
Evidence of a Bacterial Receptor for Lysozyme: Binding of Lysozyme to the Anti-σ Factor RsiV Controls Activation of the ECF σ Factor σ
All cells sense and respond to changes in their environments by transmitting information across the membrane. In bacteria, σ factors provide promoter specificity to RNA polymerase. Bacteria encode Extra-Cytoplasmic Function (ECF) σ factors, which often respond to extracellular signals. Activation of some ECF σ factors is controlled by stepwise proteolytic destruction of an anti-σ factor which is initiated by a site-1 protease. In most cases, the site-1 protease required to initiate the RIP process is thought to be the signal sensor. Here we report that the anti-σ factor RsiV, and not the site-1 protease, is the sensor for σV activation. Activation of the ECF σ factor σV is induced by lysozyme, an innate immune defense enzyme. We identify the site-1 protease as signal peptidase, which is required for general protein secretion. The anti-σ factor RsiV directly binds lysozyme. Binding of lysozyme to RsiV allows signal peptidase to cleave RsiV at site-1 and this leads to activation of σV. Thus, the anti-σ factor functions as a bacterial receptor for lysozyme. RsiV homologs from C. difficile and E. faecalis also bind lysozyme, suggesting they may utilize this receptor-ligand mechanism to control activation of σV to induce lysozyme resistance.
Published in the journal:
. PLoS Genet 10(10): e32767. doi:10.1371/journal.pgen.1004643
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004643
Summary
All cells sense and respond to changes in their environments by transmitting information across the membrane. In bacteria, σ factors provide promoter specificity to RNA polymerase. Bacteria encode Extra-Cytoplasmic Function (ECF) σ factors, which often respond to extracellular signals. Activation of some ECF σ factors is controlled by stepwise proteolytic destruction of an anti-σ factor which is initiated by a site-1 protease. In most cases, the site-1 protease required to initiate the RIP process is thought to be the signal sensor. Here we report that the anti-σ factor RsiV, and not the site-1 protease, is the sensor for σV activation. Activation of the ECF σ factor σV is induced by lysozyme, an innate immune defense enzyme. We identify the site-1 protease as signal peptidase, which is required for general protein secretion. The anti-σ factor RsiV directly binds lysozyme. Binding of lysozyme to RsiV allows signal peptidase to cleave RsiV at site-1 and this leads to activation of σV. Thus, the anti-σ factor functions as a bacterial receptor for lysozyme. RsiV homologs from C. difficile and E. faecalis also bind lysozyme, suggesting they may utilize this receptor-ligand mechanism to control activation of σV to induce lysozyme resistance.
Introduction
Cells respond to changes in their environments using signal transduction systems, which transmit information from outside the cell across the membrane to effect transcriptional responses. Regulated Intramembrane Proteolysis (RIP) is one mechanism by which cells sense and respond to changes in the environment. The RIP signal transduction system was first described as the mechanism for controlling cholesterol biosynthesis in mammals [1]. In bacteria, RIP processes regulate the activity of several alternative σ factors including multiple Extra Cytoplasmic Function (ECF) σ factors. Most RIP signal transduction systems involve sequential cleavages of a membrane-tethered protein. Following site-1 cleavage by an initial protease, a second protease cleaves the substrate within the membrane at site-2. In most cases the rate-limiting step for activation of the signal transduction system is the cleavage of the substrate at site-1 [2]. Here we describe the role of RIP in regulating the activity of the B. subtilis ECF σ factor σV in response to lysozyme.
In bacteria, σ factors combine with RNA polymerase to recognize specific promoter sequences and transcribe mRNA. ECF σ factors represent a large and diverse family of important signal transduction systems in bacteria [3]. RIP regulates the activity of several alternative σ factors including multiple ECF σ factors in the subfamily ECF01 [2], . In Escherichia coli, activation of the ECF σ factor σE is initiated by site-1 cleavage of the anti-σ factor RseA when unfolded outer membrane β-barrel proteins bind to and activate the site-1 protease DegS [4]–[7]. In Bacillus subtilis activation of the ECF σ factor σW is thought to be controlled by activation of the site-1 protease PrsW, since mutants of PrsW were isolated which resulted in constitutive cleavage of the anti-σ factor RsiW even in the absence of stress [8]. In each of these cases the site-1 protease is thought to sense the signal required for activation of these ECF σ factors.
The B. subtilis ECF σ factor, σV, belongs to the ECF30 subfamily of ECF σ factors, members of which are primarily found in firmicutes (low GC Gram-positive bacteria) [3]. A subset of the ECF30 homologs are controlled by anti-σ factors homologous to RsiV. σV is activated in response to lysozyme but not to other cell envelope stresses [9]–[11]. Lysozyme is an essential component of the host innate immune system which fights bacterial infection by cleaving cell wall saccharides and σV induces resistance to lysozyme [11]–[13]. We recently demonstrated that activation of σV requires the proteolytic destruction of the membrane tethered anti-σ factor, RsiV, in a RIP dependent mechanism [14]. This degradation requires the site-2 protease RasP [14]. RasP cleavage results in free σV which can complex with RNA polymerase and transcribe genes required for lysozyme resistance [9], [10]. Here we present evidence that the site-1 protease required for RsiV degradation is none other than signal peptidase. Signal peptidases are an essential component of the cellular secretion apparatus and are conserved from bacteria to eukaryotes. The activity of signal peptidases is not known to be regulated in response to environmental signals. We propose a model in which the sensor for cell envelope stress is the anti-σ factor RsiV. Our data indicate that RsiV is the direct receptor for lysozyme. Thus the anti-σ factor, and not the site-1 protease, is the sensor for lysozyme.
Results
Identification of the Site-1 Cleavage Site
We previously demonstrated that activation of σV required the proteolytic destruction of the anti-σ factor RsiV in a RIP dependent mechanism [14]. Upon exposure to lysozyme we could detect what appeared to be the cleaved extracellular domain of RsiV [14] (Figure 1). This suggested that the extracellular domain is removed by an unknown protease that cleaves RsiV at site-1 after treatment with lysozyme. To determine the location of the site-1 cleavage, we constructed a B. subtilis strain producing a C-terminal 6×His tagged version of RsiV (RsiV6×His). Protoplasts of this strain were then generated using lysozyme. Following this treatment, we were able to purify the RsiV6×His from the supernatant of these cells using nickel affinity resin (Figure S1). The sequence of the first 8 amino acids of the partially purified cleaved RsiV6×His was determined by Edman degradation [15]. The N-terminal sequence of the cleaved RsiV domain was (MSKIPVIG) which indicates RsiV is cleaved between A66 and M67 (Figure S1 and Figure 1A).

Disruption of Site-1 Cleavage Blocks RsiV Degradation and σV Activation
Analysis of the RsiV site-1 cleavage site revealed a canonical AXA motif suggestive of a signal peptide cleavage site. In fact subsequent analysis of RsiV in silico using SignalP [16] revealed a putative signal peptidase cleavage site between amino acids 66 and 67 of RsiV (Figure S2A). Furthermore an alignment of 185 RsiV homologs using Multiple Em for Motif Elicitation (MEME) a tool for identifying motifs in related sequences [17], reveals a highly conserved AXA motif just after their predicted transmembrane segment [18] (Figure S2B). In addition, analysis of C. difficile, E. faecalis and B. subtilis RsiV homologs using SignalP revealed the presence of a predicted signal peptidase cleavage site in each of those proteins (Figure S2A).
B. subtilis PY79 encodes five type 1 signal peptidases [19], [20]. SipS and SipT are the two major signal peptidases in B. subtilis and are redundant, but cells lacking both SipS and SipT are not viable [20]. We constructed sipS and sipT mutant strains and tested RsiV degradation and σV activation. We found that RsiV was still degraded in the absence of either SipS or SipT (Figure S3). We attempted to construct a strain to deplete signal peptidase however we were unsuccessful. Thus, to determine if disruption of cleavage at this site was sufficient to block RsiV degradation and σV activation, the alanine codon at position 66 was mutated to a tryptophan codon and tested for an effect on HEW lysozyme induced degradation. We found that while wild-type RsiV was rapidly degraded, the degradation of the RsiVA66W mutant protein was blocked even in the presence of lysozyme (Figure 1B). This suggests that the RsiVA66W mutant is unable to be cleaved at site-1. We then tested the effect of RsiVA66W mutant protein on activation of σV in response to lysozyme by measuring expression of the PsigV-lacZ reporter. In cells producing wild type RsiV, expression of PsigV-lacZ was induced 16-fold in the presence of lysozyme (Figure 1C). In contrast, in strains producing RsiVA66W there was no observable lysozyme induction of PsigV-lacZ expression. This indicates that the RsiVA66W protein is resistant to site-1 cleavage, and thus, blocks activation of σV in response to lysozyme by inhibiting RsiV degradation. In addition, a strain producing RsiVA66W is more susceptible to lysozyme than WT RsiV, due to an inability to degrade RsiV and activate σV (Table 1). Taken together this suggests that cleavage at site-1 is required for σV activation.

In the Presence of Lysozyme SipS Is Sufficient for Site-1 Cleavage of RsiV In Vitro
Our data indicate that RsiV is cleaved at a putative signal peptidase cleavage site. Since signal peptidase activity is essential in B. subtilis [20] we sought to determine if signal peptidases were directly responsible for cleavage of the anti-σ factor RsiV in vitro. The signal peptidase SipS and RsiV both contain transmembrane domains and we hypothesized that these may be important for proper control of RsiV degradation. Thus, we produced SipS and RsiV in vitro using a cell free in vitro transcription/translation system. This method has been used successfully to test the ability of other membrane proteases to cleave their substrates [21]. Briefly, mRNA of sipS and rsiV was produced using SP6 RNAP as previously described [21]. The resulting mRNA served as a translation template using wheat germ extract as a source of ribosomes. Wheat germ extract contains sufficient endogenous lipids that at least some functional membrane proteins can be produced without addition of liposomes [21]. Both SipS and RsiV were produced in sufficient quantities that they could be visualized by Coomassie staining and they are present mostly in the insoluble pellet (i.e. lipid-containing) fraction of the in vitro translation reactions (Figure S4). We combined the SipS and RsiV reactions at 1∶3 molar ratios with or without the presence of HEW lysozyme and incubated the reactions at 37°C for 6 hours. Using anti-RsiV59–285 antibodies we were unable to detect the presence of any cleavage products of 3×Flag-CBP-RsiV when it was incubated alone or in the presence of SipS (Figure 2). When 3×Flag-CBP-RsiV was incubated with SipS in the presence of HEW lysozyme we observed the production of a cleavage product and a decrease in the amount of full length 3×Flag-CBP-RsiV (Figure 2). We observed minimal cleavage when 3×Flag-CBP-RsiV was incubated in the presence of HEW lysozyme, likely due to the presence of an eukaryotic signal peptidase in the wheat germ, which also recognizes an AXA motif [22], [23] (Figure 2). The cleaved product in vitro was the same size as the cleaved product produced from RsiV6×His in vivo (Figure 2A).

We sought to determine if the A66W substitution in RsiV would block site-1 cleavage in vitro as it did in vivo. As seen in Figure 2 we observed only full length 3×Flag-CBP-RsiVA66W when 3×Flag-CBP-RsiVA66W was incubated with both SipS and HEW lysozyme (Figure 2). This suggests, in agreement with our in vivo results, that altering the signal peptidase recognition site in RsiV blocks site-1 cleavage in vitro. These data suggest that SipS is able to directly cleave RsiV and importantly this cleavage only occurs in the presence of HEW lysozyme.
RsiV Binds HEW Lysozyme
Our data suggest that RsiV is cleaved by the signal peptidase SipS in vitro only in the presence of lysozyme. However there is no peptidoglycan present in the in vitro reactions which raised the question; why is HEW lysozyme required for site-1 cleavage? We hypothesized that RsiV may directly bind HEW lysozyme. We constructed a 6×His-RsiV59–285 fusion and purified it from E. coli (Figure S5). We tested the ability of RsiV to bind HEW lysozyme using co-purification. We found that when 6×His-RsiV59–285 was bound to the nickel column HEW lysozyme was co-eluted (Figure 3A). However when HEW lysozyme was loaded on a column treated with the extract of BL21(DE3) empty vector-containing cells, we found HEW lysozyme in the flow through and wash fractions but not present in the elution fractions (Figure 3A). To confirm RsiV binds to HEW lysozyme in vivo as well as in vitro we performed a co-purification experiment using B. subtilis producing RsiV6×His. We found that when we purified cleaved RsiV6×His after treatment with lysozyme a single band with a similar size to HEW lysozyme co-eluted with RsiV6×His (Figure 3B). We confirmed that this band corresponded to HEW lysozyme by N-terminal sequencing. This suggests that RsiV binds HEW lysozyme both in vitro and in vivo.

We used Isothermal Titration Calorimetry (ITC) to confirm these observations as well as to determine the affinity of RsiV59–285 for HEW lysozyme. We found that RsiV59–285 binds to HEW lysozyme in an enthalpically-driven reaction with a Kd of 70 nM (Figure 3C; Table 2). Although it is difficult to determine the precise affinity of HEW lysozyme to peptidoglycan, because peptidoglycan is very heterogeneous, the best data suggest the Kd of PG to lysozyme is ∼50 mM [24]–[26]. This suggests that the HEW lysozyme-RsiV affinity is significantly greater than the affinity of HEW lysozyme for peptidoglycan.

Muramidase Activity Is Not Sufficient for σV Activation
Previous work found that lysozyme was able to induce σV activity. In contrast, a variety of antimicrobial compounds that inhibited peptidoglycan synthesis and damaged the cell envelope were unable to induce σV activity [9]. Since RsiV binds HEW lysozyme, we hypothesized that the protein and not the activity of HEW lysozyme was required to activate σV. Mutanolysin cleaves the same β-glycosidic bond as HEW lysozyme, but has an entirely different amino acid sequence and structure [27]. To determine if muramidase activity was sufficient for σV activation we compared the ability of HEW lysozyme, human lysozyme, and mutanolysin to induce expression of the PsigV-lacZ reporter fusion. We found that HEW lysozyme and human lysozyme, both C-type lysozymes with 58% amino acid identity and 76% similarity, produced zones of clearing (8 mm and 9 mm respectively), and induced expression of PsigV-lacZ as indicated by the blue ring around the disk (Figure 4A). We found that although mutanolysin produced a zone of clearing (7 mm) indicative of killing, it was unable to induce PsigV-lacZ expression. The lack of induction by mutanolysin suggests that muramidase activity is not sufficient for activation of σV.

Since σV is activated upon degradation of the transmembrane bound anti-σ RsiV we tested the effect of HEW lysozyme, human lysozyme, and mutanolysin, for their ability to induce RsiV degradation. We expressed rsiV from an IPTG inducible promoter and then treated cells with a sub-lethal concentration of lysozyme for 10 minutes. Using anti-RsiV59–285 antibodies a 32 kDa protein was observed by immunoblot in the absence of lysozyme, indicative of the full length RsiV (Figure 4B). In the presence of HEW lysozyme, the full-length RsiV is rapidly degraded and a smaller product, the released extracellular domain, is visible (Figure 4B). Similarly, treatment with human lysozyme also induces RsiV degradation (Figure 4B). However, treatment with mutanolysin was unable to induce degradation of RsiV (Figure 4B).
To further confirm the observation that muramidase activity was insufficient to induce RsiV degradation, we used mutanolysin to generate protoplasts of B. subtilis cells expressing rsiV. These cells were then left untreated or treated with HEW lysozyme and the status of RsiV monitored by immunoblot. We found that the protoplasts still retain full-length RsiV however upon subsequent treatment with lysozyme we observe loss of full-length RsiV (Figure 4C). This provides further evidence that muramidase activity alone is not sufficient to induce degradation of RsiV.
Muramidase Deficient Lysozyme Activates σV
Our data indicate that muramidase activity provided by mutanolysin is not sufficient to induce σV activation or RsiV degradation, thus we asked if muramidase activity was required for lysozyme-driven σV activation or RsiV degradation. It has been shown that changing human lysozyme aspartate 53 to serine abolishes muramidase activity to less than 1% [28]. Recombinant human lysozyme (R-lysozyme), and the catalytically inactive form of human lysozyme (R-lysozymeD53S) were expressed and purified from the supernatant of Pichia pastoris [29]. Muramidase activity of R-lysozyme and R-lysozymeD53S was assayed against Micrococcus lysodekticus peptidoglycan which confirmed that the R-lysozyme was active and R-lysozymeD53S was muramidase deficient (Figure S6).
We found that when purified R-lysozyme and R-lysozymeD53S were placed on a lawn of B. subtilis containing the PsigV-lacZ reporter fusion both produced a zone of induction (Figure 4A). However only the wild type R-lysozyme produced a zone of clearing (9 mm) while the R-lysozymeD53S did not produce a zone of clearing (6 mm – size of the disk) (Figure 4A). To further confirm that muramidase activity was not required for σV activation we tested the ability of the recombinant active or inactive lysozyme to induce RsiV degradation. Consistent with the zone of clearing results, treatment with human lysozyme, R-lysozyme, and R-lysozymeD52S also induce RsiV degradation (Figure 4B). However, treatment with mutanolysin was unable to induce degradation of RsiV (Figure 4B). Thus, the lack of PsigV-lacZ induction by mutanolysin and the ability of catalytically inactive R-lysozymeD53S to induce PsigV-lacZ suggests that muramidase activity is not required nor is it sufficient for activation of σV.
RsiV Binds C-Type Lysozymes HEW Lysozyme and Human Lysozyme, but Not Mutanolysin
Our data suggests that σV is activated by the C-type lysozymes HEW lysozyme and human lysozyme, but not the unrelated muramidase mutanolysin. Since RsiV can bind HEW lysozyme we sought to determine if σV activation was correlated with the ability of RsiV to bind different proteins. To test this we purified a GST-RsiV59–285 fusion protein. We then loaded 2 mg of GST-RsiV59–285 onto a glutathione column and then 2 mg of either HEW lysozyme, human lysozyme, or mutanolysin were passed over the column. The proteins were eluted and separated by SDS/PAGE (Figure 5). We found that both HEW lysozyme and human lysozyme were retained on the column when RsiV was present. This suggests that both HEW lysozyme and human lysozyme can bind RsiV59–285. In contrast, we found when mutanolysin was loaded onto the GST-RsiV-RsiV59–285 containing column mutanolysin was collected almost entirely in either the flow through and wash fractions (Figure 5). This suggests that RsiV specifically binds C-type lysozymes but not mutanolysin.

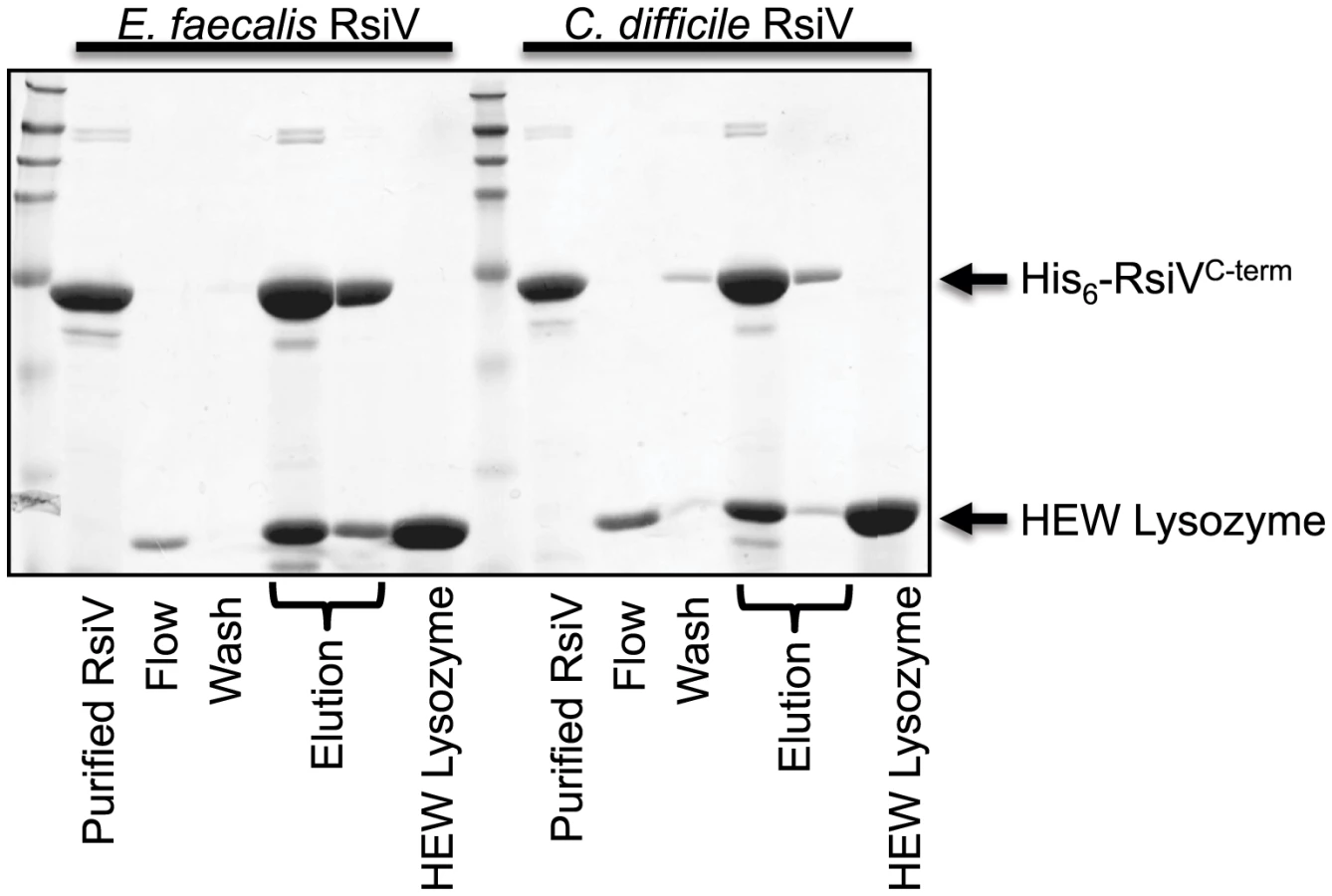
C. difficile and E. faecalis RsiV Directly Bind HEW Lysozyme
Both C. difficile and E. faecalis encode homologs of σV and RsiV and in each organism σV is activated by lysozyme and required for lysozyme resistance [11], [13], [30]. To determine if the ability of the anti-σ factors to interact with lysozyme was a conserved feature we purified the histidine-tagged extracellular domains of both C. difficile RsiV69–289 (RsiVCD) and E. faecalis RsiV72–294 (RsiVEF) and conducted binding assays. Briefly, purified C. difficile RsiVCD or E. faecalis RsiVEF protein was loaded onto a nickel column and 2 mg of HEW lysozyme passed over the column bound protein. We found that columns containing either C. difficile or E. faecalis RsiV resulted in retention of HEW lysozyme on the column. Upon elution from the column, we found that HEW lysozyme co-eluted with both RsiVCD and RsiVEF (Figure 6). This suggests that the ability of the anti-σ factor, RsiV, to bind lysozyme is a conserved feature present in RsiV homologs from other species.
Discussion
RsiV as a Receptor for C-Type Lysozyme
The primary observations of this work are that the anti-σ factor and RIP substrate RsiV acts as a sensor for the presence of lysozyme and that SipS functions as a site-1 protease. This hypothesis is supported by the following observations 1) Co-purification experiments in which RsiV binds both human and HEW lysozyme, 2) ITC showing direct binding of RsiV and HEW lysozyme, 3) RsiV cleavage in vitro at site-1 by SipS only in the presence of lysozyme, 4) Insufficiency of muramidase activity for σV activation and RsiV degradation and 5) Induction of σV activation and RsiV degradation by catalytically inactive human lysozyme. Taken together this data supports a model in which RsiV is a receptor for lysozyme.
We identify the signal peptidase SipS as a site-1 protease for the anti-σ factor RsiV. The evidence to support signal peptidase as the site-1 protease for RsiV is as follows. The site-1 cleavage site of RsiV was identified and found to resemble a signal peptide cleavage site. Mutating this cleavage site blocks RsiV degradation and σV activation. Using a cell free transcription/translation system, we demonstrate in vitro that SipS was sufficient for site-1 cleavage of RsiV only in the presence of lysozyme. Together these data indicate that signal peptidase is the site-1 protease for RsiV. The signal peptidases of B. subtilis are redundant, but cells lacking both SipS and SipT are not viable [20]. RsiV was found to be cleaved at site-1 in the absence of SipS or SipT. Thus we hypothesize that one or more of the B. subtilis signal peptidases can cleave RsiV at site-1 in vivo.
Previous work from our lab found that σV was activated only by lysozyme and not by other cell envelope stresses [9]. Here we showed that muramidase activity was not required nor was it sufficient to activate σV or induce RsiV degradation. In fact, we did not detect significant cleavage of RsiV in B. subtilis protoplasts generated by mutanolysin. However, upon addition of HEW lysozyme to these protoplasts RsiV was rapidly cleaved at site-1. In addition, the catalytically inactive form of lysozyme (R-lysozymeD53S) was still able to activate σV and degrade RsiV. Finally, cleavage of RsiV by SipS in vitro was dependent upon HEW lysozyme suggesting RsiV binding to lysozyme is required for RsiV cleavage independent of muramidase activity.
Based upon these observations we propose a model of σV activation in which RsiV is a receptor for the C-type lysozymes, HEW lysozyme and human lysozyme (Figure 7). In the absence of C-type lysozyme RsiV is in a conformation which is resistant to signal peptidase and RsiV inhibits σV activity by sequestering it to the membrane (Figure 7). In the presence of C-type lysozyme, the C-terminal domain of RsiV binds to lysozyme (Figure 7). When RsiV is bound to lysozyme, RsiV undergoes a conformational change, which allows signal peptidase to cleave RsiV at site-1 (Figure 7). This allows the site-2 protease to cleave the truncated form of RsiV leading to release of the RsiV-σV complex (Figure 7) [14]. The cytoplasmic portion of RsiV is then presumably degraded by cytosolic proteases, resulting in free σV which can complex with RNA polymerase and transcribe genes required for lysozyme resistance (Figure 7) [9], [10].

There are several examples of anti-σ factors directly sensing inducing signals, however in these cases the anti-σ factors are not degraded in RIP dependent manner. For example the anti-σ factor of Streptomyces coelicolor σR, RsrA, is responsible for sensing redox stress [31]–[33] and the anti-σ factor of Rhodobacter sphaeroides σE, ChrR, is responsible for sensing singlet oxygen [34], [35]. In E. coli regulation of the genes encoding the FecABCDE iron transport system are controlled by the iron responsive FecRI system. Evidence suggests that the anti-σ factor FecR can act indirectly as a sensor for the presence of iron-citrate [36]. However, to our knowledge this is the first example of a RIP controlled anti-σ factor acting as a receptor for the inducing signal. In the most well studied cases, the ECF σ factors that are controlled by RIP, it is the site-1 proteases that are the sensors for cell envelope damage. In particular it is clear that activation of σE in E. coli requires binding of unfolded outer membrane β-barrel proteins to the site-1 protease DegS, which allows DegS to cleave the σE anti-σ factor RseA [4], [6], [7]. In addition to DegS, the RseA binding protein, RseB, also contributes to sensing cell envelope stress [37]–[39]. In the case of σW activation in B. subtilis, isolation of mutations in the site-1 protease PrsW which result in constitutive degradation of RsiW again suggest the protease itself is the sensor for the inducing signal [8]. We found that SipS was able to cleave RsiV at site-1 only in the presence of HEW lysozyme. The enzymatic activity of signal peptidase is not known to be regulated by environmental signals. Since lysozyme binds RsiV we hypothesize that the mechanism for controlling site-1 cleavage of RsiV doesn't reside with the site-1 protease, but within the anti-σ factor itself.
A Role for Signal Peptidase in Signal Transduction
There are several examples of signal peptidases being required for production of quorum sensing signals. For example, in S. aureus signal peptidase is required for production of the quorum sensing peptide or auto-inducing peptide AIP [40]. In B. subtilis signal peptidases are required for production of the quorum sensing Phr peptides [41]. Once the Phr peptides released from the cell accumulate to sufficient quantities they are thought to directly or indirectly inhibit the Rap phosphatases which control initiation of sporulation and competence processes [42]–[44]. In each of these cases however the role of signal peptidase is not in sensing of a signal but in the production of a signal.
There is some recent evidence that signal peptidases may be involved in signal input by cleaving a sensor for detecting β-lactam antibiotics. In Staphylococcus epidermidis it has been observed that the β-lactam sensor domain of BlaR1 (a protease which degrades the transcription regulator BlaI) was released in what appears to be a signal peptidase dependent process [45]. Similarly, in Staphylococcus aureus there is recent evidence to suggest that this β-lactam sensor domain is also removed at what appears to be a putative signal peptide cleavage site [46]. However it is not yet clear what impact this processing has on the signal transduction activity of BlaR1.
σV Activation and RsiV Degradation in Other Organisms
Homologs of the σV system in E. faecalis and C. difficile are also induced by lysozyme [11], [13], [30]. In both of these organisms σV activity is inhibited by RsiV [11], [30]. We found that the RsiV homologs from both C. difficile and E. faecalis also bind HEW lysozyme, suggesting RsiV from these organisms may also sense lysozyme directly and activate using a similar mechanism. Although the site-1 protease that initiates RsiV degradation in E. faecalis or C. difficile is not known, an alignment of 185 RsiV homologs using MEME [17] reveals a highly conserved AXA motif near the predicted transmembrane domain of these RsiV homologs. Furthermore analysis of C. difficile, E. faecalis and B. subtilis RsiV homologs using SignalP [16] reveals potential signal peptidase cleavage sites in each RsiV homolog (Figure S2A). Future work will be required to see if site-1 cleavage of RsiV in other organisms is also carried out by signal peptidase.
Activation of σV in B. subtilis and E. faecalis requires cleavage of RsiV by a site-2 protease [14], [47]. In addition to cleavage of RsiV and other anti-σ factors, RasP is also able to clear signal peptidase processed signal peptides from the membrane [48]. Thus, it appears that the σV-RsiV signal transduction system has “plugged into” the signal peptide processing system. Interestingly, the σV-RsiV signal transduction system is present primarily in many, but not all firmicutes, suggesting the σV–RsiV system may be transmitted by horizontal gene transfer. Thus the ability of the anti-σ factor to act as a sensor and capitalize on an essential system present in all bacteria, may provide a mechanism to control activation of horizontally acquired ECF σ factors.
One of the surprising findings is the specific activation of σV by C-type lysozyme but not mutanolysin. In B. subtilis σV holo RNA polymerase transcribes oatA which encodes an O-acetyl transferase and in C. difficile and E. faecalis σV holo RNA polymerase transcribes a peptidoglycan deacetylase gene [9], [13], [49], [50]. Interestingly both mechanisms increase resistance to C-type lysozymes (HEW and human lysozyme) [50]–[54] but not mutanolysin [55]–[57]. Thus rather than relying on peptidoglycan damage or cell envelope stress the RsiV - σV system appears to have evolved to respond to C-type lysozyme through a receptor-ligand interaction. This interaction induces genes which provide resistance to C-type lysozyme but not to other muramidases.
An interesting question raised by the ability of B. subtilis to respond specifically to C-type lysozymes is when does B. subtilis encounter these factors. B. subtilis is often viewed simply as a soil organism however recent evidence suggests that it can colonize the intestinal tracts of a number of different organisms including Drosophila melanogaster, chickens, mice and humans [58]–[61] all of which encode C-type lysozymes [62]. Thus while B. subtilis is primarily a soil organism it may also have a more complex life associated with intestinal tract of a diverse number of organisms. It is tempting to hypothesize that lysozyme resistance could be an important trait required for colonization of the intestinal tract of higher organisms.
Materials and Methods
Strain Construction
Strains are isogenic derivatives of PY79, a prototrophic derivative of B. subtilis strain 168, and are listed in Table 3 [63]. B. subtilis competent cells were prepared by the one-step method previously described [64]. All plasmid constructs are listed in Table 4 were confirmed by DNA sequencing (University of Iowa). All oligonucleotides are listed in Table S1.


The rsiVA66W mutation was introduced onto the chromosome of PY79 by homologous recombination using the temperature sensitive plasmid pMAD [65]. To clone rsiVA66W we PCR amplified rsiVA66W plus ∼1 kb upstream with CDEP1892 and CDEP1562 and rsiVA66W+∼1 kb downstream using CDEP1561-CDEP1893. The resulting PCR products were cloned into pMAD digested with SmaI using Isothermal assembly [66]. The resulting plasmid, pCE492, was transformed into PY79 and the rsiVA66W mutation was introduced onto the chromosome by shifting temperatures as previously described [65]. The presence of the rsiVA66W mutation was confirmed by sequencing rsiV.
Site-directed mutagenesis of rsiV was performed using the QuickChange site-directed mutagenesis kit (Agilent Technologies). The rsiVA66W mutation was constructed using primer pairs CDEP1561 and CDEP1562 to generate plasmid pJH219. For IPTG-inducible expression, the rsiVA66W mutant was moved into pCE292 [11] using LR Clonase II (Invitrogen). The resulting plasmid, pJH224, places the IPTG-inducible hyper-spank (Phs) promoter upstream of rsiVA66W and was transformed into B. subtilis CDE1563 to result in JLH623.
C-terminal 6×His tagged rsiV with an optimized ribosome binding site was PCR amplified from B. subtilis using oligos CDEP1544 and CDEP1430 cloned into pEntrD-TOPO (Invitrogen) resulting in pCE363. For IPTG-inducible expression, the rsiV-6×his was moved into pCE292 using LR Clonase II resulting in pJH215. The plasmid pJH215 was transformed into CDE1563 resulting in JLH548.
A vector for a 3×Flag-CBP was constructed by PCR amplification of the gene encoding the calmodulin binding peptide (CBP) from pMZS3F with CDEP1611 and CDEP1610 [67], [68]. The resulting PCR was amplified with CDEP1612 and CDEP1610. The PCR was digested with HindIII and SphI and ligated into pDR111 digested with the same enzymes resulting in pCE417. The pCE417 was converted to a Gateway destination vector by cloning the RfC.1 cassette into pCE417 digested with Eco53kI resulting in pCE418. To construct a plasmid producing 3×Flag-cbp-RsiV+, rsiV+ was moved from pCE352 onto pCE418 using LR Clonase II resulting in plasmid pCE422.
Vectors for cell free production of RsiV and SipS production were constructed by cloning the genes on a plasmid downstream of an SP6 promoter. The sipS gene was PCR amplified from B. subtilis using CDEP1678 and CDEP1679. The PCR product was digested with AsiSI and SmaI and cloned into pEU-Flexi-His digested with AsiSI and PmiI using T4 ligase resulting in pCE448. The 3×flag-cbp-rsiV6×his gene was PCR amplified from pCE422 using CDEP1677 and CDEP1714. The PCR product was digested with AsiSI and SmaI and cloned into pEU-Flexi-His digested with AsiSI and PmiI using T4 ligase resulting in pCE455. The 3×flag-cbp-rsiVA66W 6×his for cell free production of RsiVA66W was created by QuikChange mutagenesis resulting in pCE490.
The human lysozyme open reading frame was synthesized by GenScript and the codons were optimized for expression in yeast. Using primers CDEP1888 and CDEP1889 human lysozyme was PCR amplified and cloned into P. pastoris expression vector pICZα (Invitrogen) by isothermal assembly [66] resulting in pJH326. To produce inactive lysozyme a mutation was constructed to change the codon for aspartate 53 to serine (lysozymeD53S) using primers CDEP1847 and CDEP1848 and PCR QuickChange site-directed mutagenesis (Agilent Technologies) resulting in pJH327. Expression vectors pJH326 and pJH327 were transformed into P. pastoris GS115 via the lithium chloride method (Invitrogen [69]) resulting in JLH1056 and JLH1057 respectively. Appropriate integration of the plasmids was confirmed by zeocin resistance and PCR using the primers CDEP1888 and CDEP1889.
For purification of E. faecalis RsiV, the portion of rsiV encoding the C-terminal extracellular domain (rsiVEF) was PCR amplified using CDEP1434 and CDEP1435. The PCR product was cloned into pEntrD-TOPO resulting in pJH228. To tag E. faecalis RsiV with 6×His, rsiVEF, was moved into pDEST17 using LR Clonase II resulting in pJH227.
For purification of C. difficile RsiV the portion of the rsiV encoding the C-terminal extracellular domain (rsiVCD) was PCR amplified using CDEP928 and CDEP189. The PCR product was cloned into pEntrD-TOPO resulting in pCE302. To tag C. difficile RsiV with 6×His, rsiVCD, was moved into pDEST17 using LR Clonase II, resulting in pKBW216.
To tag B. subtilis RsiV with GST, rsiV59–285, was moved from pCE458 [14] into pDEST15 using LR Clonase II resulting in pKBW204.
Construction of 2×flag-rsiV59–285 was performed by PCR amplification from B. subtilis genomic DNA using CDEP1140 and CDEP952. The resulting product was used as template in another round of PCR using CDEP950 and CDEP952 and cloned into pEntrD-TOPO, resulting in pKBW101. This was moved into the IPTG inducible 6×His expression vector pDEST17 using LR Clonase II, resulting in pKBW201.
ΔsipS::cat and ΔsipT::tet were constructed using long flanking homology PCR. Briefly sipS flanking regions were constructed by PCR amplifying with CDEP1697-CDEP1709 and CDEP1710-CDEP1700. The sipT flanking regions were constructed by PCR amplifying with CDEP1701-CDEP1711 and CDEP1712-CDEP1704. The resulting PCR products were used as primers to amplify either a chloramphenicol antibiotic cassette generated by PCR from pDG1661 using (CDEP1954-CDEP1955) or tetracycline antibiotic cassette from either pDG1515 respectively. The PCR products were transformed into B. subtilis JLH402 (amyE::Phs-rsiV+(spec) ΔsigVrsiV::kan) and confirmed by PCR resulting in JLH933 and JLH953 respectively.
Medium Supplements
Antibiotics were used at the following concentrations: chloramphenicol, 5 µg/ml; erythromycin plus lincomycin, 1 µg/ml and 25 µg/ml; kanamycin, 5 µg/ml; spectinomycin, 100 µg/ml; tetracycline, 10 µg/ml; ampicillin 100 µg/ml. The β-galactosidase chromogenic indicator 5-bromo-4-chloro-3-indolyl β-D-galactopyranoside (X-Gal) was used at a concentration 100 µg/ml. Isopropyl β-D-1-thiogalactopyranoside (IPTG) was used at a final concentration of 1 mM unless otherwise noted.
Purification of Site-1 Cleavage Product from B. subtilis
Cells producing RsiV-6×His were grown to an OD of 1 and then subcultured 1∶100 into 1L LB+IPTG (1 mM) and grown to an OD of 0.8. Cells were pelleted by centrifugation at 5000× g and frozen at −80°C. Cell pellets were thawed on ice and resuspended in 20 mL protoplast buffer (0.4 M sucrose, 10 mM potassium phosphate, 15 mM MgCl2) [70]. HEW lysozyme was added to a final concentration of 8 µg/ml and the cells were incubated at 37°C for 45 minutes. Protoplast formation was confirmed by phase contrast microscopy. Cells were pelleted by centrifugation at 5000× g and RsiV-6×His was purified from the supernatant using Ni resin (Thermo-Fisher). The resulting protein was separated by SDS PAGE and transferred to PVDF membrane (BioRad). The band containing the protein of interest was confirmed by immunoblot with anti-RsiV59–285 antibodies (Figure S1). The band of interest was cut out from the membrane and submitted for Edman degradation analysis (Iowa State University).
β-Galactosidase Activity Assays
Cultures were grown overnight in LB broth at 30°C and 20 µl were spotted onto LB agar + 10 µg/ml lysozyme. Plates were incubated at 37°C for 6 hours. Cells were harvested and resuspended in 500 µl of Z buffer (60 mM Na2HPO4, 40 mM NaH2PO4, 10 mM KCl, 1 mM MgSO4, 50 mM β-mercaptoethanol pH 7.0). Cells were transferred to a 96 well plate and optical density (OD600) determined. Cells were permeabilized by mixing with chloroform and 2% sarkosyl [9], [71]. Permeabilized cells (100 µl) were mixed with 10 mg/ml ortho-Nitrophenyl-β-galactoside (ONPG, RPI, 50 µl) and OD405 was measured over time. β-galactosidase activity units (µmol of ONP formed min−1)×103/(OD600×ml of cell suspension) were calculated as previously described [72]. Experiments were performed in triplicate. Mean and standard deviation are shown.
Lysozyme Sensitivity Assays
To determine lysozyme MIC values, B. subtilis strains were grown 16 h at 30°C and then subcultured 1∶100 in LB. Cells (100 µl) were inoculated into 100 µl of two fold serial dilutions of HEW lysozyme ranging from 200 µg/ml to 0.15 µg/ml in a round-bottom 96 well plate. The absorbance at OD600 was taken every 30 minutes using a Tecan F50 (Tecan) over a period of 20 hrs. Growth was defined as an OD600 greater than 0.1 at 14 hrs.
To determine lysozyme zones of clearing, B. subtilis strains were grown 16 h at 30°C and then diluted 1∶100 in 1.5 mL LB top agar (0.75%) containing X-Gal (100 µg/ml). Top agar was spread on solid LB+X-Gal and allowed to solidify. Whatman filter disks containing 10 µl of 10 mg/ml HEW lysozyme were placed on the top agar and incubated 16 h at 37°C.
Transcription and Cell-Free Translation Reactions
Plasmids were purified using QIAprep spin columns according to the manufacturer's instructions and resuspended in 30 µl of Milli-Q water. The concentration of the RNase-free plasmid DNA was determined by absorbance at 260 nm.
Small-scale transcription reactions were performed as previously described [21]. Briefly, the reaction mixtures were composed of 80 mM HEPES-KOH pH 7.8, 2 mM magnesium acetate, 2 mM spermidine, 10 mM DTT, 4 mM of each nucleotide triphosphate (ATP, CTP, UTP and GTP), 1.6 U/µl SP6 RNA polymerase (Promega), 1 U/µl RNasin (Promega) and 0.2 mg/mL of RNase-free plasmid DNA in a 10 µl total reaction volume. Each sample was incubated at 37°C for 4 h. The mRNA from each transcription reaction (5 µl) was mixed with 20 µl of the translation mix composed of 25% v/v wheat germ extract (WEPRO 2240 Cell free Sciences, Yokohama, Japan), 13 mM HEPES-KOH pH 7.8, 55 mM KOAc, 1.7 mM Mg(OAc)2, 0.22 mM spermidine hydrochloride, 2.2 mM DTT, 0.7 mM ATP, 0.14 mM GTP, 9 mM creatine phosphate, 0.003% NaN3, 1 mg/mL creatine kinase and 2 mM amino acids. The reaction mixtures were added to 12-kDa MWCO dialysis cups. The reservoir buffer contained all the reagents listed above except RNasin and wheat germ extract as previously described [21]. Each reaction was incubated for 16 h at room temperature.
In Vitro Protease Activity of SipS
The pellet fractions from the above-described translation reactions for SipS, RsiV (full-length), and RsiVA66W were resuspended in 5 mM MES pH 7.0, 50 mM NaCl and mixed at a 1∶3 SipS∶RsiV molar ratio. Reactions were incubated for 6 h at 37°C. The role of HEW lysozyme (0.3 mg/mL) as an activator of the RsiV cleavage was also evaluated. The cleavage of RsiV was assessed by the products observed at 26 kDa and 17 kDa and confirmed by Immunoblot analysis.
Immunoblot Analysis
Strains were grown for 16 hours in LB at 37°C. The cells were subcultured 1∶100 in LB+1 mM IPTG at 37°C and grown to an OD600 of 0.8–1. The cells were pelleted by centrifugation and resuspended in 100 µl of 2× Laemmli sample buffer and lysed by repeated sonication. Samples were electrophoresed on a 15% SDS Polyacrylamide gel (BioRad). The proteins were then blotted onto nitrocellulose. The proteins were detected by incubating with a 1∶10,000 dilution of anti-RsiV59–285 antibodies [14] or 1∶15,000 dilution of anti-σA antibodies followed by 3 washes and incubation in a 1∶10,000 dilution of goat anti-rabbit IgG (H+L) IRDye 800CW (Li-Cor) and imaged on an Odyssey CLx (Li-Cor). Quantification of band intensities was performed using Image Studio software (Li-Cor).
Expression and Purification of R-Lysozyme
P. pastoris was grown overnight in 5 ml BMGY (Buffered Glycerol Complex Medium; 100 mM potassium phosphate, pH 6.0, 1% yeast extract, 2% peptone, 1.34% Yeast Nitrogen Base, 0.0004% biotin, 1% glycerol) at 30°C and then subcultured into 300 ml BMGY overnight 30°C in 2L baffled flasks. Cultures were pelleted and washed with BMMY (Buffered Methanol Complex Medium; 100 mM potassium phosphate, pH 6.0, 1% yeast extract, 2% peptone, 1.34% Yeast Nitrogen Base, 0. 0004% biotin, 0.5% methanol) [73]. The cell pellet was resuspended in 300 ml BMMY and grown at 30°C for 5 days. Methanol to a final concentration of 5% was added each day. Cultures were pelleted by centrifugation and the supernatant was clarified by filtering through a 0.2 µm filter. The supernatant was exchanged into 50 mM sodium acetate pH 6.2, and concentrated by running the supernatant over a 3 kDa carbon fiber filter (AG Technologies Corporation). Further purification was performed by FPLC using a Capto S exchange column (GE Healthcare Life Sciences) and a 3 kDa micron filter unit (Millipore). Protein concentration was determined by reading the absorbance at 280 nm.
Measurement of Lysozyme Activity
Lysozyme activity was measured by the rate of Micrococcus lysodeikticus peptidoglycan clearing at 450 nm as previously described [74]. Briefly, M. lysodeikticus peptidoglycan was suspended to an OD600 of 0.9 and mixed with equal volumes of either buffer alone (50 mM NaOAc pH 6.2), HUM Lysozyme 20 µg/ml, R-lysozyme 20 µg/ml or R-lysozymeD52S. The rate of peptidoglycan clearing was monitored at 450 nm for 10 minutes.
Protoplast with Mutanolysin
Overnight cultures were subcultured 1∶100 in LB+1 mM IPTG and grown to an OD600 of 0.8–1. Cells were pelleted by centrifugation, washed with PBS, and resuspended in equal parts 1 M sucrose and 60 mM Tris-Cl, 4 mM MgCl2 to form a protoplast buffer [75], [76]. Mutanolysin was added to a final concentration of 2 µg/ml, and incubated shaking at 37°C for 40 minutes. Protoplast were confirmed by phase contrast microscopy. Protoplast samples were left untreated or treated with lysozyme (2 µg/ml) for 10 minutes. An equal volume of 2× Laemmli sample buffer was added to the sample before for immunoblot analysis as described.
Expression of Recombinant Proteins in E. coli
Overnight cultures of E. coli BL21λDE3 containing either pKBW201 (pDEST17-6×his-2×flag-rsiV59–285), pKBW204 (gst-rsiV59–285), pKBW216 (pDEST17-6×his-rsiVCD) or pJH227 (pDEST17-6×his-rsiVEF) were grown at 30°C in LB+ampicillin. The cell cultures were diluted 1∶100 into 500 ml of LB+ampicillin in 2 L baffled flasks and incubated at 30°C to an OD600 of 0.5–0.6. IPTG was added to a final concentration of 1 mM to induce protein production and the cultures incubated for an additional 4 hours. Cells were chilled on ice and collected by centrifugation at 5000× g. Cell pellets were stored at −80°C until time for purification.
Purification of 6×His-Tagged Proteins from E. coli
Cell pellets were thawed on ice and resuspended in 5 ml lysis buffer (50 mM Sodium Phosphate, 250 mM NaCl, 10 mM imidazole, pH 8.0) per 500 ml of initial culture volume. Cells were lysed by passaging through a Microfluidics LV1 high shear microfluidizer (Newton, MA) twice. Lysate was centrifuged at 15,000× g, for 30 minutes at 4°C, to pellet cellular debris. Cleared lysate was applied to a nickel affinity column to bind 6×His-tagged protein (Qiagen). The column was washed with 10 column volumes of wash buffer (50 mM sodium phosphate, 250 mM NaCl, 20 mM imidazole, pH 8.0). Protein was eluted with elution buffer (50 mM Sodium Phosphate, 250 mM NaCl, 250 mM imidazole, pH 8.0) and collected in 0.5 ml fractions. Samples from each fraction were analyzed by SDS-PAGE and elution fractions containing the desired protein were combined. Combined fractions were then dialyzed into lysis buffer to remove the excess imidazole. The protein was further purified with a GE Healthcare AKTA FPLC (GE Healthcare Sciences Pittsburg, PA) using a HisTrap HP nickel affinity column. Fractions containing the 6×His-2×Flag-RsiV59–285 were again combined and dialyzed into a storage buffer (25 mM Tris, 200 mM NaCl, 5% glycerol, pH 8.0) and flash frozen at −80°C until use.
Purification of GST-Tagged Proteins from E. coli
Cell pellets were thawed on ice and resuspended in 2.5 ml PBS-EW (50 mM NaH2PO4, 150 mM NaCl, 1 mM DTT, 1 mM EDTA, pH 7.2) per 250 ml of initial culture volume. Cells were lysed by 2× passage through a Microfluidics LV1 high shear microfluidizer (Newton, MA). Lysate was centrifuged at 15,000× g, for 30 minutes at 4°C, to pellet cellular debris. Cleared lysates were then passed over a Glutathione HiCap Matrix column (Qiagen Valencia, CA). The column was washed with 5 column volumes of PBS-EW. Protein was eluted with buffer TNGT (50 mM Tris, 0.4 M NaCl, 50 mM reduced L-Glutathione, 0.1% Triton-x-100, 1 mM DTT) and collected in 0.5 ml fractions. Samples from each fraction were analyzed by SDS-PAGE and elution fractions containing the desired protein were combined. Purified GST-RsiV59–285 was then dialyzed into a buffer containing 50 mM Tris-HCl pH 7.5, 200 mM NaCl and stored at 4°C until use.
Co-purification Experiments
For 6×His-tagged proteins, expression and purification was performed described above, with the following alterations. After the initial application of wash buffer 1, 2 ml of 1 mg/ml HEW lysozyme (Sigma Aldrich) was applied to the column and flow through collected. An additional 5 column volumes of wash buffer 1 was applied to remove any unbound HEW lysozyme. Elution of the proteins then proceeded as described. Samples from each fraction were mixed with an equal volume with 2× Laemmli sample buffer and analyzed on 15% SDS-PAGE gels stained with Coomassie brilliant blue. To ensure lysozyme binding was not the result of interactions with contaminating proteins from the expression strain or spurious binding to the Ni resin (Thermo-Fisher), mock-expression cells were used in a pull-down experiment as a negative control. Briefly, BL21λDE3 cells containing no plasmid were grown and processed as described. Cleared lysates from these cells were applied to a Ni affinity column in which lysozyme was then passed over. The column was washed and protein eluted under the same conditions as our normal pull-down assay.
Co-purification assays utilizing GST-tagged proteins were completed in a similar manner with the following modifications. Purified GST-tagged protein was dialyzed into PBS-EW and 2 mg of protein was applied to a Glutathione HiCap Matrix column. The column was washed with 5 column volumes of PBS-EW. 2 mg of HEW lysozyme, human lysozyme, or mutanolysin, in PBS-EW, was applied to the column. PBS-EW buffer and TNGT buffer were used as wash and elution buffers, respectively. Samples were analyzed by SDS-PAGE.
Isothermal Titration Calorimetry
The affinity of the interaction (Kd) between RsiV and HEW lysozyme was determined by isothermal titration calorimetry (ITC). Purified 6×His-2×Flag-RsiV59–285 was purified as described above. HEW lysozyme ≥98% pure was purchased from Sigma Aldrich. The proteins were co-dialyzed three times in 2 liters of 50 mM Na2HPO4, 200 mM NaCl, and pH 7.0 buffer for 8 h (each) at 4°C. Final protein concentrations as determined by absorbance at OD280 were adjusted to 6×His-2×Flag-RsiV59–285 (0.01 mM) and HEW lysozyme (0.1 mM) with filtered dialysate. The protein samples were degassed and ITC measurements recorded using a MicroCal VP-ITC System (GE Healthcare) with HEW lysozyme as the injected sample and 6×His-2×Flag-RsiV59–285 as the cell sample. 21 injections of HEW lysozyme were used, with 180 seconds spacing between events. The chamber was kept under constant stirring at 350 rpm and all experiments were performed at 25°C. The binding reaction reached saturation during the experiment and control experiments where HEW lysozyme was injected into buffer showed that the heats of dilution were constant across all injections. The constant heat of dilution, as determined by the average of the last 3–5 injections, was subtracted and the data are analyzed using the single site binding model provided in the ITC analysis package. The values for affinity, stoichiometry (n) and change in enthalpy (ΔH) and entropy (ΔS) obtained from three independent experiments were averaged and the standard deviation determined.
Supporting Information
Zdroje
1. WangX, SatoR, BrownMS, HuaX, GoldsteinJL (1994) SREBP-1, a membrane-bound transcription factor released by sterol-regulated proteolysis. Cell 77 : 53–62.
2. HoTD, EllermeierCD (2012) Extra cytoplasmic function σ factor activation. Curr Opin Microbiol 15 : 182–188 doi:10.1016/j.mib.2012.01.001
3. StarońA, SofiaHJ, DietrichS, UlrichLE, LiesegangH, et al. (2009) The third pillar of bacterial signal transduction: classification of the extracytoplasmic function (ECF) sigma factor protein family. Mol Microbiol 74 : 557–581 doi:10.1111/j.1365-2958.2009.06870.x
4. AdesSE, ConnollyLE, AlbaBM, GrossCA (1999) The Escherichia coli sigmaE-dependent extracytoplasmic stress response is controlled by the regulated proteolysis of an anti-sigma factor. Genes Dev 13 : 2449–2461.
5. AlbaBM, LeedsJa, OnufrykC, LuCZ, GrossCA (2002) DegS and YaeL participate sequentially in the cleavage of RseA to activate the sigmaE-dependent extracytoplasmic stress response. Genes Dev 16 : 2156–2168 doi:10.1101/gad.1008902
6. WalshNP, AlbaBM, BoseB, GrossCA, SauerRT (2003) OMP peptide signals initiate the envelope-stress response by activating DegS protease via relief of inhibition mediated by its PDZ domain. Cell 113 : 61–71.
7. WilkenC, KitzingK, KurzbauerR, EhrmannM, ClausenT (2004) Crystal structure of the DegS stress sensor: How a PDZ domain recognizes misfolded protein and activates a protease. Cell 117 : 483–494.
8. EllermeierCD, LosickR (2006) Evidence for a novel protease governing regulated intramembrane proteolysis and resistance to antimicrobial peptides in Bacillus subtilis. Genes Dev 20 : 1911–1922 doi:10.1101/gad.1440606
9. HoTD, HastieJL, IntilePJ, EllermeierCD (2011) The Bacillus subtilis extracytoplasmic function σ factor σV is induced by lysozyme and provides resistance to lysozyme. J Bacteriol 193 : 6215–6222 doi:10.1128/JB.05467-11
10. Guariglia-OropezaV, HelmannJD (2011) Bacillus subtilis σV confers lysozyme resistance by activation of two cell wall modification pathways, peptidoglycan O-acetylation and D-alanylation of teichoic acids. J Bacteriol 193 : 6223–6232 doi:10.1128/JB.06023-11
11. HoTD, EllermeierCD (2011) PrsW is required for colonization, resistance to antimicrobial peptides, and expression of extracytoplasmic function σ factors in Clostridium difficile. Infect Immun 79 : 3229–3238 doi:10.1128/IAI.00019-11
12. Le JeuneA, TorelliR, SanguinettiM, GiardJ-C, HartkeA, et al. (2010) The extracytoplasmic function sigma factor SigV plays a key role in the original model of lysozyme resistance and virulence of Enterococcus faecalis. PLoS One 5: e9658 doi:10.1371/journal.pone.0009658
13. HoTD, WilliamsKB, ChenY, HelmRF, PophamDL, et al. (2014) Clostridium difficile Extra-Cytoplasmic Function σ factor σV regulates lysozyme resistance and is necessary for pathogenesis in the hamster model of infection. Infect Immun IAI.01483–13–. doi:10.1128/IAI.01483-13
14. HastieJL, WilliamsKB, EllermeierCD (2013) The activity of σV, an Extra-Cytoplasmic Function σ factor of Bacillus subtilis, is controlled by a regulated proteolysis of the anti-σ factor RsiV. J Bacteriol 195 : 3135–3144 doi:10.1128/JB.00292-13
15. EDMANP (1949) A method for the determination of amino acid sequence in peptides. Arch Biochem 22 : 475.
16. PetersenTN, BrunakS, von HeijneG, NielsenH (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8 : 785–786 doi:10.1038/nmeth.1701
17. BaileyTL, BodenM, BuskeFa, FrithM, GrantCE, et al. (2009) MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37: W202–8 doi:10.1093/nar/gkp335
18. Krogha, LarssonB, von HeijneG, SonnhammerEL (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305 : 567–580 doi:10.1006/jmbi.2000.4315
19. TjalsmaH, NobackMA, BronS, VenemaG, YamaneK, et al. (1997) Bacillus subtilis Contains Four Closely Related Type I Signal Peptidases with Overlapping Substrate Specificities. J Biol Chem 272 : 25983–25992.
20. TjalsmaH, BolhuisA, van RoosmalenML, WiegertT, SchumannW, et al. (1998) Functional analysis of the secretory precursor processing machinery of Bacillus subtilis: identification of a eubacterial homolog of archaeal and eukaryotic signal peptidases. Genes Dev 12 : 2318–2331 doi:10.1101/gad.12.15.2318
21. AlyKa, BeebeET, ChanCH, GorenMa, SepúlvedaC, et al. (2013) Cell-free production of integral membrane aspartic acid proteases reveals zinc-dependent methyltransferase activity of the Pseudomonas aeruginosa prepilin peptidase PilD. Microbiologyopen 2 : 94–104 doi:10.1002/mbo3.51
22. PrehnS, WiedmannM, RapoportTA, ZwiebC (1987) Protein translocation across wheat germ microsomal membranes requires an SRP-like component. EMBO J 6 : 2093–2097.
23. PerlmanD, HalvorsonHO (1983) A putative signal peptidase recognition site and sequence in eukaryotic and prokaryotic signal peptides. J Mol Biol 167 : 391–409.
24. AmanoK, HayashiH, ArakiY, ItoE (1977) The Action of Lysozyme on Peptidoglycan with N-Unsubstituted Glucosamine Residues. Eur J Biochem 76 : 299–307.
25. DupontC, ClarkeaJ (1991) Dependence of lysozyme-catalysed solubilization of Proteus mirabilis peptidoglycan on the extent of O-acetylation. Eur J Biochem 195 : 763–769.
26. ChipmanDM, SharonN (1969) Mechanism of lysozyme action. Science 165 : 454–465.
27. LichensteinHS, HastingsAE, LangleyKE, MendiazEA, RohdeMF, et al. (1990) Cloning and nucleotide sequence of the N-acetylmuramidase M1-encoding gene from Streptomyces globisporus. Gene 88 : 81–86.
28. NashJA, BallardTNS, WeaverTE, AkinbiHT (2006) The peptidoglycan-degrading property of lysozyme is not required for bactericidal activity in vivo. J Immunol 177 : 519–526.
29. KumitaJR, JohnsonRJK, AlcocerMJC, DumoulinM, HolmqvistF, et al. (2006) Impact of the native-state stability of human lysozyme variants on protein secretion by Pichia pastoris. FEBS J 273 : 711–720 doi:10.1111/j.1742-4658.2005.05099.x
30. BenachourA, MullerC, Dabrowski-CotonM, Le BretonY, GiardJ-C, et al. (2005) The Enterococcus faecalis SigV protein is an extracytoplasmic function sigma factor contributing to survival following heat, acid, and ethanol treatments. J Bacteriol 187 : 1022–1035 doi:10.1128/JB.187.3.1022-1035.2005
31. KangJG, PagetMS, SeokYJ, HahnMY, BaeJB, et al. (1999) RsrA, an anti-sigma factor regulated by redox change. EMBO J 18 : 4292–4298 doi:10.1093/emboj/18.15.4292
32. LiW, BottrillAR, BibbMJ, ButtnerMJ, PagetMSB, et al. (2003) The Role of Zinc in the Disulphide Stress-regulated Anti-sigma Factor RsrA from Streptomyces coelicolor. J Mol Biol 333 : 461–472 doi:10.1016/j.jmb.2003.08.038
33. PagetMS, BaeJB, HahnMY, LiW, KleanthousC, et al. (2001) Mutational analysis of RsrA, a zinc-binding anti-sigma factor with a thiol-disulphide redox switch. Mol Microbiol 39 : 1036–1047.
34. GreenwellR, NamT-W, DonohueTJ (2011) Features of Rhodobacter sphaeroides ChrR required for stimuli to promote the dissociation of σE/ChrR complexes. J Mol Biol 407 : 477–491 doi:10.1016/j.jmb.2011.01.055
35. CampbellEa, GreenwellR, AnthonyJR, WangS, LimL, et al. (2007) A conserved structural module regulates transcriptional responses to diverse stress signals in bacteria. Mol Cell 27 : 793–805 doi:10.1016/j.molcel.2007.07.009
36. BraunV, MahrenS, OgiermanM (2003) Regulation of the FecI-type ECF sigma factor by transmembrane signalling. Curr Opin Microbiol 6 : 173–180 doi:10.1016/S1369-5274(03)00022-5
37. ChabaR, AlbaBM, GuoMS, SohnJ, AhujaN, et al. (2011) Signal integration by DegS and RseB governs the sigmaE-mediated envelope stress response in Escherichia coli. Proc Natl Acad Sci 108 : 2106 doi: = 10.1073/pnas.1019277108 =
38. KulpA, KuehnMJ (2011) Recognition of β-strand motifs by RseB is required for σ(E) activity in Escherichia coli. J Bacteriol 193 : 6179–6186 doi:10.1128/JB.05657-11
39. LimaS, GuoMS, ChabaR, GrossCA, SauerRT (2013) Dual molecular signals mediate the bacterial response to outer-membrane stress. Science 340 : 837–841 doi:10.1126/science.1235358
40. KavanaughJS, ThoendelM, HorswillAR (2007) A role for type I signal peptidase in Staphylococcus aureus quorum sensing. Mol Microbiol 65 : 780–798 doi:10.1111/j.1365-2958.2007.05830.x
41. StephensonS, MuellerC, JiangM, PeregoM (2003) Molecular Analysis of Phr Peptide Processing in Bacillus subtilis. J Bacteriol 185 : 4861–4871 doi:10.1128/JB.185.16.4861-4871.2003
42. JiangM, GrauR, PeregoM (2000) Differential Processing of Propeptide Inhibitors of Rap Phosphatases in Bacillus subtilis. J Bacteriol 182 : 303–310 doi:10.1128/JB.182.2.303-310.2000
43. BongiorniC, IshikawaS, StephensonS, OgasawaraN, PeregoM (2005) Synergistic regulation of competence development in Bacillus subtilis by two Rap-Phr systems. J Bacteriol 187 : 4353–4361 doi:10.1128/JB.187.13.4353-4361.2005
44. AuchtungJM, LeeCa, GrossmanAD (2006) Modulation of the ComA-dependent quorum response in Bacillus subtilis by multiple Rap proteins and Phr peptides. J Bacteriol 188 : 5273–5285 doi:10.1128/JB.00300-06
45. PowersME, SmithPa, RobertsTC, FowlerBJ, KingCC, et al. (2011) Type I signal peptidase and protein secretion in Staphylococcus epidermidis. J Bacteriol 193 : 340–348 doi:10.1128/JB.01052-10
46. LlarrullLI, TothM, ChampionMM, MobasheryS (2011) Activation of BlaR1 protein of methicillin-resistant Staphylococcus aureus, its proteolytic processing, and recovery from induction of resistance. J Biol Chem 286 : 38148–38158 doi:10.1074/jbc.M111.288985
47. VarahanS, IyerVS, MooreWT, HancockLE (2013) Eep Confers Lysozyme Resistance to Enterococcus faecalis via the Activation of the Extracytoplasmic Function Sigma Factor SigV. J Bacteriol 195 : 3125–3134 doi:10.1128/JB.00291-13
48. SaitoA, HizukuriY, MatsuoE, ChibaS, MoriH, et al. (2011) Post-liberation cleavage of signal peptides is catalyzed by the site-2 protease (S2P) in bacteria. Proc Natl Acad Sci U S A 108 : 13740–13745 doi:10.1073/pnas.1108376108
49. LaaberkiM-H, PfefferJ, ClarkeAJ, DworkinJ (2011) O-Acetylation of peptidoglycan is required for proper cell separation and S-layer anchoring in Bacillus anthracis. J Biol Chem 286 : 5278–5288 doi:10.1074/jbc.M110.183236
50. BenachourA, LadjouziR, Le JeuneA, HébertL, ThorpeS, et al. (2012) The lysozyme-induced peptidoglycan N-acetylglucosamine deacetylase PgdA (EF1843) is required for Enterococcus faecalis virulence. J Bacteriol 194 : 6066–6073 doi:10.1128/JB.00981-12
51. VollmerW, HöltjeJ (2004) The architecture of the murein (peptidoglycan) in gram-negative bacteria: vertical scaffold or horizontal layer(s)? J Bacteriol 186 : 5978–5987 doi:10.1128/JB.186.18.5978-5987.2004
52. VollmerW (2008) Structural variation in the glycan strands of bacterial peptidoglycan. FEMS Microbiol Rev 32 : 287–306 doi:10.1111/j.1574-6976.2007.00088.x
53. BeraA, HerbertS, JakobA, VollmerW, GötzF (2005) Why are pathogenic staphylococci so lysozyme resistant? The peptidoglycan O-acetyltransferase OatA is the major determinant for lysozyme resistance of Staphylococcus aureus. Mol Microbiol 55 : 778–787 doi:10.1111/j.1365-2958.2004.04446.x
54. CallewaertL, Van HerrewegheJM, VanderkelenL, LeysenS, VoetA, et al. (2012) Guards of the great wall: bacterial lysozyme inhibitors. Trends Microbiol 20 : 501–510 doi:10.1016/j.tim.2012.06.005
55. YokogawaK, KawataS, NishimuraS, IkedaY, YoshimuraY (1974) Mutanolysin, Bacteriolytic Agent for Cariogenic Streptococci: Partial Purification and Properties. Antimicrob Agents Chemother 6 : 156–165 doi:10.1128/AAC.6.2.156
56. BudzikJM, OhS-Y, SchneewindO (2008) Cell wall anchor structure of BcpA pili in Bacillus anthracis. J Biol Chem 283 : 36676–36686 doi:10.1074/jbc.M806796200
57. KobayashiK, SudiartaIP, KodamaT, FukushimaT, AraK, et al. (2012) Identification and characterization of a novel polysaccharide deacetylase C (PdaC) from Bacillus subtilis. J Biol Chem 287 : 9765–9776 doi:10.1074/jbc.M111.329490
58. CoxCR, GilmoreMS (2007) Native microbial colonization of Drosophila melanogaster and its use as a model of Enterococcus faecalis pathogenesis. Infect Immun 75 : 1565–1576 doi:10.1128/IAI.01496-06
59. CartmanST, La RagioneRM, WoodwardMJ (2008) Bacillus subtilis spores germinate in the chicken gastrointestinal tract. Appl Environ Microbiol 74 : 5254–5258 doi:10.1128/AEM.00580-08
60. TamNKM, UyenNQ, HongHA, DucLH, HoaTT, et al. (2006) The Intestinal Life Cycle of Bacillus subtilis and Close Relatives. J Bacteriol 188 : 2692–2700 doi:–10.1128/JB.188.7.2692–2700.2006
61. HongHa, KhanejaR, TamNMK, CazzatoA, TanS, et al. (2009) Bacillus subtilis isolated from the human gastrointestinal tract. Res Microbiol 160 : 134–143 doi:10.1016/j.resmic.2008.11.002
62. IrwinDM, BiegelJM, StewartC-B (2011) Evolution of the mammalian lysozyme gene family. BMC Evol Biol 11 : 166 doi:10.1186/1471-2148-11-166
63. YoungmanP, PerkinsJB, LosickR (1984) Construction of a cloning site near one end of Tn917 into which foreign DNA may be inserted without affecting transposition in Bacillus subtilis or expression of the transposon-borne erm gene. Plasmid 12 : 1–9.
64. BottKF, WilsonGa (1967) Development of competence in the Bacillus subtilis transformation system. J Bacteriol 94 : 562–570.
65. ArnaudM, ChastanetA, DébarbouilléM (2004) New vector for efficient allelic replacement in naturally nontransformable, low-GC-content, gram-positive bacteria. Appl Environ Microbiol 70 : 6887–6891 doi:10.1128/AEM.70.11.6887-6891.2004
66. GibsonDG, YoungL, ChuangR, VenterJC, HutchisonCA, et al. (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6 : 343–345 doi:10.1038/nmeth.1318
67. Babu M, Butland G, Pogoutse O, Li J, Greenblatt JF, et al.. (2009) Sequential Peptide Affinity Purification System for the Systematic Isolation and Identification of Protein Complexes from Escherichia coli. In: Reinders J, Sickmann A, editors. Methods in molecular biology (Clifton, N.J.). Methods in Molecular Biology™. Totowa, NJ: Humana Press, Vol. 564. pp. 531–537. doi:10.1007/978-1-60761-157-8.
68. ZeghoufM, LiJ, ButlandG, BorkowskaA, CanadienV, et al. (2004) Sequential Peptide Affinity (SPA) System for the Identification of Mammalian and Bacterial Protein Complexes research articles. J Proteome Res 3 : 463–468.
69. GietzRD, SchiestlRH (2007) Frozen competent yeast cells that can be transformed with high efficiency using the LiAc/SS carrier DNA/PEG method. Nat Protoc 2 : 1–4 doi:10.1038/nprot.2007.17
70. BishopDG, Op den KampJAF, Van deenenLLM (1977) The Distribution of Lipids in the Protoplast Membranes of Bacillus subtilis. Eur J Biochem 80 : 381–391.
71. GriffithKL, WolfRE (2002) Measuring beta-galactosidase activity in bacteria: cell growth, permeabilization, and enzyme assays in 96-well arrays. Biochem Biophys Res Commun 290 : 397–402 doi:10.1006/bbrc.2001.6152
72. SlauchJM, SilhavyTJ (1991) cis-acting ompF mutations that result in OmpR-dependent constitutive expression. J Bacteriol 173 : 4039–4048.
73. OkaC, TanakaM, MurakiM, HarataK, SuzukiK, et al. (1999) Human lysozyme secretion increased by alpha-factor pro-sequence in Pichia pastoris. Biosci Biotechnol Biochem 63 : 1977–1983.
74. ShugarD (1952) The measurement of lysozyme activity and the ultra-violet inactivation of lysozyme. Biochim Biophys Acta 8 : 302–309 doi:10.1016/0006-3002(52)90045-0
75. LeloupL, HaddaouiEA, ChambertR, Petit-GlatronM-F (1997) Characterization of the rate-limiting step of the secretion of Bacillus subtilis alpha-amamylase overproduced during the exponential phase of growth. Microbiology 143 : 3295–3303.
76. ChangS, CohenSN (1979) High Frequency Transformation of Bacillus subtilis Protoplasts by Plasmid DNA. Mol Gen Genet 168 : 111–115.
77. AltschulSF, MaddenTL, Schäfferaa, ZhangJ, ZhangZ, et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25 : 3389–3402.
78. PuntaM, CoggillPC, EberhardtRY, MistryJ, TateJ, et al. (2012) The Pfam protein families database. Nucleic Acids Res 40: D290–301 doi:10.1093/nar/gkr1065
79. Schutz-geschwenderA, ZhangY, HoltT, McdermittD, OliveDM, et al. (2004) Quantitative, Two-Color Western Blot Detection With Infrared Fluorescence. 800 : 1–8.
80. Ben-YehudaS, RudnerDZ, LosickR (2003) RacA, a bacterial protein that anchors chromosomes to the cell poles. Science 299 : 532–536 doi:10.1126/science.1079914
81. BlommelPG, MartinPa, WrobelRL, SteffenE, FoxBG (2006) High efficiency single step production of expression plasmids from cDNA clones using the Flexi Vector cloning system. Protein Expr Purif 47 : 562–570 doi:10.1016/j.pep.2005.11.007
82. Guérout-FleuryAM, ShazandK, FrandsenN, StragierP (1995) Antibiotic-resistance cassettes for Bacillus subtilis. Gene 167 : 335–336.
83. Guérout-FleuryAM, FrandsenN, StragierP (1996) Plasmids for ectopic integration in Bacillus subtilis. Gene 180 : 57–61.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 10
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- The Master Activator of IncA/C Conjugative Plasmids Stimulates Genomic Islands and Multidrug Resistance Dissemination
- A Splice Mutation in the Gene Causes High Glycogen Content and Low Meat Quality in Pig Skeletal Muscle
- Keratin 76 Is Required for Tight Junction Function and Maintenance of the Skin Barrier
- A Role for Taiman in Insect Metamorphosis
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy