
Canine Spontaneous Head and Neck Squamous Cell Carcinomas Represent Their Human Counterparts at the Molecular Level
Head and neck squamous cell carcinoma (HNSCC) represents the sixth leading cancer by incidence in humans; thus, developing effective therapeutic interventions is important. Although great advance has been made in our understanding of the biology of HNSCC over the past several decades, translating the research findings into clinical success has been frustratingly slow, and anticancer drug development remains a lengthy and expensive process. A significant challenge is that drug effects in current preclinical cancer models often do not predict clinical results, and there lacks translational models that can bridge the gap between preclinical research and human clinical trials. Here we report a pilot study that represents the first genome-wide characterization of spontaneously occurring HNSCCs in pet dogs. The study reveals a strong dog-human molecular homology at various levels, indicating the likelihood that spontaneous canine HNSCC molecularly represents its human counterpart. If conclusions of this pilot study are validated with a large sample size and more efforts are put into building better resource and infrastructure for canine cancer research, spontaneous canine HNSCCs could effectively serve as a much-needed translational model that bridges the gap between preclinical research and human trials.
Published in the journal:
. PLoS Genet 11(6): e32767. doi:10.1371/journal.pgen.1005277
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005277
Summary
Head and neck squamous cell carcinoma (HNSCC) represents the sixth leading cancer by incidence in humans; thus, developing effective therapeutic interventions is important. Although great advance has been made in our understanding of the biology of HNSCC over the past several decades, translating the research findings into clinical success has been frustratingly slow, and anticancer drug development remains a lengthy and expensive process. A significant challenge is that drug effects in current preclinical cancer models often do not predict clinical results, and there lacks translational models that can bridge the gap between preclinical research and human clinical trials. Here we report a pilot study that represents the first genome-wide characterization of spontaneously occurring HNSCCs in pet dogs. The study reveals a strong dog-human molecular homology at various levels, indicating the likelihood that spontaneous canine HNSCC molecularly represents its human counterpart. If conclusions of this pilot study are validated with a large sample size and more efforts are put into building better resource and infrastructure for canine cancer research, spontaneous canine HNSCCs could effectively serve as a much-needed translational model that bridges the gap between preclinical research and human trials.
Introduction
During the past several decades, great advances have been made in our understanding of the biology of head and neck squamous cell carcinoma (HNSCC) [1–8]. For example, owing to the rapid development in next-generation sequencing and other high throughput technologies, the cancer genome atlas (TCGA) [1] and others [2–4] have characterized hundreds of human HNSCC cases and discovered significantly altered genes and pathways (cell cycle, PI3K signaling, etc.). However, translating these research findings into clinical success has been frustratingly slow, and drug development remains a lengthy and expensive process [9], with costs currently estimated at over US$1 billion to bring a new drug to market [9]. With biomarker-based clinical trials not as advanced and half as many clinical trials currently available as lung or breast cancer [5], the issue is even more serious for HNSCC. One significant challenge is the lack of effective predictive models [9].
Current widely-used HNSCC models [10–12] include: 1) cell line and xenograft models; 2) genetically engineered mouse models; and 3) carcinogen-induced models. While these models have made spectacular contributions in our molecular understanding of HNSCC and are clearly indispensable, they have issues and generally do not fully represent the great complexity and heterogeneity of human HNSCC. First, cell lines and subcutaneous xenograft models lack the specific interactions between tumor cells and their native microenvironment that significantly influences carcinogenesis. Orthotopic xenograft models allow tumor development in places that are closer to the natural anatomic site [13], but the tumors still do not arise from the native head and neck epithelium, a significant issue considering the importance of cells of origin in cancer [14]. Second, genetically engineered mouse models often manipulate only one or a few major driver genes (e.g., TP53, CCND1, KRAS, AKT, etc.) [12], and normally fail to recapitulate the full alteration spectrum of human HNNCC that typically involves hundreds of genes per tumor [1–4]. Third, carcinogen-induced models such as the 7,12-dimethylbenz[a]anthracene (DMBA)–induced tumors in Syrian hamster cheek pouch do not grossly or histologically resemble human oral carcinomas [12]. Chronic administration of 4-nitroquinoline 1-oxide (4NQO), which can cause DNA adduct formation and Hras mutation, induces oral tumors in rodents [12]. However, these models do not represent the invasive pathology of human HNSCC [12]. In summary, the current widely-used HNSCC models usually fail to accurately recapitulate the full spectra of the biology, histopathology, complexity, and heterogeneity of HNSCC in humans. This, combined with the body size difference between the human and the rodents, make these models often incapable of accurately predicting the drug efficacy and toxicity in human patients [9]. Thus, HNSCC models that can bridge these models and human clinical trials are critically missing and urgently needed at present.
Spontaneously occurring HNSCCs in pet dogs may bridge this gap as they overcome many of the issues of the current HNSCC models discussed above [15–30]. First, unlike genetically-engineered or xenograft rodent models, these cancers are naturally occurring and heterogeneous, capturing the essence of human cancers. Second, as companion animals, dogs share the human environment and are exposed to many of the same carcinogens in a similar fashion, unlike carcinogen-induced cancers. Third, dogs better resemble humans in biology, e.g., with similar telomere and telomerase activities [31] and frequent occurrence of spontaneous epithelial cancers [15], than mice [32]. This further allows a closer resemblance in biology of HNSCC. Notably, the availability of a genome assembly nearly as accurate as the mouse or rat genome [33,34], in contrast to that available for another companion animal, the cat, make many analyses possible with canine cancers but not with feline cancers.
HNSCCs are relatively frequent in the dog and like their human counterparts, the oral cavity is a common site, and oral SCC is the 2nd most common oral malignant tumor in dogs [15,35–37]. The prevalence rate for nontonsillar oral SCC is estimated at 6.4–7.3 per 100,000 [15], compared to the average incidence rate of human head and neck cancers at 8.8 and 5.1 per 100,000 men and women respectively [38]. For canine tonsillar SCC, the prevalence rate varies dramatically, ranging from 91–120 per 100,000 in large cities such as London and Philadelphia to nearly none in rural areas [15]. Critically, numerous anatomic and clinical similarities of HNSCC are noted between the human and the dog [15,35]. For example, as in humans, canine HNSCCs are invasive with metastasis occurring late in disease and tumor cells spreading to the regional lymph nodes and occasionally to the lung, and local disease recurrence is common in many patients. An interesting exception to these resemblances is that canine tonsillar SCCs are more aggressive and often metastatic (73%) compared to those in other locations, which however is observed in humans. Although far from being as extensively characterized as in human HNSCCs, papillomavirus DNA is also detected in canine oral SCCs [39]. Lastly, similar treatment schemes are practiced in the two species, which include surgery, radiation therapy, and chemotherapy with agents such as piroxicam and carboplatin [40,41].
Because of the features described above, along with a large population of pet dogs (~70 million estimated in the US alone), canine HNSCC could potentially be a useful and practical model that significantly speeds up bench to bedside translation. However, a major obstacle for this realization is that canine HNSCC is markedly understudied. Our current literature search indicates that not a single canine HNSCC genome or transcriptome has ever been investigated by sequencing, microarray, or other strategies. This drastically differs from other canine cancers such as lymphoma [42,43], leukemia, and osteosarcoma [26], of which hundreds of cancer genomes have been characterized via array comparative genomic hybridization (aCGH) or molecular cytogenetic analyses. Thus, unlike the better-studied canine cancers, there are simply insufficient data to evaluate the extent of molecular similarity between canine HNSCC and its human counterpart, a key factor in determining the usefulness of canine HNSCC in basic and translational research. To address this obstacle, we set out to conduct a pilot study that represents the first genome-wide characterization of spontaneous canine HNSCC.
Results
Twelve spontaneous canine HNSCC, including nine oral SCC, cases were investigated
We characterized 12 spontaneous canine HNSCC cases by genome-wide analyses including high density aCGH and RNA-seq. These cancers come from different dog breeds, including four Labrador retrievers, three mixed breeds, and others listed in S1A Table. Among the 12 HNSCC cases, nine are oral (oSCCs) and all nontonsillar, with one from the buccal mucosa, three from the tongue, and five from the gingiva. The remaining three cases are from the nasal planum, the nostril, and the eye (the ocular adnexa). The tumors are all invasive (Fig 1A and 1B), and most are well-differentiated although some are more disorganized than others (S1A Table).
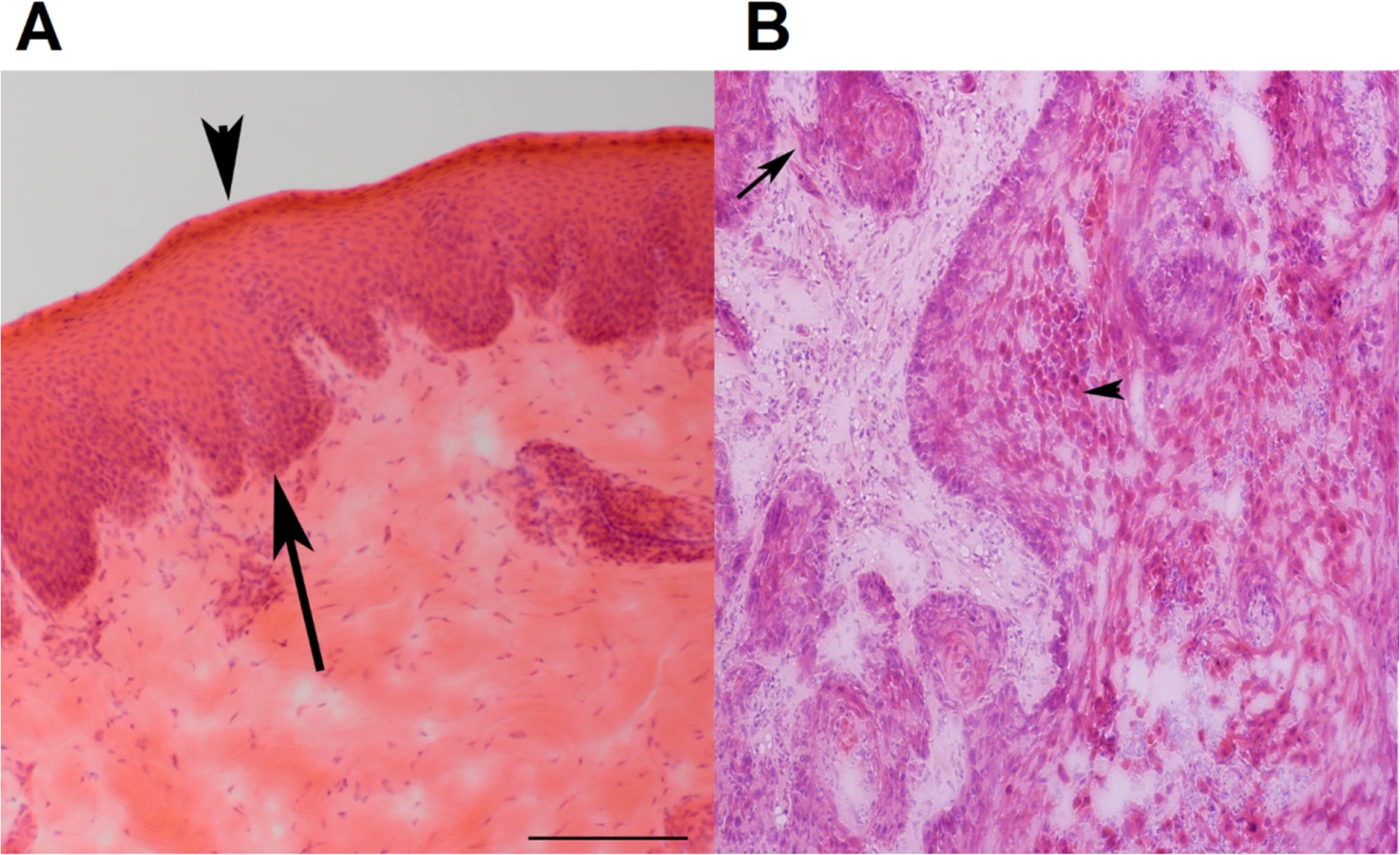
Furthermore, two cases appear to be papillomavirus-positive, based on the analysis described in later sections.
aCGH analysis reveals dog-human homology in genomic copy number abnormality
Analogous to human HNSCCs [1,2,6], aCGH analysis reveals variations in the extent of genomic copy number abnormality (CNA) among these canine tumors. Specifically, while seven canine HNSCCs harbor extensive CNAs, the remaining five tumors have hardly any CNAs in their genomes (Fig 2A and S1B Table). In some cases, the variation in the CNA prevalence is clearly related to the cancer progression stage (see tumors 419 and 419_2 in Fig 2A). In other cases however, additional factors may also have contributed. For example, tumor 1152 lacks CNAs but is at a tumor-progression stage similar to those with extensive CNAs (Fig 2A). Interestingly, among the nine oSCCs, those with a buccal mucosa or tongue location harbor significantly more CNAs than those located in the gingiva (Fig 2A and S1A and S1B Table).

As in human HNSCCs [1,2,6], both focal and broad CNAs were detected in the canine tumors (Figs 2A and S1). For example, tumor 240 harbors a focal amplification of ~10Mb located on chromosome 16, increasing the copy number of nearly all 63 genes inside by 2-fold and more (Fig 2B and S1C Table). Of these genes, 40 are also overexpressed (see later sections), among which “negative regulation of apoptosis” and “endopeptidase activity” are the most enriched functional groups. Indeed, anti-apoptosis associated genes IKBKB, POLB, SFRP1 and FNTA, as well as endopeptidase genes ADAM9 and PLAT encoded in the region are both amplified and overexpressed (S1C and S1D Table) (these genes are also recurrently amplified in human cancers including HNSCC according to cBioPortal [44] at www.cbioportal.org). Notably, 42 of the 63 genes are also significantly amplified in human HNSCC based on the TCGA study [1] (which will be examined further in a later section). These observations support that this focal amplification may have contributed to the pathogenesis of tumor 240. Meanwhile, broad events such as recurrent gain of canine chromosome 13, observed in seven tumors out of 12 total (Fig 2A), were seen as well (although other canine cancer types also harbor chromosome 13 gain [21,43]). The first 40Mb of canine chromosome 13 is syntenic to the last 48Mb of human chromosome 8q, which encodes genes including MYC and is one of the most recurrently amplified sites in human HNSCC [1,2] and other cancer types [45,46]. Consistent with published studies [21,43], these observations support common cancer drivers between the human and the dog.
In most canine HNSCCs, more amplifications than deletions were found, causing more genomic sequences and genes to be amplified than deleted (Fig 2A and S1B Table). Amplifications are, on average, also larger than deletions (S2 Fig and S1B Table). Importantly, a better correlation between the copy number status and the expression level (see later sections) was observed for amplified genes than deleted genes (Fig 2C), supporting that amplified regions harbor more cancer drivers than deleted regions. This is also consistent with human findings [2].
Many known human HNSCC genes [1,2,47–49] are also amplified/deleted in the canine tumors. Examples include recurrent amplification of known oncogene MYC; protein kinase genes MET, PTK2 (also known as FAK1) and FGFR1; apoptosis-related FADD; histone lysine methyltransferase gene WHSC1L1; NDRG1 (N-myc downstream regulated 1) which is frequently altered in human cancers including HNSCC but whose roles remain controversial [50]; cell cycle genes CCNE1, CDK6 and E2F3; and others listed in S1E Table. Notably, the most enriched functions among the amplified genes are protein kinase activity, with 15 serine/threonine kinases and 13 tyrosine protein kinases, and protease activity, with 10 serine proteases (S1E and S1G Table). This is consistent with TCGA’s human HNSCC study [1]. Examples of deleted genes in the canine tumors (Fig 2A) include cell cycle gene CDKN2A, one of the best known gene deletions in human HNSCC [1,2,6]; and protein phosphatase PTPRD and cadherin superfamily membrane FAT1, tumor suppressors [51,52] that are also frequently deleted in human HNSCC [1]. Furthermore, the most significantly enriched functions among the deleted genes are closely related to epithelial cell polarity including adhesion (18 genes), small GTPase regulator activity (19 genes), and cell junctions (11 genes) (S1F and S1G Table). This agrees with human HNSCC findings [1] and is consistent with the concept that loss of cell polarity is a hallmark of epithelial cancers such as HNSCC [53]. These observations support the dog-human molecular homology.
RNA-seq analysis reveals dog-human homology in transcriptomic alterations
To better understand alterations at the transcriptomic level, we performed RNA-seq on seven of the canine oSCCs and three matching normal tissue samples (S2A Table). The study further reveals canine tumor heterogeneity and supports dog-human molecular homologies. First, principle component analysis (PCA) separates the tumors from the normal samples (Fig 3A). More importantly, tumors 1172, 465, and 404 are distant from the other tumors in the PCA (Fig 3A), the significance of which will be discussed in later sections. Second, compared to the normal samples, the genes upregulated in the tumors are enriched in functions including: 1) cell adhesion/motility, extracellular matrix, and endopeptidase activity; 2) hypoxia and polysaccharide metabolic processes; 3) blood vessel morphogenesis and cell differentiation; and 4) immune response (Fig 3B and S2C–S2G Table). As in human cancers, these functions facilitate canine tumor cell proliferation and invasion.

We followed published strategies [2,46] to identify over/under-expressed genes. Consistent with CNA findings (Fig 2A), more genes are overexpressed than underexpressed in all tumors except tumor 404 (S2B Table). Importantly, genes recurrently overexpressed among the tumors are significantly enriched in functions associated with cell cycle (e.g., cytoskeleton, spindle, centrosome, kinetochore, etc.), protein kinase activity (e.g., PTK2, TEC, CHEK2, etc.), nucleolus, and mRNA and ncRNA processing. Recurrently underexpressed genes are, however, significantly enriched in functions related to cell junctions (e.g., 9 tight junction genes), mitochondria (e.g., respiratory chain and oxidative phosphorylation), serine protease inhibitors, and apoptosis. These functions promote cancer cell proliferation and invasion, consistent with human cancer findings [1].
Critically, we found the same genes and pathways altered in these canine cancers as reported in human HNSCCs [1,2]. For example, gene members of the receptor tyrosine kinase (RTK)/ PI3K/AKT pathway such as EGFR, PIK3CA, BRAF, and AKT1 are recurrently overexpressed among the canine tumors (Fig 3C). AKT1 is especially noteworthy because its expression level in each tumor is consistently higher than in each normal sample by 2-4-fold (Fig 3C). The PI3K/AKT pathway is indeed activated in the tumor cells as revealed by immunostaining with phospho-AKT (Fig 4A). Cell cycle is also altered, as evidenced by overexpression of multiple cyclin genes, CDK4, CDK6 and E2F1; as well as underexpression of CDKN2A and CDKN2B (Fig 3C). These pathway alterations could promote cancer cell proliferation.

Another pathway affected is TGFβ signaling, as evidenced by the recurrent overexpression of TGFB1, TGFB2, TGFBR2, TGFBR1, SMAD3 and SMAD4 in the canine tumors (Fig 3C). Other notable changes include at least 12 matrix metalloproteinase (MMP) genes, whose expression increased by hundreds to thousands fold in at least one tumor (Fig 3C). Likewise, epithelial to mesenchymal transition (EMT) genes TWIST1 and SNAI1 are also recurrently overexpressed (Fig 3C). These observations indicate EMT in the canine tumors, which is confirmed by immunofluorescent staining showing increased expression of the mesenchymal marker vimentin and decreased expression of the epithelial marker E-cadherin in the tumor cells, compared to the normal squamous cells (Fig 4B). Activation of these genes and pathways would facilitate the invasion of tumor cells into adjacent tissues. Interestingly, tumor 1172 appears to be an exception to the above observations (Figs 3C and 4B), which will be revisited in the DISCUSSION section.
Finally, two canine oSCCs appear to be canine papillomavirus (CPV)-positive, based on the detection of papillomavirus sequences, specifically CPV7 E2/E4 sequences in tumor 1172 and sequences with high homology (>90%) to human papillomavirus HPV77 E2/E4 in tumor 465, among their RNA-seq reads (S2H Table). However, unlike TCGA’s HPV-positive HNSCCs that harbor thousands of viral RNA-seq reads per sample [1], we only identified a few viral RNA-seq reads for the two canine tumors (S2H Table). This indicates that the infection is likely latent, or the CPV genome(s) infected has/have not been sequenced (we were able to download 170 HPV genomes but only 15 CPV genomes from the PaVE database at pave.niaid.nih.gov at the time of the analysis).
RNA-seq analysis reveals dog-human homologies in sequence mutation
We took advantage of RNA-seq data to examine sequence mutations in the canine samples. Briefly, to achieve more accurate mutation-finding, we utilized only coding regions with 30-300X RNA-seq read coverage, which distribute across the genome and amount to 4–6 Mb sequences in total per sample (S2I Table). The analysis again reveals dog-human homologies. First, base transitions C↔T/G↔A dominate base transversions in all samples (Fig 5A and S2I Table), indicating similar mutation mechanisms in both species. At CpG sites, the two CPV-positive canine tumors harbor significantly more transversions compared to the CPV-negative tumors (Fig 5B and S2I Table), consistent with human findings [1]. However, we did not observe a predominance of mutations at TpC sites in CPV-positive canine tumors (S2I Table), unlike the human study [1]. Second, the analysis uncovered a somatic mutation, E233K, in TP53. Similarly, consistent with human studies [3,4], genes FAT1, FAT2, UBR2, TNC and others were found to be mutated in the canine tumors (S2I Table).

As an example, Fig 5C shows the mutations found in the gene RELN in canine tumor 404, including one nonsense, five non-synonymous and one synonymous changes. Importantly, RELN is significantly mutated in human HNSCC [1,4], non-small cell lung cancer of smokers [54], and acute lymphoblastic leukemia [55]. RELN encodes an extracellular matrix glycoprotein which is known to control cell–cell interactions to regulate neuronal migration and positioning in the developing brain. Thus, alteration of RELN may contribute to tumor cell invasion and spread in both the human and the dog.
Driver-passenger discrimination via dog-human comparison for human 8q
The dog-human molecular homologies described above rationalize the use of the dog-human comparison strategy for driver-passenger discrimination as described [30] for HNSCC. Because of the small sample size of canine tumors, we tested this strategy only on human 8q, one of the most recurrently amplified regions in human HNSCC [1,2] and other cancer types. Due to interspecies genomic rearrangements, human 8q is broken into two dog chromosomal regions, which include chromosome 29 and the first 38Mb of chromosome 13 (Fig 6A). Notably, the entire human 8q is significantly amplified among TCGA’s 449 human HNSCCs (FDR < 10–6), leading to the amplification of all of its 398 genes (Figs 6A and S1). In canine tumors, however, only the chromosome 13 region is significantly altered, resulting in the amplification of 125 genes out of 210 total at FDR < 0.2. For chromosome 29 in contrast, merely 2 genes out of 188 total are amplified. These numbers significantly differ (p < 2.2×10–16) (Fig 6A). Thus, based on our strategy [30], amplified genes (125 total) on chromosome 13 are considered as driver candidate genes (DCGs), while unchanged genes on chromosome 29 (186 total) are deemed passenger candidate genes (PCGs) (Fig 6A and S1H Table).

We then performed several analyses to examine the differences between the DCGs and PCGs identified. First, a significantly better correlation between the copy number status and the mRNA expression level was observed for DCGs than PCGs, using data from TCGA [1] (Fig 6C and S1H Table). This indicates that amplification of DCGs is more functionally relevant than amplification of PCGs. Second, significantly more DCGs are mutated than PCGs, based on published human HNSCC studies [3] (p < 0.0081) (S1H Table). Lastly, well known cancer driver genes such as MYC are among the DCGs.
Conversely, we also tried the dog-human comparison strategy on the10Mb amplicon of canine tumor 240 shown in Fig 2B. In the human genome, the 10Mb region is broken into three pieces, including human chromosome 7 : 154.5–159.1Mb; chromosome 8 : 18.3–19.7Mb and 37.5–43.1Mb. Based on TCGA data [1], while 42 out of the 44 genes of chromosome 8 : 37.5–43.1Mb are significantly amplified, none of the genes of the remaining two human regions is amplified or deleted (Fig 6B). Thus, based on our strategy [30], the chromosome 8 : 37.5–43.1Mb region is more likely to harbor cancer drivers, which indeed encodes known driver gene FGFR1 [1].
Discussion
In this pilot study, we performed the first genome-wide characterization of spontaneous canine HNSCC. Although the sample size (12 cases including 9 oral) is small, the study reveals a complex alteration profile for canine HNSCC and a strong dog-human molecular homology. Our results indicate that spontaneous canine HNSCCs more accurately represent their human counterparts in various aspects than the current widely-used models. These canine cancers better model human HNSCCs in cancer cell origin and tumor microenvironment (compared to cell line and xenograft models), molecular heterogeneity and complexity (compared to genetically engineered mouse models), and histopathology and cancer biology (compared to carcinogen-induced models). If our conclusions are validated with a large sample size study, spontaneous HNSCCs in pet dogs could contribute significantly in basic and translational cancer research.
Spontaneous canine HNSCCs recapitulate many molecular features of their human counterparts
As described throughout the Results section, our genomic and transcriptomic studies support strong dog-human molecular homologies for HNSCC at various levels. For large genomic changes, the two species share a similar CNA landscape, with some tumors harboring extensive CNAs while others being nearly CNA-free [1,2] and with large amplicons that likely harbor cancer drivers discovered. Furthermore, unlike canine mammary cancers [21], no potential oncogenic chimeric fusion genes are found in these canine oSCCs, consistent with very few such genes being reported for human HNSCC [1]. At the individual gene level, numerous amplified/deleted, over/under-expressed, or mutated genes are shared between the two species (e.g., FADD amplification, CDKN2A deletion, FAT1 mutation, etc.). The altered genes are also enriched in the same functional groups, including protein kinase or protease activity for amplified/overexpressed genes, as well as cell adhesion and other epithelial polarity related functions for deleted/underexpressed genes. At the pathway level, both species show alterations in cell cycle, RTK/PI3K/AKT signaling, and TGFβ signaling and EMT.
Despite the strong homology described above, a few differences are also observed. For example, TP53 mutation is more frequent in human HNSCC [1] than in these canine tumors, which is also true for NOTCH1. This difference however could be due to our small sample size and/or using RNA-seq instead whole exome-sequencing for mutation discovery. Another discrepancy is that more canine HNSCCs appear to be CNA-free when compared to their human counterparts, which again needs to be validated with a large sample size study.
Our study indicates that canine tumors 1172 and 465 are CPV-positive but the infection is likely latent (or the CPV genome(s) infected has/have not been sequenced). Both tumors (especially tumor 1172) are distinct from other canine oSCCs in gene expression. Importantly, consistent with the finding that HPVs infect the basal layer of squamous epithelium [56], tumor 1172 exhibits features indicating basal stem cell origin. Unlike other canine oSCCs, tumor 1172 overexpresses the pluripotent marker SOX2 and at least 20 homeobox genes that are associated with embryonic morphogenesis, but does not express EMT markers in many of its tumor cells. These observations again reveal extensive heterogeneity of canine HNSCCs and a strong homology to their human counterparts.
The dominance of C↔T changes over other base substitution types indicates that deamination of C to U/T is a major sequence mutation mechanism in dogs as in humans. This result is consistent with aging being a risk factor for HNSCC development in both species [57]. Furthermore, analogous to the human [1], base transversions at CpG sites are more dominant in CVP-negative tumors than in CPV-positive tumors. However, unlike the human [1], CPV-positive tumors do not show predominance of mutations at TpC sites, which needs further study to resolve.
We hypothesize the carcinogenic mechanisms of the canine tumors based on our findings (Fig 7). Regarding cells of origin, unlike tumor 1172 discussed previously, we propose that other tumors arise from more differentiated cells of the squamous epithelium (Fig 7), because of overexpression of protease genes and EMT genes. At the genome level, we hypothesize that primary drivers include focal amplifications for tumor 240. Tumor 404 is also noteworthy, as it lacks genomic CNAs but has the largest number of underexpressed genes and appears to have a different epigenomic landscape, as shown by drastically reduced staining by 5-methylcytosine antibody (Fig 4C). While emphasizing that the significance and reasons of the observed DNA hypomethylation need further investigation, we hypothesize epigenomic aberrations as the drivers of tumor 404 (Fig 7). Finally, notable gene alterations and recurrently altered pathways are listed as cancer drivers. Fig 7 summarizes once again the complexity and heterogeneity of these canine cancers and their numerous homologies to human HNSCCs.

If validated with a large sample study, spontaneous canine HNSCCs could be used for efficient cancer driver-passenger discrimination
The dog-human molecular homologies described above indicate that canine HNSCC share similar pathogenic pathways as their human counterparts, which, if validated with a large sample study, justifies the use of our established dog-human comparison strategy [30] for driver-passenger [58] discrimination for HNSCC. Indeed, our pilot human 8q study has shown that this approach is valid. The use of canine cancers in this regard is highly significant. For example, our analysis with the copy number data of TCGA’s 449 human HNSCC cases has found over 7000 amplified/deleted genes at FDR ≤ 0.05, including those harbored by both focal amplifications/deletions and broad (chromosomal arm level) gains/losses. Among these genes, some are drivers and some are passengers. Based on our analyses, a sample size of 449 tumors is already saturating, and studying additional human tumors no longer helps in determining which amplified/deleted genes are drivers and which are passengers. However, investigating about 90 canine tumors will reduce the number of driver candidates by at least half, according to our estimation, significantly reducing the workload of downstream functional validation which is time-consuming and expensive.
If validated with a large sample size, spontaneous canine HNSCCs may bridge the gap between current preclinical models and human clinical trials
As discussed in the Induction section, a major obstacle in translational research is the lack of effective predictive models [9]. Current preclinical models, including cell culture and xenograft or genetically-induced rodent models, typically fail to represent the vast heterogeneity and complexity of human cancers and often do not predict clinical results. Thus, a cancer model that can bridge the gap between these preclinical models and human clinical trials is urgently needed.
Spontaneous canine HNSCCs could serve as such a much-needed translational model, if the dog-human molecular homologies concluded here are validated with a large sample study. Being molecularly complex and heterogeneous and recapitulating many molecular features of human HNSCC, these canine HNSCCs are more accurate representatives of their human counterparts than current cell line or rodent models. With an HNSCC incidence rate comparable to that in the human and a large pet dog population (>70 million in the US), coupled with less stringent FDA regulations governing pet clinical trials and increasingly improved resource and infrastructure in pet cancer research [18], spontaneous canine cancers have the potential to significantly accelerate the translation of basic research findings into successful clinical applications.
In summary, our study indicates that spontaneous canine HNSCCs better recapitulate the full spectra of the biology, histopathology, complexity, and heterogeneity of humans HNSCC than the current widely used models. However, we caution again that our sample size is small, and the conclusions drawn from this pilot study need to be validated with a larger sample size.
Materials and Methods
Canine tissue samples
Fresh-frozen normal and tumor tissue samples and formalin-fixed paraffin-embedded (FFPE) tissue sections of spontaneous canine HNSCCs were obtained from the Animal Cancer Tissue Repository of Colorado State University. Samples were collected from client-owned dogs that developed the disease spontaneously, under the guidelines of the Institutional Animal Care and Use Committee and with owner informed consent. The breed, age, histopathologic descriptions, and other information are provided in S1A Table. These data, along with our own H&E staining of each sample received, were reviewed by Dr. Angela E. Ellis, a canine cancer pathologist.
Tissue dissection, DNA and RNA extraction, and PCR analyses
Tissue cryosectioning, H&E staining, and cryomicrodissection were performed as described [19] to enrich for tumor cells in tumor samples and squamous epithelial cells in normal samples. Genomic DNA and RNA were then extracted from the dissected tissues using the AllPrep DNA/RNA Mini Kit (cat. no. 80204) from Qiagen. Only samples with a 260/280 ratio of ~1.8 (DNA) or ~2.0 (RNA) and showing neither degradation nor other contamination on the agarose gels were subjected to further analyses.
aCGH analyses
Canine aCGH experiments were conducted at the Florida State University Microarray Facility, with 385K canine CGH array chips from Roche NimbleGen Systems, Inc. as previously described [19]. Briefly, each array chip contains about 385,000 probes of approximately 50 bp long oligos selected from unique sequences in the canFam2 genome, providing an average resolution of one probe every 5–6 kb across the canine genome. Tumor DNA purified from the dissected tumor tissue sample and paired normal DNA purified from matching normal tissue, skin, or blood of the same dog patient (S1A Table) ware hybridized to a chip, following the manufacturer’s instructions. This was performed for each canine case except for case 573 (a male), of which a normal sample from the same dog was unavailable and hence normal DNA from another dog (female) of the same breed was used instead (S1A Table). Because of tumor 573, chromosome X was excluded from the CNA-finding to minimize artifacts.
CNAs were identified as previously described [19]. Briefly, CNAs in each tumor were detected by analyzing the log2-ratios using a software program called SEG, which we developed to more effectively decipher high density oligo aCGH data for CNA finding. As described in our previous publication [19], SEG first identified change-points at the chromosomal level via dynamic programming and then detected CNAs at the whole genome level using false discovery rate (FDR)-controlled procedure [59]. For this study, we set the desired FDR to 0.01, the total probe number cutoff to 3, and the log2-ratio mean cutoff to 0.4 for CNA discovery.
Significantly amplified/deleted genes were identified by GISTIC [60] as previously described [19,21]. For the human, level 3 data of SNP array analyses of 449 human HNSCC cases were downloaded from TCGA site (cancergenome.nih.gov/), and GISTIC [60] were used to detect amplified/deleted genes, in combination with published findings [1].
Paired-end RNA-seq
Sequencing was conducted using the Illumina platform, following the protocols from the manufacturer. RNA-seq was performed at the HudsonAlpha Institute for Biotechnology or the BGI-America, yielding 48 to 66 million paired-end sequence reads of 50bp or 49bp per sample (S2A Table).
RNA-seq data analysis
RNA-seq data analyses were performed as described [21]. Briefly, read pairs were aligned to the dog reference genome [33] canFam2 with TopHat v2.0.5 (tophat.cbcb.umd.edu). The uniquely mapped pairs were used to quantify a gene’s expression level by calculating its FPKM (fragments per kilobase of exon per million mapped fragments) value, using Cufflinks (cufflinks.cbcb.umd.edu) with default parameters and the canine gene annotation downloaded from the University of Santa Cruz (UCSC) genome site. Base substitutions were identified with VarScan2 (varscan.sourceforge.net) in coding regions with RNA-seq read coverage ranging from 30X to 300X. Over/underexpressed genes in cancers were identified as described [2,46], i.e., genes whose expression levels are outside 95% confidence internals of the expression of samples that are diploid in that gene. Differentially expressed genes between two groups of samples were identified by DESeq [61] and t-tests. Gene functional annotation and enrichment analyses were achieved using the DAVID Functional Annotation tool at david.abcc.ncifcrf.gov.
Viral RNA-seq reads were identified using a similar approach as VirusSeq (odin.mdacc.tmc.edu/~xsu1/VirusSeq.html). After aligning RNA-seq reads to the reference genome (canFam2) with TopHat v2.0.5, the unmapped reads of each sample were realigned to the HPV and CPV genomes downloaded from the PaVE database (pave.niaid.nih.gov/) with Bowtie2 v2.2.3 and BWA v0.7.10.
Immunofluorescence
Immunofluorescent staining was performed with 5-μm FFPE tissue sections as described [21,62]. Primary antibodies used include those against E-cadherin (R&D Systems; AF648), vimentin (Abcam; ab92547), phospo-AKT (Ser473) (Cell Signaling; 4060), acetyl-H4 (Millipore; 06–866), 5-methylcytosine clone 33D3 (Millipore; MABE146), and Ki67 (Life Technologies; 08–0156). Alexa Fluor488–, 647—or 594–conjugated secondary antibodies are from Jackson ImmunoResearch. Images were taken with a Zeiss LSM 710 confocal microscope.
Accession numbers
RNA-seq data have been submitted to the NCBI SRA database under accession number SRP046723. aCGH data have been submitted to the GEO database under accession number GSE61231.
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. Cancer Genome Atlas N (2015) Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 517 : 576–582. doi: 10.1038/nature14129 25631445
2. Pickering CR, Zhang J, Yoo SY, Bengtsson L, Moorthy S, et al. (2013) Integrative genomic characterization of oral squamous cell carcinoma identifies frequent somatic drivers. Cancer discovery 3 : 770–781. doi: 10.1158/2159-8290.CD-12-0537 23619168
3. Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, et al. (2011) The Mutational Landscape of Head and Neck Squamous Cell Carcinoma. Science 333 : 1157–1160. doi: 10.1126/science.1208130 21798893
4. Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, et al. (2011) Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science 333 : 1154–1157. doi: 10.1126/science.1206923 21798897
5. Hammerman PS, Hayes DN, Grandis JR (2015) Therapeutic Insights from Genomic Studies of Head and Neck Squamous Cell Carcinomas. Cancer discovery doi: 10.1158/2159-8290.CD-14-1205
6. Leemans CR, Braakhuis BJ, Brakenhoff RH (2011) The molecular biology of head and neck cancer. Nature reviews Cancer 11 : 9–22. doi: 10.1038/nrc2982 21160525
7. Bose P, Brockton NT, Dort JC (2013) Head and neck cancer: from anatomy to biology. International journal of cancer Journal international du cancer 133 : 2013–2023. doi: 10.1002/ijc.28112 23417723
8. Cantley LC, Baselga J (2011) The Era of Cancer Discovery. Cancer discovery 1 : 1–U15. doi: 10.1158/2159-8274.CD-11-0049 22586302
9. Hait WN (2010) Anticancer drug development: the grand challenges. Nature reviews Drug discovery 9 : 253–254. doi: 10.1038/nrd3144 20369394
10. Kim S (2009) Animal models of cancer in the head and neck region. Clinical and experimental otorhinolaryngology 2 : 55–60. doi: 10.3342/ceo.2009.2.2.55 19565028
11. Richmond A, Su Y (2008) Mouse xenograft models vs GEM models for human cancer therapeutics. Disease models & mechanisms 1 : 78–82.
12. Lu SL, Herrington H, Wang XJ (2006) Mouse models for human head and neck squamous cell carcinomas. Head & neck 28 : 945–954.
13. Simon C, Nemechek AJ, Boyd D, O'Malley BW Jr., Goepfert H, et al. (1998) An orthotopic floor-of-mouth cancer model allows quantification of tumor invasion. The Laryngoscope 108 : 1686–1691. 9818827
14. Visvader JE (2011) Cells of origin in cancer. Nature 469 : 314–322. doi: 10.1038/nature09781 21248838
15. Meuten DJ (2002) Tumors in domestic animals. Ames, Iowa: Iowa State University Press. xii, 788 p. p.
16. Paoloni M, Khanna C (2008) Translation of new cancer treatments from pet dogs to humans. Nature reviews Cancer 8 : 147–156. doi: 10.1038/nrc2273 18202698
17. Rowell JL, McCarthy DO, Alvarez CE (2011) Dog models of naturally occurring cancer. Trends in molecular medicine 17 : 380–388. doi: 10.1016/j.molmed.2011.02.004 21439907
18. Gordon I, Paoloni M, Mazcko C, Khanna C (2009) The Comparative Oncology Trials Consortium: using spontaneously occurring cancers in dogs to inform the cancer drug development pathway. PLoS medicine 6: e1000161. doi: 10.1371/journal.pmed.1000161 19823573
19. Tang J, Le S, Sun L, Yan X, Zhang M, et al. (2010) Copy number abnormalities in sporadic canine colorectal cancers. Genome research 20 : 341–350. doi: 10.1101/gr.092726.109 20086242
20. Youmans L, Taylor C, Shin E, Harrell A, Ellis AE, et al. (2012) Frequent alteration of the tumor suppressor gene APC in sporadic canine colorectal tumors. PloS one 7: e50813. doi: 10.1371/journal.pone.0050813 23251390
21. Liu D, Xiong H, Ellis AE, Northrup NC, Rodriguez CO, Jr., et al. (2014) Molecular homology and difference between spontaneous canine mammary cancer and human breast cancer. Cancer research doi: 10.1158/0008-5472.CAN-14-0392
22. Fenger JM, London CA, Kisseberth WC (2014) Canine osteosarcoma: a naturally occurring disease to inform pediatric oncology. ILAR journal / National Research Council, Institute of Laboratory Animal Resources 55 : 69–85. doi: 10.1093/ilar/ilu009 24936031
23. Gorden BH, Kim JH, Sarver AL, Frantz AM, Breen M, et al. (2014) Identification of Three Molecular and Functional Subtypes in Canine Hemangiosarcoma through Gene Expression Profiling and Progenitor Cell Characterization. American Journal of Pathology 184 : 985–995. doi: 10.1016/j.ajpath.2013.12.025 24525151
24. Boyko AR (2011) The domestic dog: man's best friend in the genomic era. Genome biology 12 : 216. doi: 10.1186/gb-2011-12-2-216 21338479
25. Rotroff DM, Thomas R, Breen M, Motsinger-Reif AA (2013) Naturally occuring canine cancers: powerful models for stimulating pharmacogenomic advancement in human medicine. Pharmacogenomics 14 : 1929–1931. doi: 10.2217/pgs.13.178 24279843
26. Angstadt AY, Thayanithy V, Subramanian S, Modiano JF, Breen M (2012) A genome-wide approach to comparative oncology: high-resolution oligonucleotide aCGH of canine and human osteosarcoma pinpoints shared microaberrations. Cancer genetics 205 : 572–587. doi: 10.1016/j.cancergen.2012.09.005 23137772
27. Candille SI, Kaelin CB, Cattanach BM, Yu B, Thompson DA, et al. (2007) A-defensin mutation causes black coat color in domestic dogs. Science 318 : 1418–1423. 17947548
28. Davis BW, Ostrander EA (2014) Domestic dogs and cancer research: a breed-based genomics approach. ILAR journal / National Research Council, Institute of Laboratory Animal Resources 55 : 59–68. doi: 10.1093/ilar/ilu017 24936030
29. Karyadi DM, Karlins E, Decker B, vonHoldt BM, Carpintero-Ramirez G, et al. (2013) A copy number variant at the KITLG locus likely confers risk for canine squamous cell carcinoma of the digit. PLoS genetics 9: e1003409. doi: 10.1371/journal.pgen.1003409 23555311
30. Tang J, Li Y, Lyon K, Camps J, Dalton S, et al. (2014) Cancer driver-passenger distinction via sporadic human and dog cancer comparison: a proof-of-principle study with colorectal cancer. Oncogene 33 : 814–822. doi: 10.1038/onc.2013.17 23416983
31. Nasir L, Devlin P, McKevitt T, Rutteman G, Argyle DJ (2001) Telomere lengths and telomerase activity in dog tissues: a potential model system to study human telomere and telomerase biology. Neoplasia 3 : 351–359. 11571635
32. Rangarajan A, Weinberg RA (2003) Opinion: Comparative biology of mouse versus human cells: modelling human cancer in mice. Nature reviews Cancer 3 : 952–959. 14737125
33. Lindblad-Toh K, Wade CM, Mikkelsen TS, Karlsson EK, Jaffe DB, et al. (2005) Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 438 : 803–819. 16341006
34. Ji X, Zhao S (2008) DA and Xiao-two giant and composite LTR-retrotransposon-like elements identified in the human genome. Genomics 91 : 249–258. 18083327
35. Gardner DG (1996) Spontaneous squamous cell carcinomas of the oral region in domestic animals: a review and consideration of their relevance to human research. Oral diseases 2 : 148–154. 8957928
36. Strafuss AC, Cook JE, Smith JE (1976) Squamous cell carcinoma in dogs. Journal of the American Veterinary Medical Association 168 : 425–427. 1254515
37. Hoyt RF, Withrow SJ (1984) Oral Malignancy in the Dog. J Am Anim Hosp Assoc 20 : 83–92.
38. Ragin CCR, Modugno F, Gollin SM (2007) The epidemiology and risk factors of head and neck cancer: a focus on human papillomavirus. J Dent Res 86 : 104–114. 17251508
39. Teifke JP, Lohr CV, Shirasawa H (1998) Detection of canine oral papillomavirus-DNA in canine oral squamous cell carcinomas and p53 overexpressing skin papillomas of the dog using the polymerase chain reaction and non-radioactive in situ hybridization. Vet Microbiol 60 : 119–130. 9646444
40. de Vos JP, Burm AG, Focker AP, Boschloo H, Karsijns M, et al. (2005) Piroxicam and carboplatin as a combination treatment of canine oral non-tonsillar squamous cell carcinoma: a pilot study and a literature review of a canine model of human head and neck squamous cell carcinoma. Veterinary and comparative oncology 3 : 16–24. doi: 10.1111/j.1476-5810.2005.00065.x 19379210
41. LaDueMiller T, Price GS, Page RL, Thrall DE (1996) Radiotherapy of canine non-tonsillar squamous cell carcinoma. Vet Radiol Ultrasoun 37 : 74–77.
42. Richards KL, Motsinger-Reif AA, Chen HW, Fedoriw Y, Fan C, et al. (2013) Gene profiling of canine B-cell lymphoma reveals germinal center and postgerminal center subtypes with different survival times, modeling human DLBCL. Cancer research 73 : 5029–5039. doi: 10.1158/0008-5472.CAN-12-3546 23783577
43. Thomas R, Seiser EL, Motsinger-Reif A, Borst L, Valli VE, et al. (2011) Refining tumor-associated aneuploidy through 'genomic recoding' of recurrent DNA copy number aberrations in 150 canine non-Hodgkin lymphomas. Leukemia Lymphoma 52 : 1321–1335. doi: 10.3109/10428194.2011.559802 21375435
44. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, et al. (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery 2 : 401–404. doi: 10.1158/2159-8290.CD-12-0095 22588877
45. Cancer Genome Atlas N (2012) Comprehensive molecular portraits of human breast tumours. Nature 490 : 61–70. doi: 10.1038/nature11412 23000897
46. Cancer Genome Atlas N (2012) Comprehensive molecular characterization of human colon and rectal cancer. Nature 487 : 330–337. doi: 10.1038/nature11252 22810696
47. Freier K, Knoepfle K, Flechtenmacher C, Pungs S, Devens F, et al. (2010) Recurrent copy number gain of transcription factor SOX2 and corresponding high protein expression in oral squamous cell carcinoma. Genes, chromosomes & cancer 49 : 9–16.
48. Chen Y, Chen C (2008) DNA copy number variation and loss of heterozygosity in relation to recurrence of and survival from head and neck squamous cell carcinoma: a review. Head & neck 30 : 1361–1383.
49. Wei W, Bracher-Manecke JC, Zhao X, Davies NH, Zhou L, et al. (2013) Oncogenic but non-essential role of N-myc downstream regulated gene 1 in the progression of esophageal squamous cell carcinoma. Cancer biology & therapy 14 : 164–174.
50. Melotte V, Qu XH, Ongenaert M, van Criekinge W, de Bruine AP, et al. (2010) The N-myc downstream regulated gene (NDRG) family: diverse functions, multiple applications. Faseb J 24 : 4153–4166. doi: 10.1096/fj.09-151464 20667976
51. Ortiz B, White JR, Wu WH, Chan TA (2014) Deletion of Ptprd and Cdkn2a cooperate to accelerate tumorigenesis. Oncotarget.
52. Nishikawa Y, Miyazaki T, Nakashiro KI, Yamagata H, Isokane M, et al. (2011) Human FAT1 cadherin controls cell migration and invasion of oral squamous cell carcinoma through the localization of beta-catenin. Oncology reports 26 : 587–592. doi: 10.3892/or.2011.1324 21617878
53. Royer C, Lu X (2011) Epithelial cell polarity: a major gatekeeper against cancer? Cell death and differentiation 18 : 1470–1477. doi: 10.1038/cdd.2011.60 21617693
54. Govindan R, Ding L, Griffith M, Subramanian J, Dees ND, et al. (2012) Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell 150 : 1121–1134. doi: 10.1016/j.cell.2012.08.024 22980976
55. Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, et al. (2012) The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 481 : 157–163. doi: 10.1038/nature10725 22237106
56. Blitzer GC, Smith MA, Harris SL, Kimple RJ (2014) Review of the clinical and biologic aspects of human papillomavirus-positive squamous cell carcinomas of the head and neck. International journal of radiation oncology, biology, physics 88 : 761–770. doi: 10.1016/j.ijrobp.2013.08.029 24606845
57. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, et al. (2013) Signatures of mutational processes in human cancer. Nature 500 : 415–421. doi: 10.1038/nature12477 23945592
58. Salk JJ, Horwitz MS (2010) Passenger mutations as a marker of clonal cell lineages in emerging neoplasia. Semin Cancer Biol 20 : 294–303. doi: 10.1016/j.semcancer.2010.10.008 20951806
59. Benjamini Y, Hochberg Y (1995) Controlling the False Discovery Rate—a Practical and Powerful Approach to Multiple Testing. J Roy Stat Soc B Met 57 : 289–300.
60. Beroukhim R, Getz G, Nghiemphu L, Barretina J, Hsueh T, et al. (2007) Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proceedings of the National Academy of Sciences of the United States of America 104 : 20007–20012.
61. Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome biology 11: R106. doi: 10.1186/gb-2010-11-10-r106 20979621
62. Yaping Li JX, Huan Xiong, Zhongyao Ma, Zhenghe Wang, Kipreos Edward T., Stephen Dalton and Shaying Zhao (2014) Cancer driver candidate genes AVL9, DENND5A and NUPL1 contribute to MDCK cystogenesis. Oncoscience 1 : 854–865. 25621300
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 6
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Non-reciprocal Interspecies Hybridization Barriers in the Capsella Genus Are Established in the Endosperm
- Translational Upregulation of an Individual p21 Transcript Variant by GCN2 Regulates Cell Proliferation and Survival under Nutrient Stress
- Exome Sequencing of Phenotypic Extremes Identifies and as Interacting Modifiers of Chronic Infection in Cystic Fibrosis
- The Human Blood Metabolome-Transcriptome Interface
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy