
FGFR3 Deficiency Causes Multiple Chondroma-like Lesions by Upregulating Hedgehog Signaling
Benign cartilaginous tumors, especially osteochondromas and enchondromas, are the most common primary bone tumors in humans. Several hereditary diseases are characterized by the development of cartilaginous tumors, including hereditary multiple exostoses, metachondromatosis, and enchondromatosis, which are caused by mutations in genes such as exostosin 1 and 2, tyrosine protein phosphatase non-receptor type 11, parathyroid hormone receptor 1, and isocitrate dehydrogenase 1 and 2. The proteins encoded by these genes are directly or indirectly linked to fibroblast growth factor (FGF) signaling. In addition, osteochondroma was found in several members of a family with camptodactyly, tall stature, and hearing loss syndrome, a rare inherited disorder caused by a heterozygous missense mutation in Fgfr3. In this study, we found that Fgfr3 deficiency leads to the formation of cartilaginous tumors, including osteochondromas and enchondromas, likely due to dysregulated endochondral ossification in growth plates. We also show that cartilaginous tumorigenesis in Fgfr3-deficient mice results from excessive Indian hedgehog production mediated by the activation of mitogen-associated protein kinase signaling. Based on these results, we propose a model for cartilaginous tumor development in which FGFR3 functions as a tumor suppressor. Our findings also suggest that modulation of FGFR3 and related signaling pathways is a potential therapeutic strategy for treating benign cartilaginous tumors.
Published in the journal:
. PLoS Genet 11(6): e32767. doi:10.1371/journal.pgen.1005214
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005214
Summary
Benign cartilaginous tumors, especially osteochondromas and enchondromas, are the most common primary bone tumors in humans. Several hereditary diseases are characterized by the development of cartilaginous tumors, including hereditary multiple exostoses, metachondromatosis, and enchondromatosis, which are caused by mutations in genes such as exostosin 1 and 2, tyrosine protein phosphatase non-receptor type 11, parathyroid hormone receptor 1, and isocitrate dehydrogenase 1 and 2. The proteins encoded by these genes are directly or indirectly linked to fibroblast growth factor (FGF) signaling. In addition, osteochondroma was found in several members of a family with camptodactyly, tall stature, and hearing loss syndrome, a rare inherited disorder caused by a heterozygous missense mutation in Fgfr3. In this study, we found that Fgfr3 deficiency leads to the formation of cartilaginous tumors, including osteochondromas and enchondromas, likely due to dysregulated endochondral ossification in growth plates. We also show that cartilaginous tumorigenesis in Fgfr3-deficient mice results from excessive Indian hedgehog production mediated by the activation of mitogen-associated protein kinase signaling. Based on these results, we propose a model for cartilaginous tumor development in which FGFR3 functions as a tumor suppressor. Our findings also suggest that modulation of FGFR3 and related signaling pathways is a potential therapeutic strategy for treating benign cartilaginous tumors.
Introduction
Enchondromas and osteochondromas are the most frequently occurring benign cartilaginous tumors affecting the skeleton [1,2]. The former develop as ectopic cartilaginous tissue within the bone marrow, while the latter manifest as cartilage-covered bony lesions arising on the bone surface [3]. Both tumor types can potentially undergo malignant transformation to become central or peripheral chondrosarcomas, respectively [4]. Since cartilaginous tumors are resistant to conventional chemo - and radiotherapy, surgical excision is the only treatment option [1]. A deeper understanding of the pathogenic mechanisms underlying cartilaginous tumor development is essential for the development of effective therapeutic strategies.
Cartilaginous tumors arise as a result of mutations in several genes. Hereditary multiple exostoses syndrome (HME, also called hereditary multiple osteochondromas, OMIM 133700) is associated with heterozygous loss-of-function mutations in exostosin (Ext)1 or 2, which encode Golgi-associated glycosyltransferases that mediate the polymerization of heparan sulphate (HS) chains [5,6]. Cells deficient in Ext1 or 2 fail to synthesize sufficient amounts of HS-rich proteoglycan (HSPG), which is required for the regulation of cell surface and matrix-associated signaling pathways such as Indian hedgehog (IHH), Wnt/β-catenin, fibroblast growth factor (FGF), and bone morphogenetic protein [6]. Although Ext1/2-deficient mice develop multiple bony lesions with features similar to osteochondromas in HME patients [7–10], the underlying mechanisms remain unclear. In addition, patients with enchondromatosis (Ollier disease and Maffucci syndrome, OMIM 166000) carry mutations in parathyroid hormone-related protein (PTHrP) receptor (Pthr)1 [11], while mice with the Pthr1 R150C mutation exhibit constitutive activation of HH signaling and develop multiple enchondroma-like lesions [11,12]. A somatic gain-of-function mutation in the isocitrate dehydrogenase (Idh)1/2 gene was also found to induce enchondroma formation in patients with Ollier disease and Maffucci syndrome [13–17].
Recently, the autosomal dominant disease metachondromatosis (MC, OMIM 156250) was found to be associated with heterozygous inactivating mutations in tyrosine protein phosphatase non-receptor type (Ptpn)11 [18,19], which encodes the tyrosine phosphatase SHP2, a downstream effector of receptor tyrosine kinases that activates the Ras/extracellular signal-regulated kinase (ERK) pathway [20]. MC is a rare disease characterized by enchondromas and osteochondromas [19]. The role of Ptpn11 in skeletal development and cartilaginous tumor formation has been investigated in mice [21–24]; interestingly, different mechanisms underlie the development of chondroma-like lesions in Shp2-deficient mice, enchondroma-like lesions in Pthr1 R150C mutant mice, and osteochondroma-like lesions in Ext1-deficient mice [24]. Most cartilaginous tumors are formed during skeletal development in a location adjacent to growth plates, suggesting that they arise as a result of dysregulated endochondral bone growth [1]; this is supported by the fact that the majority of the above-mentioned genes are involved in the regulation of growth plate development [24–26].
FGF receptor (FGFR)3 is a transmembrane receptor that regulates skeletal development [27]. Patients with activating FGFR3 mutations exhibit skeletal dysplasias characterized by short stature including achondroplasia (ACH, OMIM 100800), thanatophoric dysplasia I/ II (TD I, OMIM 187600 and TD II,OMIM 187601), hypochondroplasia (OMIM 146000), and severe ACH with developmental delay with acanthosis nigricans (SADDAN, OMIM 187600) [27]. Activated FGFR3 leads to impaired growth plate chondrocyte proliferation and differentiation, resulting in disordered endochondral bone growth and skeletal dysplasia in ACH/TD [28–30]. In contrast, a loss-of-function mutation in Fgfr3 in humans causes camptodactyly, tall stature, and hearing loss (CATSHL) syndrome (OMIM 610474) [31,32], and Fgfr3 deletion in mice leads to skeletal overgrowth due to enhanced proliferation of growth plate chondrocytes [33,34]. These data indicate that FGFR3 negatively regulates endochondral bone growth, and that FGFR3 mutations may involve in the development of cartilaginous tumors.
In cartilage, FGFs such as FGF18 require HSPG as a cofactor to bind to the extracellular domain of FGFR3 [35,36]. Intracellular receptor tyrosine kinase domains then recruit the SHP2–growth factor receptor-bound protein 2–son of sevenless 1 signaling complex to the cell membrane via adapter proteins, resulting in the activation of downstream effectors such as mitogen-associated protein kinase (MAPK), AKT, and signal transducer and activator of transcription [27,37,38]. FGFR3 signaling can also act as an inhibitor to regulate a negative feedback loop involving IHH and PTHrP [27]. Thus, most genes implicated in the formation of benign cartilaginous tumors are associated with FGF/FGFR3 signaling. Moreover, Colvin et al previously found that some global Fgfr3 knockout (Fgfr3-/-) mice develop ectopic pockets of hypertrophic chondrocytes below growth plates [34], and Toydemir et al reported that osteochondroma has been detected in the long bones of several members of a family with CATSHL syndrome [31]. However, the precise role of FGFR3 signaling in cartilaginous tumorigenesis is not known. In this study, we demonstrate that conditional Fgfr3 knockout (Fgfr3 cKO) mice lacking FGFR3 in chondrocytes exhibit severe enchondroma - and osteochondroma-like lesions in the skeleton, indicating that FGFR3 is critically involved in the formation of cartilaginous tumors.
Results
Morphological and histological analysis of the skeleton in Fgfr3-deficient mice
To investigate the role of FGFR3 in postnatal skeletal growth, we monitored the overall growth of Fgfr3 cKO relative to Fgfr3f/f Cre-negative control mice after tamoxifen administration. Fgfr3 cKO mice exhibited an increase in body length compared to Cre-negative mice (Fig 1A). Similarly, femur and tibia lengths increased by 13.79% and 9.32%, respectively, in mutants as compared to that in controls. The increased stature of Fgfr3 cKO mice is consistent previous findings in conventional Fgfr3 knockout mice and patients with CATSHL syndrome [31–34].

Lesions were observed around the knee joints of 12-week-old Fgfr3 cKO mice (Fig 1C, upper panels). Since these lesions appeared to be connected with bony tissue, we performed X-ray and micro-computed tomography (CT) analyses of undecalcified bone samples. The X-ray analysis showed dramatically expanded growth plates in the deformed knee joints (Fig 1C, middle panels) The broadly based sessile lesion was located near the growth plate and contained a medulla that was contiguous with the underlying bone (Fig 1C, middle panels). Micro-CT 3-dimensional (3-D) images of Fgfr3 cKO knee joints showed that the lesion surface formed a non-calcified cap (Fig 1C, lower panels). X-ray and micro-CT scans of 25-week-old Fgfr3 cKO mice revealed the progression of bony lesions (S1 Fig). We also detected multiple lesions in the wrist, an arch-like deformation of the radius, and subluxation/dislocation of the radial head in mutants (Fig 1B), which also showed heterogeneous radiodensities of costochondral junctions by X-ray (Fig 1D) and multiple bony lesions around the costal cartilage surface (Fig 1E). There were no significant lesions observed in bones with fused growth plates such as digits in these mice during the 12-month observation period (S2A Fig).
Histological analyses were carried out in order to investigate the morphological and cellular changes underlying the skeletal abnormalities in Fgfr3 cKO mice. Sections of wrist and knee joints from 12-week-old mutant mice showed multiple osteochondroma - and enchondroma-like lesions around the growth plate of the tibia (Fig 2A–2P), femur (Table 1), ulna, and radius (Fig 2Q–2X). Enchondroma-like lesions consisted of cartilaginous lobules with irregular architecture and cellular pleomorphism (Fig 2I–2L and 2U and 2V). Osteochondroma-like lesions that were structurally similar to growth plate cartilage were observed at the bone surface (Fig 2M–2P and 2W and 2X). Notably, growth plates in mutants were expanded and the columnar morphology of chondrocytes was lost (Fig 2E–2H). Multiple hypertrophic chondrocyte clusters were present in the costal cartilage of Fgfr3 cKO mice (Fig 3A and 3B). In some cases, the expansion of the chondrocyte cluster territory disrupted the integrity of the perichondrium. We also found cartilaginous islands within the trabecular bone of the ribs in mutants.



Enchondroma - and osteochondroma-like lesions were observed in several Fgfr3-deficient mouse models (Table 1). Although the incidence of chondroma-like lesions in other Fgfr3-deficient mice was relatively low, their locations and histological features were similar to what we observed in Fgfr3 cKO mice (S2B Fig). It is worth noting that those mice with conditional Fgfr3 deletion in chondrocytes had a higher incidence and greater severity of chondroma-like lesions. To determine whether wild-type chondrocytes contribute to cartilaginous tumorigenesis, cartilage tissues from chondroma-like lesions of Fgfr3 cKO mice were isolated by laser-capture microdissection (S3A Fig) and analyzed by allele-specific PCR. The lesions contained a mixture of wild-type and Fgfr3-deficient cells (S3B Fig); the fraction of Fgfr3-deficient cells ranged from 54.94% to 83.9% (S3B Fig). These results suggest that Fgfr3-null and wild-type cells interact to promote the formation of cartilaginous tumors, which can explain differences in the incidence and severity of chondroma-like lesions between conditional and global Fgfr3 knockout mice. A histological analysis revealed ectopic cartilage at the bone-ligament attachment site between the menisco-tibial ligament and meniscus in Fgfr3−/− mice (S4E–S4H Fig). This phenotype was similar to that of SHP2 cKO mice in which the protein tyrosine phosphatase non-receptor type 11 gene was deleted in fibroblast-specific protein 1-Cre-expressing fibroblasts [24]. In our study, ectopic cartilage was also observed adjacent to osteochondroma-like lesions connected to the side of growth plate cartilage in Fgfr3−/− mice (S4I–S4P Fig); however, we were unable to determine the precise anatomical location due to structural changes caused by the presence of chondroma-like lesions.
Loss of FGFR3 in mouse chondrocytes disrupts chondrocyte polarity, proliferation, differentiation, and apoptosis in growth plates
The observed chondroma-like lesions suggest that loss of Fgfr3 in chondrocytes at the postnatal stage affects the normal development and maintenance of growth plate cartilage by disrupting the coordination of chondrocyte proliferation, differentiation, and apoptosis [27]. The lesions were associated with the dysregulation of growth plate chondrocytes (Fig 2B and 2D); thus, to determine how Fgfr3 deficiency alters growth plate development and homeostasis, we performed histological and immunohistochemical analyses on the long bones of 8-week-old mice. As expected, Cre-negative mice exhibited normal growth plate organization in which chondrocytes formed distinct resting, proliferative, and hypertrophic zones (Fig 4A, left panels). In contrast, growth plates in the mutants had disorganized columns of chondrocytes stacked within the proliferative zone, with cell clusters in the resting zone and epiphysis (Fig 4A, right panels), suggesting impaired growth plate polarity. To investigate this possibility, primary cilia were detected using an anti-α-tubulin antibody. A virtual axis oriented parallel to the vertical axis of growth plates was formed by the alignment of primary cilia crossing the center of the chondrocyte column in the proliferative zone of Cre-negative mice (S5A Fig). In contrast, in Fgfr3 mutants, primary cilia of the aligned axis of chondrocytes in the growth plate were disoriented (S5B Fig). Furthermore, although most chondrocytes in chondroma-like lesions had disrupted polarity, a few formed well-organized columns with an orientation perpendicular to the boundary between the lesion and marrow cavity (S5C and S5D Fig). These data suggest that disruption of chondrocyte polarity may be responsible for the disorganization of growth plates and the formation of chondroma-like lesions in mice with absence of Fgfr3.
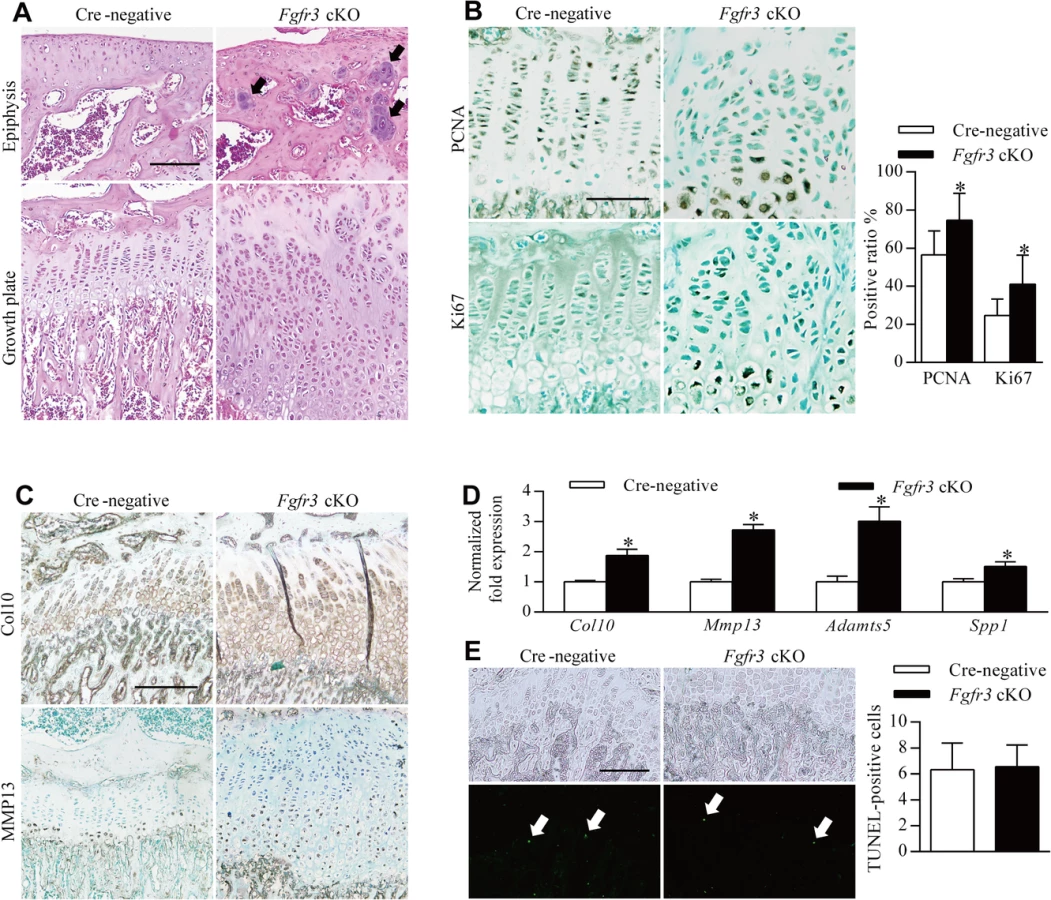
To determine whether chondrocyte proliferation was altered in growth plates of Fgfr3 cKO mice, proliferating cell nuclear antigen (PCNA) and Ki-67 expression was detected by immunohistochemistry. PCNA-positive cells were evenly distributed in the proliferative zone of growth plates in control mice (Fig 4B, upper left panel). In contrast, PCNA-(Fig 4B, upper right panel) and Ki67-positive cells (Fig 4B, lower panels) showed an irregular distribution in the proliferative and hypertrophic zones in mutants and had higher rates of proliferation (Fig 4B), consistent with observations from the growth plates of Fgfr3−/− mice [33]. Given the expansion of the hypertrophic zone in Fgfr3-deficient growth plates (Fig 2E–2H), the expression of collagen 10 and matrix metalloproteinase (MMP)13 was evaluated by immunohistochemistry to determine whether growth plate chondrocyte differentiation was induced in mutants. Immunoreactivity for these markers was increased in Fgfr3 cKO relative to control mice (Fig 4C). Furthermore, the expression of the terminal differentiation markers Mmp13, a distintegrin and metalloproteinase with thrombospondin motifs (Adamts)5, and secreted phosphoprotein (Spp)1 [24] was upregulated in Fgfr3-deficient chondrocytes (Fig 4D). These data suggest that FGFR3 negatively regulates both proliferation and early/terminal differentiation of chondrocytes in postnatal growth plates.
The role of FGFR3 in chondrocyte apoptosis remains controversial [27]; although FGFR3 overexpression was found to promote chondrocyte apoptosis in vitro [39–42], there is little in vivo evidence to support this observation [43]. We performed terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) to determine whether Fgfr3 deficiency promotes growth plate chondrocyte apoptosis. In primary chondrocytes isolated from Fgfr3 cKO mice, the proportion of TUNEL-positive cells was similar to control levels (S7 Fig). In contrast, an in vivo examination found that the proportion of TUNEL-positive cells relative to the total number of hypertrophic chondrocytes was significantly reduced in mutants as compared to control mice (Fig 4E). In addition, the osteoclast activity was considered to be involved in endochondral bone growth [44]. To investigate the potential role of osteoclast activity in pathogenesis of the skeletal phenotypes of Fgfr3 cKO mice, we performed tartrate-resistant acid phosphatase staining of bone tissue, but found no differences in growth plate osteoclast recruitment between mutant and control mice (S6A Fig). Furthermore, there were no bony lesions detected in osteoclast progenitor-specific Fgfr3 knockout mice (Fgfr3f/f; lysozymeM-Cre) by radiography (S6B Fig).
To further assess the tumorigenic properties of Fgfr3-deficient chondrocytes, we transplanted chondrocytes from Fgfr3 cKO and control mice subcutaneously into 5-week-old athymic mice. Two of five Cre-negative and four of five Fgfr3-deficient cartilage masses were detected and harvested 4 weeks after transplantation, and the Fgfr3-deficient cartilage masses were significantly larger after 4 weeks than those derived from Cre-negative cells (S8A and S8B and S8G Fig) and a histological analysis revealed larger areas of matrix-enriched cartilage in the former (S8C–S8F Fig). These data indicate that FGFR3 plays a key role in the postnatal development and homeostasis of growth plate chondrocytes, and that FGFR3 deficiency leads to increased number of disordered chondrocytes, resulting in the formation of chondroma-like lesions.
MAPK signaling is impaired while IHH expression is upregulated in Fgfr3 mutants
IHH and PTHrP signaling are important for the development and maintenance of growth plate cartilage [45]. Moreover, previous studies have shown that Pthr1 R150C mutant mice and those overexpressing Gli2, a downstream effector of IHH signaling develop enchondroma-like lesions [11], and we and others have observed that constitutively activated FGFR3 lowers the expression of Ihh mRNA in growth plate chondrocytes [28,29,46,47]. We therefore investigated whether IHH signaling is perturbed in the growth plates of Fgfr3-deficient mice by immunohistochemistry. In growth plates of Cre-negative mice, IHH expression was mainly detected within the pre-hypertrophic zone and in a few cells in the hypertrophic zone (Fig 5A, left panel). In contrast, the growth plate of Fgfr3 cKO mice consisted of disorganized chondrocytes in which IHH expression was markedly upregulated (Fig 5A, middle panel). Furthermore, strong IHH expression was observed in chondroma-like lesions in mutants (Fig 5A, right panel), implying that the dysregulation of IHH signaling is associated with cartilaginous tumorigenesis caused by Fgfr3 deficiency.

It was recently reported that SHP2 deletion in chondrocytes and chondroprogenitor cells within the groove of Ranvier results in ectopic chondrogenesis and the formation of chondroma-like lesions via MAPK-induced upregulation of IHH signaling [23,48]. Given that SHP2 acts as a downstream effector of FGFR3 signaling that modulates the MAPK pathway [27,38], we speculated that cartilaginous tumorigenesis in Fgfr3 cKO mice was caused by a mechanism similar to that of MC, which results from SHP2 deficiency. To investigate whether MAPK signaling is impaired in chondrocytes of Fgfr3 cKO mice, we evaluated MAPK activation by immunohistochemistry using an antibody against phosphorylated extracellular signal-regulated kinase (ERK). MAPK activity was detected within hypertrophic zones in Cre-negative growth plates (Fig 5B, left panel), consistent with previous observations [48]. In contrast, MAPK activity was reduced in hypertrophic zones of Fgfr3 cKO mice (Fig 5B, right panel), which was confirmed by western blot analysis of isolated chondrocytes. MAPK activation in response to FGF18 was also reduced in Fgfr3-deficient chondrocytes (Fig 5C). To determine whether the upregulation of IHH is a consequence of impaired MAPK signaling in Fgfr3 mutants, we examined the expression of genes regulating chondrogenesis by quantitative real-time (qRT-)PCR. Ihh and Pthrp mRNA levels were increased in Fgfr3-deficient chondrocytes (Fig 5D), while treatment with MAPK inhibitor (250 nM U0126 or 10 nM PD98059) also increased Ihh and Pthrp levels (Fig 5D). These results indicate that the enhanced expression of IHH in growth plates of Fgfr3 mutants is due at least in part to the suppression of MAPK signaling.
Inhibiting IHH signaling rescues the skeletal defects of Fgfr3 mutants
In the canonical IHH signaling pathway, Smoothened (Smo) dissociates from the Patched (Ptch) receptor following the binding of IHH to Ptch [49] and activates downstream effectors such as Gli transcription factors [49]. A Smo inhibitor (SMOi) such as GDC-0449 (Vismodegib/Erivedge) developed to treat basal cell carcinoma and medulloblastoma [49,50] can therefore be used to inhibit IHH signaling. Since upregulated IHH signaling is a potential mechanism underlying the cartilaginous tumorigenesis observed in Fgfr3-deficient mice, we investigated whether the process could be blocked by inhibiting IHH signaling using GDC-0449. Skeletal phenotypes after SMOi treatment were assessed by X-ray, micro-CT, and histological examination. The lower limb length of all mice was significantly reduced by SMOi treatment (Fig 6Q). X-ray analysis and micro-CT revealed joint deformation and growth plate expansion was detected by histological analyses of Fgfr3 cKO mice; however, these were alleviated by SMOi treatment (Fig 6A–6P and 6R). Importantly, there were no chondroma-like lesions in the mutants after 4 weeks of SMOi treatment (Fig 6L and 6S). The fusion of growth plates was readily observed in 8-week-old SMOi-treated Cre-negative and Fgfr3 cKO mice, but not in those receiving vehicle treatment (Fig 6I–6P). Furthermore, SMOi treatment promoted chondrocyte apoptosis in primary chondrocytes isolated from wild-type and mutant mice (S7 Fig). Chondrocytes were evenly distributed at the center of the costal cartilage and were separated from the surrounding muscle by the perichondrium in Cre-negative mice treated either with vehicle or SMOi (S9 Fig). In contrast, hypertrophic-like chondrocyte clusters were observed in the cartilage of Fgfr3 cKO mice, which were alleviated by SMOi treatment (S9 Fig). These results suggest that enhanced IHH signaling is involved in the pathogenesis of cartilaginous tumorigenesis caused by FGFR3 deficiency.

Discussion
Cartilaginous tumors are the most common primary bone tumors, with osteochondroma and enchondroma being the most prevalent benign cartilaginous lesions in humans [1]. Mutations in several genes, such as Ext1/2, Ptpn11, Pthr1, and Idh1/2 lead to cartilaginous tumorigenesis, and these genes are important in the normal development and maintenance of growth plates. Although FGFR3 plays a key role in chondrogenesis, its role in the pathogenesis of cartilaginous tumors is poorly understood; indeed, although osteochondroma is observed in several members of a family with CATSHL syndrome, there have been almost no studies examining this phenotype [31]. One reason for this is that activation of FGFR3 signaling enhances proliferation in most cell types—including fibroblasts, keratinocytes, melanocytes, epithelial cells, lymphocytes and spermatocytes—and consequently lead to cancer [51]; as such, the loss-of-function phenotype of FGFR3—which positively regulates cartilage development and may be responsible for cartilaginous tumorigenesis—may have been previously overlooked. In this study, we found that Fgfr3 deficiency impaired the normal development and homeostasis of growth plates and induced the formation of chondroma-like lesions via downregulation of MAPK and consequent upregulation of IHH signaling, (Fig 7).
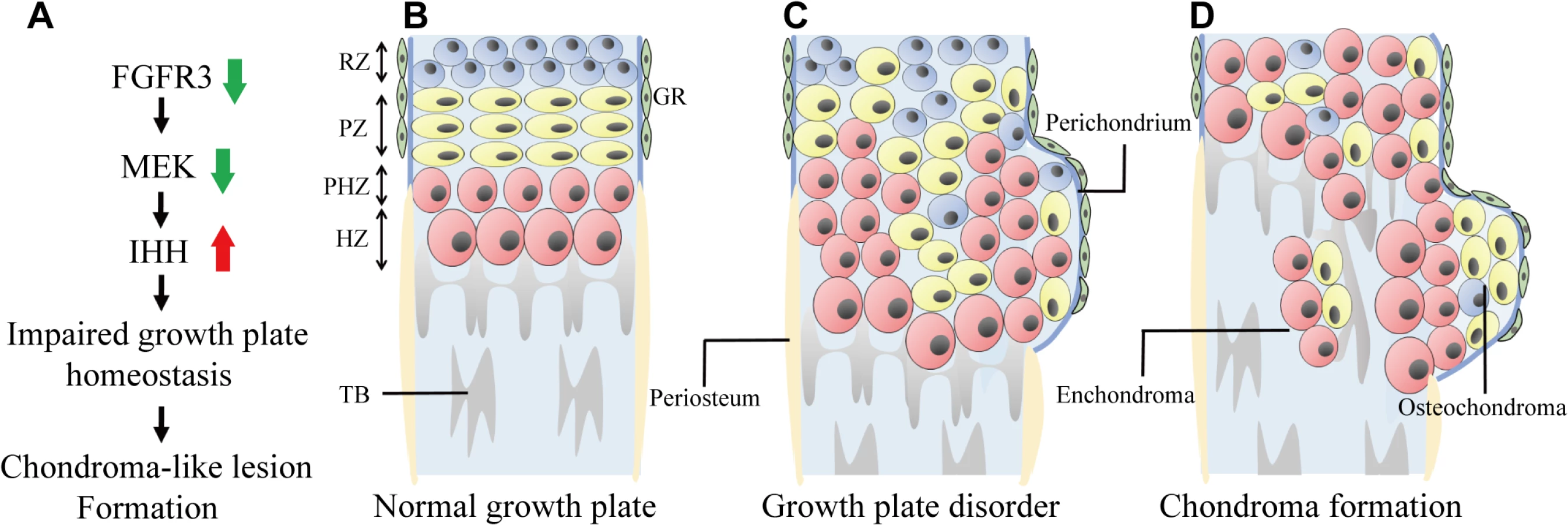
Typical osteochondroma - and enchondroma-like lesions were observed in the femur, tibia, and the distal radius/ulna of Fgfr3 mutant mice. However, these MC-like phenotypes differ from those of patients with CATSHL syndrome, who exhibit only osteochondroma [31]. A more detailed radiographic examination of these patients is needed in order to determine whether they also have enchondroma lesions.
It remains unclear how chondroma-forming cells are derived. In our study, enchondroma-like lesions in Fgfr3 cKO mice appeared to originate from disordered growth plates, which is consistent with the enchondroma formation observed in mice with Shp2 deficiency or overexpression of GLI2 or PTHR1 R150C [11,24,52]. This evidence suggests that enchondroma-forming cells arise from growth plate chondrocytes undergoing abnormal endochondral ossification. The origin of osteochondroma-forming cells is also unknown [6,53]; one possibility is that they result from dysregulated growth plate chondrocytes. Evidence in support of this comes from previous studies showing that Ext1 deficiency leads to the formation of osteochondroma-like lesions adjacent to growth plate cartilage [7–9,26]. The periosteum and perichondrium both contain chondroprogenitor cells [54,55] and are therefore also considered as potential sites of osteochondroma [10,48]. Other possibilities are that osteochondroma-forming cells are derived from the groove of Ranvier [6,23], or else the periosteum or perichondrium, which showed Cre recombinase activity in the collagen type II α1 (Col2a1)-CreERT2 mice used in this study [48]. However, since no ectopic chondrogenesis or chondroma-like lesions was observed in bones with fused growth plates (e.g., adult digits), we speculate that these lesions are mainly induced by the abnormal development and maintenance of growth plate cartilage in Fgfr3-deficient mice.
We investigated the roles of FGFR3 in the development and maintenance of growth plate chondrocytes during the postnatal stage to clarify the mechanism by which loss of Fgfr3 leads to the formation of chondroma-like growth plate lesions. Data from a recent study suggests that Shp2 deficiency induces the transition from proliferating chondrocytes to pre - or early-hypertrophic chondrocytes but delays the switch from pre - or early hypertrophic to terminal hypertrophic chondrocytes, which is thought to result in chondroma-like lesions in an MC mouse model [24]. In our study, Fgfr3 deficiency induced both early and terminal differentiation of growth plate chondrocytes in postnatal stages. We therefore propose that different mechanisms are responsible for the formation of chondroma-like lesions in Fgfr3 - and Shp2-deficient mice. The chondroma-like lesions in the former are composed of small chondrocytes surrounded by large hypertrophic chondrocytes and are located in the marrow cavity and at the bone surface adjacent to growth plates. We also observed dysregulated polarity, increased proliferation, and decreased apoptosis of growth plate chondrocytes in Fgfr3 mutants. Based on these findings, we speculate that Fgfr3 deficiency in growth plates disrupts the coordination of chondrocyte proliferation, differentiation, and apoptosis, resulting in the accumulation of abnormal chondrocytes that form lesions. Additionally, decreased osteoclast activity and vascular invasion are thought to underlie cartilaginous tumorigenesis [56]. We found that osteoclast/chondroclast recruitment around growth plates and chondroma-like lesions was unaffected in Fgfr3 cKO mice and there were no chondroma-like lesions in Fgfr3f/f; lysozyme (lys)M-Cre mice during 6 months of observation. Moreover, it was reported that despite the downregulation of vascular endothelial growth factor—a molecule required for hypertrophic chondrocyte apoptosis [57]—vascular invasion is only slightly impaired in the growth plate cartilage of Fgfr3−/− mice [43]. Therefore, changes in osteoclast/chondroclast and vascular invasion may not be involved in chondroma-like lesion formation resulting from loss of Fgfr3.
The results of this and previous studies suggest that FGFR3 signaling promotes early chondrocyte hypertrophy through SHP2 and MAPK signaling [24,27]. However, the roles of FGFR3 and ERK1/2 in terminal hypertrophy are different from that of SHP2 in growth plate chondrocytes during the postnatal stage. Although the downregulation of FGFR3, SHP2, and ERK1/2 observed in vivo induced the expansion of the growth plate hypertrophic zone, an increase in bone length was only observed in Fgfr3 - and Erk1/2-deficient mice [22,24,33,34,58]. Moreover, loss of ERK1/2 strongly inhibited chondrocyte proliferation, implying that enhancement of chondrocyte terminal hypertrophy is a major determinant of longitudinal bone overgrowth in Erk1/2-deficient mice [58,59]. These studies suggest that ERK1/2 negatively regulates growth plate chondrocyte terminal differentiation during the postnatal stage. Moreover, expression of constitutively active mitoggen-activated protein kinase kinase (MEK)1 can rescue the overgrowth of bones in Fgfr3-deficient mice by inhibiting chondrocyte differentiation [60], indicating that the MAPK pathway is activated downstream of FGFR3 signaling to regulate growth plate chondrocyte differentiation. An important remaining question is whether FGFR3 and SHP2 have MAPK-independent and-dependent roles in cartilaginous tumorigenesis.
We also speculate that cell autonomous as well as nonautonomous mechanisms are involved in the initiation and progression of cartilaginous tumors. This is supported by the fact that a recombination rate of only 6%–15% is sufficient to induce the formation of osteochondroma-like lesions in Ext1f/f mice, in which osteochondroma-like lesions contain both Ext1-null and wild-type chondrocytes [9]. A similar phenomenon was observed in patients with active Idh1 mutations and in Shp2-deficient mice [16,24], suggesting that altered paracrine signaling from mutant growth plate chondrocytes may alter homeostasis in neighboring wild-type cells. In Fgfr3−/− mice, FGFR3 is deleted in all cells, and each zone within the growth plate—especially the hypertrophic zone—is expanded [33,34]; however, the phenotype is milder than that of Fgfr3 cKO mice with mosaic Fgfr3 deletion. Moreover, the incidence of chondroma-like lesions is much lower in Fgfr3−/− than in Fgfr3 cKO mice (Table 1). One explanation for this is that chondroma formation can be initiated by Fgfr3-deficient cells in both mutants, but will be enhanced in mosaic Fgfr3 cKO mice owing to crosstalk between mutant and adjacent wild-type cells, which are not passively incorporated into lesions but may interact with mutant cells to actively promote cartilaginous tumor formation.
Previous studies have suggested that IHH upregulation contributes to metachondromatosis and that inhibiting IHH signaling suppresses cartilaginous tumorigenesis in Shp2-deficient mice [23,48]. Although we found that treatment with an IHH signaling inhibitor reduced the formation of chondroma-like lesions in Fgfr3 cKO mice, adverse secondary effects were induced, including premature growth plate fusion leading to a shorter long bone, which is similar to what was observed in mice with chondrocyte-specific deletion of IHH [61]. Other types of SMOi such as HhAntag had similar effects on the skeleton [62]. Thus, given that cartilaginous tumors occur mostly in children, caution is required when using SMOi to treat young patients with enchondroma and osteochondroma. PF-04449913 was shown to alleviate cartilaginous tumors in Shp2-deficient mice without significantly affecting bone development [23]. However, the optimal timing and dosage of IHH signaling inhibitors such as PF-04449913 must be established in order to improve treatment outcome and minimize the deleterious effects on the developing growth plate.
In conclusion, this first is the first study to demonstrate that loss of Fgfr3 leads to the downregulation of MAPK signaling and enhanced IHH expression, resulting in the formation of chondroma-like lesions, including enchondromas and osteochondromas. Based on these findings, we propose that FGFR3 has a tumor suppressor-like function in cartilage development. Fgfr3 cKO mice can thus serve as a model to dissect the roles and mechanisms of action of FGFR3 in cartilaginous tumorigenesis, which can facilitate the development of effective therapies.
Materials and Methods
Animals
Fgfr3f/f mice were previously generated by our group [63]. The Col2a1-CreERT2 [64], Col2al-Cre [65], lysM-Cre (Jackson Laboratories, Bar Harbor, ME, USA), cytomegalovirus (CMV)-Cre [66], and Fgfr3−/− [33] mice were genotyped as previously described. For inducible deletion of Fgfr3 in chondrocytes, Fgfr3f/f mice were crossed with Col2a1-CreERT2 mice to obtain Fgfr3f/+; Col2a1-CreERT2 mice, which were crossed with Fgfr3f/f mice to obtain Fgfr3f/f and Fgfr3f/+; Col2a1-CreERT2 mice. Fgfr3f/f mice were crossed with Fgfr3f/+; Col2a1-CreERT2 mice to obtain Fgfr3f/+; Col2a1-CreERT2 (Fgfr3 cKO) and Fgfr3f/f (Cre-negative) mice. Tamoxifen (1 mg/10 g body weight) was administered by intraperitoneal (i.p.) injection twice weekly for 8 weeks (for a total of 16 injections) starting 4 weeks after birth. Cre recombinase activity in the postnatal growth plate was evaluated as previously described [48]. As for Fgfr3f/+; Col2a1-CreERT2 mice, Fgfr3f/f mice were crossed with Col2al-Cre and lysM-Cre mice to generate Fgfr3f/f; Col2al-Cre and Fgfr3f/f; lysM-Cre mice, respectively. Wild-type mice were mated with the Fgfr3 f/f; CMV-Cre mice to generate Fgfr3 heterozygous mice which are expected to give 50% recombined Fgfr3 allele. Fgfr3 +/- mice were mated with the Fgfr3 +/- mice to generate Fgfr3 +/+ and Fgfr3 -/- mice. All mice were of the C3H/HeJ background. Animal experiments were performed according to protocols approved by the Laboratory Animal Welfare and Ethics Committee of the Third Military Medical University (Chongqing, China).
X-ray and micro-CT analysis
X-ray images of bony tissue were obtained using an MX-20 Cabinet X-ray system (Faxitron X-Ray, Tucson, AZ, USA). Each undecalcified specimen was scanned using a vivaCT 40 micro-CT system (Scanco Medical, Brüttisellen, Switzerland). Serial 12.5-μm 2-D and 3-D images were acquired at 70 kV and 113 mA. Constant thresholds (200) were applied to grayscale images to distinguish bone from soft tissue.
Histological assessment
Samples were fixed in 4% paraformaldehyde in 0.1 M phosphate buffer overnight, decalcified in 15% EDTA-phosphate buffered saline for 2 weeks and embedded in paraffin. Sections (5-μm thick) were stained with Safranin O/Fast Green and hematoxylin and eosin (H & E). For histomorphometric analysis, the Safranin O-positive cartilage area in 5-μm serial sections from the midsagittal region of growth plates was quantified using Image-Pro Plus 5.1 (Leeds Precision Instruments, Minneapolis, MN, USA).
Immunostaining analysis
Decalcified bone sections were deparaffinized with xylene, and endogenous peroxidase activity was quenched by treatment with 3% H2O2 for 15 min, followed by antigen retrieval by trypsinization for 10 min. Sections were then blocked with normal goat serum for 30 min and incubated at 4°C overnight with primary antibody followed by the appropriate biotinylated secondary antibody and horseradish peroxidase-conjugated streptavidin-biotin staining. Immunoreactivity was visualized with a 3,3'-diaminobenzidine tetrahydrochloride kit (ZSGB-BIO, Beijing, China) followed by counterstaining with Methyl Green. Primary antibodies against the following proteins were used: PCNA (1 : 200; BioVision, Milpitas, CA, USA), Ki67 (1 : 100; Abcam, Cambridge, MA, USA), Col10a1 (1 : 200; Abcam), MMP13 (1 : 200; Abcam), phospho-ERK (1 : 100; Cell Signaling Technology, Danvers, MA, USA), acetylated tubulin (1 : 200; Sigma-Aldrich, St. Louis, MO, USA), and IHH (1 : 100; Abcam). The number of PCNA - and Ki67-positive nuclei in three central regions of the growth plate was counted in Cre-negative and Fgfr3 cKO mice (n = 3 each).
Primary chondrocyte cultures
Primary chondrocytes were isolated from knee joint cartilage of 3-day-old mice. Dissected tissues with cartilage were first digested with 0.25% trypsinase (Gibco/Life Technologies, Carlsbad, CA, USA) at 37°C for 15 min to remove muscles, ligaments, and bone tissue. Chondrocytes were isolated from knee joints by additional digestion with 0.1% collagenase II (Gibco/Life Technologies) overnight at 37°C in a CO2 incubator. Cells were seeded in 12-well plates at a density of 2 × 105 cells/well and cultured in Dulbecco’s Modified Eagle’s Medium/F12 (1 : 1) supplemented with penicillin/streptomycin (Gibco/Life Technologies) and 10% fetal bovine serum until they reached sub-confluence. On day 3 of culturing, primary chondrocytes were treated with 1 μM 4OH-tamoxifen (Sigma-Aldrich) for 48 h. For MEK inhibitor treatment, chondrocytes were incubated in 250 nM U0126 or 10 nM PD98059 (both from Merck, Kenilworth, NJ, USA) for 24 h after treatment with and in the presence of 4OH-tamoxifen. For FGF18 treatment, 50 ng/ml FGF18 (PeproTech, Rocky Hill, NJ, USA) were added to cultures 5 and 30 min after 4OH-tamoxifen incubation.
Allotransplantation assay
Primary chondrocytes (2 × 106cells) were resuspended in 100 μl serum-free medium and mixed with an equal volume of Matrigel (BD Biosciences, Franklin Lakes, NJ, USA). The mixture was injected subcutaneously into the lower flanks of athymic nude mice (BALB/c-nu, male, 5 weeks old). Tamoxifen was administered by i.p. injection twice weekly. Chondrocyte transplants were harvested 4 weeks after transplantation.
qRT-PCR
Total RNA was extracted from primary chondrocytes with TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. All reactions were performed in a Mx3000P thermal cycler (Stratagene, Santa Clara, CA, USA) using the Two-Step QuantiTect SYBR Green RT-PCR Kit (Takara Biotechnology, Otsu, Japan) and reaction conditions were optimized for each gene by altering the annealing temperature (57°C–61°C). Each run consisted of samples for genes of interest and cyclophilin A. The forward and reverse primer sequences were as follows: cyclophilin A, 5'-CGA GCT CTG AGC ACT GGA GA-3' and 5'-TGG CGT GTA AAG TCA CCA CC-3'; Col10a1, 5'-GCA GCA TTA CGA CCC AAG AT-3' and 5'-CAT GAT TGC ACT CCC TGA AG-3'; Mmp13, 5'-CAG TTG ACA GGC TCC GAG AA-3' and 5'-CGT GTG CCA GAA GAC CAG AA-3'; Adamts5, 5'-GGA GCG AGG CCA TTT ACA AC-3' and 5'-CGT AGA CAA GGT AGC CCA CTT T-3'; Ihh, 5'-CAA TCC CGA CAT CAT CTT CA-3' and 5'-GCG GCC CTC ATA GTG TAA AG-3'; Spp1, 5'-TGG CTA TAG GAT CTG GGT GC-3' and 5'-TTG GCA GTA ATT TGC TTT TG-3'; and Pthrp, 5'-CAT CAG CTA CTG CAT GAC AAG G-3' and 5'-GGT GGT TTT TGG TGT TGG GAG-3'.
Laser capture microdissection and PCR
Tissue sections (10 μm thick) were transferred to polyethylene tetraphthalate FrameSlides (Leica, Wetzlar, Germany) and stained with H & E. Cartilage tissue in the region of chondroma-like lesions was isolated using a laser-capture microdissection system (Molecular Machines & Industries, Eching, Germany). To avoid contamination from cells of the perichondrium/periosteum or trabecular bone, the tissue was cut far away from the border between the chondroma-like lesion and surrounding tissue. DNA was extracted from cartilage tissue using the EZNA DNA extraction kit (Omega Bio-Tek, Norcross, GA, USA) and analyzed by semiquantitative PCR. The p3 and p5 primers were used to detect the unrecombined allele, whereas p1 and p5 were used to detect the recombined allele as previously described [63]. Band intensity was measured using Image Lab software (Bio-Rad Laboratories, Hercules, CA, USA). The ratio of the recombined to unrecombined allele was normalized to the level of Fgfr3 expression in heterozygous mice (taken as 1 : 1).
Western blot analysis
Chondrocytes lysates were prepared in ice-cold radioimmunoprecipitation assay buffer containing protease inhibitors (Roche Applied Science). Proteins were resolved by 10% or 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane (Millipore, Billerica, MA, USA), which was probed with antibodies against the following proteins: phospho-ERK (Cell Signaling Technology, Danvers, MA, USA), ERK (Cell Signaling Technology), and β-actin (Sigma-Aldrich). Immunoreactivity was detected by enhanced chemiluminescence (Pierce, Rockford, IL, USA).
Inhibition of Ihh signaling
For in vitro experiments, the SMOi GDC-0449 (Selleck Chemicals, Houston, TX, USA) was reconstituted in dimethyl sulfoxide (Sigma-Aldrich) and applied at a final concentration of 1 μM for 24 h. For in vivo experiments, GDC-0449 was reconstituted in 50% (w/v) 2-hydroxypropyl-β-cyclodextrin (Sigma-Aldrich) in water. Mice were injected twice weekly for 4 weeks with tamoxifen (1 mg/10 g body weight) starting from 4 weeks of age and daily with GDC-0449 (1 mg/10 g body weight) or vehicle (50% 2-hydroxypropyl-β-cyclodextrin), except on tamoxifen injection days. At 8 weeks of age, the tibia and femur were dissected for X-ray, micro-CT, and histological analyses.
Apoptosis assay
The TUNEL assay was carried out with the In Situ Cell Death Detection kit (Roche Applied Science, Pleasanton, CA, USA) according to the manufacturer’s instructions.
Statistical analysis
Results are expressed as mean ± SD. Differences between groups were analyzed with the Student’s t test. p < 0.05 was considered significant.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Bovee JV, Hogendoorn PC, Wunder JS, Alman BA (2010) Cartilage tumours and bone development: molecular pathology and possible therapeutic targets. Nat Rev Cancer 10 : 481–488. doi: 10.1038/nrc2869 20535132
2. Romeo S, Hogendoorn PC, Dei Tos AP (2009) Benign cartilaginous tumors of bone: from morphology to somatic and germ-line genetics. Adv Anat Pathol 16 : 307–315. doi: 10.1097/PAP.0b013e3181b506a1 19700940
3. Garcia RA, Inwards CY, Unni KK (2011) Benign bone tumors—recent developments. Seminars in Diagnostic Pathology 28 : 73–85. 21675379
4. Schwartz HS, Zimmerman N, Simon M, Wroble R, Millar E, et al. (1987) The malignant potential of enchondromatosis. The Journal of Bone & Joint Surgery 69 : 269–274.
5. Jennes I, Pedrini E, Zuntini M, Mordenti M, Balkassmi S, et al. (2009) Multiple osteochondromas: mutation update and description of the multiple osteochondromas mutation database (MOdb). Hum Mutat 30 : 1620–1627. doi: 10.1002/humu.21123 19810120
6. Huegel J, Sgariglia F, Enomoto-Iwamoto M, Koyama E, Dormans JP, et al. (2013) Heparan sulfate in skeletal development, growth, and pathology: The case of hereditary multiple exostoses. Dev Dyn.
7. Zak BM, Schuksz M, Koyama E, Mundy C, Wells DE, et al. (2011) Compound heterozygous loss of Ext1 and Ext2 is sufficient for formation of multiple exostoses in mouse ribs and long bones. Bone 48 : 979–987. doi: 10.1016/j.bone.2011.02.001 21310272
8. Jones KB, Piombo V, Searby C, Kurriger G, Yang B, et al. (2010) A mouse model of osteochondromagenesis from clonal inactivation of Ext1 in chondrocytes. Proc Natl Acad Sci U S A 107 : 2054–2059. doi: 10.1073/pnas.0910875107 20080592
9. Matsumoto K, Irie F, Mackem S, Yamaguchi Y (2010) A mouse model of chondrocyte-specific somatic mutation reveals a role for Ext1 loss of heterozygosity in multiple hereditary exostoses. Proc Natl Acad Sci U S A 107 : 10932–10937. doi: 10.1073/pnas.0914642107 20534475
10. Huegel J, Mundy C, Sgariglia F, Nygren P, Billings PC, et al. (2013) Perichondrium phenotype and border function are regulated by Ext1 and heparan sulfate in developing long bones: a mechanism likely deranged in Hereditary Multiple Exostoses. Dev Biol 377 : 100–112. doi: 10.1016/j.ydbio.2013.02.008 23458899
11. Hopyan S, Gokgoz N, Poon R, Gensure RC, Yu C, et al. (2002) A mutant PTH/PTHrP type I receptor in enchondromatosis. Nat Genet 30 : 306–310. 11850620
12. Tiet TD, Hopyan S, Nadesan P, Gokgoz N, Poon R, et al. (2006) Constitutive hedgehog signaling in chondrosarcoma up-regulates tumor cell proliferation. Am J Pathol 168 : 321–330. 16400033
13. Rozeman LB, Hameetman L, Cleton-Jansen AM, Taminiau AH, Hogendoorn PC, et al. (2005) Absence of IHH and retention of PTHrP signalling in enchondromas and central chondrosarcomas. J Pathol 205 : 476–482. 15685701
14. Amary MF, Bacsi K, Maggiani F, Damato S, Halai D, et al. (2011) IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J Pathol 224 : 334–343. doi: 10.1002/path.2913 21598255
15. Amary MF, Damato S, Halai D, Eskandarpour M, Berisha F, et al. (2011) Ollier disease and Maffucci syndrome are caused by somatic mosaic mutations of IDH1 and IDH2. Nat Genet 43 : 1262–1265. doi: 10.1038/ng.994 22057236
16. Pansuriya TC, van Eijk R, d'Adamo P, van Ruler MA, Kuijjer ML, et al. (2011) Somatic mosaic IDH1 and IDH2 mutations are associated with enchondroma and spindle cell hemangioma in Ollier disease and Maffucci syndrome. Nat Genet 43 : 1256–1261. doi: 10.1038/ng.1004 22057234
17. Hirata M, Sasaki M, Cairns RA, Inoue S, Puviindran V, et al. (2015) MutantIDHis sufficient to initiate enchondromatosis in mice. Proceedings of the National Academy of Sciences 112 : 2829–2834. doi: 10.1073/pnas.1424400112 25730874
18. Sobreira NL, Cirulli ET, Avramopoulos D, Wohler E, Oswald GL, et al. (2010) Whole-genome sequencing of a single proband together with linkage analysis identifies a Mendelian disease gene. PLoS Genet 6: e1000991. doi: 10.1371/journal.pgen.1000991 20577567
19. Bowen ME, Boyden ED, Holm IA, Campos-Xavier B, Bonafe L, et al. (2011) Loss-of-function mutations in PTPN11 cause metachondromatosis, but not Ollier disease or Maffucci syndrome. PLoS Genet 7: e1002050. doi: 10.1371/journal.pgen.1002050 21533187
20. Grossmann KS, Rosario M, Birchmeier C, Birchmeier W (2010) The tyrosine phosphatase Shp2 in development and cancer. Adv Cancer Res 106 : 53–89. doi: 10.1016/S0065-230X(10)06002-1 20399956
21. Bauler TJ, Kamiya N, Lapinski PE, Langewisch E, Mishina Y, et al. (2011) Development of severe skeletal defects in induced SHP-2-deficient adult mice: a model of skeletal malformation in humans with SHP-2 mutations. Dis Model Mech 4 : 228–239. doi: 10.1242/dmm.006130 21068439
22. Lapinski PE, Meyer MF, Feng GS, Kamiya N, King PD (2013) Deletion of SHP-2 in mesenchymal stem cells causes growth retardation, limb and chest deformity, and calvarial defects in mice. Dis Model Mech 6 : 1448–1458. doi: 10.1242/dmm.012849 24077964
23. Yang W, Wang J, Moore DC, Liang H, Dooner M, et al. (2013) Ptpn11 deletion in a novel progenitor causes metachondromatosis by inducing hedgehog signalling. Nature.
24. Bowen ME, Ayturk UM, Kurek KC, Yang W, Warman ML (2014) SHP2 Regulates Chondrocyte Terminal Differentiation, Growth Plate Architecture and Skeletal Cell Fates. PLoS Genet 10: e1004364. doi: 10.1371/journal.pgen.1004364 24875294
25. Kronenberg HM (2006) PTHrP and skeletal development. Annals of the New York Academy of Sciences 1068 : 1–13. 16831900
26. Sgariglia F, Candela ME, Huegel J, Jacenko O, Koyama E, et al. (2013) Epiphyseal abnormalities, trabecular bone loss and articular chondrocyte hypertrophy develop in the long bones of postnatal Ext1-deficient mice. Bone 57 : 220–231. doi: 10.1016/j.bone.2013.08.012 23958822
27. Foldynova-Trantirkova S, Wilcox WR, Krejci P (2012) Sixteen years and counting: the current understanding of fibroblast growth factor receptor 3 (FGFR3) signaling in skeletal dysplasias. Hum Mutat 33 : 29–41. doi: 10.1002/humu.21636 22045636
28. Chen L, Adar R, Yang X, Monsonego EO, Li C, et al. (1999) Gly369Cys mutation in mouse FGFR3 causes achondroplasia by affecting both chondrogenesis and osteogenesis. Journal of Clinical Investigation 104 : 1517–1525. 10587515
29. Chen L, Li C, Qiao W, Xu X, Deng C (2001) A Ser365→ Cys mutation of fibroblast growth factor receptor 3 in mouse downregulates Ihh/PTHrP signals and causes severe achondroplasia. Human molecular genetics 10 : 457–465. 11181569
30. Iwata T, Chen L, Li C, Ovchinnikov DA, Behringer RR, et al. (2000) A neonatal lethal mutation in FGFR3 uncouples proliferation and differentiation of growth plate chondrocytes in embryos. Hum Mol Genet 9 : 1603–1613. 10861287
31. Toydemir RM, Brassington AE, Bayrak-Toydemir P, Krakowiak PA, Jorde LB, et al. (2006) A novel mutation in FGFR3 causes camptodactyly, tall stature, and hearing loss (CATSHL) syndrome. Am J Hum Genet 79 : 935–941. 17033969
32. Makrythanasis P, Temtamy S, Aglan MS, Otaify GA, Hamamy H, et al. (2014) A novel homozygous mutation in FGFR3 causes tall stature, severe lateral tibial deviation, scoliosis, hearing impairment, camptodactyly and arachnodactyly. Hum Mutat.
33. Deng C, Wynshaw-Boris A, Zhou F, Kuo A, Leder P (1996) Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell 84 : 911–921. 8601314
34. Colvin JS, Bohne BA, Harding GW, McEwen DG, Ornitz DM (1996) Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nat Genet 12 : 390–397. 8630492
35. Liu Z, Xu J, Colvin JS, Ornitz DM (2002) Coordination of chondrogenesis and osteogenesis by fibroblast growth factor 18. Genes Dev 16 : 859–869. 11937493
36. Goetz R, Mohammadi M (2013) Exploring mechanisms of FGF signalling through the lens of structural biology. Nat Rev Mol Cell Biol 14 : 166–180. doi: 10.1038/nrm3528 23403721
37. Krejci P, Bryja V, Pachernik J, Hampl A, Pogue R, et al. (2004) FGF2 inhibits proliferation and alters the cartilage-like phenotype of RCS cells. Experimental cell research 297 : 152–164. 15194433
38. Krejci P, Masri B, Salazar L, Farrington-Rock C, Prats H, et al. (2007) Bisindolylmaleimide I suppresses fibroblast growth factor-mediated activation of Erk MAP kinase in chondrocytes by preventing Shp2 association with the Frs2 and Gab1 adaptor proteins. Journal of biological chemistry 282 : 2929–2936. 17145761
39. Legeai-Mallet L, Benoist-Lasselin C, Delezoide AL, Munnich A, Bonaventure J (1998) Fibroblast Growth Factor Receptor 3 Mutations Promote Apoptosis but Do Not Alter Chondrocyte Proliferation in Thanatophoric Dysplasia. Journal of Biological Chemistry 273 : 13007–13014. 9582336
40. L'Hote CG, Knowles MA (2005) Cell responses to FGFR3 signalling: growth, differentiation and apoptosis. Exp Cell Res 304 : 417–431. 15748888
41. Harada D, Yamanaka Y, Ueda K, Nishimura R, Morishima T, et al. (2007) Sustained phosphorylation of mutated FGFR3 is a crucial feature of genetic dwarfism and induces apoptosis in the ATDC5 chondrogenic cell line via PLCgamma-activated STAT1. Bone 41 : 273–281. 17561467
42. Krejci P, Prochazkova J, Smutny J, Chlebova K, Lin P, et al. (2010) FGFR3 signaling induces a reversible senescence phenotype in chondrocytes similar to oncogene-induced premature senescence. Bone 47 : 102–110. doi: 10.1016/j.bone.2010.03.021 20362703
43. Amizuka N, Davidson D, Liu H, Valverde-Franco G, Chai S, et al. (2004) Signalling by fibroblast growth factor receptor 3 and parathyroid hormone-related peptide coordinate cartilage and bone development. Bone 34 : 13–25. 14751559
44. Michigami T (2013) Regulatory mechanisms for the development of growth plate cartilage. Cell Mol Life Sci 70 : 4213–4221. doi: 10.1007/s00018-013-1346-9 23640571
45. Long F, Ornitz DM (2013) Development of the endochondral skeleton. Cold Spring Harb Perspect Biol 5: a008334. doi: 10.1101/cshperspect.a008334 23284041
46. Naski MC, Wang Q, Xu J, Ornitz DM (1996) Graded activation of fibroblast growth factor receptor 3 by mutations causing achondroplasia and thanatophoric dysplasia. Nat Genet 13 : 233–237. 8640234
47. Li C, Chen L, Iwata T, Kitagawa M, Fu X-Y, et al. (1999) A Lys644Glu substitution in fibroblast growth factor receptor 3 (FGFR3) causes dwarfism in mice by activation of STATs and ink4 cell cycle inhibitors. Human molecular genetics 8 : 35–44. 9887329
48. Kim HK, Feng GS, Chen D, King PD, Kamiya N (2013) Targeted disruption of Shp2 in chondrocytes leads to metachondromatosis with multiple cartilaginous protrusions. J Bone Miner Res.
49. Xie J, Bartels CM, Barton SW, Gu D (2013) Targeting hedgehog signaling in cancer: research and clinical developments. OncoTargets and therapy 6 : 1425. doi: 10.2147/OTT.S34678 24143114
50. Rudin CM, Hann CL, Laterra J, Yauch RL, Callahan CA, et al. (2009) Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. New England Journal of Medicine 361 : 1173–1178. doi: 10.1056/NEJMoa0902903 19726761
51. Krejci P (2014) The paradox of FGFR3 signaling in skeletal dysplasia: Why chondrocytes growth arrest while other cells over proliferate. Mutation Research/Reviews in Mutation Research 759 : 40–48. doi: 10.1016/j.mrrev.2013.11.001 24295726
52. Ho L, Stojanovski A, Whetstone H, Wei QX, Mau E, et al. (2009) Gli2 and p53 cooperate to regulate IGFBP-3 - mediated chondrocyte apoptosis in the progression from benign to malignant cartilage tumors. Cancer Cell 16 : 126–136. doi: 10.1016/j.ccr.2009.05.013 19647223
53. Jones KB, Pacifici M, Hilton MJ (2014) Multiple Hereditary Exostoses (MHE): Elucidating the Pathogenesis of a Rare Skeletal Disorder Through Interdisciplinary Research. Connective tissue research: 1–26.
54. De Bari C, Dell'Accio F, Luyten FP (2001) Human periosteum-derived cells maintain phenotypic stability and chondrogenic potential throughout expansion regardless of donor age. Arthritis Rheum 44 : 85–95. 11212180
55. Ito Y, Fitzsimmons JS, Sanyal A, Mello MA, Mukherjee N, et al. (2001) Localization of chondrocyte precursors in periosteum. Osteoarthritis Cartilage 9 : 215–223. 11300744
56. Superti-Furga A, Spranger J, Nishimura G (2012) Enchondromatosis revisited: new classification with molecular basis. Am J Med Genet C Semin Med Genet 160C: 154–164. doi: 10.1002/ajmg.c.31331 22791316
57. Zelzer E, Mamluk R, Ferrara N, Johnson RS, Schipani E, et al. (2004) VEGFA is necessary for chondrocyte survival during bone development. Development 131 : 2161–2171. 15073147
58. Sebastian A, Matsushita T, Kawanami A, Mackem S, Landreth GE, et al. (2011) Genetic inactivation of ERK1 and ERK2 in chondrocytes promotes bone growth and enlarges the spinal canal. J Orthop Res 29 : 375–379. doi: 10.1002/jor.21262 20922792
59. Matsushita T, Chan YY, Kawanami A, Balmes G, Landreth GE, et al. (2009) Extracellular signal-regulated kinase 1 (ERK1) and ERK2 play essential roles in osteoblast differentiation and in supporting osteoclastogenesis. Molecular and cellular biology 29 : 5843–5857. doi: 10.1128/MCB.01549-08 19737917
60. Murakami S, Balmes G, McKinney S, Zhang Z, Givol D, et al. (2004) Constitutive activation of MEK1 in chondrocytes causes Stat1-independent achondroplasia-like dwarfism and rescues the Fgfr3-deficient mouse phenotype. Genes Dev 18 : 290–305. 14871928
61. Maeda Y, Nakamura E, Nguyen MT, Suva LJ, Swain FL, et al. (2007) Indian Hedgehog produced by postnatal chondrocytes is essential for maintaining a growth plate and trabecular bone. Proc Natl Acad Sci U S A 104 : 6382–6387. 17409191
62. Brechbiel JL, Ng JM, Curran T (2011) PTHrP treatment fails to rescue bone defects caused by Hedgehog pathway inhibition in young mice. Toxicologic pathology 39 : 478–485. doi: 10.1177/0192623311399788 21411723
63. Su N, Xu X, Li C, He Q, Zhao L, et al. (2010) Generation of Fgfr3 conditional knockout mice. International journal of biological sciences 6 : 327. 20582225
64. Zhu M, Chen M, Lichtler A, O'Keefe R, Chen D (2008) Tamoxifen-inducible Cre-recombination in articular chondrocytes of adult Col2a1-CreERT2 transgenic mice. Osteoarthritis and Cartilage 16 : 129–130. 17888690
65. Yang G, Sun Q, Teng Y, Li F, Weng T, et al. (2008) PTEN deficiency causes dyschondroplasia in mice by enhanced hypoxia-inducible factor 1alpha signaling and endoplasmic reticulum stress. Development 135 : 3587–3597. doi: 10.1242/dev.028118 18832389
66. Arango NA, Lovell-Badge R, Behringer RR (1999) Targeted mutagenesis of the endogenous mouse Mis gene promoter: in vivo definition of genetic pathways of vertebrate sexual development. Cell 99 : 409–419. 10571183
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 6
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Non-reciprocal Interspecies Hybridization Barriers in the Capsella Genus Are Established in the Endosperm
- Translational Upregulation of an Individual p21 Transcript Variant by GCN2 Regulates Cell Proliferation and Survival under Nutrient Stress
- Exome Sequencing of Phenotypic Extremes Identifies and as Interacting Modifiers of Chronic Infection in Cystic Fibrosis
- The Human Blood Metabolome-Transcriptome Interface
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy