
Independent Neuronal Origin of Seizures and Behavioral Comorbidities in an Animal Model of a Severe Childhood Genetic Epileptic Encephalopathy
Childhood epilepsy syndromes, such as the early epileptic encephalopathies (EE’s) encompass seizure disorders that manifest early and negatively impact or completely block developmental progression. Recently, mutations in DNM1 (dynamin 1) have been implicated in two EE syndromes, Lennox-Gastaut Syndrome and Infantile Spasms. Dynamin 1 is a large multimeric protein that is critical for electro-chemical communication between neurons. To understand the relationship between severe seizures and the cognitive and behavioral developmental outcomes in DNM1 patients, we focus on “fitful” mice that carry a mutation in the dynamin 1 gene. Fitful mice have an EE disorder that is highly reminiscent of the documented human patients. Here, we describe genetic manipulations in the mice that allow us to determine that the seizure activity has independent cellular origins from the developmental and behavioral consequences. This separation confirms that the seizures do not cause the severe developmental delay and abnormal behaviors in this animal model and further suggests that any treatments aimed at controlling the seizures per se may not be effective for some of the most acute neurobehavioral symptoms in these patients.
Published in the journal:
. PLoS Genet 11(6): e32767. doi:10.1371/journal.pgen.1005347
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005347
Summary
Childhood epilepsy syndromes, such as the early epileptic encephalopathies (EE’s) encompass seizure disorders that manifest early and negatively impact or completely block developmental progression. Recently, mutations in DNM1 (dynamin 1) have been implicated in two EE syndromes, Lennox-Gastaut Syndrome and Infantile Spasms. Dynamin 1 is a large multimeric protein that is critical for electro-chemical communication between neurons. To understand the relationship between severe seizures and the cognitive and behavioral developmental outcomes in DNM1 patients, we focus on “fitful” mice that carry a mutation in the dynamin 1 gene. Fitful mice have an EE disorder that is highly reminiscent of the documented human patients. Here, we describe genetic manipulations in the mice that allow us to determine that the seizure activity has independent cellular origins from the developmental and behavioral consequences. This separation confirms that the seizures do not cause the severe developmental delay and abnormal behaviors in this animal model and further suggests that any treatments aimed at controlling the seizures per se may not be effective for some of the most acute neurobehavioral symptoms in these patients.
Introduction
Epileptic encephalopathies (EE’s) encompass a group of seizure disorders that impact overall development including cognitive, sensory and motor progress with severe developmental consequences and comorbidities. Recently, de novo missense mutations in DNM1 have been implicated in two EE’s, Lennox-Gastaut Syndrome and Infantile Spasms. Dynamin 1 (DNM1) is a large GTPase necessary for synaptic vesicle recycling and endocytosis at the presynapse.
Currently, at least eight patients have been documented with mutations in DNM1 [1–3]. This number is likely to grow based on the increasing efficacy of genetic diagnosis of EE by genome sequencing. Affected children suffer from infantile spasms that progress to seizures, global developmental delay, profound intellectual disability, lack of speech and general hypotonia. All patients carry de novo heterozygous missense variants which are most likely dominant negative based on in vitro studies [4]. Studies of “fitful” mice, which carry a spontaneous missense mutation in the Dnm1 gene, have demonstrated a dominant negative effect of the mutant protein in vitro and the mice display seizures and neurosensory defects [5].
The fitful mouse model recapitulates many of the features of the DNM1 EE syndrome. The homozygous mice have early onset seizures, ataxia and general decline in overall activity and health, succumbing to a seizure and demise before four weeks of age. The fitful mutation resides in an alternatively spliced exon and wildtype Dnm1 is produced from the alternate exon (Fig 1). Thus, the homozygous mice are analogous to the heterozygous patients with respect to having both wildtype and mutant dynamin 1 expressed. While a Dnm1 knockout mouse has been studied [6], neither the heterozygous nor homozygous mice have seizures and therefore do not recapitulate the syndrome observed in humans, further suggesting that both the EE-causing human DNM1 variants and fitful are gain-of-function mutations.
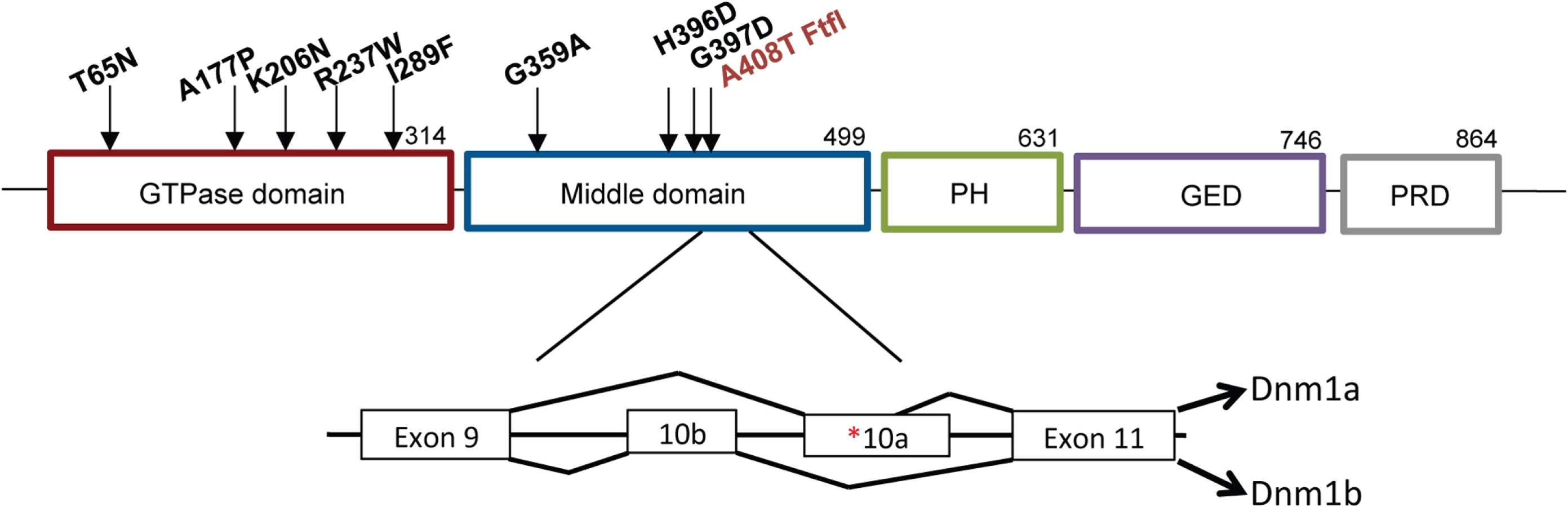
Homozygous fitful mice (Dnm1Ftfl/Ftfl) are severely affected: they exhibit a progressive ataxic movement disorder, typically lethal tonic-clonic seizures and die before four weeks of age [5]. Fitful heterozygotes (Dnm1Ftfl/+) are less severe with no movement disorder and non-lethal tonic-clonic seizures that have an onset at about two to three months of age [5]. Heterozygous Dnm1 knockout mice (Dnm1null/+) do not have any overt phenotype, whereas homozygous knockout mice exhibit perinatal lethality with a general failure to thrive, but neither genotype has seizures [6]. Compound heterozygous mice expressing the fitful mutation in combination with the null allele have been described previously (Dnm1Ftfl/null; [5]). The onset and appearance of lethal seizures is very similar between fitful homozygotes and compound heterozygotes, but their locomotor deficit manifests in a slightly different phenotype; the compound heterozygotes are not ataxic, although they exhibit a slight tremor and hunched appearance that increases continually in severity until death. See Table 1 for a summary of the dynamin 1 alleles referred to in this study.

In a given genetic disease it can be very difficult to distinguish syndromic comorbidities due to pleiotropy from those resulting from damage attributable to the seizures themselves–both are plausible (reviewed in [8,9]). In order to make the distinction for DNM1 epileptic encephalopathy, we engineered a conditional Dnm1Ftfl mouse model of DNM1 EE. Observations of Dnm1Ftfl/flox mice in combination with various neuronal subpopulation specific cre strains demonstrate unique seizure phenotypes and establish the separation of comorbidities from seizures.
Results
Generation of a conditional Dnm1Ftfl/null EE activating mouse line
In order to study the fitful mice more closely, we made a less severe conditional model by combining the dominant-negative Ftfl allele (Dnm1Ftfl/+) with a Dnm1 floxed allele (Dnm1flox/+; Fig 2, [7]). The floxed allele has loxP sites flanking exons 2–4 of Dnm1 and upon recombination using cre recombinase technology the exons are deleted resulting in a frameshift and the Dnm1 null allele (Fig 2). Mice carrying the Dnm1Ftfl and the intact floxed allele (Dnm1Ftfl/flox) are phenotypically identical to fitful heterozygous mice (Table 1). When a ubiquitous cre recombinase is used to delete the floxed allele, the resulting mice are referred to as Dnm1Ftfl/null and phenotypically resemble the compound heterozygous Dnm1Ftfl/null germline mice (Table 1). Further and importantly, this allelic combination provides a genetically manipulatable system in which the severe Dnm1Ftfl/Ftfl homozygous or compound heterozygous Dnm1Ftfl/null genotype can be recapitulated in specific neuronal populations using cell specific cre recombinase expression (Fig 2). Thus the contribution of particular neuronal populations to the Dnm1 fitful severe phenotype can be assessed. It is worth noting that cells that do not express the cre remain Dnm1Ftfl/flox heterozygous, the milder fitful genotype which exhibits a non-lethal seizure disorder with late onset.

We used several different cre recombinase-expressing mouse lines to determine the outcome of dominant negative fitful expression in specific neuronal cell populations. Table 2 details the cre mouse lines used for this study and the timing and expression in the brain according to published data. Further, to confirm the timing and expression of these lines in our hands, we crossed the various cre lines to a reporter line with a floxed-stop red fluorescent protein variant tdTomato [10]. Fig 3 shows representative sections of hippocampus, cortex and cerebellum from cre line mice crossed to tomato reporter lines. As Gad2-cre is expressed widely in inhibitory neurons [11], these mice have the most extensive reporter expression of the inhibitory lines in the tissues examined (Fig 3A and 3B). In the Pvalb-cre mice, which express cre in parvalbumin positive inhibitory neurons, there is sparse hippocampal expression in the dentate gyrus region with greater expression in the cortex and high expression in the cerebellum (Fig 3A and 3B). The Sst-cre is expressed in somatostatin positive inhibitory cells and the mice show the expected patterns in the dentate gyrus within cells residing in the hilus and projecting onto granule cells, with high expression in the cortex and discrete cells, such as Purkinje cells, in the cerebellum as well (Fig 3A). Cort-cre is expressed in cortistatin positive inhibitory neurons and the mice have expression in the cortex and hippocampus, but not the cerebellum (Fig 3A). Dlx5/6-cre expresses in inhibitory neurons of the forebrain and we see very little to no expression in the cerebellum (Fig 3A) or brainstem. Emx1-cre is highly expressed in the majority of excitatory neurons in the hippocampus and cortex, but not outside the forebrain (Fig 3A; [12]).


The extent of deletion of the floxed allele was assayed at the protein level. The Dnm1Ftfl allele is expressed in the entire brain of the Dnm1Ftfl/flox animals, therefore only a decrease in Dnm1 abundance, not a complete deletion, can be detected. We detect a decrease approaching 50% in overall Dnm1 abundance in protein from cortical lysates of Emx1-cre;Dnm1Ftfl/flox animals compared to wildtype littermates (S1 Fig), indicating that the floxed allele is being deleted from a majority of cells.
Dnm1Ftfl/null EE phenotype in inhibitory neurons leads to hyperexcitability and premature death
Dnm1Ftfl/+ and Dnm1flox/flox mice were crossed to Gad2-cre recombinase lines and then intercrossed to generate Gad2-cre;Dnm1Ftfl/flox compound heterozygous mice. Gad2-cre recombinase expresses in the majority of GABAergic inhibitory neurons [11]. The loss of Dnm1Ftfl heterozygosity in inhibitory neurons resulted in early onset lethal seizures with age at death ranging from postnatal 15–27 days old (P15-P27; Fig 4). Notably, the resulting mutants developed normally until two weeks of age and did not display any obvious neurosensory or locomotor deficits of the type that are always observed in Dnm1Ftfl/Ftfl homozygotes. These results demonstrate that selective GABAergic neuron deletion of wildtype Dnm1 in the presence of the fitful allele is sufficient for seizure generation, but not the most overt locomotor comorbidities.

Graded seizure onset and lethality corresponds to the range of Dnm1 deletion
We reasoned that activating the EE genotype in a smaller subset of interneurons, such as the parvalbumin positive interneurons, would result in a less severe phenotype than activation in the majority of interneurons as above. Parvalbumin expressing interneurons represent about 40% of GABAergic interneurons and include fast spiking neurons that play a role in perisomatic inhibition of pyramidal neurons and cortical network oscillations [15–17]. Pyramidal cells are the primary excitatory neurons of the cortex and hippocampus. However, observations of seizure onset and lethality in the Pvalb-cre;Dnm1Ftfl/flox mice revealed a phenotype similar in severity to the Gad2-cre;Dnm1Ftfl/flox compound heterozygotes. The mice have early onset lethal seizures with a 91% fatality rate before P28 (Fig 4). We have video recorded these mice to confirm the seizure as the initiating event. The few mice that survived past 4 weeks of age succumbed to seizures within the next couple of weeks. In general, the young mice had no overt behavioral comorbidities. The few older surviving mice had visible locomotor dysfunction that appeared as a full body tremor or shakiness upon walking.
Somatostatin expressing (Sst) interneurons comprise about 30% of GABAergic neurons and have increased morphological and connective diversity than parvalbumin expressing neurons, but they also have a more discrete population [18,19]. In contrast to Pvalb-cre;Dnm1Ftfl/flox compound heterozygotes, Sst-cre;Dnm1Ftfl/flox mice have a variable seizure onset ranging from very early in life at P15 to later in life at P40 and older. Nevertheless, almost all of the mice have a lethal seizure event; 22% succumb before 4 weeks old and surviving mice ultimately have a lethal seizure before six months of age (Fig 4). Using the Cort-cre line which expresses in a limited amount of cortistatin positive interneurons [11], we see a longer lifespan for the mutants but 47% do succumb to seizures by six months of age (Fig 4). This suggests a graded seizure onset and lethality corresponding to the extent of Dnm1 depletion and also points to network-specific effects that may contribute to decreased inhibition and the seizure disorder. We note that while this timing does overlap with the normal onset of seizures in germline Dnm1Ftfl/+ heterozygotes, those seizures are never lethal.
Diversity of EEG phenotypes from the Dnm1 defect
We performed electroencephalography (EEG) analysis on the first Pvalb-cre;Dnm1Ftfl/flox mouse that survived beyond 3 weeks of age. EEG recordings from this mouse showed frequent, distinctive 5 Hz spike-wave discharges (SWDs) when the mouse was not moving (Fig 5A). It had on average 230 SWD episodes/hour of almost one second duration in general, with some extraordinary episodes of over 2 minutes. The antiepileptic drug, ethosuximide, completely blocked all SWD activity for a period of 15 minutes post injection. Of the other cre mice tested, only the Sst-cre;Dnm1Ftfl/flox mice showed any abnormal EEG activity including high amplitude poly-spike and slow spike-waves that end abruptly followed by a quiet baseline (Fig 5B). These data show the diversity of the EEG phenotypes arising from the dynamin 1 defect when spatial etiologies can be dissected systematically.

Forebrain-specific activation of the Dnm1Ftfl/null EE phenotype
The lethality of the seizures suggests that the hindbrain/brainstem is involved. The brainstem region, and circuitry that initiates in the cortex and connects through the mid/hindbrain (including the cerebellum) to the brainstem, is important to regulate cardiovascular, respiratory and central nervous system autonomous functions. Malfunction of this system is suspected to underlie sudden unexpected death in epilepsy (SUDEP; [20]). To test the hypothesis that disruption of neurotransmission in the hindbrain/brainstem is sufficient for the lethality of the seizures, we used the forebrain GABAergic neuron specific cre recombinase Dlx5/6-cre [21] to remove Dnm1 from a subset of forebrain inhibitory neurons and specifically leave the hindbrain intact. We expected the mice to have non-lethal seizures, but the Dlx5/6-cre;Dnm1Ftfl/flox mice experienced early onset lethal seizures, much like the Gad2-Dnm1Ftfl/flox mice (Fig 4). Before seizure onset mice appeared healthy, but 100% died by P16-P28. This suggests that acute deletion of Dnm1 from the hindbrain is not required for lethality and that the seizure generates in the forebrain and most likely propagates through the rest of the brain and the brainstem. This may exemplify a primary defect that has secondary consequences.
Activation of the Dnm1Ftfl/null EE phenotype in glutamatergic neurons is not sufficient for severe lethal seizures
Deletion of wildtype Dnm1 from cortical and hippocampal excitatory neurons with Emx1-cre [12] results in mice that are viable and fertile with normal lifespans (Fig 4). In contrast to GABAergic Dnm1 compound heterozygotes (Gad2-cre;Dnm1Ftfl/flox or Pvalb-cre;Dnm1Ftfl/flox mice), these mice do not have early onset lethal seizures and also have normal EEG (Fig 5C). The mice do have later onset non-lethal seizures (68% of mice; n = 38) that begin between the ages of 53–146 days old. This is consistent with the fitful heterozygous phenotype and is expected due to the non-cre expressing neurons maintaining the heterozygous genotype (Dnm1Ftfl/flox). Also, Emx1-cre;Dnm1Ftfl/+ heterozygous littermate mice (without the floxed allele) have a similar seizure phenotype with onset at 57–182 days of age (70% of mice; n = 30). Initial observations suggested that the compound heterozygous mice (Emx1-cre;Dnm1Ftfl/flox) exhibit several abnormalities which preceded seizure onset. We noted that they fail to thrive when weaned onto normal mouse chow solid diet (requiring a gel-based solid instead), are visibly smaller than littermates, have postural abnormalities, are hyperactive and demonstrate repetitive behavior in the form of stereotypical running around the perimeter of their homecage. Weights of adult mice show that the mutants are on average 20–30% smaller than control littermates (Emx1-cre;Dnm1flox/+ vs Emx1-cre;Dnm1Ftfl/flox; Fig 6, top). Analysis of posture using a dynamic weight bearing assay demonstrated that the mutants distribute more weight on their hindpaws than the forepaws relative to sex-matched controls (Emx1-cre;Dnm1flox/+ vs Emx1-cre;Dnm1Ftfl/flox; Fig 6, bottom) or to fitful heterozygous comparison mice (Dnm1Ftfl/+ vs Emx1-cre;Dnm1Ftfl/flox; Fig 6, bottom) confirming the altered posture observed. Neither the Emx1-cre;Dnm1flox/+ nor Emx1-cre;Dnm1Ftfl/+ littermates had these abnormalities. Table 3 summarizes the overall seizure and behavioral phenotypes observed in the six different cre lines documented above.


Emx1-cre;Dnm1Ftf/flox mice show abnormal locomotor, exploratory and repetitive behavior
To examine the neurobehavioral phenotypes observed and any potential subtle differences in the Emx1-cre;Dnm1Ftf/flox compound heterozygotes, mice were evaluated in a behavioral testing battery. We used littermate controls that carried the Emx1-cre and the Dnm1flox allele, but lacked the Dnm1Ftfl allele (Emx1-cre;Dnm1flox/+) and also a comparison group that carried the Dnm1Ftfl allele, but not the Emx1-cre (Dnm1Ftfl/+). This allowed us to compare the Emx1-cre compound heterozygotes to controls as well as fitful heterozygotes to ask if the behavioral deficits were due to off target effects of the cre, or due to the heterozygous fitful allele independent of neuron specific deletion of the wildtype allele. The mice were tested in a comprehensive battery and there were no significant differences observed for mutants compared to controls for several tests such as social approach, spontaneous alteration and grip strength. Furthermore, several test results were confounded by the hyperactivity demonstrated in the mutants (see below). The forced swim test, novel spatial recognition, rotarod and gait assessment were inconclusive due to the hyperactivity and/or repetitive behavior of the mutants.
In the open field, male and female Emx1-cre;Dnm1Ftfl/flox mice demonstrated significant increases in total distance traveled relative to sex-matched control and fitful heterozygous mice confirming the observed hyperactivity (Fig 7A). Both male and female mutant mice also demonstrated significant reductions in rearing behavior, as measured by vertical beam breaks (Fig 7B). The reduced rearing may be confounded by, but is also consistent with, the increased hyperactivity and indicates altered exploratory activity. Interestingly, mutant males demonstrated a significant increase in stereotypical rotational behavior relative to control males, which was not observed in females (Fig 7C). This increase in rotational behavior is consistent with anecdotal observations of the repetitive running behavior observed in the homecage and confirms the repetitive behavior of the mutants. Male and female mutants also demonstrated significant disruptions in wheel running activity, both total time spent running and speed, over a 48-hour period relative to sex-matched controls (Fig 7D). Mice were also evaluated for cognitive phenotypes, but all tasks except for one were confounded by the hyperactivity observed in the mutants. The spontaneous alternation task was successfully carried out and demonstrated intact hippocampal working memory in the mutants compared to controls and fitful heterozygous mice (S2 Fig).

Altered anxiety and depressive related behaviors in Emx1-cre;Dnm1Ftfl/flox mice
Emx1-cre compound heterozygotes exhibited increases in time spent at the perimeter of the open field relative to sex-matched controls which is indicative of an anxiogenic-like phenotype (Fig 8A). Male and female mutant mice also demonstrated significant increases in immobility times relative to sex-matched controls in the tail suspension test (Fig 8B), indicative of a depressive-like behavior.

Altered dopaminergic and glutamatergic response in Emx1-cre;Dnm1Ftfl/flox mice
Stimulation of locomotor activity by amphetamine was exacerbated in both male and female mutant Emx1-cre;Dnm1Ftfl/flox mice relative to controls immediately post drug administration (Fig 9, top). Challenge with the NMDA antagonist MK801 also resulted in an exacerbated increase in locomotor activity in mutant males compared to male controls, while there was no significant difference in female mice across genotypes (Fig 9, bottom).

Discussion
The large GTPase DNM1 has recently been confirmed as an EE gene. DNM1 patients have seizures that begin early in life and usually manifest as infantile spasms. These patients exhibit developmental, cognitive, behavioral and neurological deficits that do not appear to respond to traditional therapy.
Seizure onset in DNM1 patients ranges from 2–13 months of age and usually presents with infantile spasms. The seizure type manifests in various forms as the patient ages, ranging from absence seizures to generalized tonic clonic seizures. EEG recordings in these patients reveal varied epileptiform discharges initiating as hypsarrhythmia, and evolving from slow generalized spike-wave discharges to paroxysmal fast activity [1,3]. Patients have not responded well to antiepileptic therapy, with the majority being resistant. Interestingly, two patients, with DNM1 middle domain mutations, have had seizure control on the ketogenic diet while two other patients, with GTPase domain mutations, had no relief on the diet [1,3]. Commonly, the patients were reported to have hypotonia with one child presenting with ataxia and mild tremor. All patients have severe intellectual disability and developmental delay. One child is reported to have autism spectrum disorder [3]. For two patients, evidence of developmental delay was noted before the onset of epilepsy suggesting an effect of DNM1 variants on neurodevelopment unrelated to seizure activity [3].
Fitful mice recapitulate the disorder documented in DNM1 patients and exhibit an early onset seizure syndrome and comorbid behavioral alterations. Recently, Dhindsa et al [4] characterized several human DNM1 variants at the cellular level. Noticeably, the human G359A variant, located in the same domain as fitful (middle; see Fig 1), had a cellular phenotype consistent with fitful when expressed in Cos-7 cells. Both mutations conferred a decrease in endocytosis, a reticular dynamin 1 distribution and inefficient oligomerization [4,5]. The other human variants studied (A177P, K206N; Fig 1), located in the GTPase domain and expected to disrupt GTPase activity, had decreased endocytosis, altered localization and vesicle abnormalities when overexpressed in cells [4]. While some of the human variants and fitful reside in different domains of dynamin 1, they all significantly decrease endocytosis suggestive of a dominant-negative effect. It is plausible that the GTPase domain and middle domain mutations cause dynamin 1 protein dysfunction by different mechanisms (GTPase activity versus oligomerization) that ultimately result in defective dynamin 1 activity and inefficient endocytosis. In the case of the patients, the dominant negative effect of DNM1 results in a generally similar overall phenotype suggesting a similar pathway, the most probable mechanism being decreased or inefficient endocytosis resulting in synaptic vesicle pool depletion. Inhibitory neurons are more sensitive to a lack of Dnm1 possibly due to their tonic activity [22] and lack of vesicle replenishment in these neurons would cause decreased inhibition and network hyperactivity.
Many brain disorders, including the epileptic encephalopathies as well as Fragile X syndrome, Rett syndrome, tuberous sclerosis, Lennox-Gastaut syndrome and West syndrome (reviewed in [23,24]), manifest early in life and are accompanied by spasms and/or seizures that are usually intractable. The high level of cognitive and behavioral anomalies that co-occur in these patients with syndromes of varying genetic etiologies are suggestive of disruptions during brain development. Patients exhibit developmental, cognitive, behavior and neurological deficits that often do not respond to therapies and may lead to early death. The relentless seizure activity is correlated with, and generally assumed to contribute to, the progressive cognitive decline. This may not be the case at least for DNM1 encephalopathy, suggested by the fact that two of the DNM1 patients became seizure free on the ketogenic diet but still have the above mentioned syndromic features and developmental delay was noted in two patients even before the onset of epilepsy. Using the fitful mice as a model of the severe DNM1 EE, we have demonstrated clear separation of the seizure etiology from several major behavioral and neurological comorbidities. This strongly supports the conclusion that amelioration of the seizures is unlikely to curtail the severe behavioral deficits.
Wildtype Dnm1 deletion from the majority of Dnm1Ftfl inhibitory neurons is sufficient to cause early onset epilepsy and premature death, without other obvious behavioral impairment. The seizure etiology varies depending on the amount of neurons that have deleted wildtype dynamin 1, but also on the specific type of neuron or circuitry. Deleting wildtype Dnm1 from the majority of interneurons results in 100% loss of viability before weaning age in inhibitory specific compound heterozygotes. Sparse interneuron deletion results in occasional adult onset seizures that are rarely lethal as observed with Cort-cre. Interestingly, parvalbumin expressing interneurons which account for about 40% of GABAergic neurons appear to have a very important role in controlling excitation as deletion of wildtype Dnm1 from these neurons has very severe consequences. Comparatively, the compound heterozygous fitful mutation expressed in somatostatin positive interneurons (about 30% of GABAergic neurons) leads to a less severe disorder. Notably, we observed differences in EEG recordings - indicating alterations in seizure pathology - from Dnm1Ftfl/flox mice crossed to the Pvalb and the Sst cre driver lines. These suggest that there may be network-specific effects influencing the magnitude and scope of discrete components of the seizure disorder. This type of circuitry malfunction, while beyond the scope of this study, will be critical to dissect in the future.
Conversely, impaired synaptic function due to Dnm1 mutation in excitatory neurons results in significant behavioral impairment prior to the emergence of seizures. This suggests that at least several major behavioral comorbidities are not explicitly caused by the seizures, thus separating their etiology. These mice are visibly smaller, hyperactive, repetitive and have reduced rearing (exploratory) activity in their cages. Interestingly, hyperactivity and lower exploratory activity have been reported for autism spectrum disorder (ASD) patients [25] and repetitiveness is a core characteristic [26,27]. The excitatory Dnm1 deleted mice also displayed decreased motivation, increased depression and anxiousness. Due to the hyperactivity in the mutants, it precluded the ability to evaluate their cognitive phenotype without ruling out locomotor confounds in tests of learning and memory with the exception of spontaneous alternation task which demonstrated intact hippocampal working memory (see supplemental data). Taken together, the observations suggest that presynaptic dysfunction of the excitatory neurons leads to behavioral alterations associated with psychiatric comorbidities, such as documented for ASD and hyper-reactivity to glutamatergic and dopaminergic blockade.
It seems likely that there could also be behavioral abnormalities associated with the GABAergic neuron phenotypes, perhaps even ones that appear to be normal in the excitatory compound heterozygotes. However, the challenge is that the very severely affected mice do not live long enough to begin behavioral assays with them. An option for the future will be to analyze the behavior of the GABAergic compound heterozygotes that live longer using the inhibitory cres with graded expression, such as the Sst-cre or Cort-cre.
There are limitations to the genetic system utilized in this study. The most obvious limitation being the difficulty in quantitating the actual extent and specificity of deletion of the floxed allele. We have addressed this by analyzing the Emx1-cre;Dnm1Ftfl/flox line reasoning that this cre expresses in the majority of glutamatergic cells in the brain and we should be able to observed a change in Dnm1 levels in such a large population of cells. Indeed, we do see a 50% decrease in Dnm1 levels in these mice, indicating a deletion of one Dnm1 allele. Further, as a correlate, we observe a graded seizure phenotype in the mice that corresponds to the amount of inhibitory neurons that we are targeting for deletion. While indirect, this observation supports the concept that the floxed allele is deleting in a predicted proportion of the neurons.
Another potential caveat regards the degree to which one can interpret the clinical significance of genetically manipulating a subset of neurons in a model system of a human disorder where the defect presumably occurs in all neurons that express the gene. For example, in some mouse models such as the Dravet Syndrome SCN1a mutants, disrupting the gene in individual populations of neurons does not replicate the human phenotype, suggesting that an intact inhibitory/excitatory circuit is required for the full disorder [28]. Although the small number of reported DNM1 patients have a uniformly severe disorder, we do not know the degree of de novo variant mosaicism in these patients. It is difficult to know whether even partial DNM1 genotypes can also lead to severe outcomes or whether mosaic outcomes would be milder as has been observed in some developmental excitability disorders, such as X-linked Rett Syndrome [29]. In our case, to varying degrees all inhibitory neuron cre mice displayed the lethal seizure phenotypes including sparser Sst - and Cort-cre, demonstrating that widespread dynamin-1 dysfunction is not required for severe seizures at least. Regardless, while we cannot know formally that severe seizures observed in Dnm1Ftfl/flox inhibitory cre conditional mutants have the same network pathophysiology as those that occur in the germline condition, such uncertainty does not affect our key observation that overtly impaired excitatory conditional mutants do not have a severe seizure disorder of any kind.
Wildtype Dnm1 deletion from Dnm1Ftfl inhibitory neurons is sufficient to cause early onset epilepsy and premature death, without other obvious behavioral impairment. Wildtype Dnm1 deletion from Dnm1Ftfl excitatory neurons results in behavioral impairment without seizures, strongly suggesting that the behavioral comorbidities are not caused by the seizures in the germline mutant mice, with clear implications for human DNM1 patients. Together these results suggest that preventing seizures per se in Dnm1 fitful mice, and potentially EE patients, may not prevent all of the severe comorbidities.
Materials and Methods
Generation and maintenance of mouse lines
All mice were housed and procedures performed with approval of Institutional Animal Care and Use Committee (IACUC). All mice were obtained from The Jackson Laboratory, maintained in a room with a 14 hour light on/10 hour light off cycle, and given free access to LabChow meal and water.
The fitful mice, B6.SZ, arose at The Jackson Laboratory (Bar Harbor, ME) as a spontaneous mutation on the C57BL/6J inbred strain in 2000 [5]. Dnm1tm2.1Pdc/J mice contain loxP sites flanking exons 2–4 of the Dnm1 gene (Fig 2; [7]). Mice obtained from The Jackson Laboratory were backcrossed and maintained on a C57BL/6J background. We refer to mice with this allele as the floxed mice, or Dnm1flox/flox. Homozygous floxed mice are viable, fertile and normal in size with no obvious phenotypic abnormalities. Mouse lines obtained from JAX: STOCK Gad2tm2(cre)Zjh/J (backcrossed to C57BL/J 6 generations), B6;129P2-Pvalbtm1(cre)Arbr/J, STOCK Ssttm2.1(cre)Zjh/J (backcrossed to C57BL/J 6 generations), STOCK Corttm1(cre)Zjh/J (backcrossed to C57BL/J 5 generations), STOCK Tg(Dlx6a-cre)1Mekk/J (backcrossed fully to C57BL/J), B6.129S2-Emx1tm1(cre)Krj/J (backcrossed fully to C57BL/6), B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J.
Genotyping
Genotypes were determined by PCR using tail DNA. Genotyping Dnm1Ftfl (fitful) was done as previously [5]. The Dnm1tm2.1Pdc/J allele was PCR amplified with specific primers (condF, 5’CAGCTGGGTATAATGAGGCCTCATC3’; condR, 5’GCATGGGTGCAC TCACATACACAA3’). Presence of cre in the various strains was determined by PCR amplification with generic cre primers (oIMR1084, 5’GCGGTC TGGCAGTAAAAACTA C3’; oIMR1085, 5’GTGAAACAGCATTGCTGTCACTT3’; oIMR7338, 5’CTAGGCCACAGAATTGAAAGATCT3’; oIMR7339, 5’GTAGGTGGAAAT TCTAGCATCATCC3’).
Immunohistochemistry
Mice were anesthetized with tribromoethanol, perfused with 0.1M PBS then 4% paraformaldehyde in 0.1M PBS and postfixed overnight. Sagittal sections of 60–70 μm thick were cut on a vibratome. Sections were blocked and permeabilized for 4.5 hours in PBS 1% BSA 4% Goat serum and 2.3% Triton-X, then incubated in primary antibody (see dilutions below) overnight at 4 degrees Celsius while rocking. Sections were then washed and incubated in 1 hour in secondary antibody (goat anti-mouse conjugated to AlexaFluor 488; Invitrogen), 1 : 1000 in PBS 1% BSA 4% Goat serum and 0.3% Triton-X. After another wash, sections were stained for 5 minutes with DAPI (1 : 1000 in PBS), washed again, and mounted on Shandon Colorfrost Plus slides (Thermo Scientific) with Fluorogel (Electron Microscopy Sciences). Confocal images were taken on a Leica SP5 microscope.
Reverse-transcription PCR
Total RNA was prepared from brains with Trizol (Invitrogen) following the manufacturer's suggested conditions and protocol. RNA (0.5μg) was reverse transcribed with AMV reverse transcriptase (Promega). cDNA was diluted and amplified for 25 cycles at an annealing temperature of 55°C with primers spanning Dnm1 exons 9 to 12 (Dnm1ex9F, 5′-GAACTGCGA AGGGAGATCAG-3′; Dnm1ex12R, 5′-GGTCACAATTCGCTCCATCT-3′).
Western blot
Protein extracts were made in IP lysis buffer (10 mM HEPES pH 7.5, 137 mM NaCl, 0.4% NP-40, 10% glycerol) with Complete-mini proteinase inhibitor mix (Roche) added fresh. Extracts were quantified using the Bradford reagent (Bio-Rad). Extracts (50 μg protein) were diluted in Laemmli buffer, incubated at 95°C for 5 minutes, resolved by SDS-PAGE and transferred to nitrocellulose membrane. All membrane blotting steps were carried out in TBS plus Tween (TBST). Blots were blocked in 5% milk, then incubated at RT with primary antibody (for specific dilutions see below) for 4 hours, HRP-conjugated secondary antibody (1 : 15000) for 1h and visualized with Luminata Forte (Millipore). Membranes were incubated with Restore Western blot stripping buffer (Thermo Scientific) at room temperature for 10 minutes while shaking to remove antibodies for subsequent hybridization. Primary antibodies used were dynamin-1 (1 : 3000; Thermo Scientific, PA1-660), actin (1 : 1000; Abcam, ab3280). Secondary antibodies used were HRP anti-mouse (Thermo Scientific), HRP anti-rabbit (BioRad).
Spontaneous seizure analysis and 24-hour video recording
Mice were observed for age-of-onset, frequency and severity in their homecage for handling evoked seizures during weekly cage changing for at least 175 days. 24-hour recording was done using infrared-video on isolated mice in a chamber.
EEG
Adult mice aged between 6 and 8 weeks were anesthetized with tribromoethanol (400 mg/kg i.p.). Small burr holes were drilled (1 mm anterior to the bregma and 2 mm posterior to the bregma) on both sides of the skull 2 mm lateral to the midline. Four teflon-coated silver wires were soldered onto the pins of a microconnector (Mouser electronics, Texas). The wires were placed between the dura and the brain and a dental cap was then applied. The mice were given a post-operative analgesic of carprofen (5 mg/kg subcutaneous) and allowed a minimum 48 h recovery period before recordings. Differential amplification recordings were recorded between all four electrode pairs, providing 6 channels for each subject. Mice were connected to the EEG Stellate Lamont Pro-36 programmable amplifier (Lamont Medical Instruments, Madison, WI) for a 24-hour period with accompanying video recording. EEG data were recorded with Stellate Harmonie software (Stellate Systems, Inc., Montreal, Canada). The antiepileptic drug, ethosuximide (Sigma), was dissolved in saline and administered by intraperitoneal injection at a dose of 200mg/kg. EEG and video recordings were continued for a further hour.
Behavioral testing
Adult male and female Emx1-cre;Dnm1flox/+ (control), Dnm1Ftfl/+ (fitful heterozygotes) and Emx1-cre;Dnm1Ftfl/flox (mutant) mice (n = 7–8 per sex, per genotype) were evaluated in a battery of behavioral tests. Order of testing was as follows: Open field, tail suspension, dynamic weight bearing, wheel running, amphetamine stimulation, MK-801 stimulation. Behavioral tests were conducted by trained technicians blind to genotype. All testing was performed during the light cycle with at minimum 1 day of rest between tests and 1 week between pharmacological challenges. Sexes were independently analyzed due to the sexually dimorphic behaviors. A one-way ANOVA within sex followed by a Dunnett’s post-hoc test with wildtype as control was utilized to compare data for tests defined by one factor. A two-way repeated measures ANOVA (treatment x time) with a Bonferroni post-hoc analysis was computed for tests involving more than one treatment and multiple timepoints/trials. Explicit use of these measurements is stated in the figure legends.
Spontaneous open field activity
Mice were acclimated to an anteroom adjacent to the testing room for a minimum of 60 min. Subjects were individually placed into open field arenas (40 x 40 x 40 cm; Omnitech Electronics, Inc, Columbus, OH USA) housed in sound attenuated chambers with chamber lighting ~ 500 lux. Distance traveled (cm), vertical activity, perimeter and center time (sec) and rotational behavior were recorded via infrared beams (Versamax Software; Omnitech Electronics, Inc.) over the course of a 30 min period.
Tail suspension test
Mice were acclimated to the testing room for a 60 min period under ambient lighting conditions (~ 20 lux). During the test, subjects are suspended by their tails with laboratory tape to a flat metal bar connected to a force transducer (Med-Associates, St. Albans, VT USA) for a 6 minute testing period. Immobility time is automatically calculated by Med Associates software. A significant number of female Emx1-cre;Dnm1flox/+ mice (N = 3 of 8), one Dnm1Ftfl/+ female (N = 1 of 8) and one Emx1-cre;Dnm1Ftfl/flox female (N = 1 of 8) were excluded from the assay due to tail climbing which is common in C57Bl/6J mice in this assay [30].
Dynamic weight bearing
Weight bearing and postural abnormalities were evaluated in the Dynamic Weight Bearing Apparatus (EB-Instruments, Pinellas Park, FL USA). Subjects were weighed and acclimated to the testing room for a 60 min habituation period. Following acclimation, mice were individually placed in the chamber and allowed to move freely within the apparatus for a 3–5 min test periodDuring the data capture, the raw data for each paw is synchronized with the images from a video camera and the averaged values are recorded with a sampling rate of 10Hz. Paw surface area (mm2) was normalized to body weight.
Wheel running
Mice were individual housed in a new cage with ad libitum food and water, and a running wheel equipped with a wireless transponder (Med-Associates, St. Albans, VT USA). Mice were left undisturbed throughout the 48 hour recording period. Data were analyzed for distance traveled (revolutions per minute; rpm) and time spent running (min).
Amphetamine and MK-801 stimulated locomotor activity
Mice were habituated to the open field arenas for a 90 min session under ambient lighting (~50 lux). The arenas used for this test were the same open field arenas previously used to evaluate spontaneous activity (as described above). At the conclusion of the 90 min session, mice were challenged with amphetamine (1 mg/kg, i.p.), immediately returned to the chamber, and behavior was recorded for an additional 60 min period. One week following amphetamine challenge, mice were subjected to an acute challenge with the NMDA antagonist MK-801 (0.178 mg/kg, sc) following identical procedures used for the amphetamine challenge. Test compounds: d-amphetamine hemisulfate and (+) MK-801 hydrogen maleate were purchased from Sigma (St. Louis, MO USA) and dissolved fresh in NaCl. All drug concentrations were corrected for % active moiety of the free base and administered at an injection volume of 10 ml/kg.
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. EuroEPINOMIC-RES Consortium EPGP, Epi4K Consortium. De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am J Hum Genet. 2014;95 : 360–370. doi: 10.1016/j.ajhg.2014.08.013 25262651
2. Deciphering Developmental Disorders Study. Large-scale discovery of novel genetic causes of developmental disorders. Nature. 2015;519 : 223–228. doi: 10.1038/nature14135 25533962
3. Allen AS, Berkovic SF, Cossette P, Delanty N, Dlugos D, Eichler EE, et al. De novo mutations in epileptic encephalopathies. Nature. 2013;501 : 217–221. doi: 10.1038/nature12439 23934111
4. Dhindsa RS, Bradrick SS, Yao XY, Heinzen EL, Petrovski S, Krueger BJ, et al. Epileptic encephalopathy causing DNM1 mutations impair synaptic vesicle endocytosis. Neurol Genet. 2015;1: e4.
5. Boumil RM, Letts VA, Roberts MC, Lenz C, Mahaffey CL, Zhang ZW, et al. A missense mutation in a highly conserved alternate exon of dynamin-1 causes epilepsy in fitful mice. PLoS Genet. 2010;6.
6. Ferguson SM, Brasnjo G, Hayashi M, Wolfel M, Collesi C, Giovedi S, et al. A selective activity-dependent requirement for dynamin 1 in synaptic vesicle endocytosis. Science. 2007;316 : 570–574. 17463283
7. Ferguson SM, Raimondi A, Paradise S, Shen H, Mesaki K, Ferguson A, et al. Coordinated actions of actin and BAR proteins upstream of dynamin at endocytic clathrin-coated pits. Dev Cell. 2009;17 : 811–822. doi: 10.1016/j.devcel.2009.11.005 20059951
8. van Rijckevorsel K. Cognitive problems related to epilepsy syndromes, especially malignant epilepsies. Seizure. 2006;15 : 227–234. 16563807
9. Wagner GP, Zhang J. The pleiotropic structure of the genotype-phenotype map: the evolvability of complex organisms. Nat Rev Genet. 2011;12 : 204–213. doi: 10.1038/nrg2949 21331091
10. Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nature Neurosci. 2010;13 : 133–140. doi: 10.1038/nn.2467 20023653
11. Taniguchi H, He M, Wu P, Kim S, Paik R, Sugino K, et al. A resource of Cre driver lines for genetic targeting of GABAergic neurons in cerebral cortex. Neuron. 2011;71 : 995–1013. doi: 10.1016/j.neuron.2011.07.026 21943598
12. Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JL, Jones KR. Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J Neurosci. 2002;22 : 6309–6314. 12151506
13. Hippenmeyer S, Vrieseling E, Sigrist M, Portmann T, Laengle C, Ladle DR, et al. A developmental switch in the response of DRG neurons to ETS transcription factor signaling. PLoS Biol. 2005;3: e159. 15836427
14. Potter GB, Petryniak MA, Shevchenko E, McKinsey GL, Ekker M, Rubenstein JL. Generation of Cre-transgenic mice using Dlx1/Dlx2 enhancers and their characterization in GABAergic interneurons. Mol Cell Neurosci. 2009;40 : 167–186. doi: 10.1016/j.mcn.2008.10.003 19026749
15. Bartos M, Vida I, Jonas P. Synaptic mechanisms of synchronized gamma oscillations in inhibitory interneuron networks. Nature Rev Neurosci. 2007;8 : 45–56.
16. Markram H, Toledo-Rodriguez M, Wang Y, Gupta A, Silberberg G, Wu C. Interneurons of the neocortical inhibitory system. Nature Rev Neurosci. 2004;5 : 793–807.
17. McBain CJ, Fisahn A. Interneurons unbound. Nature Rev Neurosci. 2001;2 : 11–23.
18. Dutton SB, Makinson CD, Papale LA, Shankar A, Balakrishnan B, Nakazawa K, et al. Preferential inactivation of Scn1a in parvalbumin interneurons increases seizure susceptibility. Neurobiol Dis. 2012;49C: 211–220.
19. Rudy B, Fishell G, Lee S, Hjerling-Leffler J. Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Dev Neurobiol. 2010;71 : 45–61.
20. Tolstykh GP, Cavazos JE. Potential mechanisms of sudden unexpected death in epilepsy. Epilepsy Behav. 2013;26 : 410–414. doi: 10.1016/j.yebeh.2012.09.017 23305781
21. Monory K, Massa F, Egertova M, Eder M, Blaudzun H, Westenbroek R, et al. The endocannabinoid system controls key epileptogenic circuits in the hippocampus. Neuron. 2006;51 : 455–466. 16908411
22. Hayashi M, Raimondi A, O'Toole E, Paradise S, Collesi C, Cremona O, et al. Cell - and stimulus-dependent heterogeneity of synaptic vesicle endocytic recycling mechanisms revealed by studies of dynamin 1-null neurons. PNAS. 2008;105 : 2175–2180. doi: 10.1073/pnas.0712171105 18250322
23. Brooks-Kayal A. Molecular mechanisms of cognitive and behavioral comorbidities of epilepsy in children. Epilepsia. 2011;52 Suppl 1 : 13–20. doi: 10.1111/j.1528-1167.2010.02906.x 21214535
24. Nieh SE, Sherr EH. Epileptic encephalopathies: new genes and new pathways. Neurotherapeutics: the journal of the American Society for Experimental NeuroTherapeutics. 2014;11 : 796–806.
25. Pierce K, Courchesne E. Evidence for a cerebellar role in reduced exploration and stereotyped behavior in autism. Biol Psychiatry. 2001;49 : 655–664. 11313033
26. World Health Organization (1992) The ICD-10 Classification of Mental and Behavioural Disorders. Geneva, Switzerland.
27. American Psychiatric Association (1994) Diagnostic and Statistical Manual of Mental Disorders. Washington D.C.
28. Ogiwara I, Iwasato T, Miyamoto H, Iwata R, Yamagata T, Mazaki E, et al. Nav1.1 haploinsufficiency in excitatory neurons ameliorates seizure-associated sudden death in a mouse model of Dravet syndrome. Hum Mol Gen. 2013;22 : 4784–4804. doi: 10.1093/hmg/ddt331 23922229
29. Youssoufian H, Pyeritz RE. Mechanisms and consequences of somatic mosaicism in humans. Nat Rev Genet. 2002;3 : 748–758. 12360233
30. Mayorga AJ, Lucki I. Limitations on the use of the C57BL/6 mouse in the tail suspension test. Psychopharmacol (Berl). 2001;155 : 110–112.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 6
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Non-reciprocal Interspecies Hybridization Barriers in the Capsella Genus Are Established in the Endosperm
- Translational Upregulation of an Individual p21 Transcript Variant by GCN2 Regulates Cell Proliferation and Survival under Nutrient Stress
- Exome Sequencing of Phenotypic Extremes Identifies and as Interacting Modifiers of Chronic Infection in Cystic Fibrosis
- The Human Blood Metabolome-Transcriptome Interface
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy