
Necrotic Cells Actively Attract Phagocytes through the Collaborative Action of Two Distinct PS-Exposure Mechanisms
Necrosis is a type of cell death often caused by cell injury and is linked to human diseases including neuron degeneration, stroke, and cancer. Necrotic cells undergo distinct morphological changes, including swelling, before being engulfed and degraded by engulfing cells. The clearance of necrotic cells from animal bodies is important for wound healing and for preventing harmful inflammatory and autoimmune responses. However, the mechanisms by which necrotic cells are removed remain elusive. We study the recognition of necrotic neurons in the nematode C. elegans. There is a common belief that the plasma membrane of necrotic cells are ruptured, allowing the detection of phosphatidylserine (PS), a so-called “eat me” signal molecule, by specific transmembrane receptors on the surface of engulfing cells. Contrary to this belief, we found that necrotic neurons actively present PS to their outer surface through two parallel molecular mechanisms, one of which is shared by cells undergoing apoptosis, a “cell suicide” event, whereas the other is unique to necrotic cells. Ca2+-influx, a key factor that triggers necrosis, is implicated in activating a unique PS-scramblase. Our findings reveal novel necrotic cell-specific “eat me” signal-exposure mechanisms and indicate that cells that die through different mechanisms (necrosis and apoptosis) utilize both common and unique mechanisms to attract engulfing cells. They further demonstrate that C. elegans is an effective model system for studying the fate of necrotic cells.
Published in the journal:
. PLoS Genet 11(6): e32767. doi:10.1371/journal.pgen.1005285
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005285
Summary
Necrosis is a type of cell death often caused by cell injury and is linked to human diseases including neuron degeneration, stroke, and cancer. Necrotic cells undergo distinct morphological changes, including swelling, before being engulfed and degraded by engulfing cells. The clearance of necrotic cells from animal bodies is important for wound healing and for preventing harmful inflammatory and autoimmune responses. However, the mechanisms by which necrotic cells are removed remain elusive. We study the recognition of necrotic neurons in the nematode C. elegans. There is a common belief that the plasma membrane of necrotic cells are ruptured, allowing the detection of phosphatidylserine (PS), a so-called “eat me” signal molecule, by specific transmembrane receptors on the surface of engulfing cells. Contrary to this belief, we found that necrotic neurons actively present PS to their outer surface through two parallel molecular mechanisms, one of which is shared by cells undergoing apoptosis, a “cell suicide” event, whereas the other is unique to necrotic cells. Ca2+-influx, a key factor that triggers necrosis, is implicated in activating a unique PS-scramblase. Our findings reveal novel necrotic cell-specific “eat me” signal-exposure mechanisms and indicate that cells that die through different mechanisms (necrosis and apoptosis) utilize both common and unique mechanisms to attract engulfing cells. They further demonstrate that C. elegans is an effective model system for studying the fate of necrotic cells.
Introduction
Cell death during animal development and under pathological conditions is important for removing unwanted cells that are often harmful. Necrosis and apoptosis are two morphologically distinct types of cell death events. Whereas cells undergoing apoptosis display features such as cytoplasm shrinkage, chromatin condensation, nuclear DNA fragmentation, and well-maintained plasma membrane integrity, necrotic cells display cell and organelle swelling, excessive intracellular membranes, and the eventual rupture of intracellular and plasma membranes (reviewed in [1,2]). Necrosis is most frequently observed during cell injury, and is closely associated with diseases such as stroke, neurodegeneration, chronic inflammation, and cancer [3–7]. Although necrosis was historically considered an uncontrolled cell death event caused by acute damage, recent discoveries made in multiple organisms demonstrated that in addition to injury-induced necrosis, cells possess genetic pathways that specifically trigger necrosis in response to extracellular or intracellular stimuli (reviewed in [8–11]). For instance, tumor necrosis factor (TNF) induces a necrosis pathway executed through Ser/Thr kinases [10]. In addition, hyperexcitation of neurons or glial cells induced by the massive release of neurotransmitters or constitutively active ion channels cause excitotoxic necrosis [7,12,13]. Unlike apoptosis, which relies on caspase-mediated death-triggering mechanisms, known necrosis-triggering pathways appear to be independent of caspase-activities (reviewed in [8,14]). On the other hand, like apoptotic cells, in many cases necrotic cells have been observed to be engulfed by phagocytes [15,16]. Efficient clearance of necrotic cells from animal bodies helps to resolve the wounded area; furthermore, cell-corpse removal is essential for reducing harmful inflammatory and auto-immune responses induced by the contents of necrotic cells [15,17]. It is currently unclear how necrotic cells expose the “eat me” signal molecules on their surfaces to attract engulfing cells.
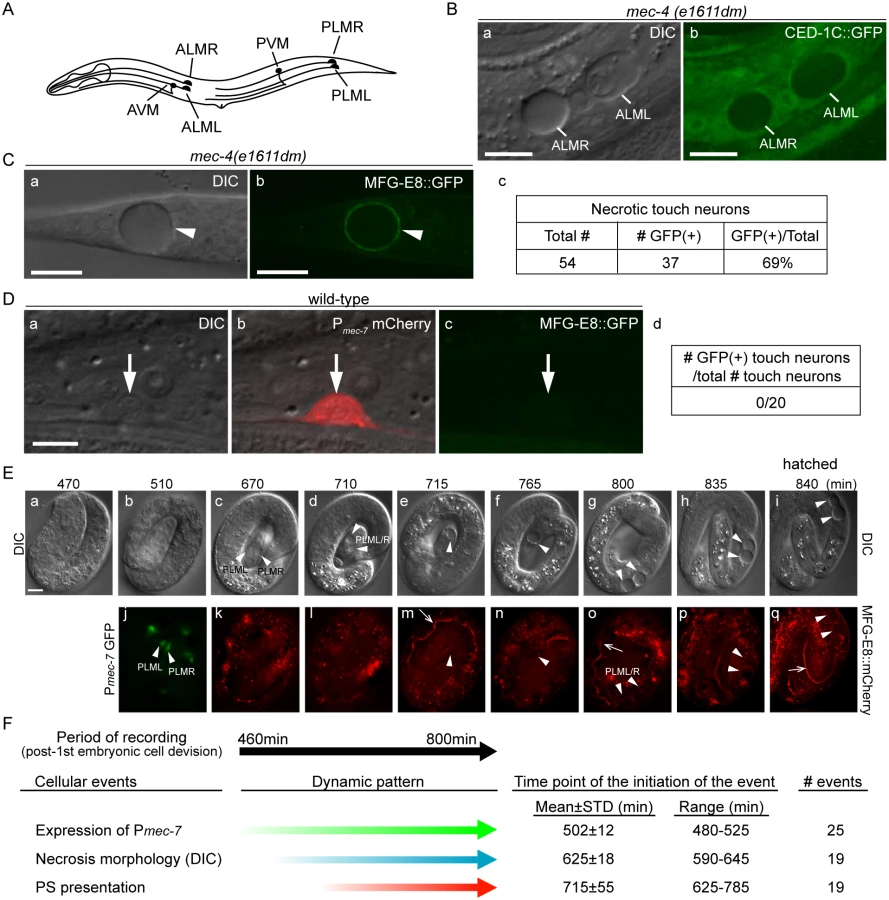
Besides being an excellent model organism for studying the mechanisms of apoptosis and the removal of apoptotic cells [18], the soil nematode Caenorhabditis elegans has also been established as a model for studying necrosis [8,13]. In C. elegans, a number of mutations in the subunits of ion channels, the acetylcholine receptor, and trimeric GTPases induce specific neurons to undergo necrotic cell death that mimics the excitotoxic necrosis, which occurs during stroke, trauma, and neurodegenerative disorders in humans (reviewed in [8]). In particular, specific mutations in multiple genes trigger the necrosis of six mechanosensory (touch) neurons (AVM, PVM, ALML/R and PLML/R) required to sense gentle mechanical stimuli along the body wall [19–21]. Dominant (dm) mutations in mec-4, which encodes a core subunit of a multimeric, mechanically gated sodium channel belonging to the DEG/ENaC family specifically expressed in the touch neurons, lead to hyperactive channel conductivity of Na+ and Ca2+ and induce these neurons to undergo necrosis [19,22]. In mec-4(dm) mutants, the six dying neurons swell to many times their original sizes and develop cytoplasmic vacuoles and large membranous whorls, and are easily distinguishable from living or apoptotic cells under Differential Interference Contrast (DIC) optics by their giant sizes (Fig 1) [16,21]. This type of cell death does not require CED-3 caspase activity [23], and is instead triggered by the influx of Ca2+ into the cytoplasm [22,24]. Despite their distinct modes of triggering cell death, the seven ced genes needed for the engulfment of apoptotic cells are also required for the efficient removal of necrotic touch neurons [25], indicating the presence of certain common recognition and engulfment mechanisms for dying cells. On the other hand, the distinct cellular features observed during macrophage engulfment of necrotic mammalian cells imply that unique pathways exist to clear necrotic and apoptotic cells [26].
Phosphatidylserine (PS), a membrane phospholipid, is a known “eat me” signal presented on the surface of apoptotic cells to directly or indirectly attract phagocytic receptors such as C. elegans CED-1, Drosophila Draper, and mammalian Tim4 and BAI1, leading to the initiation of their engulfment [27–31]. In living cells, PS is almost exclusively localized to the inner leaflet of the plasma membrane, at least partially due to an ATP-dependent aminophospholipid translocase activity that selectively returns PS and PE (phosphatidylethanolamine) from the outer to the inner leaflet [32–34]. During the early stage of apoptosis, PS is detected on the outer leaflet, suggesting a process of trans-bilayer redistribution [32,33]. Phospholipid scramblases, by catalyzing the random, bi-directional “flip-flop” of phospholipids across the membrane bilayer, could potentially counter the aminophospholipid translocase activity [35]. The mouse transmembrane protein 16F (TMEM16F) was recently found to act as a novel Ca2+-activated phospholipid scramblase [36]. However, TMEM16F does not seem to be involved in exposing PS on apoptotic cell surfaces [37]. On the other hand, mouse Xk-related protein 8 and CED-8, its C. elegans homolog, mediate PS exposure in response to apoptotic stimuli [38,39]. These results suggest that different phospholipid scramblases function in different cell types and in response to different stimuli. In addition, the mammalian ATP-binding-cassette transporter A1 (ABCA1) has been implicated in the translocation of PS from the inner to the outer leaflet [40,41], although evidence to the contrary also exists [42].
Previously, using milk-fat-globule EGF8 (MFG-E8::GFP), a GFP-tagged, secreted PS reporter, we have detected the presentation of PS specifically on the surface of apoptotic cells during animal development [43]. We have further identified two alternative mechanisms that promote PS exposure in apoptotic somatic and germ cells, respectively [43]. The PS exposure on apoptotic cell surface during embryonic development, which is necessary for their engulfment, relies on the function of C. elegans CED-7, a homolog of mammalian ABCA1 transporters [43].
Considering the insights that have been made to understand how apoptotic cells are recognized and removed, the mechanisms by which necrotic cells are engulfed remain poorly defined. In particular, it is unclear whether necrotic cells are capable of the active presentation of “eat me” signaling molecules such as PS to attract engulfing cells. Rather, it was assumed that PS was detected on necrotic cell surfaces due to the rupture of necrotic cell membranes [44]. The work reported here establishes that necrotic C. elegans touch neurons actively present PS on their outer surfaces while maintaining plasma membrane integrity. It further defines two mechanisms that act in parallel to promote the exposure of PS on necrotic cell surfaces, one that is shared with apoptotic somatic cells, and another that is unique to necrotic touch neurons.
Results
Necrotic touch neurons actively externalize phosphatidylserine on their surfaces
The dominant mutant allele e1611dm of C. elegans mec-4 results in the necrotic death of six touch neurons, which swell to several times of their original diameter (Fig 1A and 1B(a)), displaying a morphology distinct from somatic apoptotic cells, which undergo cytoplasmic shrinkage and nuclear condensation [19,21,45–47]. Previously, necrotic AVM and PVM were observed inside the hypodermis [16]. To visualize the engulfment status of all necrotic touch neurons, we expressed Pced-1 ced-1C::gfp, which produced a GFP reporter that is distributed evenly in the cytoplasm of engulfing cell types, including hypodermal cells [48]. We detected all six necrotic touch neurons as dark holes embedded inside the GFP-labeled engulfing cells in newly hatched mec-4(e1611dm) larva (Fig 1B), establishing that necrotic touch neurons are engulfed by hypodermal cells.
To examine whether necrotic cells expose PS on their cell surfaces, we expressed mfg-e8::gfp, a secreted PS reporter [43], in mec-4(e1611dm) mutant worms. In newly hatched L1 larvae, we detected GFP specifically enriched on the surface of necrotic touch neurons (Fig 1C). In contrast, when mfg-e8::gfp was expressed in mec-4(+) worms, no fluorescence was detected on the surface of living touch neurons (Fig 1D). These results suggest that PS is present specifically on the surface of cells that undergo necrosis.
Previously, it was generally believed that necrosis caused prominent plasma membrane rupture [1,2]. If that is the case, it is possible that MFG-E8::GFP molecules penetrate through the plasma membrane and associate with the PS molecules on the inner leaflet of the plasma membrane. To distinguish whether the enriched MFG-E8 signal is a result of PS exposure on the outer or inner surfaces of necrotic cells, we examined whether the necrotic touch neurons observed in the mec-4(e1611dm) mutants lost plasma membrane integrity. We observed the localization of GFP or mRFP reporters expressed specifically in touch neurons under the control of the mec-7 promoter (Pmec-7) [49] in mec-4(e1611dm) worms and found that the fluorescent signals were exclusively retained inside necrotic cells (S1A (a, b, e, f) Fig). In parallel, secreted GFP (ssGFP) reporters, which are tagged with a signal sequence from SEL-1 [50] and expressed specifically from hypodermal (Pcol-10) and body wall muscle (Pmyo-3) cells [28,51], two types of cells that neighbor the touch neurons, were not observed inside touch neurons (S1A (c, d, g, h) Fig). These GFP signals were detected inside coelomocytes, mobile cells that possess high endocytic activity (S1B Fig), indicating that they are indeed secreted into the close proximity of the touch neurons. No plasma membrane penetration of the touch-neuron reporter or neighboring-cell reporters was observed during a 36-hr observation period from the appearance of necrotic cell morphology in embryos to the mid-L4 stage. The above lines of evidence indicate that the plasma membrane of necrotic touch cells is not permeable to GFP or mRFP molecules (which are of sizes between 25 and 27 kD). Thus, it is unlikely that the same plasma membrane would be permeable to MFG-E8::GFP, which is substantially larger (78 kD). Furthermore, when wild-type and mec-4(e1611dm) worms were stained with propidium iodide, a small molecular weight (MW = 688 Da) fluorescent dye that is not permeable across the intact plasma membrane (Materials and Methods), we did not observe propidium iodide signal in the living or necrotic cells (S1C Fig). The only propidium iodide signal observed came from the intestinal track, inside which were ingested propidium iodide-stained bacteria cells (S1C Fig). Together, the above results indicate that, against the common belief that necrotic cells passively expose PS through plasma membrane rupture and in this manner attract engulfing cells, the C. elegans necrotic touch neurons maintain cell integrity and actively expose PS, which may function as a specific “eat me” signal, on their surfaces.
Using a live-cell recording protocol that we established for touch neurons (Materials and Methods), we monitored the dynamics of an MFG-E8::mCherry reporter during embryogenesis. Among the six touch neurons, four are born during mid embryogenesis, including PLML and PLMR, which were reported to arise at approximately 510 min post the 1st embryonic cell division (the 1st-cleavage), whereas AVM and PVM were reported to be born at the L1 larval stage [46,47]. At hatching, PLML and PLMR should have existed for 290 min. We observed that the enrichment of PS on necrotic PLML and PLMR was a gradual process after necrosis was initiated at a morphological level (Fig 1E and 1F) (S1 Movie). Among the following three events, (1) the differentiation of touch neurons, which is indicated by the expression of Pmec-7 gfp, (2) the swelling of touch neurons undergoing necrosis, which is visible under DIC optics, and (3) the exposure of PS on the outer surface, indicated by the enrichment of MFG-E8::mCherry, cell differentiation occurs the earliest, initiating approximately 480–525 min after the 1st cleavage (Fig 1E(b, j) and 1F) (S1 Movie). On average 120 min later, the swelling morphology of PLML/R starts to develop, indicating that the constitutively active Na+/Ca2+ channel starts to initiate necrosis. The first time point when the enrichment of PS is detected on PLML/R surface varies; yet in all 19 cases monitored, it occurs after the initial appearance of the necrotic morphology observed by DIC (S1 Movie). Subsequently, the PLML/R surface MFG-E8::mCherry intensity continues to increase until the time point of hatching (Fig 1E(p and q)). Our observations established the order of the initiation of these three events, and further suggest a causal relationship between the initiation of necrosis and the exposure of PS.
Multiple types of necrotic neurons expose PS on their surfaces
To determine whether PS-externalization is a general phenomenon occurring to different types of neurons that undergo necrosis, we monitored PS enrichment on non-touch neurons. u662, a gain-of-function mutation in deg-3, which encodes a subunit of an acetylcholine receptor ion channel [20], causes the necrosis of the six touch neurons and a few additional sensory and inter-neurons through hyper-activation of the acetylcholine receptor ion channel [20]. Cells undergoing necrosis in mec-4 and deg-3 dominant mutants display the same distinct morphology [19,20,52,53]. In deg-3(u662) animals, we detected PS on the surface of necrotic neurons, including touch neurons and other types of neurons (S2 Fig). This result suggests that PS-exposure is a general feature of neurons induced to undergo necrosis through excitotoxicity.
CED-1 recognizes necrotic cells and initiates necrotic-cell engulfment
CED-1 is a phagocytic receptor that is localized on the surface of several types of cells, including all engulfing cell types and clusters around apoptotic cells in response to the neighboring “eat me” signals [28,43]. To determine the efficiency of necrotic cell clearance in the ced-1(e1735); mec-4(e1611dm) double mutants, we chose to score the dynamic presence of necrotic PLML and PLMR, two touch neurons in the tail that undergo necrosis during mid-embryogenesis, throughout all larval stages. The rationale is that the longer a necrotic touch cell persists during larval development, the less efficient its removal process must be. The same scheme was used to score the efficiency of necrotic cell clearance throughout this report (Materials and Methods). We found the ced-1(e1735) null mutation greatly reduced the efficiency of necrotic cell removal (Fig 2A), consistent with a previous report [25]. Prior results indicate that the extracellular domain of CED-1 is responsible for recognizing the surface feature(s) of apoptotic cells. We analyzed two truncated forms of CED-1 for their ability to recognize necrotic cells by monitoring GFP-tagged truncated forms expressed in larval hypodermal cells, which engulf necrotic touch neurons. We first found that CED-1ΔC::GFP, a truncated form of CED-1 (Fig 2B) that remains bound to the plasma membrane of the engulfing cells, is highly enriched on the phagocytic cup or phagosomal surface (Fig 2C and 2E) in comparison to other regions of the plasma membrane of the same cell. Quantification of GFP fluorescence intensity on phagocytic cups or phagosomes is on average 3.1 times of that on other plasma membrane regions of the same cell (Fig 2E(d)). We next found that CED-1Ex::GFP, a truncated and secreted form of CED-1 (Fig 2B), was specifically enriched on the surfaces of necrotic cells (Fig 2D). These results indicate that the extracellular domain of CED-1 directly recognizes necrotic cells, and that the high level of CED-1ΔC::GFP detected on the engulfing cell membrane region around necrotic cells is not merely a result of necrotic touch neurons being embedded inside hypodermal cells.

The extracellular domain of CED-1 directly associates with PS
Since necrotic cells, like apoptotic cells, specifically expose PS on their surfaces, and because CED-1 recognizes both necrotic and apoptotic cells in vivo, we tested whether PS could act as a ligand for CED-1. The extracellular domain of CED-1 was expressed as a fusion protein to glutathione S-transferase (GST) (CED-1-GST) in an insect cell expression system, affinity purified by glutathione-sepharose chromatography (Fig 3A), and tested for its binding affinities to PS in vitro (Materials and Methods). We first employed an Enzyme-Linked ImmunoSorbent Assay (ELISA)-like reaction to test the interaction between CED-1-GST and PS or phosphatidylcholine (PC). The CED-1 protein displayed efficient association with PS but not PC, in a dose-dependent manner (Fig 3B). We next examined this association in a surface plasmon resonance assay [29]. In this assay, CED-1-GST was applied onto channels of the HPA chip, on which equivalent numbers of PS-containing liposomes and PC-only liposomes had been immobilized (S3 Fig). We found that the values in response units after the injection of CED-1-GST to the channel with PS-containing liposome were higher than those obtained for the control channel (S3B Fig). These two assays indicate that the extracellular domain of CED-1 interacts directly with PS. Furthermore, free PS-containing liposomes, but not PC-liposomes, could efficiently compete with PS-containing liposomes coated on the well for binding to CED-1-GST in the ELISA-like reactions (Fig 3C), suggesting that CED-1 specifically recognizes PS as a component of membrane bilayer. In the ELISA-like assay, binding between CED-1-GST with two other phospholipids, phosphatidylethanolamine (PE) and phosphatidylinositol (PI), was also detected (Fig 3D). All these results indicate that the extracellular domain of CED-1 directly associates with phospholipids containing an amino group or negative charge, including PS, and support the hypothesis that PS serves as a ligand for CED-1 for the recognition of necrotic and apoptotic cells.

CED-7 is essential for PS exposure on the surface of necrotic cells
Among the seven engulfment ced genes that act in two parallel pathways, ced-7, which encodes a member of the ABC transporter family (Fig 4A), is the only one required for the presentation of PS on the outer surface of apoptotic cells in C. elegans embryos [43]. The ced-7(n1996) null mutation severely impairs the removal of necrotic touch neurons (Fig 4C). We found that among the null or strong loss-of-function mutations of six ced genes, only the ced-7(n1996) mutation significantly reduced the percentage of necrotic touch neurons exhibiting surface PS (Fig 4D). These results indicate that CED-7 is essential for PS exposure on necrotic touch neurons. In support of this conclusion, a CED-7::GFP reporter expressed in touch neurons (Pmec-7 ced-7::gfp) is observed on the surface of necrotic touch neurons in mec-4(e1611dm) background (Fig 4B), consistent with a role of CED-7 in PS-flipping from one leaflet of the plasma membrane to the other.
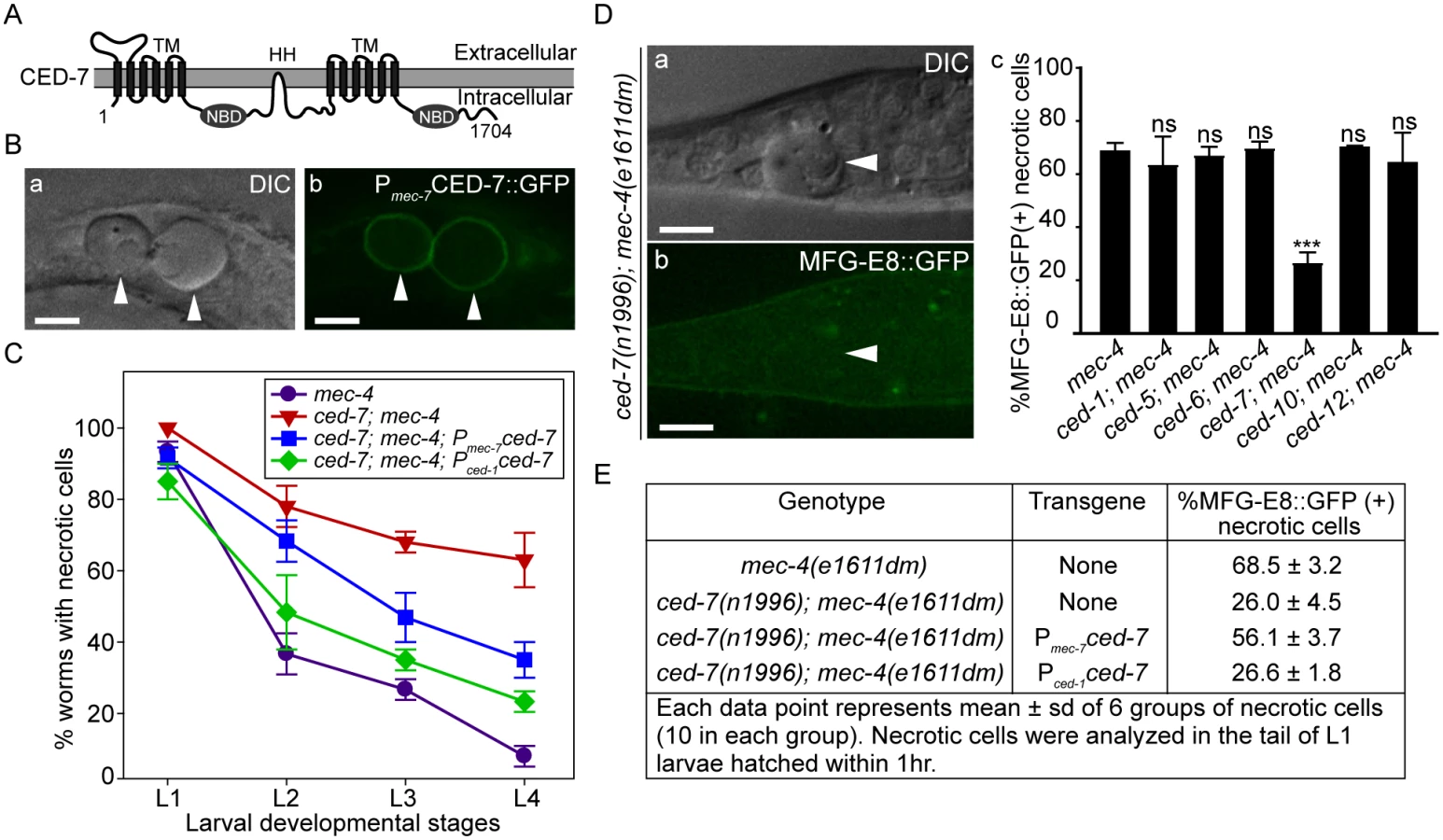
CED-7 is broadly expressed in all cells [54]. To determine whether ced-7 functions to promote necrotic cell removal in the engulfing cells or in touch neurons, we tested the effect of cell type-specific expression of ced-7 in the rescue of ced-7 mutant phenotypes. The ced-7(n1996) null mutation severely delays necrotic cell removal, resulting in the persistent existence of necrotic touch neurons in more than 60% of L4 larvae (Fig 4C). Expression of ced-7 in either touch neurons (Pmec-7 ced-7) or neighboring engulfing cells (Pced-1 ced-7) each partially rescued the necrotic-cell removal defect (Fig 4C), indicating that the functions of CED-7 in necrotic and engulfing cells both contribute to the efficient removal of necrotic cells. We further found that the specific expression of ced-7 in touch neurons but not in neighboring engulfing cells primarily rescued the PS exposure defect of ced-7(n1996) mutants (Fig 4E). This result clearly indicates that the touch cell-specific role of CED-7 is responsible for promoting PS exposure.
ANOH-1, the C. elegans homolog of mammalian TMEM16F plays a specific role for the exposure of PS on the surfaces of necrotic cells
Mammalian TMEM16F, a multispan transmembrane domain protein, is a Ca2+-activated phospholipid scramblase that triggers PS exposure in response to Ca2+-influx [36]. Given that the mec-4(dm)-induced touch neuron necrosis is mediated by Ca2+ influx [22,24], we examined whether ANOH-1, a close C. elegans homolog of TMEM16F (Figs 5B and S4), mediated PS exposure when necrosis occurred. We analyzed an anoh-1(tm4762) deletion allele (www.wormbase.org) for the removal of dying cells. The tm4762 allele carries a 202-bp deletion that results in a frameshift and a premature stop codon after amino acid 17 of the predicted ANOH-1b open reading frame and removes the start codon of the alternatively-spliced ANOH-1a open reading frame (S4 and S5B(b) Figs) (also see the next section), presumably generating a null allele. In anoh-1(tm4762) mutant embryos, the numbers of apoptotic cells are the same as that displayed in wild-type embryos at 5 different embryonic developmental stages (Fig 5C). Furthermore, the dynamics of the engulfment and degradation processes of three individual apoptotic cells, C1, C2, and C3, are normal comparing to wild-type embryos (Fig 5E), using previously established protocols [48,55]. Similarly, the number of apoptotic germ cells are virtually the same in wild-type and anoh-1(tm4762) mutant adult hermaphrodites at four time points (Fig 5D). These results indicate that anoh-1, unlike ced-7, is not involved in the removal of apoptotic cells. In contrast, in anoh-1(tm4762); mec-4(e1611dm) double mutant animals, the removal of necrotic touch neurons is siginificantly delayed: at L1 and L2 stages, the mean numbers of persistent necrotic PLML/R in anoh-1(tm4762) background are approximately 1.6-fold and 1.5-fold of that in wild-type larvae, respectively, whereas at L3 and L4 stages, the mean numbers are not significantly different from wild-type animals (Fig 5F). These results strongly suggest that anoh-1 specifically contributes to efficient removal of necrotic but not apoptotic cells.

In wild-type and anoh-1 mutant animals, we further measured the MFG-E8::GFP signal intensity and calculated the ratio of GFP intensity on necrotic PLML/R to that in an equivalent area of the neighboring live cells (Materials and Methods). Lack of PS enrichment on the surface of necrotic cells will result in a ratio of approximately 1.0. We first quantified the PS signal intensity at the early L1 stage, when the necrotic cell removal defect displayed by the anoh-1 mutants was the most prominent among all four larval stages (Fig 5F). The anoh-1(tm4762) mutation significantly inhibits PS enrichment on necrotic touch neurons, reducing the median value of this ratio from 1.34 in the wild-type animals to 1.13 (Fig 6B and 6C). This result indicates a unique function of ANOH-1 in promoting PS exposure on necrotic cell surfaces. At the young L2 larval stage (15–16 hrs after hatching), the average relative PS signal intensity value increases from the young L1 value in both the wild-type (from 1.36 to 1.71) and anoh-1(tm4762) mutant (1.20 to 1.51) strains (S6B Fig), probably as a result of the continuous accumulation of the GFP signal on the surface of necrotic neurons,. The PS signal increase in anoh-1 mutants could explain the reduced removal defect at later developmental stages (Fig 6A).

ANOH-1 acts in touch neurons to promote the removal of necrotic touch neurons
Based on its gene structure, anoh-1 is predicted to encode two splice variants, anoh-1a and anoh-1b (Fig 5A). The predicted ANOH-1b protein carries an additional 18 residues at the amino-terminus, as compared to the predicted ANOH-1a protein (S4 Fig). Using RT-PCR (Materials and Methods), we detected the existence of the anoh-1b transcript in whole worm extracts (S5B Fig). To determine which of the two splice variants and in which cell type anoh-1 is functional in the removal of necrotic cells, we individually expressed anoh-1a and anoh-1b in touch neurons (Pmec-7) or engulfing cells (Pced-1), in the anoh-1(tm4762); mec-4(e1611dm) background (Fig 5F and 5G). To monitor the subcellular localization of each isoform, anoh-1a and anoh-1b were each tagged with gfp. Among the four expression constructs tested, only anoh-1b, when expressed in touch neurons (Pmec-7 anoh-1b::gfp), rescued the necrotic cell removal defect of these mutants (Fig 5F and 5G). In addition, Pmec-7 anoh-1b::gfp also leads to the recovery of PS exposure on necrotic neuron surfaces (S7 Fig). These results indicate that anoh-1b is the functional form and that it acts in necrotic touch neurons to promote their removal.
To understand the function of the amino-terminal 18 amino acids present in ANOH-1b, which are absent from the predicted ANOH-1a protein, we characterized the subcellular localization of each form, as N - or C-terminal GFP-tagged proteins, expressed in touch neurons. GFP::ANOH-1b and ANOH-1b::GFP are both localized to the plasma membrane, consistent with the hypothesized role of ANOH-1 in promoting PS on cell surface (Fig 5H(a, b, e, and f)). In contrast, GFP::ANOH-1a and ANOH-1a::GFP are both observed inside touch neurons, enriched on the nuclear surface (Fig 5H(c, d, g, and h)). These results suggest that the plasma membrane localization is essential for the function of ANOH-1 in necrotic cell-removal.
To determine the expression pattern of anoh-1b, we cloned the 617 bp DNA fragment immediately 5’ to Exon 1 of anoh-1b, and tentatively regard this fragment as the Panoh-1b promoter. The nuclear localization sequence (NLS)-tagged GFP signal produced by Panoh-1b is expressed in touch neurons (Figs 5I and S5C(l, o, r)). This result supports the touch neuron-specific function of ANOH-1 (the b isoform) in promoting PS-exposure. In addition, by comparing the expression patterns of Panoh-1b NLS-GFP and a pan-neuronal expression reporter Prab-3 dsRed [56], we observed strong Panoh-1b activity in many neurons in the head and tail (S5C Fig). This result is consistent with a previous report detecting anoh-1 expression in sensory neurons [57]. In addition, anoh-1 expression is also observed in intestinal cells as previously reported [57] and pharyngeal muscles (S5C (g, h, i) Fig).
ANOH-1 acts in a parallel pathway to CED-7 to regulate PS exposure
To investigate the functional relationship between CED-7 and ANOH-1 in the clearance of necrotic cells, we first compared the numbers of persistent necrotic cells throughout larval developmental in anoh-1(tm4762) ced-7(n1996) double mutants and in anoh-1 or ced-7 single mutant animals, in the mec-4(e1611dm) mutant background. We observed that inactivating ced-7 causes a stronger necrotic cell removal defect than that of anoh-1 (Fig 6A). Furthermore, inactivating both ced-7 and anoh-1 results in an enhanced necrotic cell removal defect starting at the L2 stage (Fig 6A), suggesting that CED-7 and ANOH-1 might perform partially parallel functions during removal. We further quantified the PS signal intensity at the early L1 stage. Inactivating both ced-7 and anoh-1 further reduces the PS signal on the surface of necrotic PLML/R, which is already significantly reduced by the ced-7 or anoh-1 single mutations compared to the wild-type background (Fig 6B and 6C) or the ced-6 mutation, which delays necrotic cell removal but does not affect PS exposure (S8 Fig). The above results suggest that CED-7 and ANOH-1 both contribute to the PS-externalization activity; furthermore, they may do so through partially parallel pathways. At the L2 stage, when the necrotic corpse-removal and PS-exposure phenotypes of anoh-1 mutants are greatly reduced, the PS-exposure phenotype of the anoh-1 ced-7 double mutants was similar to that of ced-7 single mutants (S6 Fig), again suggesting that inactivating anoh-1 delays but does not block necrotic-cell removal.
CED-8 makes a minor contribution to the removal of necrotic touch neurons
We examined whether a loss-of-function mutation of ced-8, which is proposed to encode a phospholipid scramblase essential for PS-exposure on the surface of apoptotic cells, also affects the removal of necrotic touch neurons. In a ced-8(n1891) mutant allele, a strong loss-of-function allele that carries a nonsense mutation, truncating CED-8 (458aa) after aa 219 [58], there is a significant necrotic touch neuron removal defect at the L1 larval stage (Fig 7A). However, this defect was not observed in any of the later larval stages (Fig 7A), suggesting that CED-8 merely delayed but did not block necrotic cell removal. We further analyzed the functional relationship between anoh-1 and ced-8. At the L1 stage, the anoh-1(tm4762); ced-8(n1891) double mutants display a more severe necrotic cell-removal defect as compared to each single mutant (Fig 7B). These results suggest that anoh-1 and ced-8 act in parallel and thus the necrotic cell removal function of ced-8 is independent of anoh-1.

Discussion
Active externalization of PS on the surface of necrotic cells
The presentation and function of PS on the surface of cells undergoing necrosis have previously been overlooked, primarily because of the long-existing notion that these cells encounter injury and lose plasma membrane integrity [44,59,60]. However, in recent years, accumulating evidence has demonstrated that in addition to cell injury, necrosis could also be induced by genetic programs (reviewed in [9]). In short, multiple molecular mechanisms exist that induce and execute necrosis [9,14]; thus all necrotic cells do not necessarily lose plasma membrane integrity. The loss of membrane integrity of necrotic cells in culture, where there are no phagocytes to engulf them, does not necessarily represent what happens inside animal bodies, where engulfing cells target dying cells at early stages of their death [16,61] (our own observations). On the other hand, those necrosis events that occur inside animal bodies were rarely examined for plasma membrane integrity. In the case of neuronal excitotoxic necrosis, electron microscopic studies of rat brains and C. elegans touch neurons reported the swelling of necrotic cells and the presence of surrounding phagocytes, yet no loss of plasma membrane integrity [16,62]. By monitoring signals elicited from GFP or mRFP reporters either inside or outside necrotic cells and by incubating worms with propidium iodide, a small molecule dye that is not plasma membrane permeable, we observed that the necrotic C. elegans touch neurons induced by excitotoxicity maintain plasma membrane integrity throughout embryonic and larval development. These results indicate that the common notion that necrotic cells lose plasma membrane integrity is not necessarily true for all kinds of necrotic cells at all developmental stages, and further suggest that for PS to be present on the surface of necrotic touch neurons, an active PS exposure mechanism must exist. Further genetic studies reported here revealed that at least two separate PS-exposure activities act to promote PS exposure on the surface of necrotic cells.
PS as a common “eat me” signal elicited by necrotic and apoptotic cells and a ligand for phagocytic receptor(s)
PS is an evolutionarily conserved “eat me” signal exposed by apoptotic cells in metazoan organisms ranging from simple to complex and it is implicated in recruiting engulfing cells [27,29,43,63–65]. Here we report the active exposure of PS on the surface of necrotic touch neurons. The fact that both necrotic and apoptotic cells expose PS on their surfaces implies the existence of a conserved dying cell-recognition mechanism. Indeed, we further discovered the novel function of phagocytic receptor CED-1 in recognizing necrotic cells in addition to apoptotic cells. The extracellular domain of CED-1 (CED-1Ex) alone, without the transmembrane or intracellular domains, is sufficient for associating to the surface of necrotic cells, suggesting an extracellular ligand-receptor interaction. In support of this hypothesis, we have detected direct and selective in vitro interaction between CED-1Ex and acidic phospholipids including PS. On the other hand, CED-1Ex does not display affinity to phosphatidylcholine (PC), a neutral phospholipid without a net charge and is most abundant on the outer surface of the plasma membrane [32]. Consistent with this conclusion, Draper, the Drosophila orthologue of CED-1, also directly associates with PS exposed to the surface of apoptotic cells [29]. Together, the in vivo and in vitro observations indicate that the direct interaction between the CED-1 family phagocytic receptors and PS is an important mechanism that brings together phagocytes and their target cells, regardless of whether these cells die of apoptosis or necrosis.
As an “eat me” signal, PS is also known to attract phagocytic receptors via an indirect mechanism. Secreted bridging molecules such as mouse MFG-E8 bring dying and engulfing cells together by interacting simultaneously with both PS and phagocytic receptors [66]. C. elegans TTR-52, a transthyretin-like secreted protein, has been implicated as a bridging molecule that links PS on apoptotic cells to CED-1 on engulfing cell surfaces [67]. Our finding, together with that reported by Wang et al (2010) [67], indicate that direct and indirect interactions between PS and CED-1 provide two distinct molecular mechanisms to support the recognition of dying cells by CED-1.
The phosphatidylserine receptor (PSR) protein family was originally identified as PS receptors that promote apoptotic cell-removal yet was later reported to function in other developmental processes and possess several biochemical activities that are in conflict with the proposed role as PS receptors [68–71]. Recently, it was reported that C. elegans PSR-1 displayed an in vitro PS-binding affinity [72]. In vivo, psr-1 mutants display weak defects in apoptotic - and necrotic-cell removal [72,73]. PSR-1 might thus contribute to the recognition of dying cells in addition to CED-1.
CED-7 represents a common PS-exposure mechanism utilized by both apoptotic and necrotic cells
In C. elegans, CED-7, a member of the ABC transporter family, was known to regulate PS exposure by apoptotic cells [43]. Mouse ABCA1 was also reported to participate in PS redistribution during apoptotic cell clearance [40,74]. Our discovery of CED-7 as a key factor in promoting the externalization of PS by necrotic touch neurons further demonstrates that the presentation of the “eat me” signal shares conserved mechanism(s) during different types of cell death. We further discovered that CED-7 has two distinct functions, one in necrotic and the other in engulfing cells. How CED-7 acts in necrotic cells to promote PS exposure remains to be elucidated. CED-7 is ubiquitously expressed. There thus must be dying cell-specific mechanisms that activate CED-7. Whether the CED-7 activation mechanisms are common or distinct in necrotic and apoptotic cells remains unknown. Moreover, the engulfing cell-specific function of CED-7 is a mystery and requires further investigation. Previous research suggests that engulfing cells might also externalize PS and that ABC transporters might be involved in this event [40,75]. The function of this event remains to be clarified.
A novel neuronal-specific PS-exposure mechanism represented by a Ca2+-dependent PS scramblase
Our observations indicate that ANOH-1, the C. elegans homolog of mammalian TMEM16F, functions in necrotic neurons to promote PS exposure. ANOH-1 is primarily expressed in neurons, including touch neurons [57] (Figs 5I and S5C). These lines of evidence indicate a cell type-specific function of ANOH-1 to facilitate necrotic-cell removal.
The vertebrates TMEM16 family of proteins, also known as anoctamins, are divided into two subfamilies based on two distinct Ca2+-dependent biochemical activities: Cl- channels and lipid scramblases [37,76]. In addition, TMEM16F possesses both biochemical activities [36,77]. Mammalian TMEM16F promotes cellular PS exposure in response to Ca2+ ionophore yet not to apoptotic stimuli [37]. The Ca2+-activated phospholipid scramblase activity of the TMEM16 subfamily provides an important clue towards revealing a necrosis-specific PS-exposure mechanism (Fig 7C). As an evolutionarily conserved feature, Ca2+ influx is known to be an effective trigger of the excitotoxic death of mammalian neurons [78]. For example, the activation of the NMDA receptor upon binding to excessive glutamate elicits an initial rise of cytoplasmic calcium that induces a subsequent calcium-dependent calcium release from the ER [12,79,80]. Elevation of cytoplasmic Ca2+ is also a critical trigger for excitotoxic necrosis of neurons in C. elegans, including that of touch neurons and other types of neurons [24,81]. Particularly in touch neurons, the dominant mutation in MEC-4, a subunit of a multimeric, mechanically gated DEG/ENaC channel, leads to an increased influx of Ca2+, resulting in necrosis [19,22] (Fig 7C). We propose that in touch neurons that undergo Ca2+-activated necrosis, Ca2+ further acts as an activating factor for the PS-exposure activity of ANOH-1 (Fig 7C). This Ca2+-dependent PS-exposure mechanism might apply to multiple kinds of necrotic neurons including but not limited to mechanosensory neurons. Moreover, the possibility remains that CED-7 or CED-8 might also be activated by Ca2+ in necrotic neurons (Fig 7C). As the disruption of Ca2+ homeostasis is closely associated with neuron degeneration conditions [82], the work reported here has a broader application in understanding the physiological role of the clearance of many kinds of degenerative neurons resulted from pathological conditions or aging.
Multiple activities contribute to PS exposure on the surface of necrotic cells
Our finding that the anoh-1 ced-7 double mutants display more severe defects in PS-exposure and necrotic cell-removal than each single mutant alone suggests that ANOH-1 and CED-7 together provide the necessary activities for efficient PS-exposure on necrotic touch neurons. One possible model is that they act in two independent and partially redundant pathways. The common function of CED-7 in both necrotic and apoptotic cells and the necrotic cell-specific function of ANOH-1 in facilitating PS-exposure have established that cells die of different mechanisms employ both common and unique molecular activities to present a common “eat me” signal. Given that a necrotic C. elegans neuron possesses a surface area many times of that of an apoptotic cell, the cooperation of multiple molecular activities such as those represented by CED-7 and ANOH-1, are likely essential for the efficient and timely exposure of PS on the cell surface at a level high enough to attract engulfing cells (Fig 7C). We further found that CED-8, a homolog of the mammalian phospholipid scramblase Xk8 [38,39], also made a modest contribution to the removal of necrotic cells. ced-8 and anoh-1 act in two independent pathways to promote PS exposure. Currently, it is unknown whether CED-8 facilitates PS-exposure to the surface of necrotic cells or whether CED-8 acts in necrotic cells; moreover, the functional relationship between ced-7 and ced-8 is unknown. CED-8 might represent a third pathway that is in parallel to both the CED-7 and the ANOH-1 pathways (Fig 7C).
Materials and Methods
Mutations, strains, and transgenic arrays
C. elegans was grown at 20°C as previously described [83] unless indicated otherwise. The N2 Bristol strain was used as the wild-type strain. Mutations are described in [84] and by the Wormbase (http://www.wormbase.org) unless noted otherwise: LGI, ced-1(e1735), ced-12(n3261); LGII, enIs46[Pmec-7 ced-7 and punc-119(+)]; LGIII, ced-7 (n1996), ced-6 (n2095), anoh-1(tm4762), unc-119(ed3); LGIV, ced-5(n1812), ced-10(n1993); LGV, unc-76(e911), deg-3(u662); LG X, ced-8(n1891), mec-4(e1611dm). The tm4762 allele was generated and provided by the National Bioresource Project of Japan and was outcrossed twice prior to analysis. The precise location of nucleotide deletion has been confirmed by allele-sequencing. Integrated transgenic arrays used are as follows: LGII, ttTi5605[mos] [85]; LGV, enIs33[Pdyn-1 mfg-e8::gfp and punc-76(+)] [43].
Extrachromosomal arrays were generated by microinjection [86] of plasmids with coinjection marker punc-76(+) [87] into strains carrying the unc-76(e911) mutant. Transgenic animals were isolated as non-Unc animals.
We obtained a single-copy insertion of Pmec-7 ced-7 in chromosome II in the ttTi5605 locus using the MosSCI insertion method [85], in strain EG4322 (ttTi5605; unc-119(ed3)) [86]. The transgenic array also carries the C. briggsae unc-119(+) genomic DNA that rescues the unc-119(ed3) phenotype. The single-copy insertion of the transgenic array into anticipated locus was confirmed by single-worm PCR analysis.
Plasmid construction
The Pmec-7 mrfp (pZL08) and Pmec-7 mCherry constructs were generated by replacing GFP in Pmec-7 gfp (pPD117.01, a gift from Andrew Fire) with mrfp [88] or mCherry [89]. Pcol-10 ssGFP is a secreted GFP reporter expressed by hypodermal cells under the control of Pcol-10, the promoter for col-10. It is generated by replacing the myo-3 promoter (Pmyo-3) in the Pmyo-3 ssGFP reporter [90] with Pcol-10, a gift from V. Ambros [28]. Pced-1 ced-7 was constructed by placing the 5.1kb CED-7 cDNA [54] behind the Pced-1 promoter [28]. Pmec-7 ced-7 and Pmec-7 ced-7::gfp were constructed by replacing Pced-1 from Pced-1 ced-7 with the Sph-1-ClaI fragment of Pmec-7 from pPD117.01, respectively. The Pmec-7 ced-7/unc-119(+) construct for single-copy MosSCI insertion was generated by cloning the Pmec-7 ced-7 fragment into the BssHII and SpeI sites of plasmid CFJ151 [85].
The anoh-1a cDNA was amplified from total RNA from mixed-stage C. elegans population through RT-PCR and cloned into pPD117.01 to generate Pmec-7 gfp::anoh-1a and Pmec-7 anoh-1a::gfp. The anoh-1b genomic-cDNA chimeric fragment was constructed by ligating exon 1 and intron 1 of anoh-1b genomic DNA with anoh-1a cDNA, and similarly cloned into pPD117.01 to generate Pmec-7 gfp::anoh-1b and Pmec-7 anoh-1b::gfp. Pced-1 anoh-1a::gfp and Pced-1 anoh-1b::gfp were constructed by replacing Pmec-7, respectively, with Pced-1 from pZZ829 [91]. The 617bp 5’ UTR of anoh-1b together with the first 297bp of exon 1 of the anoh-1b isoform was PCR-amplified from N2 worm extracts and cloned into pPD95.69 (a gift from Andy Fire), a promoter-less vector carrying a gfp cDNA tagged with a SV40 nuclear localization signal (NLS::gfp), between SphI and XmaI sites to generate Panoh-1b NLS::gfp, which allowed us to identify the cells in which anoh-1b was expressed.
RNA preparation for RT-PCR
Total RNA was isolated from mixed-stage C. elegans population using Trizol extraction with column purification (Qiagen, Inc.). cDNA was synthesized using the iScript cDNA Synthesis Kit (BIO-RAD, Inc.).
Assays detecting in vitro interaction between CED-1 and phospholipids
The cDNA encoding the extracellular region of CED-1 tagged with GST at its C-terminus (CED-1-GST) was expressed in insect Sf9 cells using Bac-to-Bac Baculovirus Expression System, a baculovirus-based vector system, (Life Technologies Japan, Tokyo, Japan), and the resulting protein was affinity-purified by glutathione-Sepharose chromatography (GE Healthcare Japan, Tokyo, Japan), essentially as described previously [92]. An ELISA-like solid-phase binding reaction was conducted virtually according to the published procedure [93]. In brief, varying amounts of CED-1-GST or GST, the latter serving as a negative control, were added to the wells of a 96-well culture container surface-coated with phospholipids and incubated for 1 h at room temperature. The wells were washed, successively incubated with anti-GST antibody (Millipore, Inc.) and anti-mouse IgG antibody conjugated with horseradish peroxidase, and then subjected to a colorimetric reaction with o-phenylenediamine followed by the measurement of OD490. An assay for surface plasmon resonance was done with Biacore 3000 (GE Healthcare Japan) using the HPA chip pre-bound by liposomes, as described previously [29]. Liposomes were prepared using PC alone (PC-only liposome) or a mixture of PC and PS at a molar ratio of 7 : 3 (PS-containing liposome) as described previously [94]. Phospholipids were purchased from Avanti Polar Lipids (Alabaster, USA).
DIC microscopy
According to the previous reports, among the six touch neurons, ALML and ALMR are born at ~450 min post-1st cleavage, PLML and PLMR are born at ~510 min post 1st-cleavage, whereas AVM and PVM are born at ~9 hrs after hatching, at the L1 larval stage [46,47]. To determine the efficiency of necrotic cell clearance during all four larval stages, we chose to score the presence of necrotic PLML and PLMR, two touch neurons in the tail. L1, L2, L3, and L4 larvae were staged as larvae collected within 1 hr, 15–16 hrs, 24–25 hrs, and 33–34 hrs after embryos hatching, respectively. The total number of necrotic touch neurons in the tail of 10 worms was scored, and the mean of three repeats was calculated. The number of apoptotic cells were scored in embryos of different stages, in the head of young L1 larvae hatched within 1 hr, and in the gonad of adult hermaphrodites 48 hrs after the mid-L4 stage as described [48].
Fluorescence microscopy
Olympus IX70-Applied Precision DeltaVision microscope equipped with Photometris Coolsnap digital camera and Applied Precision Softworx 5.0 software was used to acquire serial Z-stacks of fluorescence images at 0.5 μm intervals and to deconvolve these images of embryos and larvae [48]. To quantify the MFG-E8::GFP signal intensity on the surface of necrotic cells (In), following deconvolution of z-stack images using the Applied Precision Softworx software, the necrotic cell surface was outlined by two closed polygons and the signal intensity in the area of the bigger polygon was subtracted with that of the smaller polygon. To normalize the signal intensity, the same two polygons were placed in the area neighboring the necrotic cell and the background fluorescence intensity (Ib) was measured using the formula similar to that applied to necrotic cell surface. The relative signal intensity (Ir) of MFG-E8::GFP enriched on the necrotic cell surface is represented as In/Ib. For each data point, at least 40 necrotic cells were quantified.
To monitor the dynamics of PS presentation during the necrosis of touch neurons in embryos via time-lapse recording, embryos were mounted on an agar pad on a glass slide in M9 solution [48]. The starting point of recording was at 460 min-post the 1st cleavage (the 1st embryonic cell division), when an embryo reached the 2-fold stage. Recording was performed in 5-min interval until the embryo hatched. At each time point, a Z-stack of images composed of 40 serial Z sections at 0.5 μm/section were captured. Since embryos continue to move inside the eggshell, PLML and PLMR were followed by monitoring both the touch neuron reporter Pmec-7 GFP and the distinct swelling morphology of necrotic cells.
For propidium iodide staining, mixed-stage worms were washed off plate using Hanks’ balanced salt solution buffer (HBSS buffer; with calcium and magnesium, Fisher Scientific) containing 10 μM propidium iodide and incubated for 2 hrs [95]. Worms were subsequently washed three times using HBSS buffer and mounted on an agar pad on a glass slide in 30 mM sodium azide for microscopic observation. Olympus IX70-Applied Precision DeltaVision microscope was used to acquire serial Z-stacks at 0.5 μm interval. Excitation and emission wavelengths used are ~540 and ~590 nm, respectively.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Golstein P, Kroemer G (2007) Cell death by necrosis: towards a molecular definition. Trends Biochem Sci 32 : 37–43. 17141506
2. Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, et al. (2009) Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ 16 : 3–11. doi: 10.1038/cdd.2008.150 18846107
3. Jacobson MD, Bergeron L (2002) Cell death in the nervous system. In: Jacobson MD, McCarthy N, editors. Apoptosis, the molecular biology of programmed cell death: Oxford University Press. pp. 278–301.
4. Yamashima T (2004) Ca2+-dependent proteases in ischemic neuronal death: a conserved 'calpain-cathepsin cascade' from nematodes to primates. Cell Calcium 36 : 285–293. 15261484
5. Challa S, Chan FK (2010) Going up in flames: necrotic cell injury and inflammatory diseases. Cell Mol Life Sci 67 : 3241–3253. doi: 10.1007/s00018-010-0413-8 20532807
6. Whelan RS, Kaplinskiy V, Kitsis RN (2010) Cell death in the pathogenesis of heart disease: mechanisms and significance. Annu Rev Physiol 72 : 19–44. doi: 10.1146/annurev.physiol.010908.163111 20148665
7. Noch E, Khalili K (2009) Molecular mechanisms of necrosis in glioblastoma: the role of glutamate excitotoxicity. Cancer Biol Ther 8 : 1791–1797. 19770591
8. Vlachos M, Tavernarakis N (2010) Non-apoptotic cell death in Caenorhabditis elegans. Dev Dyn 239 : 1337–1351. doi: 10.1002/dvdy.22230 20108319
9. McCall K (2010) Genetic control of necrosis—another type of programmed cell death. Curr Opin Cell Biol 22 : 882–888. doi: 10.1016/j.ceb.2010.09.002 20889324
10. Zhou W, Yuan J (2014) Necroptosis in health and diseases. Semin Cell Dev Biol.
11. Moquin D, Chan FK (2010) The molecular regulation of programmed necrotic cell injury. Trends Biochem Sci 35 : 434–441. doi: 10.1016/j.tibs.2010.03.001 20346680
12. Mody I, MacDonald JF (1995) NMDA receptor-dependent excitotoxicity: the role of intracellular Ca2+ release. Trends Pharmacol Sci 16 : 356–359. 7491714
13. Driscoll M, Gerstbrein B (2003) Dying for a cause: invertebrate genetics takes on human neurodegeneration. Nat Rev Genet 4 : 181–194. 12610523
14. Galluzzi L, Kepp O, Krautwald S, Kroemer G, Linkermann A (2014) Molecular mechanisms of regulated necrosis. Semin Cell Dev Biol 35 : 24–32. doi: 10.1016/j.semcdb.2014.02.006 24582829
15. Krysko DV, D'Herde K, Vandenabeele P (2006) Clearance of apoptotic and necrotic cells and its immunological consequences. Apoptosis 11 : 1709–1726. 16951923
16. Hall DH, Gu G, Garcia-Anoveros J, Gong L, Chalfie M, Driscoll M (1997) Neuropathology of degenerative cell death in Caenorhabditis elegans. J Neurosci 17 : 1033–1045. 8994058
17. Poon IK, Hulett MD, Parish CR (2010) Molecular mechanisms of late apoptotic/necrotic cell clearance. Cell Death Differ 17 : 381–397. doi: 10.1038/cdd.2009.195 20019744
18. Metzstein MM, Stanfield GM, Horvitz HR (1998) Genetics of programmed cell death in C. elegans: past, present and future. Trends Genet 14 : 410–416. 9820030
19. Driscoll M, Chalfie M (1991) The mec-4 gene is a member of a family of Caenorhabditis elegans genes that can mutate to induce neuronal degeneration. Nature 349 : 588–593. 1672038
20. Treinin M, Chalfie M (1995) A mutated acetylcholine receptor subunit causes neuronal degeneration in C. elegans. Neuron 14 : 871–877. 7718248
21. Chalfie M, Sulston J (1981) Developmental genetics of the mechanosensory neurons of Caenorhabditis elegans. Dev Biol 82 : 358–370. 7227647
22. Bianchi L, Gerstbrein B, Frokjaer-Jensen C, Royal DC, Mukherjee G, Royal MA, et al. (2004) The neurotoxic MEC-4(d) DEG/ENaC sodium channel conducts calcium: implications for necrosis initiation. Nat Neurosci 7 : 1337–1344. 15543143
23. Ellis HM, Horvitz HR (1986) Genetic control of programmed cell death in the nematode C. elegans. Cell 44 : 817–829. 3955651
24. Xu K, Tavernarakis N, Driscoll M (2001) Necrotic cell death in C. elegans requires the function of calreticulin and regulators of Ca(2+) release from the endoplasmic reticulum. Neuron 31 : 957–971. 11580896
25. Chung S, Gumienny TL, Hengartner MO, Driscoll M (2000) A common set of engulfment genes mediates removal of both apoptotic and necrotic cell corpses in C. elegans. Nat Cell Biol 2 : 931–937. 11146658
26. Krysko DV, Denecker G, Festjens N, Gabriels S, Parthoens E, D'Herde K, et al. (2006) Macrophages use different internalization mechanisms to clear apoptotic and necrotic cells. Cell Death Differ 13 : 2011–2022. 16628234
27. Ravichandran KS (2010) Find-me and eat-me signals in apoptotic cell clearance: progress and conundrums. J Exp Med 207 : 1807–1817. doi: 10.1084/jem.20101157 20805564
28. Zhou Z, Hartwieg E, Horvitz HR (2001b) CED-1 is a transmembrane receptor that mediates cell corpse engulfment in C. elegans. Cell 104 : 43–56.
29. Tung TT, Nagaosa K, Fujita Y, Kita A, Mori H, Okada R, et al. (2013) Phosphatidylserine recognition and induction of apoptotic cell clearance by Drosophila engulfment receptor Draper. J Biochem 153 : 483–491. doi: 10.1093/jb/mvt014 23420848
30. Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S (2007) Identification of Tim4 as a phosphatidylserine receptor. Nature 450 : 435–439. 17960135
31. Park D, Tosello-Trampont AC, Elliott MR, Lu M, Haney LB, Ma Z, et al. (2007) BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature 450 : 430–434. 17960134
32. Balasubramanian K, Schroit AJ (2003) Aminophospholipid asymmetry: A matter of life and death. Annu Rev Physiol 65 : 701–734. 12471163
33. Vance JE, Steenbergen R (2005) Metabolism and functions of phosphatidylserine. Prog Lipid Res 44 : 207–234. 15979148
34. Segawa K, Kurata S, Yanagihashi Y, Brummelkamp TR, Matsuda F, Nagata S (2014) Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science 344 : 1164–1168. doi: 10.1126/science.1252809 24904167
35. Sims PJ, Wiedmer T (2001) Unraveling the mysteries of phospholipid scrambling. Thromb Haemost 86 : 266–275. 11487015
36. Suzuki J, Umeda M, Sims PJ, Nagata S (2010) Calcium-dependent phospholipid scrambling by TMEM16F. Nature 468 : 834–838. doi: 10.1038/nature09583 21107324
37. Suzuki J, Fujii T, Imao T, Ishihara K, Kuba H, Nagata S (2013) Calcium-dependent phospholipid scramblase activity of TMEM16 protein family members. J Biol Chem 288 : 13305–13316. doi: 10.1074/jbc.M113.457937 23532839
38. Suzuki J, Denning DP, Imanishi E, Horvitz HR, Nagata S (2013) Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science 341 : 403–406. doi: 10.1126/science.1236758 23845944
39. Chen YZ, Mapes J, Lee ES, Skeen-Gaar RR, Xue D (2013) Caspase-mediated activation of Caenorhabditis elegans CED-8 promotes apoptosis and phosphatidylserine externalization. Nat Commun 4 : 2726. doi: 10.1038/ncomms3726 24225442
40. Hamon Y, Broccardo C, Chambenoit O, Luciani MF, Toti F, Chaslin S, et al. (2000) ABC1 promotes engulfment of apoptotic cells and transbilayer redistribution of phosphatidylserine. Nat Cell Biol 2 : 399–406. 10878804
41. Alder-Baerens N, Muller P, Pohl A, Korte T, Hamon Y, Chimini G, et al. (2005) Headgroup-specific exposure of phospholipids in ABCA1-expressing cells. J Biol Chem 280 : 26321–26329. 15905177
42. Williamson P, Halleck MS, Malowitz J, Ng S, Fan X, Krahling S, et al. (2007) Transbilayer phospholipid movements in ABCA1-deficient cells. PLoS One 2: e729. 17710129
43. Venegas V, Zhou Z (2007) Two alternative mechanisms that regulate the presentation of apoptotic cell engulfment signal in Caenorhabditis elegans. Mol Biol Cell 18 : 3180–3192. 17567952
44. Zong WX, Thompson CB (2006) Necrotic death as a cell fate. Genes Dev 20 : 1–15. 16391229
45. Hedgecock EM, Sulston JE, Thomson JN (1983) Mutations affecting programmed cell deaths in the nematode Caenorhabditis elegans. Science 220 : 1277–1279. 6857247
46. Sulston JE, Horvitz HR (1977) Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev Biol 56 : 110–156. 838129
47. Sulston JE, Schierenberg E, White JG, Thomson JN (1983) The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev Biol 100 : 64–119. 6684600
48. Li Z, Lu N, He X, Zhou Z (2013) Monitoring the clearance of apoptotic and necrotic cells in the nematode Caenorhabditis elegans. Methods Mol Biol 1004 : 183–202. doi: 10.1007/978-1-62703-383-1_14 23733578
49. Hamelin M, Scott IM, Way JC, Culotti JG (1992) The mec-7 beta-tubulin gene of Caenorhabditis elegans is expressed primarily in the touch receptor neurons. EMBO J 11 : 2885–2893. 1639062
50. Grant B, Greenwald I (1997) Structure, function, and expression of SEL-1, a negative regulator of LIN-12 and GLP-1 in C. elegans. Development 124 : 637–644. 9043078
51. Okkema PG, Harrison SW, Plunger V, Aryana A, Fire A (1993) Sequence requirements for myosin gene expression and regulation in Caenorhabditis elegans. Genetics 135 : 385–404. 8244003
52. Hong K, Driscoll M (1994) A transmembrane domain of the putative channel subunit MEC-4 influences mechanotransduction and neurodegeneration in C. elegans. Nature 367 : 470–473. 8107806
53. Treinin M, Gillo B, Liebman L, Chalfie M (1998) Two functionally dependent acetylcholine subunits are encoded in a single Caenorhabditis elegans operon. Proc Natl Acad Sci U S A 95 : 15492–15495. 9860996
54. Wu Y, Horvitz HR (1998a) The C. elegans cell corpse engulfment gene ced-7 encodes a protein similar to ABC transporters. Cell 93 : 951–960. 9635425
55. Shen Q, He B, Lu N, Conradt B, Grant BD, Zhou Z (2013) Phagocytic receptor signaling regulates clathrin and epsin-mediated cytoskeletal remodeling during apoptotic cell engulfment in C. elegans. Development 140 : 3230–3243. doi: 10.1242/dev.093732 23861060
56. Choi J, Richards KL, Cinar HN, Newman AP (2006) N-ethylmaleimide sensitive factor is required for fusion of the C. elegans uterine anchor cell. Dev Biol 297 : 87–102. 16769048
57. Wang Y, Alam T, Hill-Harfe K, Lopez AJ, Leung CK, Iribarne D, et al. (2013) Phylogenetic, expression, and functional analyses of anoctamin homologs in Caenorhabditis elegans. Am J Physiol Regul Integr Comp Physiol 305: R1376–1389. doi: 10.1152/ajpregu.00303.2012 24049119
58. Stanfield GM, Horvitz HR (2000) The ced-8 gene controls the timing of programmed cell deaths in C. elegans. Mol Cell 5 : 423–433. 10882128
59. Denecker G, Vercammen D, Steemans M, Vanden Berghe T, Brouckaert G, Van Loo G, et al. (2001) Death receptor-induced apoptotic and necrotic cell death: differential role of caspases and mitochondria. Cell Death Differ 8 : 829–840. 11526436
60. Krysko DV, Brouckaert G, Kalai M, Vandenabeele P, D'Herde K (2003) Mechanisms of internalization of apoptotic and necrotic L929 cells by a macrophage cell line studied by electron microscopy. J Morphol 258 : 336–345. 14584035
61. Elliott MR, Ravichandran KS (2010) Clearance of apoptotic cells: implications in health and disease. J Cell Biol 189 : 1059–1070. doi: 10.1083/jcb.201004096 20584912
62. Hajos F, Garthwaite G, Garthwaite J (1986) Reversible and irreversible neuronal damage caused by excitatory amino acid analogues in rat cerebellar slices. Neuroscience 18 : 417–436. 3526173
63. van den Eijnde SM, Boshart L, Baehrecke EH, De Zeeuw CI, Reutelingsperger CPM, Vermeij-Keers C (1998) Cell surface exposure of phosphatidylserine during apoptosis is phylogenetically conserved. Apoptosis 3 : 9–16. 14646513
64. Wang X, Wang J, Gengyo-Ando K, Gu L, Sun CL, Yang C, et al. (2007) C. elegans mitochondrial factor WAH-1 promotes phosphatidylserine externalization in apoptotic cells through phospholipid scramblase SCRM-1. Nat Cell Biol 9 : 541–549. 17401362
65. Zullig S, Neukomm LJ, Jovanovic M, Charette SJ, Lyssenko NN, Halleck MS, et al. (2007) Aminophospholipid translocase TAT-1 promotes phosphatidylserine exposure during C. elegans apoptosis. Curr Biol 17 : 994–999. 17540571
66. Hanayama R, Tanaka M, Miwa K, Shinohara A, Iwamatsu A, Nagata S (2002) Identification of a factor that links apoptotic cells to phagocytes. Nature 417 : 182–187. 12000961
67. Wang X, Li W, Zhao D, Liu B, Shi Y, Chen B, et al. (2010) Caenorhabditis elegans transthyretin-like protein TTR-52 mediates recognition of apoptotic cells by the CED-1 phagocyte receptor. Nat Cell Biol 12 : 655–664. doi: 10.1038/ncb2068 20526330
68. Schlegel RA, Williamson P (2007) P.S. to PS (phosphatidylserine)—pertinent proteins in apoptotic cell clearance. Sci STKE 2007: pe57. 17940275
69. Chen B, Liu Q, Ge Q, Xie J, Wang ZW (2007) UNC-1 regulates gap junctions important to locomotion in C. elegans. Curr Biol 17 : 1334–1339. 17658257
70. Hong X, Zang J, White J, Wang C, Pan CH, Zhao R, et al. (2010) Interaction of JMJD6 with single-stranded RNA. Proc Natl Acad Sci U S A 107 : 14568–14572. doi: 10.1073/pnas.1008832107 20679243
71. Webby CJ, Wolf A, Gromak N, Dreger M, Kramer H, Kessler B, et al. (2009) Jmjd6 catalyses lysyl-hydroxylation of U2AF65, a protein associated with RNA splicing. Science 325 : 90–93. doi: 10.1126/science.1175865 19574390
72. Yang H, Chen YZ, Zhang Y, Wang X, Zhao X, Godfroy JI 3rd, et al. (2015) A lysine-rich motif in the phosphatidylserine receptor PSR-1 mediates recognition and removal of apoptotic cells. Nat Commun 6 : 5717. doi: 10.1038/ncomms6717 25564762
73. Wang X, Wu YC, Fadok VA, Lee MC, Gengyo-Ando K, Cheng LC, et al. (2003) Cell corpse engulfment mediated by C. elegans phosphatidylserine receptor through CED-5 and CED-12. Science 302 : 1563–1566. 14645848
74. Rigot V, Hamon Y, Chambenoit O, Alibert M, Duverger N, Chimini G (2002) Distinct sites on ABCA1 control distinct steps required for cellular release of phospholipids. J Lipid Res 43 : 2077–2086. 12454269
75. Mapes J, Chen YZ, Kim A, Mitani S, Kang BH, Xue D (2012) CED-1, CED-7, and TTR-52 regulate surface phosphatidylserine expression on apoptotic and phagocytic cells. Curr Biol 22 : 1267–1275. doi: 10.1016/j.cub.2012.05.052 22727702
76. Pedemonte N, Galietta LJ (2014) Structure and function of TMEM16 proteins (anoctamins). Physiol Rev 94 : 419–459. doi: 10.1152/physrev.00039.2011 24692353
77. Yang H, Kim A, David T, Palmer D, Jin T, Tien J, et al. (2012) TMEM16F forms a Ca2+-activated cation channel required for lipid scrambling in platelets during blood coagulation. Cell 151 : 111–122. doi: 10.1016/j.cell.2012.07.036 23021219
78. Wojda U, Salinska E, Kuznicki J (2008) Calcium ions in neuronal degeneration. IUBMB Life 60 : 575–590. doi: 10.1002/iub.91 18478527
79. Sattler R, Tymianski M (2001) Molecular mechanisms of glutamate receptor-mediated excitotoxic neuronal cell death. Mol Neurobiol 24 : 107–129. 11831548
80. Mattson MP, LaFerla FM, Chan SL, Leissring MA, Shepel PN, Geiger JD (2000) Calcium signaling in the ER: its role in neuronal plasticity and neurodegenerative disorders. Trends Neurosci 23 : 222–229. 10782128
81. Mano I, Driscoll M (2009) Caenorhabditis elegans glutamate transporter deletion induces AMPA-receptor/adenylyl cyclase 9-dependent excitotoxicity. J Neurochem 108 : 1373–1384. doi: 10.1111/j.1471-4159.2008.05804.x 19054279
82. Mattson MP (2007) Calcium and neurodegeneration. Aging Cell 6 : 337–350. 17328689
83. Brenner S (1974) The genetics of Caenorhabditis elegans. Genetics 77 : 71–94. 4366476
84. Riddle DL, Blumenthal T, Meyer BJ, Priess JR, editors (1997) C. elegans II. Plainview, NY: Cold Spring harbor Laboratory Press.
85. Frokjaer-Jensen C, Davis MW, Hopkins CE, Newman BJ, Thummel JM, Olesen SP, et al. (2008) Single-copy insertion of transgenes in Caenorhabditis elegans. Nat Genet 40 : 1375–1383. doi: 10.1038/ng.248 18953339
86. Jin Y (1999) Transformation. In: Hope IA, editor. C elegans, a practical approach. Oxford: Oxford University Press. pp. 69–96.
87. Bloom L, Horvitz HR (1997) The Caenorhabditis elegans gene unc-76 and its human homologs define a new gene family involved in axonal outgrowth and fasciculation. Proc Natl Acad Sci U S A 94 : 3414–3419. 9096408
88. Campbell RE, Tour O, Palmer AE, Steinbach PA, Baird GS, Zacharias DA, et al. (2002) A monomeric red fluorescent protein. Proc Natl Acad Sci U S A 99 : 7877–7882. 12060735
89. Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY (2004) Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol 22 : 1567–1572. 15558047
90. Fares H, Greenwald I (2001) Genetic analysis of endocytosis in Caenorhabditis elegans: coelomocyte uptake defective mutants. Genetics 159 : 133–145. 11560892
91. Yu X, Odera S, Chuang CH, Lu N, Zhou Z (2006) C. elegans Dynamin mediates the signaling of phagocytic receptor CED-1 for the engulfment and degradation of apoptotic cells. Dev Cell 10 : 743–757. 16740477
92. Kuraishi T, Nakagawa Y, Nagaosa K, Hashimoto Y, Ishimoto T, Moki T, et al. (2009) Pretaporter, a Drosophila protein serving as a ligand for Draper in the phagocytosis of apoptotic cells. EMBO J 28 : 3868–3878. doi: 10.1038/emboj.2009.343 19927123
93. Kawasaki Y, Nakagawa A, Nagaosa K, Shiratsuchi A, Nakanishi Y (2002) Phosphatidylserine binding of class B scavenger receptor type I, a phagocytosis receptor of testicular sertoli cells. J Biol Chem 277 : 27559–27566. 12016218
94. Shiratsuchi A, Umeda M, Ohba Y, Nakanishi Y (1997) Recognition of phosphatidylserine on the surface of apoptotic spermatogenic cells and subsequent phagocytosis by Sertoli cells of the rat. J Biol Chem 272 : 2354–2358. 8999945
95. Jain PT, Chang SH, Gutry PP, Berezesky IK, Trump BF (1993) The relationship between [Ca2+]i and cell death using an in vivo model: a study using the ced-1 mutant strain of C. elegans. Toxicol Pathol 21 : 572–583. 8052804
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 6
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Non-reciprocal Interspecies Hybridization Barriers in the Capsella Genus Are Established in the Endosperm
- Translational Upregulation of an Individual p21 Transcript Variant by GCN2 Regulates Cell Proliferation and Survival under Nutrient Stress
- Exome Sequencing of Phenotypic Extremes Identifies and as Interacting Modifiers of Chronic Infection in Cystic Fibrosis
- The Human Blood Metabolome-Transcriptome Interface
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy