
Exome Sequencing of Phenotypic Extremes Identifies and as Interacting Modifiers of Chronic Infection in Cystic Fibrosis
Whole exome and whole genome sequencing provide the opportunity to test for associations between expressed traits and genetic variants that cannot be tested with chip technology, particularly variants that are too rare to be included on chips designed for genome-wide association analysis. We used exome sequencing to identify variants in CAV2 and TMC6 that modify the age-of-onset of chronic Pseudomonas aeruginosa infection among children with cystic fibrosis, and validated our findings in a large cohort of children with cystic fibrosis. For a fixed number of study participants, it is known that the extreme phenotypes design provides greater statistical power than a random sampling design. In the extreme phenotypes design, one compares the frequency of a given set of genetic variants in one extreme of age-of-onset (early onset) to that in the other extreme (late onset). Here, we employed an alternative design that compares genetic frequencies in exomes sampled from one extreme to that among exomes from a large set of controls. We show that this design confers substantially greater statistical power for discovery of CAV2 and TMC6 and provide general conditions under which this single extreme versus control design is more powerful than the extreme phenotypes design.
Published in the journal:
. PLoS Genet 11(6): e32767. doi:10.1371/journal.pgen.1005273
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005273
Summary
Whole exome and whole genome sequencing provide the opportunity to test for associations between expressed traits and genetic variants that cannot be tested with chip technology, particularly variants that are too rare to be included on chips designed for genome-wide association analysis. We used exome sequencing to identify variants in CAV2 and TMC6 that modify the age-of-onset of chronic Pseudomonas aeruginosa infection among children with cystic fibrosis, and validated our findings in a large cohort of children with cystic fibrosis. For a fixed number of study participants, it is known that the extreme phenotypes design provides greater statistical power than a random sampling design. In the extreme phenotypes design, one compares the frequency of a given set of genetic variants in one extreme of age-of-onset (early onset) to that in the other extreme (late onset). Here, we employed an alternative design that compares genetic frequencies in exomes sampled from one extreme to that among exomes from a large set of controls. We show that this design confers substantially greater statistical power for discovery of CAV2 and TMC6 and provide general conditions under which this single extreme versus control design is more powerful than the extreme phenotypes design.
Introduction
Cystic fibrosis (CF) (MIM219700) is a life shortening, monogenic condition caused by mutations in cystic fibrosis transmembrane conductance regulator (CFTR) [1]. CF is associated with dysfunction in multiple exocrine organs (e.g. pancreas, intestine, liver, lung), but the primary cause of morbidity and mortality is progressive obstructive lung disease associated with persistent endobronchial infection and neutrophilic inflammation [2,3]. Specifically, individuals with cystic fibrosis are at high risk for Pseudomonas aeruginosa (P. aeruginosa) airway infection, with a typical overall pattern of progression from first acquisition, to more frequent infections, to chronic infection and then to chronic mucoid P. aeruginosa. Eventually, the airways of ~80% of adult patients are chronically infected with P. aeruginosa.
P. aeruginosa airway infection in individuals with CF is associated with increased morbidity and reduced survival [4–6]. In a large registry-based study, any history of P. aeruginosa airway infection at or before age 2 years was associated with reduced lung function, increased frequency of pulmonary exacerbations and reduced survival [4–6]. Onset of mucoid P. aeruginosa is associated with a sharp decline in lung function [7,8] and even higher mortality [9]. Collectively, these observations support a causal relationship between frequent P. aeruginosa infection and morbidity and mortality in CF. Current CF treatment guidelines focus on early detection and attempted eradication of P. aeruginosa airway infection prior to establishment of chronic infection [10]. Aggressive acute and maintenance antibiotic treatment of P. aeruginosa infection in individuals with CF in the U.S. has coincided with a rise in the median predicted survival from 28 to 38 years [11].
The age-of-onset of chronic P. aeruginosa infection of the lungs in individuals with CF varies widely, and the heritability for age-of-onset of chronic/persistent P. aeruginosa infection of the airway, independent of CFTR genotype, has been estimated to be 0.85 (on a scale of 0 to 1) [12]. Identification of host genetic factors could help delineate sub-populations of individuals for more aggressive monitoring and treatment; identify new targets to facilitate development of therapeutics; and lead to a better understanding of the pathophysiology of P. aeruginosa infection in CF. Additionally, finding modifiers of P. aeruginosa airway infection in CF could provide a model for studies to find disease modifiers of other Mendelian conditions and perhaps for complex traits, as well. To this end, an intense search to find genetic modifiers of airway P. aeruginosa infection in CF has been underway over the past decade.
Variants in MBL2 and SCL9A3 have been associated with first acquisition of airway P. aeruginosa in children with CF [13], [14], and nominally significant associations between age-of-onset of chronic P. aeruginosa infection have been reported recently for other lectin pathway genes: FNC1, FNC2 and MASP3 [15]. More recently, we used exome sequencing and an extreme phenotypes design to discover two variants in DCTN4 associated with early onset of chronic P. aeruginosa airway infection [16]. However, even collectively, these variants explain only a small fraction of the genetic variance of risk for P. aeruginosa airway infection, indicating that other genetic modifiers of P. aeruginosa airway infection remain to be found.
To find variants/pathways that influence risk of early chronic P. aeruginosa infection in individuals with cystic fibrosis, we used a study design in which a single phenotypic extreme is compared to a control population. In contrast, in an extreme phenotypes study design, frequencies of variants in the two opposite s of the same phenotype in a study population are directly compared. In the extreme phenotypes design, power depends both on the group sizes and on the difference in variant frequencies between extremes (i.e., the empirical effect size). However under certain genetic architectures, most or all of the effect size can be due to enrichment (or depletion) of casual variants in only one extreme relative to the entire study population. In such cases, comparison of the single enriched (or depleted) extreme to the entire study population will increase statistical power.
We sampled CF individuals from each of two extremes of phenotype, generated exome data for each sample, and then compared each extreme separately to a large control exome data set in order to discover genetic variants associated with the phenotypic expression in each CF sample. One extreme comprised 85 individuals with cystic fibrosis and extreme early onset chronic P. aeruginosa infection; the other comprised 65 individuals with extreme late onset P. aeruginosa (S1 Fig). Study individuals were drawn from the Early Pseudomonas Infection Control (EPIC) Observational Study and the Genetic Modifier Study (GMS; a member of the North American CF Gene Modifier Consortium [17],[18]). The large control data set included 3,239 individuals of European ancestry who were ascertained to study non-lung disease phenotypes as part of the NHLBI Exome Sequencing Project (ESP). In both the extreme early onset P. aeruginosa vs. control population and the extreme late onset P. aeruginosa vs. control population comparisons, we discovered a gene associated with time to chronic P. aeruginosa infection in individuals with CF. Specifically, we found a variant in CAV2, encoding caveolin-2, was associated with increased age-of-onset (i.e., protective) and variants in TMC6, encoding transmembrane-like channel 6 were associated with decreased age-of-onset (i.e., deleterious). Both of these results were subsequently replicated in a large cohort of independent samples from the Early Pseudomonas Infection Control (EPIC) [19] study cohort. Furthermore, we demonstrate that the statistical power to find both genes was greater under the single extreme vs. control population design than under the extreme phenotypes design. Together, these findings provide proof-of-principle that use of exome sequencing and a single extreme phenotype vs. control population design can identify genetic modifiers of Mendelian conditions and may be generalizable to the search for rare variants underlying complex, common diseases.
Results
CAV2 is associated with extreme early onset P. aeruginosa airway infection
A per-gene comparison between the extreme early onset chronic P. aeruginosa group (n = 85) and the control group (n = 3,239) using the optimal sequence kernel association test with small sample adjustment (aSKAT-O)[20] revealed significant differences between the frequencies of variants in two genes, CFTR (p<10–16) and CAV2 (p = 1.1x10-6) (Fig 1A). Examination of the eight (nonsynonymous) variants included in the CAV2 by-gene test showed that the aSKAT-O signal was likely due entirely to imbalance in frequency between groups for the single common variant (rs8940; p.(Q130E), MAF = 0.19 in European Americans (EA) per the NHLBI Exome Variant Server); the seven other variants had a cumulative MAF < 0.2%. This prediction was confirmed by a per-variant analysis in which the result for rs8940 replicated the by-gene result for CAV2, p = 6.7 x 10–7, for rs8940, which occurs at a highly conserved site (Genetic Evolutionary Rate Profiling (GERP) score = 5.9).

We attempted to validate the association between early age-of-onset of chronic P. aeruginosa and CAV2 rs8940 by screening an additional 643 unrelated EPIC participants using the Infinium Human Exome BeadChip and manual genotyping of rs8940 among individuals for whom the exome chip genotyping was not performed (S1 Table). Association between the presence of at least one rs8940 minor allele and age-of-onset of chronic P. aeruginosa infection was tested using the Cox proportion hazards model stratified on age at entry to the EPIC study [16] The model was adjusted for CFTR risk group [21], number of bacterial cultures tested for each individual, presence of a DCTN4 risk allele [16], age at enrollment and its interaction with rs8940. This model revealed a significant inverse association (p = 0.01; HR = 0.53, 95% CI = [0.32–0.88]) between age-of-onset of chronic P. aeruginosa infection and presence of at least one minor allele of rs8940 (Fig 1B and S2 Table). This effect was stronger among younger enrollees (p = 0.01 for rs8940 x enrollment age interaction.) Similar results were obtained after adjustment for ancestry using principal components (PCs) on the subset of 608 individuals with exome or genome-wide genotype chip data available (p = 0.0054; HR = 0.48, 95% CI = [0.29, 0.81]) (S2 Table).
CAV2 is located ~1 Mb from CFTR and higher than expected frequencies of CAV2 rs8940 alternate alleles were found in individuals with various CFTR mutations that cause CF, including F508del-CFTR, N1303K, G542X and W1282X (S3 Table), resulting in an association between CFTR mutation and CAV2 rs8940. Because age of acquisition and onset of chronic P. aeruginosa infection in CF might vary by CFTR mutation, and because CFTR mutations were associated with CAV2 rs8940, there is potential for the association between rs8940 and time to chronic P. aeruginosa infection to be due to confounding. To remove any possible confounding by CFTR mutation, we repeated the Cox model analysis restricted to individuals homozygous for F508del-CFTR (n = 312); the results were essentially unchanged (p = 0.027; HR = 0.44, 95% CI = [0.21–0.91]). Even stronger results were obtained when the analysis of F508del-CFTR homozygotes was restricted to the individuals with self-declared European ancestry and adjusted for PCs (n = 286) (p = 0.0087; HR = 0.35, 95%CI = [0.16–0.77]) (S2 Table), but the difference in HR among the F508del-CFTR homozygous group and remaining individuals did not reach statistical significance (p = 0.13). These findings support a strong association between presence of the CAV2 rs8940 alternate allele and protection against earlier and more severe P. aeruginosa infection, an association that is independent of the CFTR mutation.
TMC6 is associated with extreme late onset P. aeruginosa airway infection
We next performed a per-gene comparison between the extreme late onset chronic P. aeruginosa airway infection extreme (n = 65) and the ESP control group above using aSKAT-O and found that the frequencies of variants in CFTR (p<1x10-10) and TMC6 (p = 9.5x10-7) differed significantly between the two groups (Fig 2A). Examination of variants in TMC6 in the late onset extreme vs. control group revealed a complex pattern of differences among the 36 non-synonymous variants contributing to the by-gene test (S4 Table). Specifically, the late onset group had fewer rare variants (cumulative observed MAF = 0.007 vs. 0.014), a higher observed MAF for two (rs12449858, rs2748427) of three common variants (rs34712518, rs12449858, rs2748427 with MAFs of 0.06, 0.09, and 0.19 per EVS, respectively) in TMC6 and a lower derived allele frequency at rs3471258. Based on these observations, we next performed a validation analysis in which we collapsed all of the rare variants into one score (i.e., TMC6 rare variant score) while the three common variants were analyzed individually. Thus we performed a total of four tests in the validation analysis using the Cox model as above for the CAV2 validation.
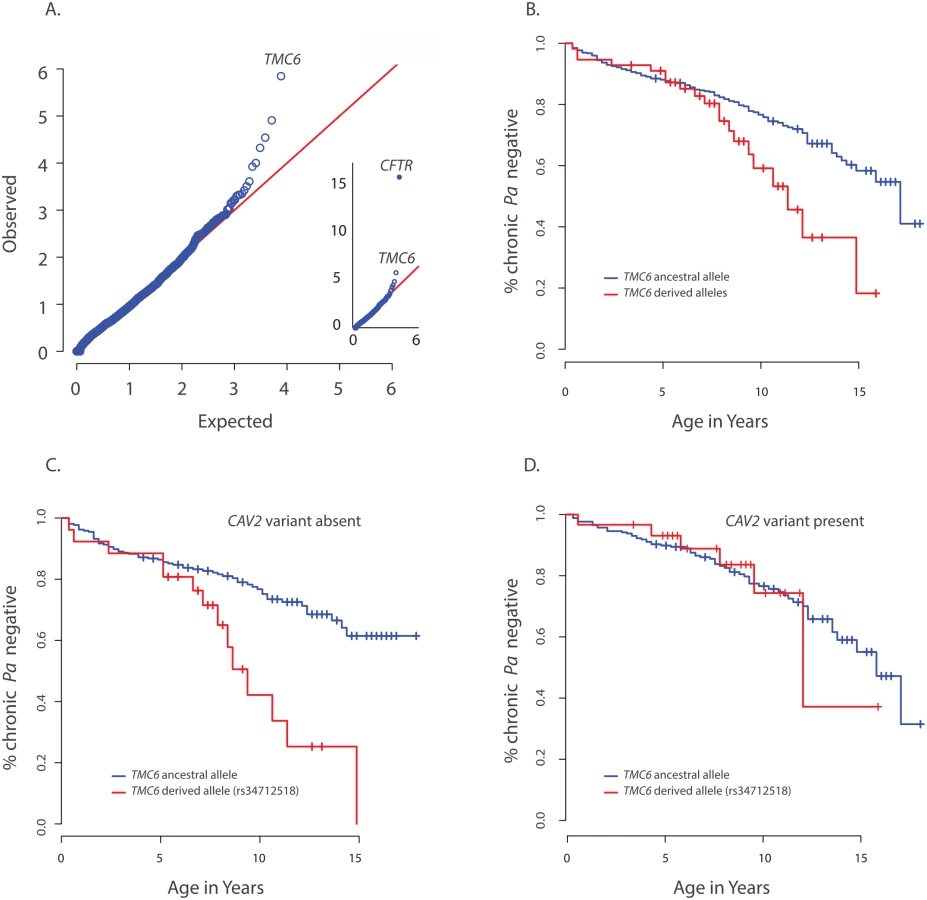
The TMC6 validation analysis was performed using data from 580 unrelated EPIC participants genotyped using the Illumina Exome chip. The presence of one or more derived alleles at rs34712518 was significantly associated with age-of-onset of chronic P. aeruginosa infection in the primary test of association (p = 0.012, HR = 1.8, 95% CI = [1.3–2.8], p< 0.05 after correction for the four tests). The Kaplan Meier plot comparing age-of-onset of chronic P. aeruginosa between TMC6 rs34712518 variant groups showed a striking divergence in risk with progressing age (Fig 2B), indicating the hazard ratio is dependent on age (i.e. the hazards are not proportional over ages) and that an age-allele interaction should be fitted in the Cox model for a more accurate description of the effect. At age 7 years, the HR was 4.5 (p = 0.01, [1.4–12.6]), and at 10 years, the HR was 5.2 (p = 0.00046, [2.1–43.0]) (S5 Table). Variant rs12449858 was not significantly associated with age-of-onset of chronic P. aeruginosa (p = 0.12, HR = 0.7, 95% CI = [0.44, 1.1]), nor was the collapsed rare variant score (p = 0.10, HR = 0.38, 95% CI = [0.12–1.2]).
The derived allele of TMC6 rs2748427 also was marginally associated with age-of-onset of chronic P. aeruginosa infection (p = 0.027, HR = 1.4 95% CI = [1.03–1.8], p = 0.08 after multiple-test correction). However, there was a strong association between the derived allele counts of rs3412518 and rs2748427 (Pearson correlation = 0.5; p<2x10-16), and individuals with a derived rs2748427 allele but not a derived rs34712518 allele showed no independent risk of chronic P. aeruginosa infection (HR = 1.18, p = 0.29, [0.86–1.6]). This suggests that the variant with a lower MAF, rs34712518 (MAF = 0.06; p.(G191D)) is associated with higher risk while the more common variant, rs2748427, is not independently associated with increased risk. The result was similar after adjusting for PCs and limiting the analysis to F508del-CFTR homozygotes (S5 Table). There were no significant interactions with enrollment age nor with chronological age in either case.
We tested for interactions between risk variants found in DCTN4, CAV2, and TMC6. No significant interaction was identified between DCTN4 and either CAV2 or TMC6. In contrast, we did find a significant interaction between the derived rs3412518 allele in TMC6 and the rs8920 CAV2 allele (p = 0.007). Specifically, by age 10, children with the derived rs3412518 allele in TMC6 without the protective CAV2 allele were at much higher risk of chronic P. aeruginosa infection (p = 2.6x10-5, HR = 12.1, [3.8–38.8] (Fig 2C), while children with the derived rs3412518 allele in TMC6 with the protective CAV2 variant had no significant excess risk of chronic P. aeruginosa infection at this same age (p = 0.41, HR = 2.1, [0.38–11.3]) (Fig 2D and S5 Table). When the test for the TMC6-CAV2 interaction was limited to the F508del-CFTR group, the test was again significant (p = 0.03) with an estimated HR = 29.1 at age 10 for F508del-CFTR homozygous children with both the TMC6 and CAV2 deleterious alleles (95% CI = [4.1, 204.8], n = 129, S3 Fig) compared to HR = 2.6 (95% CI = [0.3, 23.1], n = 144) for those with the TMC6 deleterious allele and a protective (derived) CAV2 allele.
CAV2 and TMC6 are associated with lung function
Because acquisition of P. aeruginosa is associated with worse lung function over time, strong modifiers of P. aeruginosa infection may also be associated with lung function. Accordingly, we tested for association between CAV2 rs8940 and TMC6 rs34712518 cystic fibrosis-specific percentiles of forced expiratory volume at 1 second (FEV1). Among 572 children with lung function data beyond age 7 years, an age at which lung function studies are considered reliable, the presence of one or more CAV2 rs8940 derived alleles was associated with a 5.5 percentile point increase in lung function (p = 0.001) (Fig 3A), while presence of one or more TMC6 rs34712518 alleles was associated with an 8.0 percentile point decrease in CF-specific FEV1 (p = 0.01) (Fig 3B). Both of these effects were in the direction predicted based on their association with time to chronic P. aeruginosa infection. No interaction effect between variants was detected for the FEV1 outcome.

Power of the single extreme phenotype vs. control population design
We sought to understand to what extent discovery of these associations statistical power afforded by using the single extreme vs. control population design instead of an extreme phenotypes design. We performed a detailed power study for TMC6, using simulations based on the observed association between TMC6 and age-of-onset of chronic P. aeruginosa airway infection in the validation data set. The results not only show explicit power gains but the methods also serve as a model for power calculations for other studies. Specifically, the distributions of event times for CF individuals with and without the rs3412518 alternate allele were well described by different Weibull distributions (Fig 4A). The Weibull distribution is widely used in time-to-event analysis because of its flexibility and interpretability [22]. We calculated the expected frequency of the TMC6 rs3412518 alternate allele as a function of age among children who had not yet reached chronic P. aeruginosa airway infection by that age (Fig 4B). The results show a striking depletion of the rs3412518 alternate allele among CF individuals who were older and free of chronic P. aeruginosa airway infection. This is expected, intuitively, for a modestly rare allele that confers high risk: individuals carrying the alternate allele will succumb earlier to chronic P. aeruginosa airway infection and will be under-represented among individuals free of P. aeruginosa at older ages.

The probability of carrying a TMC6 rs3412518 alternate allele can be calculated for children who became chronically infected with P. aeruginosa at a particular age to obtain the TMC6 allele distributions among our extreme samples for the observed ages in each extreme group (using the same methods that were used to generate Fig 4B). New samples, conditional on the observed ages, were drawn from the Weibull-based distributions, and the power of aSKAT-O to detect a difference between groups was calculated for our sample sizes. The result was an approximately 40% increase in power across a range of test sizes for the single extreme phenotype vs. control design relative to the extreme phenotypes design for TMC6 rs3412518 (Fig 5A). We also calculated the power based on a sample size of 150 (i.e., all available exomes originating from one extreme) in the late onset chronic P. aeruginosa airway infection extreme vs. population control and observed exceptional power for the single extreme vs. control design (99%) when all exomes were devoted to the single extreme (Fig 5A, red line). This is consistent with the increasing differences in the population MAF and the allele frequency in the late age-of-onset extreme (Fig 4B, red dotted line versus blue line, respectively.) In a separate power analysis, we drew samples from the observed distributions of variants over TMC6 (S4 Table) to better represent a by-gene analysis. Power gains were even greater under this scenario for the single extreme vs. control design relative to the extreme phenotypes design (S5B Fig). Doubling the sample size of the extreme in a single extreme vs. control design provides greater power than the extreme phenotypes design even when the allele frequency of the causal variant in both extremes differs from the population frequency.

Discussion
We used a single extreme phenotype vs. control population study design to discover variants in TMC6 and CAV2 associated with age-of-onset of chronic P. aeruginosa airway infection in individuals with CF. Examination of the variants within CAV2 and TMC6 allowed us to tailor our validation analysis to provide additional information on the role of specific variants that could not be uncovered in the by-gene discovery analysis. We found that CAV2 rs8940 was associated with protection against chronic P. aeruginosa airway infection and TMC6 rs34712518 was associated with earlier age-of-onset of chronic P. aeruginosa airway infection. Among individuals with both variants in the validation set, the protective effect of CAV2 rs8940 nullified the deleterious effect of TMC6 rs34712518 (i.e., a significant interaction was present rather than an additive effect). Consistent with these results, we also found a significant difference in lung function between children with and without CAV2 rs8940 and TMC6 rs34712518. Notably, the FEV effect size for TMC6 rs34712518 is of the same magnitude as that for risk group 1 vs. risk group 2 CFTR genotype groups in this cohort (no functional CFTR versus some residual function, respectively, in risk groups 1 and 2 [21]). Together, these findings suggest that both CAV2 and TMC6 are strong modifiers of age-of-onset of chronic P. aeruginosa airway infection in persons with CF. Neither of these discoveries were possible using the extreme phenotypes (extreme vs. extreme) analysis with our samples, with p-values for both genes being greater than 0.001 under that design.
The observation that CAV2 is a modifier of age-of-onset of chronic P. aeruginosa airway infection in individuals with CF is strongly supported by functional studies of caveolin-2 and its dimeric complement, caveolin-1. Caveolin-2 is one of a family of three structural proteins of caveolae, flask-shaped invaginations on the surface of lung epithelium and myeloid cells (Zaas 2009–1)[23]. Caveolins contribute to host defenses by regulating lipid-raft-mediated endocytosis and play a substantial role in the inflammatory response to infection and protein trafficking in the lung epithelium of individuals with CF [24,25]. In models of invasive infection, P. aeruginosa co-opts the host endocytotic machinery of lung epithelial cells where it replicates. In a variety of human cell lines, caveolin-2 forms a heterodimer with caveolin-1, which after infection with P. aeruginosa, co-localizes with CFTR and P. aeruginosa [26]. Knock-out of CAV2 in murine epithelial cells prevents P. aeruginosa invasion [27]. Notably, CAV2 is among the genes identified by GeneGO in conjunction with the Cystic Fibrosis Foundation in the MetaMiner-CF database and network tool as potential modifier genes in CF based on its role in endosome formation in lung epithelial cells [28]. PolyPhen2 classifies the CAV2 rs8940 variant as “probably damaging,” with a score of 0.997, while the Combined Annotation-Dependent Depletion (CADD) score puts this variant among the 2% of SNVs most likely to be pathogenic [29] (CADD-PHRED score = 17.1)(http://cadd.gs.washington.edu/download, V1.1).
The role of lung epithelial cell invasion in chronic P. aeruginosa airway infection in persons with CF remains unknown [30]. However, invasion of host epithelial cells at some point in the development of chronic P. aeruginosa airway infection is suggested by the presence of anti-Pseudomonal antibodies early in life among persons with CF infected with P. aeruginosa [31] and a spike in anti-P. aeruginosa antibody titers in persons with CF prior to diagnosis of chronic P. aeruginosa airway infection [7,32]. This suggests that variants in CAV2 might reduce early P. aeruginosa invasion of lung epithelia, thereby reducing the risk of chronic P. aeruginosa airway infection. Alternatively, co-expression of caveolin-2 with F508del-CFTR in murine cell lines can rescue mutant CFTR expression on the cell surface [33]. Isoforms of caveolin-2 might rescue mis-folded CFTR proteins (e.g., F508del-CFTR) resulting in increased cell surface expression of mutant CFTR and improved lung defenses against P. aeruginosa. While the mechanism(s) by which CAV2 rs8940 influences risk of chronic P. aeruginosa airway infection in persons with CF remains to be determined, our results are consistent with the known functional roles of caveolins in response to P. aeruginosa infection in CF models and highlight caveolin-2 as a potential therapeutic target to block P. aeruginosa invasion.
TMC6 encodes a highly conserved, integral transmembrane protein that interacts with zinc transporter 1 to influence intracellular zinc concentrations. Mutations in TMC6 underlie epidermodysplasia verruciformis, a condition in which persons are unusually sensitive to development of skin lesions caused by invasion human papilloma virus (HPV) [34]. One postulated mechanism is that variants in TMC6 dysregulate HPV replication via an effect on AP-1 transcription [35]. AP-1 plays a cooperative role in the P. aeruginosa-dependent induction of IL-8, a key mediator in human bronchial epithelial cells and the lung pathology of persons with CF [36]. In addition, an AP-1 transcription factor binding site is located in the promoter of DEFB1, which encodes beta-defensin-2 [37], another important cytokine involved in protection against P. aeruginosa in persons with CF. Accordingly, variants in TMC6 may influence the regulation of AP-1 and its downstream effectors, ultimately modifying the response to P. aeruginosa. Moreover, HPV gains entry to cells via both clathrin-dependent and caveolin-dependent endocytosis [38,39]. TMC6 could affect HPV invasion at the point of caveolin-mediated endocytosis, functionally linking TMC6 and CAV2 and providing a basis for the observed interaction between TMC6 and CAV2 on risk of chronic P. aeruginosa airway infection. These results provide a compelling reason to pursue functional studies of the associated variants. However, it should be kept in mind that causal variants may in LD with the discovery variants.
We identified a strong interaction between risk variants in CAV2 and TMC6. This finding highlights two considerations for studies aimed at identifying risk variants for complex traits. First, to increase the power of association studies for novel risk variants, it might be necessary to test for interactions with known genetic risk variants. This is feasible from a multiple-testing standpoint, given the relatively small numbers of known risk variants. Second, the possibility that a substantial portion of the “missing heritability” might be due to interactions among risk variants should emphasize recent attention to development and use of methods for identifying significant interactions in the face of multiple-testing in discovery analyses, a difficult but important problem.
We used a single extreme phenotype vs. control population design to gain power over an extreme phenotypes design to identify genes with differences in variant distributions between groups. Our power studies based on modeling of observed distributions for variants in TMC6 revealed that use of this strategy resulted in substantial power gains over the extreme phenotypes design. The power gain for identifying the association between age-of-onset of chronic P. aeruginosa airway infection and TMC6 variants derives from the observation that the late onset chronic P. aeruginosa airway extreme was depleted of TMC6 risk variants, while the frequency of these risk variants in the extreme early onset chronic P. aeruginosa airway infection did not differ substantially from the control population. Indeed, since the TMC6 risk variant has little effect until age 5 to 7 years, selecting children with age-of-onset before age 7 years did not enrich for this risk variant. That risk variants were depleted in the population of “survivors” (children with later age-of-onset in this setting) is an important consideration for studies to discover risk variants for childhood-onset conditions such as CF because the direct implication is that cohorts of adults can be underpowered to detect risk variants that affect the phenotype early in life.
The relative power of a single extreme phenotype vs. control population compared to an extreme phenotypes design will depend on the genetic architecture underlying the phenotype studied. In either design, selecting extreme individuals in at least one “case” arm of the study will result in enrichment of causal variants in that arm relative to both population controls and to the opposite extreme, a fact that can be demonstrated by Bayes Theorem, but the degree of relative enrichment (effect size) might differ between designs. Given N samples in each extreme, the extreme phenotypes design will usually be more powerful than a single extreme phenotype vs. control for situations in which both extremes exhibit differences in variant distributions relative to population controls, regardless of how large the sample size is for the control population. This is due to the diminishing “rate of return” in power as the control population sample size increases while the “case” sample size stays fixed at N. On the other hand, we found no realistic situation in which an extreme phenotypes design with N samples per arm had better power than a 2N single extreme vs. control population design. The implication is that investigators with access to control exomes/genomes might elect to allocate resources entirely to one phenotypic extreme so as to maximize power. This is fortuitous because only one extreme can be defined well or is of greater biomedical interest for many conditions (e.g., high blood pressure, risk of early stroke, etc.). Consequently, we think the single extreme phenotype vs. control population design is likely to be a desirable approach, particularly for adult-onset common diseases.
Generating exome/genome data from large control populations is expensive and as additional exomes/genomes become publically available, investigators will increasingly rely on public repositories of sequence data (e.g., dbGaP) for discovery studies. Such studies should always be validated with appropriate independent samples, but attention should still be given to avoiding false positives at the discovery stage. Our extreme and control exomes were sequenced contemporaneously at the same lab, but factors such as differences in targets, sequencing platforms, variant calling, etc. can introduce bias (i.e., batch effects) that result in confounding. Correcting for these confounders is critical and results based on the use of such population controls that lack explicit description of how these were managed should be interpreted with caution. Extensive considerations on the use sequence data from population controls are provided by Dercach, et al [40], along with methods for potential bias correction. Additionally, the large imbalance in samples sizes of the groups compared in the extreme phenotype vs. control population design must be considered. With very different sample sizes, tests that are based on the mean and variance of the test statistic for large samples (“second order asymptotics”, which is by far the most common measure of operating characteristics of a test) can fail to provide reliable p-values with test sizes that are too large (p-values that are too small). The aSKAT-O test with its “small sample adjustment” provided a correction for this problem. Permutation tests, including Fisher’s exact test, also retain reliability with imbalanced sample sizes but suffer from lower power, challenges with covariate adjustment and difficulty in computational implementation over the entire exome compared to aSKAT-O. Nevertheless, as data from thousands of exomes / genomes that can serve as controls become available to the scientific community, we think the extreme phenotype vs. population controls will become an increasingly popular study design.
Materials and Methods
Ethics statement
This research was approved by the Seattle Children's Hospital IRB (approval numbers 12974 and 11686). All study participants provided written informed consent or assent for the use of their DNA in studies aimed at identifying genetic risk variants for cystic fibrosis (CF) and for broad data sharing. Institutional certification was obtained for each sample in which exome sequencing was performed to allow deposition of phenotype and genotype data in the database for Genotypes and Phenotypes (dbGaP) and BAM files in the NCBI short-read archive.
Exome sequencing
QC of sample DNA, library production, exome capture, clustering and sequencing
Detailed methods are given in Emond et al [16]. Initial quality control (QC) performed on all samples included sample quantification (PicoGreen), sex determination and genotype fingerprinting (using Illumina BeadXpress). Approximately 3.5 ug of genomic DNA was used library construction steps; sample shotgun libraries were captured for exome enrichment using one of three in-solution capture products: CCDS 2008 (~26 Mb), Roche/Nimblegen SeqCap EZ Human Exome Library v1.0 (~32 Mb; Roche Nimblegen EZ Cap v1) or EZ Cap v2 (~34 Mb). Using the automated Illumina cBot cluster station, non-multiplexed samples were processed in batches of eight (one for each lane of the flow-cell). Hybridization was followed by cluster generation via bridge PCR, and enriched libraries were sequenced on either an Illumina GAIIx or HiSeq2000.
Read mapping, variant calling, and filtering
Detailed methods are provided in Emond et al [16]. Single nucleotide variants (SNVs) were called using the UMAKE pipeline at University of Michigan, which allowed all samples to be analyzed simultaneously, both for variant calling and filtering. BAM files were summarized using BWA, refined by duplicate removal, recalibration, and indel re-alignment. We excluded all reads that were not confidently mapped (Phred-scaled mapping quality < 20) from further analysis. To avoid PCR artifacts, we clipped overlapping ends in paired reads. We then computed genotype likelihoods for exome targeted regions and 50 flanking bases, accounting for per base alignment quality (BAQ) using samtools [41]. Variable sites and their allele frequencies were identified using glfMultiples, and a support vector machine (SVM) classifier was used flag probable false-positive variant sites. SNVs at HapMap polymorphic sites and Omni 2.5 array polymorphic sites in the 1000 Genomes project data were flagged as likely true positives. A total of 1,908,614 SNVs passed the SVM filter, with an overall transversion to transition ratio (Ts/Tv) of 2.84. After the initial SNV calls were generated, we re-examined the VCF files and applied filters considering total read depth, the number of individuals with coverage at the site, the fraction of variant reads in each heterozygote, the ratio of forward and reverse strand reads for reads carrying reference and variant alleles, and the average position of variant alleles along a read. CFTR genotypes from the exome sequencing results were in agreement with the clinical genotype data.
Selection of phenotypic extremes
The definition of chronic P. aeruginosa infection was chosen to parallel the “Leeds” criterion suggested by Lee et al. within the sampling frame of the EPIC study. Routine bacterial cultures were scheduled to be taken during quarterly visits (once every 3-months). We defined an individual to have reached the chronic endpoint if s/he had at least two positive P. aeruginosa culture-quarters during any one-year period. Individuals in the analysis set had a median of 3.5 culture-quarters per year.
Selection of extreme individuals
Individuals with extreme phenotypes were selected from the EPIC study, which was open to CF-affected individuals of any CFTR genotype, and from GMS, which included only individuals with the F508del-CFTR homozygous genotype. The early age-of-onset group comprised all individuals of European ancestry in either EPIC or GMS who had consented/assented for deposition of exome data to dbGaP and who had reached the chronic infection endpoint by age 7 at the time of exome sequencing (ancestry was determined using principal components analysis). Age 7 years demarks the earliest quartile of onset ages among EPIC DNA Collection study individuals (median age-of-onset of chronic P. aeruginosa airway infection = 14.3 years among the 1322 participants from EPIC DNA Collection study). Four individuals with age-of-onset later than 7 years but less than the median were included in the early onset extreme in order to make use of available sequencing, resulting in a total of 11 early onset individuals from GMS and 75 from EPIC. The median age-of-onset among the early extreme was 2.6 years.
EPIC individuals for the late onset extreme (n = 46 successful exomes) were selected from among the oldest individuals who were still free of chronic P. aeruginosa airway infection at the time of selection of the exome sequencing sample. The 19 individuals selected for the late onset extreme from the GMS were free of chronic P. aeruginosa airway infection until at least age 18 years, with one person free until age 58 years. One person in the late onset extreme became chronic at age 14.4 years after being selected for sequencing; otherwise, no person in the late onset extreme became chronically infected before age 21. The number of quarters with positive P. aeruginosa culture results among the early onset extreme was 70 times greater than that for the late onset extreme (905 vs 13) despite the longer observation times for the latter. The percentage of individuals in the early and late extremes who were identified by newborn screening (NBS) was 38.9% and 6.5%, respectively, compared to 21.6% for the EPIC Observational cohort overall [42]). Because NBS has a protective effect on acquisition of Pa [42], a larger percent of NBS individuals in the early-onset extreme makes this sample even more extreme in terms of Pa risk than it would be with a smaller percentage of NBS-identified individuals. Pancreatic enzyme use was similar in both extremes (93.4% early onset individuals ever used pancreatic enzymes, compared to 94.6% of the late onset individuals.)
Two sample test by gene for exome data
The adjusted SKAT-O method described Lee et al. [20] was used to obtain p-values for each gene. Non-synonymous variants annotated to a given gene according to the RefSeq/NCBI 37/Hg19 model were included in each by-gene test. The small-sample adjustment was critical for the discovery analysis, as the imbalance in sample sizes resulted in marked overdispersion of the qq-plots (spuriously low p-values; S2 Fig) for tests without a small sample correction. Each by-gene test was adjusted for PC1, PC2 and PC3 from a principal components decomposition of the entire set of exome data after applying the QC filters described above; any control group individuals that could be potential ancestral outliers were liberally trimmed using smartPCA from the EIGENSOFT package (http://www.hsph.harvard.edu/alkes-price/software/). Powers for the extreme phenotypes design (n = 65 vs. n = 85) and the extreme phenotype vs. control large set design were calculated prior to the analysis assuming an elevated frequency of risk variants in one extreme and not the other; powers specific to TMC6 were calculated post hoc by modeling the time-to-event distributions with Weibull distributions and by using the observed MAFs of variants in TMC6.
Cox proportional hazards validation analyses
Censored data methods (Kaplan-Meier survival curves and the Cox proportional hazards (PH) model) were used to in the validation analysis to estimate age-of-onset of chronic P. aeruginosa airway infection and to test for differences between genetic groups in the associated hazard ratio (HR) for onset of chronic P. aeruginosa infection. The Cox PH model test has maximal statistical power when hazards are proportional, but the test size remains valid when the PH assumption is violated, providing an a priori parsimonious test choice in this time-to-event scenario. All models were stratified into five groups according to quintiles of enrollment age, because enrollment criteria for the validation set included being free of chronic P. aeruginosa airway infection, making it invalid to compare children enrolled at later ages (selected to be free of chronic P. aeruginosa airway infection) to those enrolled at earlier ages. Heuristically, stratification in the Cox model results in making comparisons within strata without the need for proportional hazards across strata, and then combines the results from different strata for an overall test statistic [43]. While over-stratification can result in a loss of power, failure to stratify could possibly lead to confounding, which is the more important consideration here. All models included enrollment age, the number of observations on study (in order to adjust for less sensitivity for detection of the end-point in individuals with fewer observations) and an indicator for CFTR risk group 2 (to adjust for severity of the CFTR dysfunction)[21]. The two group classification system of McKone et al is a parsimonious method for a complex set of genotypes that works well in practice: risk group 2 genotypes include those with at least one functional group IV or V allele, are thought to have residual CFTR function, correlate highly with pancreatic sufficient disease and have a generally milder course of disease [21], and was significantly associated with age-of-onset of chronic P. aeruginosa in all models (p ≤ 0.01). Sex/gender, pancreatic enzyme use, identification by newborn screening and F508del-CFTR homozygous genotype were not significant predictors of age-of-onset, did not affect variant HR estimates and were not included in the final models. Race was not included in the model because there were not enough individuals on non-European ancestry to attain convergence of the estimation algorithm. However, the models were fitted for individuals of European ancestry only to determine whether any significant effects were driven by the few African American individuals in the analysis (S1 Table). Further, principal components were constructed for the subset of validation individuals for whom chip data were available (N = 608), and PCs 1, 2, and 3 were used to adjust for possible confounding by ancestry among this subset of CF individuals. To even further avoid potential ancestral confounding, models were fitted to the subset of F508del-CFTR homozygous individuals, both with and without PC adjustment, to ensure that residual confounding by happenstance imbalance in CFTR-mutation severity among CAV2 or TMC6 groups did not account for the observed associations.
Lung function analysis
As an additional validation analysis, association between discovered variants and lung function was tested. All EPIC participants underwent lung function tests starting at age 6 years of age as part of routine care, generally on a quarterly basis. Forced expiratory volume in 1 second (FEV1) is regarded as the most informative measure of lung function among CF individuals, and CF-specific percentile equations have been developed for lung function studies in CF. We tested for association between mean CF-specific FEV1 and TMC6 and CAV2 variants using generalized estimating equations (GEE) to account for correlation of measurements within subjects after adjusting for age and CFTR clinical risk group. Five hundred seventy-two children had variant calls and lung function measurements at age 7 and beyond (lung function measurements before age 7 not used due to the “learning” effect and potential unreliable measurements before age 7) with a median of 23 lung function measurements and a median of 4.4 years of observation time (min = .5, max = 12).
Calculation of principal components
Principal components (PCs) were calculated for the 557 individuals with exome chip data using ancestry informative markers on the chip. These PCs were used to adjust for potential confounding by population stratification in the validation analysis. In addition, we sought to increase the sample size for the validation analysis with PC adjustment and to compare exome-based PC adjusted with AIMs-based PC adjustment by using genotype information from two different sources. Among the validation individuals who were genotyped for CAV2 manually and who did not have chip data (n = 86), most (n = 51) had 353 ancestry informative markers (AIMs) available from the Illumina Golden Gate AIM (“fingerprint”) chip used in another study, and n = 354 individuals had data from both chips. Among subjects who had PCs for both the exome chip and the fingerprint chip, the linear correlation between PCs1 from either source was very high (r = 0.91) with higher correlation among individuals not clustering with the main group (r = 0.98). This indicates that one set of PCs can be converted to the other via a linear transformation for the purposes of identifying ancestral group. We regressed the exome chip PCs on the fingerprint chip PCs to obtain a linear conversion equation by which to convert PCs from the fingerprint chip to the scale of those for the exome chip. We converted PC1 for the 53 individuals without exome chip data to the exome chip scale for use in adjusting for ancestry in the validation analyses for sample size of 608. We examined all three subsets with PC data available: PCs from exome chip only, PCs from fingerprint chip only; and PCs from combined sources.
Supporting Information
Zdroje
1. Accurso FJ, Rowe SM, Clancy JP, Boyle MP, Dunitz JM, et al. (2010) Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N Engl J Med 363 : 1991–2003. doi: 10.1056/NEJMoa0909825 21083385
2. Chmiel JF, Konstan MW (2007) Inflammation and anti-inflammatory therapies for cystic fibrosis. Clin Chest Med 28 : 331–346. 17467552
3. Gibson RL, Burns JL, Ramsey BW (2003) Pathophysiology and management of pulmonary infections in cystic fibrosis. Am J Respir Crit Care Med 168 : 918–951. 14555458
4. Lee TW, Brownlee KG, Conway SP, Denton M, Littlewood JM (2003) Evaluation of a new definition for chronic Pseudomonas aeruginosa infection in cystic fibrosis patients. J Cyst Fibros 2 : 29–34. 15463843
5. Emerson J, Rosenfeld M, McNamara S, Ramsey B, Gibson RL (2002) Pseudomonas aeruginosa and other predictors of mortality and morbidity in young children with cystic fibrosis. Pediatr Pulmonol 34 : 91–100. 12112774
6. Proesmans M, Balinska-Miskiewicz W, Dupont L, Bossuyt X, Verhaegen J, et al. (2006) Evaluating the "Leeds criteria" for Pseudomonas aeruginosa infection in a cystic fibrosis centre. Eur Respir J 27 : 937–943. 16707392
7. Li Z, Kosorok MR, Farrell PM, Laxova A, West SE, et al. (2005) Longitudinal development of mucoid Pseudomonas aeruginosa infection and lung disease progression in children with cystic fibrosis. JAMA 293 : 581–588. 15687313
8. Demko CA, Byard PJ, Davis PB (1995) Gender differences in cystic fibrosis: Pseudomonas aeruginosa infection. J Clin Epidemiol 48 : 1041–1049. 7775991
9. Henry RL, Mellis CM, Petrovic L (1992) Mucoid Pseudomonas aeruginosa is a marker of poor survival in cystic fibrosis. Pediatr Pulmonol 12 : 158–161. 1641272
10. Doring G, Taccetti G, Campana S, Festini F, Mascherini M (2006) Eradication of Pseudomonas aeruginosa in cystic fibrosis patients. Eur Respir J 27 : 653. 16507869
11. Zemanick ET, Accurso FJ (2014) Cystic fibrosis transmembrane conductance regulator and pseudomonas. Am J Respir Crit Care Med 189 : 763–765. doi: 10.1164/rccm.201402-0209ED 24684355
12. Green DM, Collaco JM, McDougal KE, Naughton KM, Blackman SM, et al. (2012) Heritability of respiratory infection with Pseudomonas aeruginosa in cystic fibrosis. J Pediatr 161 : 290–295 e291. doi: 10.1016/j.jpeds.2012.01.042 22364820
13. Dorfman R, Sandford A, Taylor C, Huang B, Frangolias D, et al. (2008) Complex two-gene modulation of lung disease severity in children with cystic fibrosis. J Clin Invest 118 : 1040–1049. doi: 10.1172/JCI33754 18292811
14. Dorfman R, Taylor C, Lin F, Sun L, Sandford A, et al. (2011) Modulatory effect of the SLC9A3 gene on susceptibility to infections and pulmonary function in children with cystic fibrosis. Pediatr Pulmonol 46 : 385–392. doi: 10.1002/ppul.21372 20967843
15. Haerynck F, Van Steen K, Cattaert T, Loeys B, Van Daele S, et al. (2012) Polymorphisms in the lectin pathway genes as a possible cause of early chronic Pseudomonas aeruginosa colonization in cystic fibrosis patients. Hum Immunol 73 : 1175–1183. doi: 10.1016/j.humimm.2012.08.010 22940091
16. Emond MJ, Louie T, Emerson J, Zhao W, Mathias RA, et al. (2012) Exome sequencing of extreme phenotypes identifies DCTN4 as a modifier of chronic Pseudomonas aeruginosa infection in cystic fibrosis. Nat Genet 44 : 886–889. doi: 10.1038/ng.2344 22772370
17. Drumm ML, Konstan MW, Schluchter MD, Handler A, Pace R, et al. (2005) Genetic modifiers of lung disease in cystic fibrosis. N Engl J Med 353 : 1443–1453. 16207846
18. Wright FA, Strug LJ, Doshi VK, Commander CW, Blackman SM, et al. (2011) Genome-wide association and linkage identify modifier loci of lung disease severity in cystic fibrosis at 11p13 and 20q13.2. Nat Genet 43 : 539–546. doi: 10.1038/ng.838 21602797
19. Treggiari MM, Rosenfeld M, Mayer-Hamblett N, Retsch-Bogart G, Gibson RL, et al. (2009) Early anti-pseudomonal acquisition in young patients with cystic fibrosis: rationale and design of the EPIC clinical trial and observational study'. Contemp Clin Trials 30 : 256–268. doi: 10.1016/j.cct.2009.01.003 19470318
20. Lee S, Emond MJ, Bamshad MJ, Barnes KC, Rieder MJ, et al. (2012) Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am J Hum Genet 91 : 224–237. doi: 10.1016/j.ajhg.2012.06.007 22863193
21. McKone EF, Goss CH, Aitken ML (2006) CFTR genotype as a predictor of prognosis in cystic fibrosis. Chest 130 : 1441–1447. 17099022
22. Lawless JF (2003) Statistical Models and Methods for Lifetime Data. Hoboken, New Jersey and Canada: John Wiley & Sons.
23. Zaas DW, Swan ZD, Brown BJ, Li G, Randell SH, et al. (2009) Counteracting signaling activities in lipid rafts associated with the invasion of lung epithelial cells by Pseudomonas aeruginosa. J Biol Chem 284 : 9955–9964. doi: 10.1074/jbc.M808629200 19211560
24. Zaas DW, Swan Z, Brown BJ, Wright JR, Abraham SN (2009) The expanding roles of caveolin proteins in microbial pathogenesis. Commun Integr Biol 2 : 535–537. 20195460
25. Wright JM, Joseloff E, Nikolsky Y, Serebriyskaya T, Wetmore D (2010) Interactions between an inflammatory response to infection and protein trafficking pathways favor correction of defective protein trafficking in Cystic Fibrosis. Bioinformation 5 : 228–233. 21364822
26. Bajmoczi M, Gadjeva M, Alper SL, Pier GB, Golan DE (2009) Cystic fibrosis transmembrane conductance regulator and caveolin-1 regulate epithelial cell internalization of Pseudomonas aeruginosa. Am J Physiol Cell Physiol 297: C263–277. doi: 10.1152/ajpcell.00527.2008 19386787
27. Zaas DW, Duncan MJ, Li G, Wright JR, Abraham SN (2005) Pseudomonas invasion of type I pneumocytes is dependent on the expression and phosphorylation of caveolin-2. J Biol Chem 280 : 4864–4872. 15545264
28. Wright JM, Nikolsky Y, Serebryiskaya T, Wetmore DR (2009) MetaMiner (CF): a disease-oriented bioinformatics analysis environment. Methods Mol Biol 563 : 353–367. doi: 10.1007/978-1-60761-175-2_18 19597794
29. Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, et al. (2014) A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46 : 310–315. doi: 10.1038/ng.2892 24487276
30. Singh PK, Schaefer AL, Parsek MR, Moninger TO, Welsh MJ, et al. (2000) Quorum-sensing signals indicate that cystic fibrosis lungs are infected with bacterial biofilms. Nature 407 : 762–764. 11048725
31. Burns JL, Gibson RL, McNamara S, Yim D, Emerson J, et al. (2001) Longitudinal assessment of Pseudomonas aeruginosa in young children with cystic fibrosis. J Infect Dis 183 : 444–452. 11133376
32. Carlsson M, Eriksson L, Pressler T, Kornfalt R, Mared L, et al. (2007) Autoantibody response to BPI predict disease severity and outcome in cystic fibrosis. J Cyst Fibros 6 : 228–233. 17166780
33. Trzcinska-Daneluti AM, Ly D, Huynh L, Jiang C, Fladd C, et al. (2009) High-content functional screen to identify proteins that correct F508del-CFTR function. Mol Cell Proteomics 8 : 780–790. doi: 10.1074/mcp.M800268-MCP200 19088066
34. Lazarczyk M, Dalard C, Hayder M, Dupre L, Pignolet B, et al. (2012) EVER proteins, key elements of the natural anti-human papillomavirus barrier, are regulated upon T-cell activation. PLoS One 7: e39995. doi: 10.1371/journal.pone.0039995 22761942
35. Lazarczyk M, Favre M (2008) Role of Zn2+ ions in host-virus interactions. J Virol 82 : 11486–11494. doi: 10.1128/JVI.01314-08 18787005
36. Bezzerri V, d'Adamo P, Rimessi A, Lanzara C, Crovella S, et al. (2011) Phospholipase C-beta3 is a key modulator of IL-8 expression in cystic fibrosis bronchial epithelial cells. Journal of immunology 186 : 4946–4958. doi: 10.4049/jimmunol.1003535 21411730
37. Harder J, Meyer-Hoffert U, Teran LM, Schwichtenberg L, Bartels J, et al. (2000) Mucoid Pseudomonas aeruginosa, TNF-alpha, and IL-1beta, but not IL-6, induce human beta-defensin-2 in respiratory epithelia. American journal of respiratory cell and molecular biology 22 : 714–721. 10837369
38. Smith JL, Campos SK, Ozbun MA (2007) Human papillomavirus type 31 uses a caveolin 1 - and dynamin 2-mediated entry pathway for infection of human keratinocytes. J Virol 81 : 9922–9931. 17626097
39. Day PM, Lowy DR, Schiller JT (2003) Papillomaviruses infect cells via a clathrin-dependent pathway. Virology 307 : 1–11. 12667809
40. Derkach A, Chiang T, Gong J, Addis L, Dobbins S, et al. (2014) Association analysis using next-generation sequence data from publicly available control groups: the robust variance score statistic. Bioinformatics 30 : 2179–2188. doi: 10.1093/bioinformatics/btu196 24733292
41. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, et al. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25 : 2078–2079. doi: 10.1093/bioinformatics/btp352 19505943
42. Rosenfeld M, Emerson J, McNamara S, Thompson V, Ramsey BW, et al. (2012) Risk factors for age at initial Pseudomonas acquisition in the cystic fibrosis epic observational cohort. J Cyst Fibros 11 : 446–453. doi: 10.1016/j.jcf.2012.04.003 22554417
43. Fleming TR, Harrington, David P (2005) Conting Processes and Survival Analysis. Hoboken, New Jersey: John Wiley and Sons.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 6
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Non-reciprocal Interspecies Hybridization Barriers in the Capsella Genus Are Established in the Endosperm
- Translational Upregulation of an Individual p21 Transcript Variant by GCN2 Regulates Cell Proliferation and Survival under Nutrient Stress
- Exome Sequencing of Phenotypic Extremes Identifies and as Interacting Modifiers of Chronic Infection in Cystic Fibrosis
- The Human Blood Metabolome-Transcriptome Interface
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy