentiation is usually associated with increased susceptibility to Fas-mediated apoptosis [28]. By quantifying annexin V binding, we observed that the susceptibility of splenic CD4 T cells to undergo apoptosis, upon exposure to exogenous FasL, increased significantly at days 28 and 250 pi (Fig. 3F, upper panel, P<0.05). Interestingly, the surface expression of CD95 (Fas receptor) was found to parallel the susceptibility to FasL-mediated death (Fig. 3F, upper panel, P<0.05). As expected, no significant alterations were found in the sensitivity of blood and LNs CD4 T cells to FasL-mediated apoptosis or in their CD95 expression (Fig. 3F, middle and lower panels). Despite the differentiation and increased sensitivity of splenic CD4 T cells to FasL-mediated apoptosis, splenic, blood or LN CD4 T cells of infected macaques were not more prone to apoptotic death in the absence of the apoptotic stimulus (Fig. 3F). This suggested the inexistence of death-receptor signaling in vivo, prompting us to analyses the levels of FasL in infected macaques. In agreement, neither the serum FasL levels, nor its splenic or LN transcript were found increased after infection (Fig. S2A–C). Globally, the results presented point to an early expansion and differentiation of the splenic CD4 T cell pool towards an effector memory phenotype after L. infantum infection.
A Th1-polarized cytokinic profile is induced in the spleen early after infection, but converts to an IL10-dominated environment during the chronic phase
We evaluated the gene expression levels of a panel of cytokines and transcription factors in total splenic mononuclear cells (SMCs). The qPCR analysis demonstrated a biphasic response with induction of Th1-related transcripts during the acute phase that converted to an IL10-enriched environment during the chronic phase (Fig. S3A–I). In agreement with previous studies [3], [8], we did not observe any modification in the transcript levels of the genes encoding the Th2-related cytokines IL-4 and Il-13 or the Th2 master regulator GATA-3 (Fig. S3D–F).
Given the prominent expansion and differentiation of CD4 T cells in the spleen of L. infantum-infected macaques (Fig. 3D–E), we evaluated the expression of the same genes in sorted splenic CD4 T cells. We detected significant 3 - and 2.5-fold elevations for IFNG and TBX21 (T-bet) transcripts, respectively and a non-significant 2-fold increase in TNF expression at day 28 pi (Fig. 4A–B, P<0.05), indicating CD4 T cells as a source of these Th1-associated factors during the acute phase. This molecular signature was found to be transient and the expression of IFNG and TBX21 declined during the chronic phase (Fig. 4A–B). As before, no significant modifications were observed in the transcript levels of Th2-associated transcripts, in sorted splenic CD4 T cells, even though non-significant 2-fold increases in IL4 and GATA3 occurred at day 28 pi (Fig. 4D–F).

Emerging evidence has associated increased lymphoid expression of the immunosuppressive IL-10 and TGF-β cytokines as underlying factors responsible for parasite persistence and chronicity in leishmaniasis [29]–[31]. We observed a significant 2.5-fold increase in the IL10 transcript in sorted splenic CD4 T cells at day 250 after infection (P<0.05), with no changes in the expression of TGFB1 (Fig. 4H). Moreover, we failed to detect any significant modification in the expression levels of FOXP3, the master transcription factor for Treg differentiation, suggesting a FoxP3 negative phenotype for the splenic IL-10-producing CD4 T cells during VL in NHP, as previously suggested in both experimental mice models and natural human infections [31]–[33].
In LNs, we failed to detect any significant changes in the mRNA levels of Th1 or Th2-related cytokines or transcription factors in both total and sorted CD4 T cells during infection (Fig. 4A–F and Fig. S3A–F). Interestingly, a significant 2-fold increase on the expression of IL10 occurred at day 250 pi in total LN cells (Fig. S3G, P<0.05), concomitant with the parasite multiplication in these organs (Fig. 1B). We further observed a marginally non-significant increase in the transcript of TGFB1 during the chronic phase (Fig. S3H, P = 0.058). However, in sorted LN CD4 T cells none of these transcripts underwent significant induction at the chronic phase (Fig. 4G–H). Finally, we observed an increase, albeit not-statistical, in the FOXP3 transcript both in total LN cells and sorted CD4 T cells (Fig. 4H–I and Fig. S3H–I). An environment enriched in IL-10 and TGF-β may provide a safe niche for the parasite, hence explaining the ramping increase in parasite load in LNs during chronic infection.
L. infantum infection of rhesus macaques induces the production of non-specific IgG concomitant with defective B cell maturation and curtailed development of germinal centers
To further characterize the immune events in L. infantum-infected rhesus macaques we quantified the serum levels of total and L. infantum-specific IgM and IgG (Fig. 5A–B). A non-significant increase in total serum IgM occurred during acute infection, with the levels falling back to steady state by day 60 pi (Fig. 5A). This was paralleled by a significant increase of L. infantum-specific IgM up to the 11th day pi (Fig. 5A), which subsequently decreased to pre-infection levels. The total levels of serum IgG were significantly elevated at every time point evaluated after infection (Fig. 5B), demonstrating that the classical hypergammaglobulinemia observed during VL [1] rapidly develops upon infection. Nevertheless, this does not reflect the development of a sustained L. infantum-specific IgG response, as only modest increases were observed in the serum levels of L. infantum-specific IgG, and these were confined to the early stages of infection (Fig. 5B). Importantly, during the chronic phase, while total IgG remained high, the levels of L. infantum-specific IgG returned to pre-infection levels, indicating a weak production of parasite-specific IgG.

Given that the spleen was colonized by parasites during both early and late stages of infection (Fig. 1C) and that it is the major organ of B cell differentiation [34], we sought to explore in detail the splenic B cell dynamics during the course of infection. No significant variations occurred in the total number of splenic B cells during infection (Fig. 5C). Next, we discriminated four distinct B cell populations, based on their differentiation status, [35], [36] and followed their dynamics throughout infection. A significant decrease in the percentage of naïve (CD21+CD27−) splenic B cells was observed at days 28 and 250 after infection (P<0.05; Fig. 5D–E). This decrease was accompanied by increases in the frequencies of immature (CD21−CD27−) and effector memory (CD21−CD27+) B cells (Fig. 5D–E). Interestingly, during the chronic phase only the immature population remained significantly elevated when compared with the level at t = 0 (P<0.05), while the effector memory population contracted (Fig. 5D–E). Finally, no significant variations occurred in the resting memory (CD21+CD27+) subset of B cells throughout infection (Fig. 5D–E).
Loss of CD45RA expression in B cells is an early marker of B cell activation and differentiation into an Ig-secreting cell [37], [38]. Indeed, we observed a significant increase in the percentage of splenic B cells having lost CD45RA expression after infection (Fig. 5F). We thus addressed the expression of CD45RA in the previously defined B cell subsets. In the naïve and resting memory B cell subsets, no significant loss of CD45RA was observed throughout infection (not shown). On the other hand, a marked downregulation of CD45RA occurred after infection in the immature and effector memory subsets (Fig. 5G–H). Overall, among the total splenic B cell pool, L. infantum infection induced a significant increase in the frequency of immature/activated B cells (here defined as CD21−CD27−CD45RA−) that persisted during chronic infection. In contrast, the activated/memory (CD21−CD27+CD45RA−) B cell population peaked at day 28 pi but declined at the chronic phase. We found a significant positive correlation between the levels of L. infantum-specific serum IgG and the frequency of CD21−CD27+CD45RA− splenic B cells (Rs = 0.9291, P = 0.0001) (Fig. 5I), but not with the percentages of CD21−CD27−CD45RA− B cells (Fig. 5I). Thus, our results suggest that a failure to maintain the activated memory B cell population may underlie the poor production of parasite-specific IgG.
As increased expression of CD27 in splenic B cells is considered an indication of GC experience [39], we performed tissue immunofluorescence in splenic sections retrieved from naïve, acutely and chronically-infected monkeys to explore the dynamics of GC development during infection (Fig. 6). The visualization of B and T cell areas as well as GC morphology and numbers during the course of infection was achieved by multiparametric analysis of splenic sections stained for CD3, CD20, IgD and Ki-67 [40]. The number and relative size of the GCs observed in naïve animals increased shortly after infection (Fig. 6A–B and 6E–F), peaking by one month after parasite inoculation (Fig. 6C, 6E–F and S4). At this time point, the increase in GC size was clear as noted by the enlarged central area harboring Ki-67+ proliferating cells and the peripheral localization of IgD+ naïve B cells that are excluded from the ongoing GC reaction (Fig. 6C). At the chronic phase, we observed a moderate remodeling of splenic architecture with less defined T and B cell areas (Fig. 6D). Additionally, we observed an increased number of Ki-67+ cells scattered throughout both B and T cells areas, suggestive of widespread immune activation. Overall, the decrease in the frequency of splenic activated memory B cells (CD21−CD27+CD45RA−) at the chronic phase (Fig. 5H) was paralleled by a decrease in the number and average size of germinal centers (Fig. 6D and 6E).

Contrasting with the spleen, in lymph nodes, B cell differentiation was negligible with essentially all cells maintaining a naïve CD21+CD27−CD45RA+ phenotype throughout infection (Fig. S5). Accordingly, tissue immunofluorescence and GC quantification revealed no relevant changes in the number, size or morphology of lymph node-associated GCs (Fig. S6).
L. infantum infection induces transient expansion of a CXCR5+BCL6+ splenic CD4 T cell population associated with parasite-specific antibody production
The shortened duration of the Leishmania-specific antibody response associated with defective differentiation of the splenic B cell pool and curtailed development of germinal centers (Fig. 5) prompted us to explore the dynamics of Tfh-associated factors in the spleen of L. infantum-infected rhesus macaques. The transcript levels of CXCR5 and BCL6 were significantly augmented in sorted splenic CD4 T cells at day 28 pi, with CXCR5 levels remaining elevated during the chronic phase (Fig. 7A, P<0.05), while no variation was observed in expression of the PDCD1 gene (PD-1; Fig. 7A). We further observed a 5-fold increase in the transcript levels of IL21 in sorted splenic CD4 T cells at day 28 pi (Fig. 7A, P<0.05) followed by a decrease in the chronic phase. Interestingly, the serum levels of IL-21 were found persistently elevated, from the 11th day after infection until the chronic phase (Fig. S7A) and qPCR analysis of SMCs revealed increased abundance of the IL21 transcript as early as day 11 pi (Fig. S7B), thus suggesting that additional populations may produce IL-21 in the early acute phase as well as in the chronic phase.

Flow cytometric analysis indicated a significant increase in the frequency of splenic CD4 T cells expressing CXCR5 at days 28 (P<0.01) and 250 pi (P<0.05) and of Bcl-6 at day 28 pi (P<0.05; Fig. 7B–C). However, no significant variations were observed in surface PD-1 expression throughout infection (Fig. 7B–C), consistent with qPCR analysis. Additionally, the percentage of splenic CD4 T cells expressing both CXCR5 and Bcl-6 peaked at day 28 pi and decreased at chronic infection (P<0.05; Fig. 7B and D). Interestingly, we failed to detect any significant variation in the expression of PD-1 among the CXCR5+ or CXCR5+Bcl-6+ CD4 T cell populations (Fig. 7B–D). Significant positive correlations were found when plotting the frequency of the DP CXCR5+Bcl-6+ splenic CD4 T cell population against the activated memory splenic B cell population (Fig. 7E, Rs = 0.8091, P = 0.0039) and the serum levels of L. infantum-specific IgG (Fig. 7F, Rs = 0.8055, P = 0.0039).
To gain additional insight into the spatial dynamics of Tfh differentiation, splenic tissue sections were stained for CD4, PD-1 and CxCR5 (Fig. 8). In our hands, no commercially available Bcl-6 antibody was able to detect the protein by immunofluorescence in rhesus macaques (not shown). As expected, in naïve macaques very few CD4 T cells were seen infiltrating CxCR5 areas that define the B cell follicle (Fig. 8A). By day 11 pi, and paralleling the development of GCs (Fig. 6B), an increased number of CD4 T cells were present inside the follicle, with a few expressing PD-1 in addition to CxCR5 (Fig. 8B). CD4 T cells progressively accumulated inside follicles as acute infection progressed. By one month after parasite inoculation, hence at the peak of the GC response, increased numbers of CD4 T cells could be visualized in B cell areas (Fig. 8C). Interestingly, some of the follicle-infiltrating CD4 T cells were expressing PD-1, but not CxCR5 and, conversely, follicle-associated CD4 T cells expressing CxCR5 did not express PD-1 (Fig. 8C and Fig. 8E). Spatially, CD4+ PD-1+ CxCR5− cells occupied a more central position in the follicle, relative to the peripheral localization of CD4+ CxCR5+ PD-1− cells (Fig. 8C and Fig. 8E). Finally, and consistent with the dynamics of GC development, CxCR5+ areas were largely devoid of CD4 T cells by day 250 pi, despite continuous parasite presence (Fig. 8D).

The expression of the Tfh classical markers CxCR5 and PD-1 in lymph node CD4 T cells increased progressively after infection at the transcript (Fig. S8A) and protein levels (Fig. S8B–D), though without statistical value. Expression of Bcl-6 in lymph node CD4 T cells was negligible (S8B–D) and no changes were observed at the transcript level (Fig. S8A). Tissue immunofluorescence revealed the presence of some CD4 T cells in follicular areas, particularly at one month after infection and during the chronic phase. The majority of these cells appeared to express PD-1 but not CxCR5, with only a minute number expressing both surface markers (Fig. S9C–E).
Overall, our results point to a model in which L. infantum infection induces an acute expansion of a CXCR5+Bcl-6+ CD4 T cell population in the spleen associated with the production of parasite-specific IgG. The kinetics of expansion and contraction of the double positive CxCR5+Bcl-6+ CD4 T cell population closely paralleled the development and resolution of germinal centers in the spleen. Hence, we provide a detailed description of the immune events underlying the suboptimal parasite-specific humoral response in rhesus macaques infected with L. infantum.
Discussion
The lack of efficient vaccines or immune therapies for human VL highlights the need for alternative animal models able to complement the extensive research performed in rodents over the past 30 years. In this study, we show that L. infantum infection of rhesus macaques drives an early expansion and differentiation of splenic CD4 T cells with a Th1 molecular signature that is concomitant with parasite containment in visceral compartments. Furthermore, we provide evidence of the lack of a robust Tfh response throughout infection, which possibly underlies the poor and short-lived production of Leishmania-specific antibodies. Finally, the emergence of an immunosuppressive environment may facilitate parasite dissemination and/or growth in additional niches during chronic infection. In a general manner, the non-human primate model introduced here confirmed some previous observations made in murine models and, more importantly, in human patients. Particularly, the expansion of the CD4 T cell pool associated with a Th1 polarization during the acute phase and the chronic persistence of the parasite associated with augmented expression of IL-10. More importantly, we provided evidence that defects in B cell and T follicular helper differentiation comprise the mechanistic basis for the occurrence of hypergammaglobulinemia and inefficient specific humoral response during VL.
The acute stage of infection is characterized by rapid parasite clearance from the blood and BM, with parasite containment in the spleen and liver. Studies in mice evidenced that half of the intravenously inoculated parasites are eliminated in splenic phagocytes within 24 hours after infection [41]. We propose that a rapid decrease in parasite numbers may occur in a similar manner in rhesus macaques, providing large quantities of parasite antigens to initiate an adaptive response. Indeed, we observed that the acute phase of infection is characterized by expansion and differentiation of splenic CD4 T cells towards effector memory phenotypes associated with increased levels of Th1-related factors (IFN-γ and T-bet). This differentiation of the splenic T cell pool is consistent with increased susceptibility to FasL-mediated apoptosis. However, in the absence of FasL, these cells are not prone to die. Thus, the higher levels of T cell apoptosis previously reported in VL [42] may be the consequence of T cell differentiation/activation processes rather than being the direct cause of pathogenesis. In LNs, which were minimally colonized during the acute phase, no changes were observed in the extent of T cell differentiation and of level of apoptosis. Altogether, our results denote a mobilization of the immune system early after infection associated with elimination or at least containment of the parasite in visceral organs.
In the chronic phase, we observed increased parasite loads in the spleen and liver, associated with signs of hepatocellular damage (elevated serum levels of ALT), as well as evidence of parasite growth in previously low-colonized organs such as the LNs. These changes were associated with an immune context distinct from the one observed during the acute phase. Splenic CD4 T cells maintained an effector-memory phenotype, but shifted from an IFN-γ-producing phenotype towards enrichment in IL-10. The expression of the latter has long been associated with chronicity and disease progression in VL [43], [44]. Importantly, conventional Th1 cells have recently been identified as the main source of IL-10 during VL, in a regulatory mechanism that presumably becomes operative to avoid excessive damage associated with pro-inflammatory cytokine secretion [31]–[33], [45].
In striking contrast with the spleen, the increased parasite load detected in the LNs during the chronic phase does not result in the differentiation of the CD4 T cell pool, a phenotype that can be ascribed to the presence of immunosuppressive cytokines - IL-10 and TGF-β. Accordingly, the anergic behavior of lymph node CD4 T cells during chronic infection was shown, in a hamster model of VL, to result from the activity of macrophage-derived TGF-â [4]. Thus, our results are strongly suggestive of compartmentalized immune responses, underlying the complexity of the immune response in NHPs, which is reminiscent of VL in humans.
A dysfunctional humoral immune response has long been recognized in VL [46], [47]; however the mechanisms behind such dysregulation remain poorly studied. In L. infantum-infected rhesus macaques we observed a dramatic increase in the circulating levels of total IgG. Such increased is not paralleled by the serum levels parasite-specific IgG, which peak at one month after infection and decrease thereafter. While the decline in the levels of specific IgG may reflect the loss of the GC and Tfh responses, as discussed below, it may also be a consequence of accelerated decay in the serum of the infected animals. Although we have not examined the occurrence of proteinuria, there is ample evidence of renal involvement in VL associated with tubular and glomerular damage that may lead to accelerated clearance [48]. Splenic B cells are activated and exhibit differentiation towards two main subsets: an immature subset that persists throughout infection and an activated memory subset that peaks at day 28 and declines in the chronic phase. Tissue imaging revealed that splenic germinal centers initially develop, peak in number and size by one month after infection, but ultimately fail to be maintained at the chronic phase, hence closely paralleling the kinetics of memory B cell frequency. Interestingly, B cell differentiation in the lymph nodes was minimal. Additionally, we detected a non-significant increase in the number of lymph node-associated germinal centers, suggesting a minor contribution of these organs to the production of parasite-specific IgG.
The transient expansion of a splenic CD4 T cell population expressing CXCR5 and Bcl-6 correlates with the emergence of activated memory B cells and the levels of parasite-specific IgG. Concomitant with the expansion of the CXCR5+Bcl-6+ CD4 T cell population is an increase in the transcript levels of IL21 in splenic CD4 T cells. IL21 mediates production of high affinity antibodies and also plays an essential role in the differentiation of plasma and memory B cells [49]. Interestingly, we observed that the serum levels of IL-21 remained elevated during the chronic phase, despite the decline in the IL21 transcript in splenic CD4 T cells, suggesting that additional immune populations may produce the transcript. Moreover, elevated serum levels of IL-21 are a biomarker for the risk of developing autoimmunity [50] and the presence of autoantibodies is a recurrent finding in VL patients [51], [52].
GC-associated Tfh cells have classically been defined by their follicular localization and simultaneous expression of CXCR5, Bcl-6 and PD-1 [23]. We observed the transient expansion of CD4 T cells expressing CXCR5 and Bcl-6, but lacking PD-1. Interestingly, a recent report has identified IL-21-producing, Bcl-6+PD-1low CD4 T cells, located at the interface between the T cell zone and the follicle, as providers of B cell help in T-dependent extra-follicular B cell responses [53]. These cells were named pre-GC Tfh cells as they appear early after immunization and are progressively replaced by Bcl-6+PD-1hi CD4 T cells that locate within GCs [53]. Furthermore, recent studies have proposed that full expression of the Tfh differentiation program depends on cognate interactions between primed CD4 T cells and antigen-activated B cells [25], [54], [55]. In this sense, B cells play a crucial role for the survival of Tfh cells and commitment to the Tfh lineage [25]. In the absence of B cells, Tfh cells are still developed, albeit in significantly lower numbers that fail to express PD-1 [54]. We observed an augmentation in the serum level of total, but not specific, IgG as early as day 7 pi, as well as the expansion of an immature/activated splenic B cell population detected at day 11. Thus, it is conceivable that L. infantum infection induces an early skewing of the B cell pool favoring the inappropriate differentiation of plasma cells from low-affinity B cells, and preventing the entry of a sufficient number of activated B cells in the follicle to sustain the Tfh response. Indeed, several Leishmania-derived factors have been identified as polyclonal activators of B cells [56], [57] and, in a murine intradermal model of VL, an early polyclonal B cell response was associated with disease progression [17]. Similarly, infection of mice with the related parasite Trypanosoma cruzi induces a massive extra-follicular splenic B cell response associated with the production of non-specific antibodies [58]. In this sense, the CXCR5+Bcl-6+PD-1− CD4 T cell population that we detect at the end of the acute phase may represent a pre-GC-Tfh state that does not mature to a bona fide Tfh population due to the lack of cognate interactions with B cells. It is however worth referring that confocal imaging revealed the presence of some CxCR5+ PD1+ CD4 T cells inside B cell areas, at early time points after infection, suggesting that some bona fide Tfh cells might engage into the GC pathway, even though their numbers appear compromised. We also observed that some follicle-associated CD4 T cells expressed PD-1 but not CxCR5, particularly by one month after infection in the spleen, and at the chronic phase in lymph nodes. Given their clearly atypical phenotype, considering the current definition of Tfh cells, further studies would be required to completely elucidate their nature.
Interestingly, the results we present here concerning L. infantum infection are clearly distinct from recent findings regarding SIV/HIV infections, in which a pathological accumulation of Tfh cells occurs that accounts for the abnormalities in the B cell compartment observed during infection [59]–[61].
In the chronic phase, the decline of CXCR5+Bcl-6+ CD4 T cells may also be related to increased IL-10, as it was shown to regulate the expression of Bcl-6 and IL-21 in CD4 T cells [62]. Additionally, IL-10 enhances proliferation of activated human B lymphocytes and induces secretion of high amounts of immunoglobulin [63]. Thus, an IL-10-enriched environment combined with the early skewing of the B cell response may represent a biased environment that precludes maintenance of a Tfh response and production of specific-IgG, while sustaining the production of non-specific antibodies. We could not unfortunately provide definitive evidence for the presence of extrafollicular plasmablasts in the spleen due the absence of a clear phenotypic definition in non-human primates. Nevertheless, the global picture points to a humoral response dominated by low-affinity or irrelevant antibodies produced by polyclonally or extrafollicularly-activated B cells. Although the contribution of specific antibodies to a protective response against an intracellular pathogen such as Leishmania remains under debate, some studies have suggested that specific antibodies are required for an efficient uptake of the parasite [20], and protection, in an experimental vaccine against L. infantum [64]. Furthermore, the formation of a functional germinal center and IL-21 production were associated with lesion resolution in a model of cutaneous leishmaniasis [65]. Thus, one may envision that the loss of parasite-specific antibodies observed in the chronic phase may facilitate parasite dissemination and promote chronicity.
In conclusion, we used here a NHP model to decipher the immune events associated with parasite establishment and chronicity in VL. Our results indicate that despite the differentiation of effector memory CD4 T cells in the main parasitized organs early after infection, the establishment of an IL-10 enriched environment in the chronic phase and the absence of a fully maturated and sustained Tfh response may participate in the immunodeficiency associated with VL chronicity.
Materials and Methods
Animal, parasites and infections
Eleven colony-outbred young adult (3–5 kg) rhesus macaques (Macaca mulatta) of Chinese origin, seronegative for STLV-1 (Simian T Leukemia Virus type-1), SRV-1 (type D retrovirus), herpes-B viruses and SIVmac were used in this study. A group was left as non-infected control (n = 3) and the remaining animals were inoculated intravenously via the saphenous vein with 2×107 stationary-phase L. infantum promastigotes (clone MHOM/MA/67/ITMAP-263) per kg of body weight. Subgroups of infected animals were euthanized at three time points after infection covering both acute and chronic phases (n = 2 for days 11 and 28 pi and n = 4 for day 250 pi). Peripheral blood and internal organs (axillary and inguinal lymph nodes, spleen, liver and bone marrow) were recovered for cellular analysis. Blood sampling was performed at additional time points before and after infection. For each blood-sampling point, a hemogram was elaborated using a LH750 hematology analyzer (Beckman Coulter).
Parasite quantification
DNA was extracted from cell pellets of blood or organs using the QIAamp DNA Mini Kit (QIAGEN). A TaqMan-based qPCR assay for detection and quantification of L. infantum kinetoplastid DNA was adapted from a described protocol [27]. Reaction mixtures were composed of ABI TaqMan PCR 2× (Applied Biosystems), 375 nM of direct primer (CTTTTCTGGTCCTCCGGGTAGG), 375 nM of reverse primer (CCACCCGGCCCTATTTTACACCAA), 250 nM of hydrolysis probe (5′FAM-TTTTCGCAGAACGCCCCTACCCGC-3′TAMRA) and 100 ng of sample DNA. Thermocycling settings consisted of one hold of 10 min at 95°C followed by a two-step temperature (95°C for 15 s and 60°C for 60 s) over 40 cycles in an ABI Prism 7900 HT (Applied Biosystems). A standard curve was established corresponding to a range of 50.000 to 0.01 parasites.
Sample normalization was performed by quantifying a host gene (macaque albumin), in 10 µL parallel reactions consisting of SYBR Green ROX Mix 2× (Thermo Scientific), 100 nM of forward primer (CCATTGGTGAGACCAGAGGT), 100 nM of reverse primer (GAGGCAGGCAGCTTTATCAG), 100 ng of DNA and the same thermal profile used for parasite quantification. A calibration curve ranging from 10.000 to 0.1 cells was established and parasite load expressed as the number of parasites per million of host cells.
Quantification of serum analites
Quantification of ALT, CRP, C3, C4 and total IgG and IgM were all performed on an AutoAnalyzer (PRESTIGE 24i, PZ Cormay S.A.). The detailed protocols employed are described in the Supporting Material and Methods section.
ELISA for L. infantum-specific immunoglobulins
The relative titters of L. infantum-specific antibodies in the serum of infected macaques were quantified adapting a protocol described elsewhere [66]. Briefly, 96-well plates were coated overnight with 10 µg/mL of soluble axenic amastigote Leishmania antigen (prepared as described before [66]) and blocked with 200 µL of PBS/low-fat-milk 5%/FCS 5%. Sera from individual macaques were analyzed at a 1∶200 dilution. Horseradish peroxidase-conjugated anti-macaque IgG (1∶5,000) and anti-macaque IgM (1∶10,000) was then added to each well and the tetramethyl benzidine substrate solution was used to detect antigen-specific antibody by absorbance at 492 nm.
Immunophenotyping
Fresh cell suspensions were prepared from macaque spleen and LNs (a pool of axillary and inguinal LNs). Peripheral blood was collected to EDTA-coated tubes. Cells were stained with a panel of monoclonal antibodies. The fluorochrome-conjugated antibodies used are provided in Supporting Table 1 (Table S1). After lysing erythrocytes in a hypotonic solution, fifty thousand events corresponding to mononuclear cells were acquired in a Cytomics FC500 (Beckman Coulter) and further analyzed using FlowJo software (Tree Star, Inc.). Intracellular Bcl-6 staining was performed after fixing and permeabilizing the cells with the FoxP3 staining buffer set (eBiosciences).
Ex-vivo apoptosis
Peripheral blood was recovered to heparin-coated tubes. PBMCs and SMCs were isolated by density gradient centrifugation using LymphoPrep (PAA Laboratories). PBMCs, SMCs and LN cells were cultured overnight in complete media in the presence of FasL, 100 ng/ml, or vehicle (control). The percentage of apoptotic CD4 T cells was determined by flow cytometry after surface staining with FITC-labeled annexin-V combined with surface staining for CD4 and CD3, as described previously [67].
Quantitative-PCR
Approximately 500,000 CD4 T cells from SMCs or LN cell suspensions were sorted using a FACS Aria II cell sorter (BD Biosciences), lysed in RLT buffer (RNeasy Micro Kit, QIAGEN) and stored at −80°C until further use. A similar number of total SMCs or total LN cells were lysed and stored. RNA was purified and reverse transcribed using the AffinityScript QPCR cDNA synthesis kit (Stratagene). Gene expression was analyzed by qPCR in 10 µL reactions, using 100 ng of cDNA. The thermal profile consisted of a hold of 15 min at 95°C, followed by 40 cycles of denaturation (95°C, 15 sec), annealing (60°C, 30 sec) and extension (72°C, 30 sec). Ct values were normalized by quantifying the levels of two macaque reference genes, GAPDH and RPS14 and results expressed as fold change in gene expression relative to non-infected samples. Macaque-specific primers were designed using the AutoPrime software. A list of sequences, gene accession numbers and predicted amplicon size of the oligonucleotides used is provided in Table S2. The obtained sizes for the PCR products are depicted in Fig. S10.
Immunofluorescence confocal microscopy of tissue sections
Optimal cutting temperature compound (OCT)-embedded tissues (spleen and peripheral lymph nodes) were sectioned (7.5 µm thickness) in a frozen cryostat and stored unfixed at −80°C until use. A double fixation procedure was employed and consisted of 4% PFA (15 minutes at room temperature) followed by acetone (20 minutes at −20°C). Slides were saturated in blocking solution (5% normal goat serum, 0.3% triton X-100) for 1 hour at RT. Fluorochrome-conjugated antibodies were diluted in antibody dilution buffer (1% BSA, 0.3% triton X-100) and incubated overnight with tissue sections at 4°C. Table S1 provides detailed information on the antibodies used for tissue immunofluorescence. After washing, slides were mounted with antifade mounting medium. Sections were imaged in a Zeiss LSM 710 confocal microscope. Tiled Z-stacks were acquired with a 20× objective and stitched using the Image J stitching plugin [68]. Average intensity projections were obtained from the stitched tiles using built-in Image J tools. Images were further analyzed and processed using Image J and Adobe Photoshop.
Quantification of germinal centers by tissue immunohistochemistry
Splenic and peripheral lymph node sections were fixed in 4% PFA (15 minutes at RT) and saturated in blocking solution. Sections were incubated for one hour at room temperature with Ki-67 antibody (clone MIB-1, 1/50 dilution in antibody incubation buffer). After washing, sections were incubated with HRP-coupled secondary antibody for 1 hour at RT and the 3,3-diaminobenzidine (DAB) substrate added for revelation. Germinal centers were identified as Ki-67+ cell aggregates and manually counted under a magnifying glass coupled to a digital camera and normalized to the number of GCs per 50 mm2 of area. For determination of germinal center area, micrographs at 40× magnification were acquired and the average area of each GC, defined by Ki-67 staining, was quantified manually using Image J. Two micrographs from two distinct animals per time point were analyzed and the results pooled. Representative micrographs are show in Figure S4.
Statistical analysis
Statistics were performed with the GraphPad Prism 5 software. Data is presented as means ± SEM. A one-way analysis of variance (ANOVA) followed by a Bonferroni's post hoc test was employed for comparison between naïve and infected animals at different time points after infection. A Spearman's rank test was employed for correlations.
Ethics statement
All the animal experiments described in the present study were conducted at the MIRcen platform according to the European Union guidelines for the handling of laboratory animals (http://ec.europa.eu/environment/chemicals/lab_animals/home_en.htm). The animal care and use protocol issued by the IACUC/ethics committee (MIRcen, CAJ–10–30) that approved the study.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. MurrayHW, BermanJD, DaviesCR, SaraviaNG (2005) Advances in leishmaniasis. Lancet 366 : 1561–1577.
2. KayeP, ScottP (2011) Leishmaniasis: complexity at the host-pathogen interface. Nat Rev Microbiol 9 : 604–615.
3. NylenS, SacksD (2007) Interleukin-10 and the pathogenesis of human visceral leishmaniasis. Trends Immunol 28 : 378–384.
4. BanerjeeR, KumarS, SenA, MookerjeeA, MukherjeeP, et al. (2011) TGF-beta-regulated tyrosine phosphatases induce lymphocyte apoptosis in Leishmania donovani-infected hamsters. Immunol Cell Biol 89 : 466–474.
5. NietoA, Dominguez-BernalG, OrdenJA, De La FuenteR, Madrid-ElenaN, et al. (2011) Mechanisms of resistance and susceptibility to experimental visceral leishmaniosis: BALB/c mouse versus syrian hamster model. Vet Res 42 : 39.
6. KenneyRT, SacksDL, GamAA, MurrayHW, SundarS (1998) Splenic cytokine responses in Indian kala-azar before and after treatment. J Infect Dis 177 : 815–818.
7. KarpCL, el-SafiSH, WynnTA, SattiMM, KordofaniAM, et al. (1993) In vivo cytokine profiles in patients with kala-azar. Marked elevation of both interleukin-10 and interferon-gamma. J Clin Invest 91 : 1644–1648.
8. GotoH, PriantiMG (2009) Immunoactivation and immunopathogeny during active visceral leishmaniasis. Rev Inst Med Trop Sao Paulo 51 : 241–246.
9. McCallLI, ZhangWW, MatlashewskiG (2013) Determinants for the development of visceral leishmaniasis disease. PLoS Pathog 9: e1003053.
10. BrenchleyJM, VintonC, TabbB, HaoXP, ConnickE, et al. (2012) Differential infection patterns of CD4+ T cells and lymphoid tissue viral burden distinguish progressive and nonprogressive lentiviral infections. Blood 120 : 4172–4181.
11. HurtrelB, PetitF, ArnoultD, Muller-TrutwinM, SilvestriG, et al. (2005) Apoptosis in SIV infection. Cell Death Differ 12 Suppl 1 : 979–990.
12. MicciL, CervasiB, EndeZS, IrieleRI, Reyes-AvilesE, et al. (2012) Paucity of IL-21-producing CD4(+) T cells is associated with Th17 cell depletion in SIV infection of rhesus macaques. Blood 120 : 3925–3935.
13. PorrozziR, PereiraMS, TevaA, VolpiniAC, PintoMA, et al. (2006) Leishmania infantum-induced primary and challenge infections in rhesus monkeys (Macaca mulatta): a primate model for visceral leishmaniasis. Trans R Soc Trop Med Hyg 100 : 926–937.
14. GrimaldiGJr (2008) The utility of rhesus monkey (Macaca mulatta) and other non-human primate models for preclinical testing of Leishmania candidate vaccines. Mem Inst Oswaldo Cruz 103 : 629–644.
15. EvansTG, KrugEC, WilsonME, VasconcelosAW, de AlencarJE, et al. (1989) Evaluation of antibody responses in American visceral leishmaniasis by ELISA and immunoblot. Mem Inst Oswaldo Cruz 84 : 157–166.
16. SmeltSC, CotterellSE, EngwerdaCR, KayePM (2000) B cell-deficient mice are highly resistant to Leishmania donovani infection, but develop neutrophil-mediated tissue pathology. J Immunol 164 : 3681–3688.
17. DeakE, JayakumarA, ChoKW, Goldsmith-PestanaK, DondjiB, et al. (2010) Murine visceral leishmaniasis: IgM and polyclonal B-cell activation lead to disease exacerbation. Eur J Immunol 40 : 1355–1368.
18. BankotiR, GuptaK, LevchenkoA, StagerS (2012) Marginal zone B cells regulate antigen-specific T cell responses during infection. J Immunol 188 : 3961–3971.
19. ScottP, NatovitzP, SherA (1986) B lymphocytes are required for the generation of T cells that mediate healing of cutaneous leishmaniasis. J Immunol 137 : 1017–1021.
20. WoelbingF, KostkaSL, MoelleK, BelkaidY, SunderkoetterC, et al. (2006) Uptake of Leishmania major by dendritic cells is mediated by Fcgamma receptors and facilitates acquisition of protective immunity. J Exp Med 203 : 177–188.
21. ZotosD, TarlintonDM (2012) Determining germinal centre B cell fate. Trends Immunol 33 : 281–288.
22. LutherSA, MaillardI, LuthiF, ScarpellinoL, DiggelmannH, et al. (1997) Early neutralizing antibody response against mouse mammary tumor virus: critical role of viral infection and superantigen-reactive T cells. J Immunol 159 : 2807–2814.
23. MaCS, DeenickEK, BattenM, TangyeSG (2012) The origins, function, and regulation of T follicular helper cells. J Exp Med 209 : 1241–1253.
24. OnabajoOO, GeorgeJ, LewisMG, MattapallilJJ (2013) Rhesus macaque lymph node PD-1(hi)CD4+ T cells express high levels of CXCR5 and IL-21 and display a CCR7(lo)ICOS+Bcl6+ T-follicular helper (Tfh) cell phenotype. PLoS One 8: e59758.
25. CrottyS (2011) Follicular helper CD4 T cells (TFH). Annu Rev Immunol 29 : 621–663.
26. MontesCL, Acosta-RodriguezEV, MerinoMC, BermejoDA, GruppiA (2007) Polyclonal B cell activation in infections: infectious agents' devilry or defense mechanism of the host? J Leukoc Biol 82 : 1027–1032.
27. MaryC, FarautF, LascombeL, DumonH (2004) Quantification of Leishmania infantum DNA by a real-time PCR assay with high sensitivity. J Clin Microbiol 42 : 5249–5255.
28. KrammerPH, ArnoldR, LavrikIN (2007) Life and death in peripheral T cells. Nat Rev Immunol 7 : 532–542.
29. PaganAJ, PetersNC, DebrabantA, Ribeiro-GomesF, PepperM, et al. (2012) Tracking antigen-specific CD4(+) T cells throughout the course of chronic Leishmania major infection in resistant mice. Eur J Immunol 43(2): 427–38.
30. AndersonCF, OukkaM, KuchrooVJ, SacksD (2007) CD4(+)CD25(−)Foxp3(−) Th1 cells are the source of IL-10-mediated immune suppression in chronic cutaneous leishmaniasis. J Exp Med 204 : 285–297.
31. NylenS, MauryaR, EidsmoL, ManandharKD, SundarS, et al. (2007) Splenic accumulation of IL-10 mRNA in T cells distinct from CD4+CD25+ (Foxp3) regulatory T cells in human visceral leishmaniasis. J Exp Med 204 : 805–817.
32. OwensBM, BeattieL, MooreJW, BrownN, MannJL, et al. (2012) IL-10-producing Th1 cells and disease progression are regulated by distinct CD11c(+) cell populations during visceral leishmaniasis. PLoS Pathog 8: e1002827.
33. StagerS, MaroofA, ZubairiS, SanosSL, KopfM, et al. (2006) Distinct roles for IL-6 and IL-12p40 in mediating protection against Leishmania donovani and the expansion of IL-10+ CD4+ T cells. Eur J Immunol 36 : 1764–1771.
34. MebiusRE, KraalG (2005) Structure and function of the spleen. Nat Rev Immunol 5 : 606–616.
35. MoirS, FauciAS (2009) B cells in HIV infection and disease. Nat Rev Immunol 9 : 235–245.
36. TitanjiK, VeluV, ChennareddiL, Vijay-KumarM, GewirtzAT, et al. (2010) Acute depletion of activated memory B cells involves the PD-1 pathway in rapidly progressing SIV-infected macaques. J Clin Invest 120 : 3878–3890.
37. CaldwellCW, MartyLM, FeldbushTL (1991) Expression of the low Mr isoform of CD45 (CD45RO) in B-cell non-Hodgkin's lymphomas. Clin Immunol Immunopathol 58 : 377–384.
38. JacksonSM, HarpN, PatelD, ZhangJ, WillsonS, et al. (2007) CD45RO enriches for activated, highly mutated human germinal center B cells. Blood 110 : 3917–3925.
39. BerkowskaMA, DriessenGJ, BikosV, Grosserichter-WagenerC, StamatopoulosK, et al. (2011) Human memory B cells originate from three distinct germinal center-dependent and -independent maturation pathways. Blood 118 : 2150–2158.
40. CumontMC, DiopO, VaslinB, ElbimC, ViolletL, et al. (2008) Early divergence in lymphoid tissue apoptosis between pathogenic and nonpathogenic simian immunodeficiency virus infections of nonhuman primates. J Virol 82 : 1175–1184.
41. EngwerdaCR, KayePM (2000) Organ-specific immune responses associated with infectious disease. Immunol Today 21 : 73–78.
42. EidsmoL, WoldayD, BerheN, SabriF, SattiI, et al. (2002) Alteration of Fas and Fas ligand expression during human visceral leishmaniasis. Clin Exp Immunol 130 : 307–313.
43. HeinzelFP, SadickMD, MuthaSS, LocksleyRM (1991) Production of interferon gamma, interleukin 2, interleukin 4, and interleukin 10 by CD4+ lymphocytes in vivo during healing and progressive murine leishmaniasis. Proc Natl Acad Sci U S A 88 : 7011–7015.
44. GhalibHW, PiuvezamMR, SkeikyYA, SiddigM, HashimFA, et al. (1993) Interleukin 10 production correlates with pathology in human Leishmania donovani infections. J Clin Invest 92 : 324–329.
45. O'GarraA, VieiraP (2007) T(H)1 cells control themselves by producing interleukin-10. Nat Rev Immunol 7 : 425–428.
46. ColleJH, Truffa-BachiP, ChedidL, ModabberF (1983) Lack of general immunosuppression during visceral Leishmania tropica infection in BALB/c mice: augmented antibody response to thymus-independent antigens and polyclonal activation. J Immunol 131 : 1492–1495.
47. Campos-NetoA, Bunn-MorenoMM (1982) Polyclonal B cell activation in hamsters infected with parasites of the genus Leishmania. Infect Immun 38 : 871–876.
48. Agenor Araujo Lima VerdeF, Araujo Lima VerdeF, De Francesco DaherE, Martins Dos SantosG, Saboia NetoA, et al. (2009) Renal tubular dysfunction in human visceral leishmaniasis (Kala-azar). Clin Nephrol 71 : 492–500.
49. LuthjeK, KalliesA, ShimohakamadaY, BelzGT, LightA, et al. (2012) The development and fate of follicular helper T cells defined by an IL-21 reporter mouse. Nat Immunol 13 : 491–498.
50. JonesJL, PhuahCL, CoxAL, ThompsonSA, BanM, et al. (2009) IL-21 drives secondary autoimmunity in patients with multiple sclerosis, following therapeutic lymphocyte depletion with alemtuzumab (Campath-1H). J Clin Invest 119 : 2052–2061.
51. LouzirH, Belal-KacemiL, SassiA, LaouiniD, Ben IsmailR, et al. (1994) Natural autoantibodies, IgG antibodies to tetanus toxoid and CD5+ B cells in patients with Mediterranean visceral leishmaniasis. The Leishmania Study Group. Clin Exp Immunol 95 : 479–484.
52. ArgovS, JaffeCL, KruppM, SlorH, ShoenfeldY (1989) Autoantibody production by patients infected with Leishmania. Clin Exp Immunol 76 : 190–197.
53. LeeSK, RigbyRJ, ZotosD, TsaiLM, KawamotoS, et al. (2011) B cell priming for extrafollicular antibody responses requires Bcl-6 expression by T cells. J Exp Med 208 : 1377–1388.
54. KerfootSM, YaariG, PatelJR, JohnsonKL, GonzalezDG, et al. (2011) Germinal center B cell and T follicular helper cell development initiates in the interfollicular zone. Immunity 34 : 947–960.
55. JohnstonRJ, PoholekAC, DiToroD, YusufI, EtoD, et al. (2009) Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science 325 : 1006–1010.
56. Cordeiro-Da-SilvaA, BorgesMC, GuilvardE, OuaissiA (2001) Dual role of the Leishmania major ribosomal protein S3a homologue in regulation of T - and B-cell activation. Infect Immun 69 : 6588–6596.
57. RicoAI, GironesN, FresnoM, AlonsoC, RequenaJM (2002) The heat shock proteins, Hsp70 and Hsp83, of Leishmania infantum are mitogens for mouse B cells. Cell Stress Chaperones 7 : 339–346.
58. BermejoDA, Amezcua VeselyMC, KhanM, Acosta RodriguezEV, MontesCL, et al. (2011) Trypanosoma cruzi infection induces a massive extrafollicular and follicular splenic B-cell response which is a high source of non-parasite-specific antibodies. Immunology 132 : 123–133.
59. XuY, WeatherallC, BaileyM, AlcantaraS, De RoseR, et al. (2013) SIV Infects Follicular Helper CD4 T cells in Lymphoid Tissues During Pathogenic Infection of Pigtail Macaques. J Virol 87 : 3760–73.
60. PetrovasC, YamamotoT, GernerMY, BoswellKL, WlokaK, et al. (2012) CD4 T follicular helper cell dynamics during SIV infection. J Clin Invest 122 : 3281–3294.
61. LindqvistM, van LunzenJ, SoghoianDZ, KuhlBD, RanasingheS, et al. (2012) Expansion of HIV-specific T follicular helper cells in chronic HIV infection. J Clin Invest 122 : 3271–3280.
62. Chacon-SalinasR, Limon-FloresAY, Chavez-BlancoAD, Gonzalez-EstradaA, UllrichSE (2011) Mast cell-derived IL-10 suppresses germinal center formation by affecting T follicular helper cell function. J Immunol 186 : 25–31.
63. RoussetF, GarciaE, DefranceT, PeronneC, VezzioN, et al. (1992) Interleukin 10 is a potent growth and differentiation factor for activated human B lymphocytes. Proc Natl Acad Sci U S A 89 : 1890–1893.
64. SilvestreR, Cordeiro-Da-SilvaA, SantaremN, VergnesB, SerenoD, et al. (2007) SIR2-deficient Leishmania infantum induces a defined IFN-gamma/IL-10 pattern that correlates with protection. J Immunol 179 : 3161–3170.
65. Gibson-CorleyKN, BoggiattoPM, BockenstedtMM, PetersenCA, WaldschmidtTJ, et al. (2012) Promotion of a functional B cell germinal center response after Leishmania species co-infection is associated with lesion resolution. Am J Pathol 180 : 2009–2017.
66. SilvestreR, SantaremN, CunhaJ, CardosoL, NietoJ, et al. (2008) Serological evaluation of experimentally infected dogs by LicTXNPx-ELISA and amastigote-flow cytometry. Vet Parasitol 158 : 23–30.
67. LaforgeM, Campillo-GimenezL, MonceauxV, CumontMC, HurtrelB, et al. (2011) HIV/SIV infection primes monocytes and dendritic cells for apoptosis. PLoS Pathog 7: e1002087.
68. PreibischS, SaalfeldS, TomancakP (2009) Globally optimal stitching of tiled 3D microscopic image acquisitions. Bioinformatics 25 : 1463–1465.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 4
- Koronavirus hýbe světem: Víte jak se chránit a jak postupovat v případě podezření?
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
Nejčtenější v tomto čísle
- The 2010 Cholera Outbreak in Haiti: How Science Solved a Controversy
- , , , Genetic Variability: Cryptic Biological Species or Clonal Near-Clades?
- Efficient Parvovirus Replication Requires CRL4-Targeted Depletion of p21 to Prevent Its Inhibitory Interaction with PCNA
- An Overview of Respiratory Syncytial Virus
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy