
An Integrated Cell Purification and Genomics Strategy Reveals Multiple Regulators of Pancreas Development
Discovery of specific pancreas developmental regulators has accelerated in recent years. In contrast, the global regulatory programs controlling pancreas development are poorly understood compared to other organs or tissues like heart or blood. Decoding this regulatory logic may accelerate development of replacement organs from renewable sources like stem cells, but this goal requires identification of regulators and assessment of their functions on a global scale. To address this important challenge for pancreas biology, we combined purification of normal and mutant cells with genome-scale methods to generate and analyze expression profiles from developing pancreas cells. Our work revealed regulatory gene sets governing development of pancreas progenitor cells and their progeny. Our integrative approach nominated multiple pancreas developmental regulators, including suspected risk genes for human diabetes, which we validated by phenotyping mutant mice on a scale not previously reported. Selection of these candidate regulators was unbiased; thus it is remarkable that all were essential for pancreatic islet development. Thus, our studies provide a new heuristic resource for identifying genetic functions underlying pancreas development and diseases like diabetes mellitus.
Published in the journal:
. PLoS Genet 10(10): e32767. doi:10.1371/journal.pgen.1004645
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004645
Summary
Discovery of specific pancreas developmental regulators has accelerated in recent years. In contrast, the global regulatory programs controlling pancreas development are poorly understood compared to other organs or tissues like heart or blood. Decoding this regulatory logic may accelerate development of replacement organs from renewable sources like stem cells, but this goal requires identification of regulators and assessment of their functions on a global scale. To address this important challenge for pancreas biology, we combined purification of normal and mutant cells with genome-scale methods to generate and analyze expression profiles from developing pancreas cells. Our work revealed regulatory gene sets governing development of pancreas progenitor cells and their progeny. Our integrative approach nominated multiple pancreas developmental regulators, including suspected risk genes for human diabetes, which we validated by phenotyping mutant mice on a scale not previously reported. Selection of these candidate regulators was unbiased; thus it is remarkable that all were essential for pancreatic islet development. Thus, our studies provide a new heuristic resource for identifying genetic functions underlying pancreas development and diseases like diabetes mellitus.
Introduction
The pancreas is a vital internal organ with exocrine and endocrine functions. The exocrine pancreas is composed of acinar cells that secrete digestive enzymes into a branched network of bicarbonate-secreting duct cells. Endocrine cells form clusters called islets of Langerhans that secrete hormones such as insulin, glucagon, pancreatic polypeptide, somatostatin, and ghrelin produced, respectively, by beta cells, alpha cells, PP cells, delta cells and a transient population of epsilon cells [1]. Classical genetic approaches revealed that exocrine and endocrine cells develop from a common multipotent progenitor that expresses the transcription factors Sox9 [2] [3], Pdx1 [4], and Ptf1a [5]. Through mouse embryonic development, Sox9+ multipotent progenitors generate endocrine progenitors that express the basic helix-loop-helix (bHLH) transcription factor Neurog3 [6], which produce all pancreatic endocrine cells [7]. Although these approaches have revealed much about individual factors that regulate pancreatic development [8], we have yet to understand the regulatory logic underlying pancreas formation [9].
Genome-scale approaches to organ development can provide unbiased views of the genetic interactions regulating transient cell populations such as multipotent progenitor cells and their lineage-restricted progeny. However, to decipher the regulatory logic that culminates in successful organogenesis, gene expression in distinct, developing cell types must be acquired. Then, algorithms that analyze expression patterns across multiple cell types can be executed. Such integrated efforts have been used successfully for studies of hematopoiesis [10] [11]. We postulate that similar approaches to identify gene sets orchestrating formation and maturation of pancreas cells should advance islet beta cell-replacement efforts in diabetic patients. However, the application of such an approach to solid organs like the pancreas has proven difficult. In particular, crucial transient cell subsets like multipotent pancreatic progenitors and endocrine precursors represent a small fraction of cells in the developing pancreas – making analysis of these cells challenging. Thus, while prior studies used cell fractionation and genome-scale analysis of gene expression to advance understanding of pancreas development [12] [13] [14] [15] none was able to comprehensively assess purified progenitors and their endocrine or exocrine progeny from multiple developmental stages. Moreover, most previous studies were limited to pair-wise comparisons of wild-type cells in the endocrine lineage, thereby precluding powerful inferences from mutant analysis, and none prioritized or validated multiple candidate regulators by phenotyping mutant mice.
Here, we used a combination of cell sorting and transgenic cell labeling to purify and profile twelve pancreatic cell types at specific stages of development and used methods to optimize RNA quantification from relatively small numbers of cells. Our data set encompasses multipotent Sox9+ pancreatic progenitors, Neurog3+ endocrine progenitors, fetal and adult alpha cells and beta cells, and exocrine cells including fetal acinar cells and adult duct cells. Statistical comparisons demonstrate the highly reproducible quality of gene expression profiles obtained. We found that iterative probabilistic modeling, optimized with data on established pancreatic regulators, succeeded in nominating and ranking scores of novel candidate regulators and their functions. We validated a subset of these predictions using mutant Neurog3 mice and by phenotyping pancreas development in appropriate mutant mice. This comprehensive, integrated effort with discrete, operationally-defined populations of purified fetal and adult pancreatic cells provides gene expression profiling at higher resolution than previously achieved, identifies new regulators of pancreas development that are validated in vivo, and elucidates new elements of the regulatory logic underlying development of the endocrine and exocrine pancreas.
Results
Purification and gene expression profiling of fetal and adult pancreatic cells
To dissect developmental mechanisms of pancreatic development and maturation, we adopted a strategy using staged mice, FACS purification of specific cell subsets, genome-scale gene expression profiling coupled to bioinformatics analysis, and validation using mutant mice (Figure 1A). Using a combination of surface markers and transgenic reporter mice, we isolated 12 cell populations and profiled gene expression using GeneChip microarrays (Figure 1B; Methods). These included embryonic day (E) 11 cells enriched for Sox9+ multipotent pancreatic progenitors [3], E15 pancreatic ‘progenitors’ enriched for the markers Sox9 and CD24 [16] [17], E15 Neurog3+ endocrine progenitors enriched for CD133 and CD49f [16], E15 acinar cells, Glucagon+ alpha cells from postnatal day (P) 1 and 8–12 weeks, fetal and adult beta cells from E15, E17, P1, P15, and 8–12 weeks, and duct cells from 8–12 weeks. To our knowledge, comparative analysis of this range of mouse pancreatic cell types and developmental stages has not been reported.
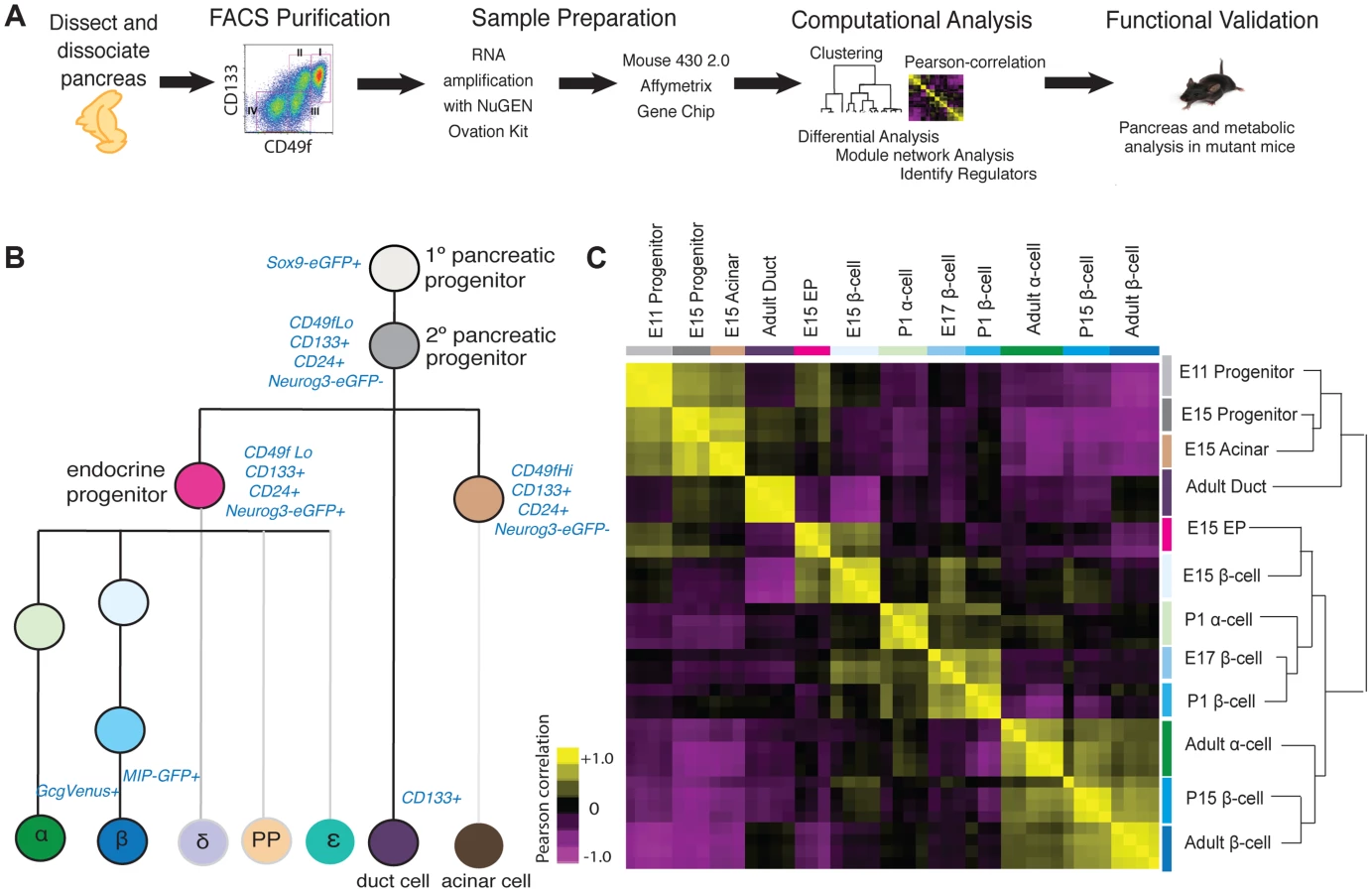
To assess the quality and reproducibility of replicate cell isolations, RNA collection and gene expression profiles, we obtained the Pearson correlation coefficient of pairwise-comparisons between samples and performed unsupervised hierarchal clustering. This analysis revealed tight clustering of biological replicates for each cell subset isolated (Figure 1C). We verified the expression of established pancreatic markers and developmental regulators [9] for each specific cell type profiled, using microarray (Figure S1A), or with quantitative PCR for a subset of ultra-abundant mRNAs encoding proteins like insulin and glucagon, which can saturate microarray probes (Figure S1B, S1C). Sox9+ E11 and E15 pancreatic progenitor cells were enriched for expression of expected mRNAs encoding Sox9, cMyc, and Onecut1. Likewise we confirmed that adult ductal cells were enriched for expression of Muc1, Sox9, Onecut1 and Hes1 mRNA (Figure S1A). At multiple stages, purified beta cells were highly enriched for mRNAs encoding Pdx1, Insulin and Glucokinase, while alpha cells expressed expected markers, including Pax6, MafB, Arx, and Glucagon (Figure S1A). Thus, appropriate scaling of mouse collections overcame inherently low numbers of fetal pancreatic cell subsets to generate a unique, coherent set of highly reproducible gene expression data sets spanning multiple pancreatic cell lineages and developmental stages.
Pearson correlation and unsupervised hierarchical clustering analysis revealed grouping of cell types and their gene expression based on developmental stage, and exocrine or endocrine function. Undifferentiated fetal pancreatic progenitors from E11 clustered closest to E15 progenitor cells and E15 acinar cells (Figure 1C). E15 Neurog3+ endocrine progenitors clustered closely with fetal alpha cells and beta cells, forming a cluster distinct from fetal progenitor, ductal and acinar cells. Adult duct cell gene expression clustered with that of E11 and E15 pancreatic progenitors cells, instead of other adult cells, likely reflecting the postulated origins of pancreatic progenitors from primitive fetal ductal cells [18]. Unexpectedly, we did not observe clustering by endocrine cell type; rather, we observed clustering of postnatal and adult beta cells with adult alpha cells, and close clustering of fetal beta cells with neonatal alpha cells (Figure 1C). Pair-wise differential expression analysis (Table S1) and unsupervised hierarchical clustering analysis with over 30 adult mouse tissues [19] supported this conclusion (Figure S2). As described below, this similarity likely reflects common functions of mature adult alpha cells and beta cells as nutrient-responsive cells that produce, process and secrete peptide hormones—functions distinct from those in fetal alpha and beta cells.
Identifying distinct gene sets in pancreas progenitor and exocrine development
The clustering of cell types in our Pearson correlation analysis (Figure 1C) indicated that specific cell types expressed distinct genes. To investigate this further, we used module mapping [20], which determines if a set of genes associated with or governing a specific biological function is significantly enriched or depleted within a sample (see Methods). As expected, module mapping revealed gene sets enriched in E11 pancreatic progenitors that remained enriched in E15 pancreatic progenitors, fetal acinar cells, and duct cells (Group I; Figure 2). This included gene sets regulating cell proliferation, cell fate commitment, branching morphogenesis and gland development; together, these functions reflect the known proliferative capacity and differentiation potential of these cell populations [21]. In addition, we observed that progenitor and duct cells shared modules with fetal endocrine cells (Group I; Figure 2), suggesting common genetic regulatory features. These commonalities are consistent with recent findings indicating a latent potential in pancreatic acinar or duct cells for conversion into endocrine cells [22], [23]. To identify gene sets enriched or uniquely expressed in E11 and E15 Sox9+ pancreatic progenitors, acinar or duct cells, we (1) obtained the gene signatures for each cell type (Figure S3; Table S2; see Methods for gene signature criteria), and (2) identified differentially expressed genes (Figure S4; Tables S2, S3, S4). This analysis revealed that E11, E15 progenitors and E15 acinar cells shared distinct but overlapping gene signatures (Figure S3A). Genes in these signatures included many transcriptional regulators, including Bcl11a, and genes involved in RNA processing and translation such as Spin2, Rpp40, and Rpl23, whose possible roles have not been previously noted in pancreas development. Thus, our analysis identified genes and gene sets that are expressed during pancreatic progenitor and exocrine cell development.

Identifying gene sets in endocrine progenitors and their fetal and adult endocrine progeny
Maturation of defining beta cell functions, such as glucose sensing and insulin secretion, increases from fetal through post-natal stages [7] [24]; however, a comprehensive analysis of beta cell gene expression from fetal to adult stages has not, to our knowledge, been reported in mice. Likewise, little is known about gene expression changes accompanying maturation of alpha cells [25]. Module mapping revealed gene sets initially expressed in Sox9+ pancreatic progenitor cells and Neurog3+ endocrine progenitors (including the terms cell proliferation and cell fate commitment) that were maintained in fetal beta and alpha cells but extinguished in adult endocrine cells (Group I; Figure 2). A second group of gene sets (with terms like cell adhesion, angiogenesis, hormone activity and eye development) was expressed after the Neurog3+ stage in alpha and beta cells. Strikingly, nearly all these modules were transiently downregulated in P15 beta cells and lost in adult alpha cells, but maintained in adult beta cells (Group II; Figure 2). These findings are consistent with prior studies showing that at early stages of development (E15, E17, and P1) immature alpha and beta cells are establishing neurovascular connections [26] [27], proliferating [28] [29], and developing components necessary for hormone synthesis, processing or secretion [30]. A third group of modules (associated with terms like voltage-gated ion channel activity, exocytosis, synapse, and calcium ion homeostasis) was expressed initially at the Neurog3+ stage then maintained throughout endocrine cell development (Group III; Figure 2). Thus, consistent with the clustering pattern of endocrine cells, we identified many gene sets that were shared between alpha and beta cells.
Next, we sought to identify distinguishing gene sets and signatures between alpha and beta cells. Module mapping revealed that adult beta cells (compared to alpha cells) maintained dozens of distinct gene sets (linked to terms like cell adhesion, calcium ion binding, eye development and G-protein coupled receptor activity; Groups II; Figure 2). These findings are consistent with established roles of GPCRs and calcium transients in regulating adult beta-cell proliferation, maturation and physiological regulation [31]. Our analysis similarly revealed distinct gene signatures between differentiated alpha and beta cells (Figure S3A, S3B; Table S2). Differentially expressed genes enriched in postnatal beta cells included Cldn8, C1qb, and Gdf3, while Fap, Ctxn2, and Mctp2 were highly expressed in adult alpha cells (Figure S4; Table S4). Collectively, this work provides a useful resource for exploring gene regulation and development of islet beta and alpha cells (see below).
Iterative module network analysis (IMNA) identifies regulators of pancreas development
After obtaining genes and gene sets that were differentially expressed during pancreas development, we sought to identify the regulatory logic governing their expression. Mutations in loci encoding transcription factors constitute an important group of risk factors for diabetes mellitus and pancreatic malformations in humans [32] [33]. Thus, we focused on identifying regulatory networks governed by transcription factors. To do this we adapted the module network function in Genomica, a probabilistic algorithm that groups genes based on co-expression patterns (modules) and predicts the regulators that might control such gene co-expression (Figure 3A; [34] [35]). To identify regulators, we first analyzed 1642 genes expressed in the developing pancreas that encode transcription factors (TF) or DNA-binding factors [36] [37] [38]. We optimized our parameters with a ‘training’ set of 82 established pancreatic regulators (Methods; [9]). Because Genomica is constrained to choose one group of regulators per gene set - a recognized limitation [39] - we employed an Iterative Module Network Analysis (IMNA) approach, in which we identified candidate regulators after multiple iterations (‘runs’) of the module network program. We then systematically varied the number of modules and runs (Figure 3B; Figure S5; Methods), and found that 100 iterations of 75 modules identified 99% (81/82) of established transcriptional regulators of pancreas development that included Neurog3, Arx, Glis3, Pdx1, Isl1, Fev and Myt1 (Figure 3D; Table S5). The quality of predictions did not improve with more iterations or modules (Methods). To determine the validity of our outputs, we ranked candidate regulators based on their frequency of occurrence across all iterations (Figure 3D) and performed Gene Set Enrichment Analysis on these ranked regulators (GSEA; Figure 3C; [40]). This analysis revealed that established pancreatic regulators ranked significantly higher in the list indicating that the top-ranked predictions were likely to be true regulators (Figure 3C, 3D; Table S5). One highly-ranked candidate regulator was Bcl11a, a gene previously linked by human GWAS studies to increased type 2 diabetes (T2D) risk [41] [42] [43]. Of 26 loci encoding DNA-binding factors that have been linked to diabetes risk, we found that 22 (85%, P = 1.38×10−8) were expressed in the developing pancreas, and 21 of these were highly ranked by IMNA as possible regulators (Figure 3E, 3F). This provided unique evidence for roles of these diabetes risk genes in regulating pancreas development and led us to establish analytic methods to identify and prioritize transcriptional regulators for further in vivo testing (see below).

Neurog3 encodes a bHLH transcription factor with essential roles in the endocrine pancreas [44], and there is intensive interest in identifying downstream targets and functions of Neurog3 during pancreas development. Genomica module network analysis identified sets of candidate Neurog3 target genes and further predicted these to be activated (n = 327) or repressed (n = 263) by Neurog3 (Figure 4A; Figure S6A; see Methods). Genes predicted to be induced by Neurog3 included known targets such as Pax4, Rfx6, Nkx2.2, Snail2 and Insm1, as well as novel candidates like Etv1 and Runx1t1 (Figure 4A). We did not detect known regulators of pancreas development among the set of genes predicted to be repressed by Neurog3, an area not previously well-characterized [9]; thus, we prioritized analysis of the gene set predicted to be activated by Neurog3. Functional enrichment analysis for biological processes through DAVID (Figure 4E) predicted roles for these Neurog3 targets in processes including RNA biosynthesis and transcription, protein transport, localization and secretion, catabolic processes, cell cycle control and chromatin organization. These functional categories were corroborated independently by in vivo testing (see below). Thus, the module network algorithm readily identified both established and previously unrecognized Neurog3-dependent gene regulatory programs and target genes governing pancreatic endocrine development.

In vivo validation of genes regulated by Neurog3
To validate predictions from IMNA, we initially focused on analyzing gene expression changes associated with the loss of Neurog3 in vivo. We purified Neurog3-null cells from the pancreata of E15 Neurog3eGFP/eGFP mutant embryos [45] [46], an approach not previously reported [47] [48] [49]. Of the 6367 differentially expressed, 3188 were downregulated and 3179 were upregulated by the loss of Neurog3 expression (Table S6). These included both known targets such as Pax4, Rfx6, Nkx2.2, Snail2 and Insm1, as well as predicted novel targets such as Etv1 and Runx1t1 (Figure 4A). Expression analysis of dissected mouse fetal pancreas by quantitative PCR confirmed that Runx1t1 and Etv1 mRNA were significantly decreased upon the loss of Neurog3 (Figure 4B) and upregulated upon adenoviral misexpression of Neurog3 in pancreatic epithelial cells (Figure 4C; detailed in Methods). Moreover, immunostaining detected Runx1t1 protein in a subset of pancreatic Neurog3+ cells at E15, which was lost in Neurog3-null epithelium (Figure 4D).
Remarkably, 87% of genes predicted by IMNA to be activated by Neurog3 (Figure 5A; P = 1.05×10−191) and 73% of targets predicted to be repressed by Neurog3 (Figure S6A; P = 7.02×10−102) were validated by our expression profiling of Neurog3+ control and mutant Neurog3-null cells. Functional enrichment analysis of Neurog3-activated targets identified by expression profiling indicated roles in transcription, mRNA processing, protein transport and secretion, cell morphogenesis, catabolic processes and chromatin organization (Figure 4F). These categories matched well with those predicted by our independent module network analysis (Figure 4E). Similarly, functional enrichment analysis of biological roles of predicted Neurog3-repressed targets matched those identified by expression profiling (compare Figure S6B, S6C). In summary, this in vivo mutant mouse analysis substantially validated specific predictions made by IMNA about Neurog3 target genes and their Neurog3-dependent biological functions.

Functional validation of previously unrecognized pancreas developmental regulators in vivo
To assess and validate candidate transcriptional regulators nominated by IMNA, we chose to assess in vivo functions of genes (1) highly ranked by IMNA (Table S5), (2) predicted to regulate target genes or functions involved in pancreas development (Figure S7), (3) without known roles in pancreas development at the initiation of these studies, and (4) with available mutant mouse alleles [50] [51] [52] [53]. These included Prdm16, Etv1, Runx1t1, and Bcl11a (Table S5). During pancreas development, mRNAs encoding each of these factors were more abundant in Neurog3+ endocrine progenitors than in Sox9+ pancreatic progenitors or beta cells (Figure 5A, 5B, 5D, 5G). This suggested possible roles for each factor in islet development. We also analyzed Gfi, a transcriptional regulator expressed in the fetal pancreas (Figure S8E–G), which has an established role in hematopoietic development [54] [55], but not nominated by IMNA as a regulator of pancreas development.
Prdm16 encodes a transcriptional regulator and histone methyltransferase [56]. Our gene expression studies showed that Prdm16 is highly expressed in Sox9+ pancreatic progenitor cells and Neurog3+ endocrine progenitors, then maintained at lower levels in alpha cells and beta cells (Figure 5A). IMNA predicted that Prdm16 regulates expression of Arx (Figure S7A). Supporting these findings, we recently reported that homozygous null mutation of Prdm16 leads to impaired development of pancreatic islets [17]. This included inappropriately increased expression of Arx, increased alpha cell and PP cell numbers (a known outcome of Arx misexpression; [57]), and disrupted beta cell development [17]. Thus, our work supports the prediction that Prdm16 is required for pancreas development in vivo.
Bcl11a encodes a zinc-finger transcription factor involved in hematopoiesis [53] but without known roles in pancreas development. IMNA nominated Bcl11a as a candidate regulator of pancreas development, and predicted new target genes Ins2, Glucagon and Ppy (Figure S7B). Remarkably, we observed reduced mRNA expression of each of these genes in FACS-purified endocrine cells from homozygous null mutant Bcl11a−/− mice (Figure 5C). We did not find significant changes in allocation of fetal islet cell subsets in Bcl11a mutants (Figure S8C), and lethality at P1 precluded further phenotyping. Thus, in vivo analysis confirmed a requirement for Bcl11a in endocrine development.
Etv1 (also known as Er81) encodes a transcription factor involved in neurogenesis and maturation of neural cells [50], but has no known function in pancreas development. Etv1 mRNA levels were reduced in Neurog3eGFP/eGFP null pancreata (Figure 4B), indicating that Etv1 is a direct or indirect target of Neurog3 and might have roles in islet cell development. Consistent with this possibility, we observed decreased levels of mRNAs encoding islet cell hormones, including a significant reduction of Pancreatic polypeptide mRNA in Etv1−/− mutant pancreas (Figure 5E). Likewise, morphometry of P1 null Etv1−/− pancreas revealed severely reduced islet PP cell mass (Figure 5F). Thus, in vivo analysis confirmed a requirement for Etv1 in pancreatic islet development.
Runx1t1 (also called Eto or Mtg8; [51]) encodes a transcription factor related to the Drosophila runt protein, and mutations in this gene have been linked to blood, lung and breast neoplasia [58] [59]. We detected Runx1t1 and Neurog3 co-expression in fetal pancreatic epithelial cells (Figure 4D). Runx1t1 mRNA was reduced in homozygous Neurog3 mutant pancreas (Figure 4B), and Runx1t1 protein was undetectable in homozygous null Neurog3eGFP/eGFP mutant cells (Figure 4D), supporting the view that Neurog3 regulates Runx1t1 expression. IMNA analysis indicated Runx1t1 regulates Pancreatic polypeptide (Figure S7D). Analysis of pancreas development in mice lacking Runx1t1, which expire at birth, revealed increased mRNA levels of Pancreatic polypeptide and Ghrelin expression (Figure 5H). Together with findings of significant islet cell hyperplasia in Runx1t1 null mutants (P. Pauerstein, C.B. and S.K.K., in preparation), our analysis confirmed an essential role for Runx1t1 in pancreas development.
Gfi1 encodes a transcriptional regulator [60] and is expressed in fetal pancreas (Figure S8E) but was not nominated by IMNA as a regulator of pancreas development. Consistent with this prediction, we did not detect disrupted islet development or glucose regulation in mice lacking Gfi1, despite exhaustive systematic phenotyping (Figure S8F, S8G). Thus, our rigorous integration of developmental, genomic and bioinformatic approaches identified four candidate regulators of pancreas development, and mutant mouse analysis confirmed that all four were also required in vivo.
Discussion
Elucidating the regulatory interactions underlying global transcriptional programs that control development of solid organs like the pancreas has been a challenge. Classical and recent studies have advanced our understanding of the cellular origins, genetics, morphogenesis, and cell lineage relationships in the developing pancreas, and have identified features of transient pancreatic progenitors or lineage-specific endocrine progenitors [8]. However these and other fetal pancreatic cell subsets are generated in relatively small numbers, hampering prior comprehensive genomic-scale efforts to dissect pancreas development. Here we combined several powerful approaches – including cell sorting, transgenic cell labeling, genomic-scale expression profiling, bioinformatics, and targeted mutagenesis in mice – to identify elements comprising genetic regulatory hierarchies in the developing pancreas. This effort has revealed both the complexity and structural framework of transcriptional programs underlying pancreas cell differentiation and maturation, and provides a strategy for similar studies in other solid organs.
Purification of cell subsets from defined genetic mouse strains by flow cytometry ([16] [17], this study) generated highly-reproducible gene expression profiles of a dozen pancreatic cell subsets – a degree of comprehensiveness unprecedented in prior studies. This innovation permitted deconvolution of gene expression profiles into co-expressed and co-regulated genes. We found that many gene sets were re-used in multiple lineages and stages. These findings are reminiscent of the general gene regulatory circuitry identified during hematopoiesis [10]. For example, immature alpha cell and beta cells from fetal or neonatal pancreas shared common gene sets that clustered distinctly from those in mature alpha and beta cells from adult pancreas. This feature likely reflects commonalities of mature alpha and beta cells as nutrient-responsive cells that produce, process and secrete polypeptide hormones, and corroborate similarities of gene regulation observed in adult human alpha and beta cells [61]. Global similarities of gene expression in adult alpha and beta cells shown here are also consistent with recent findings that in specific experimental settings, adult alpha cells may acquire beta cell features [62] [63] or vice-versa [64]. Identification of gene sets controlling function of mature beta cells may foster progress in producing replacement beta cells from renewable stem cell sources for diabetic patients [65]. For example, gene sets regulating calcium ion transport or responsiveness were enriched in adult beta cells, consistent with studies showing that calcium-dependent signaling pathways regulate beta cell maturation in mice and humans [31] [66]. Stimulation of calcium-responsive pathways, such as calcineurin/NFATc signaling, can enhance functional maturation of beta cells [31]. Thus, our reference data sets should prove useful for advancing efforts to produce or replace beta cells in diabetes.
Compared to the endocrine cell lineage or exocrine acinar cells, little is known about the genetic programs defining pancreatic exocrine duct cells [21]. Pearson correlation identified clustering between adult ductal cells and fetal pancreatic cells, including endocrine progenitor cells. This indicated that regulatory programs maintaining adult ductal cell gene expression and fate are unexpectedly similar to those in transient oligopotent fetal cell subsets, as suggested by unsupervised clustering with 30 mouse tissues. Thus, our study provides support for strategies focused on ‘reprogramming’ duct cells into other desired fates, including insulin-producing cells [67] [23] and could accelerate use of somatic cell reprogramming for therapeutic aims.
Human genetic studies have revealed that transcription factors have major roles in the pathogenesis of pancreatic malformation, including agenesis and diabetes mellitus [32] [68]; thus, we focused here on elucidating previously unrecognized transcriptional regulators required for pancreas development. Our general strategy was to exploit co-variance of transcription factors in gene sets and the cellular states they might regulate. Iterative use of a gene expression-based probabilistic program identified known regulators with high efficiency and predicted new regulatory functions for scores of transcription factors. IMNA identified known and novel regulators of endocrine development (see below), including a subset of transcription factors previously implicated by human GWAS in type 2 diabetes risk [41], [43]. We also noted that the frequency of detecting regulators of exocrine differentiation, like Mist1, was lower (Table S5). This likely reflects the lower representation of gene sets from differentiated exocrine cell types (2/12 cell subsets purified and analyzed here) compared to endocrine cell subsets. Therefore, further studies of gene regulation in subsets of purified fetal pancreatic exocrine cells could therefore likely identify additional exocrine pancreatic regulators.
To validate and assess the biological significance of predictions based on gene expression and module analysis and to control for variables introduced by our FACS-based approach, we analyzed relevant mutant mouse strains, including Neurog3 mutants. This combined approach proved to be a powerful way to test, for example, predictions of Neurog3 target gene expression, and to functionally validate transcriptional regulators identified by IMNA. Bcl11a, Runx1t1, Prdm16 and Etv1 encode transcription factors not previously linked to roles in pancreas development when these studies began. Prior studies had revealed crucial roles for Bcl11a in regulating blood development [69] [53] and diseases [70] [71], and for Runx1t1 in midgut development [51] and neoplasias of blood, lung and breast [58] [59]. Prdm16 has well characterized functions in adipogenesis [72] [73], leukemia pathogenesis [74], and neuronal stem cells maintenance [75]. Etv1/Er81 is an established regulator of fetal neuronal development [50] whose mis-expression leads to cancer pathogenesis in diverse tissues [76]. Independent genetic screens in our group, concurrent with studies here, identified roles for Prdm16 in regulating allocation of pancreatic islet cells in development [17].
Strikingly, after unbiased selection of these four candidate regulators, analysis of mouse strains harboring targeted mutations in Etv1, Prdm16, Bcl11a and Runx1t1 here or in recent studies from our group [17] revealed defects of pancreas development in all of the mutants. Though expressed in islet development, Gfi1, an established regulator of myeloid and enteric development [54] [77] [78], did not meet criteria of a regulator through our bioinformatic analysis. Accordingly, intensive investigation revealed no detectable phenotypes in pancreas development or glucose control in mice lacking Gfi1. Thus, our integrated approach accurately predicted essential regulators of islet development, demonstrating the robustness of our cell purification and gene expression profiling. This level of functional validation with mutant mouse phenotyping is, to our knowledge, unprecedented for integrative genomic approaches to pancreas development. Clearly, additional in-depth phenotypic studies, including in mice permitting conditional or pancreas-specific gene targeting, could prove valuable for understanding the molecular roles of these factors in pancreas development. Improved methods to isolate, purify and analyze cognate human pancreatic cell subsets [79] should enable an analogous integrative approach to identify factors regulating human pancreatic development.
Endoderm-derived epithelial cells and their progeny accomplish the vital physiological functions of the adult pancreas, and in other gastrointestinal organs. Thus, in these initial investigations we focused on deciphering the transcriptional hierarchies underlying epithelial cell development in the pancreas. However, prior studies have revealed that non-epithelial cells, including vascular endothelium, neuronal cells, and mesenchyme-derived signals control basic aspects of pancreas development [7] [80]. Thus, a complete deconstruction of pancreas development will require assessments, akin to those described here, of gene expression data from additional important cell subsets. Likewise additional data from epigenetic, genome-scale ChIP-Seq, proteomics and enhancer analyses [81] need to be integrated into the regulatory frameworks described here. The coordinated developmental, cellular, molecular and computational approaches described here should provide a paradigm for identifying genes and circuitry underlying development and postnatal maturation in other visceral organs, as well as assessment of regenerated pancreatic cell types.
Methods
Animals
All animal studies were approved by Stanford University and performed in accordance with Stanford University Animal Care and Use guidelines. Discomfort of animals was limited to that which was unavoidable in the conduct of scientifically valuable research. Analgesic, anesthetic, and tranquilizing drugs used where indicated and where appropriate to minimize discomfort and pain.
Mice harboring the Sox9-eGFP BAC transgene were obtained from Mutant Mouse Regional Resource Center, University of California at Davis [82]. Because of eGFP perdurance, Sox9-eGFP+ cells in Sox9-eGFP mice contain a mixture of Sox9+ and Sox9neg progeny. However, the percent of Sox9neg cells is low [17]. Neurog3eGFP transgenic mice were a kind gift from Drs. Guoqiang Gu and Douglas Melton [13]. Neurog3 knock-in reporter mice were a kind gift from Dr. Klaus Kaestner [45] [46] and provided by Dr. O. Cleaver. Mouse Insulin Promoter (MIP)-GFP mice were a gift from Dr. M. Hara (University of Chicago, Chicago, IL; [83]) and maintained in a CD-1 background. Glucagon-Venus mice were a gift from Dr. Fiona Gribble [84]. Runx1t1 mutant mice were rederived from MRC Harwell (Stock number FESA:000373). Etv1 mutant mice were a gift from Dr. Thomas Jessell and provided by Dr. Julia Kaltschmidt (Memorial Sloan Kettering Institute). Bcl11a mutant mice were derived from Bcl11a floxed mice [53] by crossing with a Cre deleter strain (CMV-Cre), obtained from Jackson Laboratories (Stock number 006054). Prdm16 mutant mice were obtained from Jackson Laboratories (Stock number 013100). Gfi1 mutant mice are described in [54]. Genotyping follows published methods. Mice were mated overnight and checked for plugs. Noon on the day of vaginal plug appearance was counted as embryonic day 0.5 (E0.5).
Cell sorting strategy and flow cytometry
Pancreata were obtained at E11, E15, E17, postnatal day (P) 1, P15, and 8–12 weeks of age. To obtain E11 Sox9+ pancreatic progenitors, we dissected the dorsal pancreas from Sox9-eGFP reporter embryos and dissociated with TrypLE Express (Invitrogen, Carlsbad, CA) at 37°C for 5 min and triturated. TrypLE was neutralized with 10% (v/v) FBS in PBS [17]. Approximately 200 pancreata were dissected to obtain four replicates of Sox9+ pancreatic progenitors. E15 pancreatic progenitors, Neurog3+ endocrine progenitors, and acinar cells were collected using a combination of cell surface markers and transgenic cell labeling. This approach was used to collect hormoneneg Neurog3+ endocrine progenitors because GFP perdurance labels their hormone+ descendants. Briefly, the Neurog3eGFP transgenic pancreata were dissected and visually assessed for GFP expression. GFP+ pancreata were pooled and dissociated with 0.05% Trypsin EDTA at 37°C for 8 min and triturated (GFPneg pancreata were collected for FACS gating purposes). Trypsin EDTA was neutralized with 10%(v/v) FBS in 10 mM EGTA, PBS [16] and the cells were treated for 15 min in a blocking solution composed of FACS buffer (2% fetal bovine serum in PBS, 10 mM EGTA was supplemented for experiments of E15 pancreas) containing 300 ng/ml rat IgG (Jackson ImmunoResearch, West Grove, PA). We used the following primary antibodies: biotin anti-CD133 (13A4, 1∶100; eBioscience, San Diego, CA), Pacific Blue anti-CD24 (M1/69, 1∶100; BioLegend, San Diego, CA), and PE anti-CD49f (GoH3, 1∶50; R&D, Minneapolis, MN). Streptavidin-APC (1∶200; eBioscience) was used to visualize biotinylated antibodies. Gating was performed in accordance to Sugiyama [16]. The antibody combinations are shown in Figure 1B. E15 Neurog3-null cells were collected similarly from GFP+ Neurog3eGFP/eGFP knock-in embryos. A total of 405 pancreata were dissected to collect each replicate of Neurog3+ endocrine progenitors. Each replicate comprises ∼15,000 cells.
Beta cells and alpha cells were obtained from MIP-GFP and Glucagon-Venus reporter mice, respectively. Beta cells were collected from E15, E17, postnatal day (P) 1, P15, and 8–12 week-old male mice, while alpha cells were collected at P1 and 8–12 week-old male mice. E15 and E17 MIP-GFP+ pancreata were dissociated with 0.05% Trypsin EDTA for 8–10 min at 37°C and triturated from 300 pancreata and 240 pancreata, respectively. P1 MIP-GFP+ and P1 Glucagon-Venus+ pancreas were dissociated with 1 mg/ml collagenase (Sigma-Aldrich; C01030) for 8 minutes, followed by mixing, spinning and further dissociation with 1 mg/ml dispase for 8 minutes at 37°C from approximately 230 pancreata. P15 and adult pancreata were dissociated by standard intraductal ligation and digestion with 1 mg/ml collagenase [31]. Each replicate consisted of at least 3 male mice. Beta cells from the stages E15, E17 and P1 are termed ‘fetal’ beta cells, while beta cells from P15 and adult mice are ‘postnatal’ beta cells based on their ability to couple glucose detection with insulin secretion [85]. Adult duct cells were collected using APC anti-CD133 (1∶100; BioLegend, San Diego, CA). To exclude blood cell contamination from our sorts we used cell-surface markers Ter119 and CD45 (1∶100; Biosciences), which label erythroid cells and leukocytes, respectively. Live-dead cell exclusion was performed with 10 µg/mL Propidium Iodide (PI; Sigma) or 10 µg/mL Aqua (L34957; Invitrogen).
Cell sorting was performed in FACS Aria I and II machines fitted with a 100 uM nozzle using DIVA software (BD Biosciences, San Jose, CA). FACS data were analyzed by using FlowJo software (Tree Star, San Carlos, CA). Cells were collected in 10% fetal bovine serum in PBS and processed for RNA collection (for acinar cells, 2% fetal bovine serum in PBS supplemented with 10 mM EGTA). Cell death did not exceed 30% per sample. All cell types were collected in at least triplicate from a minimum of 15,000 cells per sample.
Molecular biology
Total mRNA was isolated using the Arcturus PicoPure kit (Applied Biosystems) for all microarray samples. For quantitative PCR analysis, whole pancreas mRNA at E18 or P1 was collected by homogenizing each pancreas in 1.5 ml of RLT buffer and RNA was extracted using the Qiagen RNAeasy Micro kit (Qiagen). cDNA synthesis was performed with Ambion Retroscript kit. Quantitative PCR studies were performed using an ABI7500 system, Applied Biosystems (Foster City, CA). Replicates were processed independently, and each cDNA was tested in duplicate. Expression level was normalized to beta-actin. Information about primer and probe sets is available upon request.
Microarray data preprocessing, normalization and clustering
RNA quality was accessed with Agilent's BioAnalyzer (Stanford PAN facility). 50 ng to 100 ng of mRNA with a RNA integrity number (RIN) score >9 were amplified with NuGen Ovation Kit V2 (NuGEN) and fragmented and labeled with the Biotin and fragmentation labeling kit (NuGEN) following manufacturer's protocol. Hybridization and image analysis processing was done in accordance to the Stanford PAN facility. The Affymetrix Mouse 430 2.0 GeneChip was used.
For gene expression analysis, arrays were RMA normalized using justRMA package in R. After normalization, probes with raw expression value of 100 in all arrays were filtered out—leaving a total of 23,093 probes. For each expressed probe, expression values were log2-transformed, and mean-centered across all the conditions before pair-wise Pearson correlation was performed. Unsupervised hierarchical clustering and array clustering of pair-wise Pearson correlation was performed using Cluster 3.0 [86]. This dendrogram was overlaid with the Pearson correlation plot (Figure 1C).
Unsupervised hierarchical clustering analysis with Microarray data from over 30 adult mouse tissues was downloaded from NIH GEO, with accession number GSE1133 [19]. The data discussed in this publication have been deposited in NCBI's Gene Expression Omnibus [87] and are accessible through GEO Series accession number GSE54374 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE54374).
Module mapping
The module mapping function of Genomica [34] was executed to identify gene sets that are enriched or depleted in each sample. Here, we entered gene ontology terms belonging to biological processes (BP-GO terms) from mouse and the mean-centered normalized expression values for all the arrays (23,093 probes). We used the default settings of the program: P-value of 0.05, FDR correction of 0.05, and the hierarchical agglomerative (correction centered) clustering method. Probes with an expression level > = 1 (on a log2 scale) were considered upregulated, while probes with an expression level < = −1 (on a log2 scale) were considered downregulated. We displayed the average expression of gene hits from each enriched gene set (based on a log2 scale of mean-centered values). GO gene sets of biological functions were downloaded from website DAVID and imported into the Genomica Software [20] [34].
Gene signatures and pair-wise comparisons
Gene signatures for each cell type were calculated using Student t-test comparing signals in the arrays of a particular cell-type versus the rest of the arrays. The genes selected as ‘signature genes’ met four parameters: P-value< = 0.001, FDR< = 0.05, log2 fold change > = 1, and standard deviation < = 0.5 of arrays in the same cell type. FDR correction was estimated using the p.adjust package in R. Negative gene signatures were selected using a log2 fold change < = −1, P-value< = 0.001, FDR< = 0.05, and standard deviation < = 0.5 of arrays in the same cell type. Similar methods were applied for transcription factor signatures. For each gene signature, we performed functional enrichment analysis of Gene Ontology terms related to biological processes (GOTERM_BP_FAT) through DAVID. BP-GO terms were considered significant if FDR>0.05. DAVID default settings were used.
To obtain differentially expressed genes between two cell types we used the Student t-test where the obtained P-values were adjusted for multiple testing using the p.adjust function of R with Benjamini-Hochberg method (adjusted P<0.05). The log2 fold change difference between each representative cell type was calculated by averaging the transformed probe signals in the arrays of the first cell type and subtract that from the second cell type. Fetal endocrine cells included E15 beta cells, E17 beta cells, P1 beta cells, and P1 alpha cells. Postnatal endocrine cells include P15 beta cells, Adult beta cells, and Adult alpha cells.
IMNA expression-based prediction of regulators, their target genes and functions
We identified candidate regulators and their regulation programs using the Module Networks algorithm in the Genomica software [10] [34]. Genomica detects modules of co-expressed genes (gene sets) and their shared regulatory programs. A regulation program is a small set of genes whose expression is predictive of the expression level of the module genes using a decision (regression) tree structure. Given the expression values of a pool of candidate regulator genes, a set of modules and their associated regulation programs are automatically inferred by an iterative procedure. This procedure searches for the best gene partition into modules and for the regulation program of each module while optimizing a target function. The target function is the Bayesian score derived from the posterior probability of the model (see [34] for a detailed description of the algorithm). The program requires two input lists: 1) list of potential regulators as determined by the user and 2) normalized mean-centered expression data for the samples of interest. We compiled a list of 1642 mouse transcription factors (TF) or TF components from various sources [36], [37] [38]. Our list of input ‘regulators’ was a filtered list of TFs and TF components that were expressed in our arrays (total of 3310 expressed probes based on our list of 1642 candidate TFs). Our list of input ‘expression values’ consisted of the normalized and mean-centered values for all 12 cell populations (described above; 23,093 probes). We applied the default settings and set the maximum tree depth to 5.
We optimized the maximum number of modules by counting the number of known regulators predicted. The list of known pancreatic regulators (82 total) was compiled from [9] and literature searches on all predicted regulators in pubmed. Genomica identifies the sets of candidate regulators that are most predictive of the expression pattern for each module (co-expressed genes). We tested the quality of its predictions by setting: 25, 50, 75, and 100 modules. After a single run, 100 modules identified 25% of known regulators of pancreas development. Because this number was low and because single runs were inherently unstable, we reasoned that multiple iterations may yield better predictions. We tested the optimal number of iterations counting the number of known regulators after 1, 20, 40, 60, 80, and 100 iterations for each module number. Reproducibility was determined by ranking the frequency that each candidate regulator is identified after each run and by performing Gene set enrichment analysis (GSEA; [40]; Figure S5) of true positives (known regulators). We determined that 100 iterations of 75 modules was the best setting; it identified 99% of known pancreatic regulators and provided the best GSEA enrichment score. Increasing the number of iterations to 110 or 120 predicted the same number of known pancreatic regulators but worsened the enrichment score. To obtain the GSEA enrichment score of diabetes risk factor genes, we compiled a total of 72 risk factor genes from [42] and [88]. Then we performed GSEA on factors that were transcription factors or DNA-binding proteins (26/72 genes) and were expressed in the pancreas (22/26 TFs genes). Using our list of 22 risk factor genes we obtained the enrichment score relative to the ranked list from our IMNA approach as described above.
The predicted targets and functions for a subset of candidate regulators were determined by extracting the modules that each regulator was predicted to regulate. All the modules that were positively regulated were grouped (comprising of a minimum of 5 modules per regulator). This was similarly done for modules that a regulator was predicted to repress. We obtained the predicted biological functions for each regulator by performing functional enrichment analysis on each list of genes through DAVID. BP-GO terms were considered significant if FDR>0.2. DAVID default settings were used.
To validate the predicted targets and functions of Neurog3, we performed Student's t-test to obtain differentially expressed genes between E15 Neurog3+ endocrine progenitors and E15 Neuorg3-null cells. We obtained ∼8000 probes that were differentially expressed with a 2-fold difference (4604 probes enriched in E15 endocrine progenitors and 4217 probes enriched in Neurog3-null cells). We compared this list to targets predicted by Genomica. This analysis yielded an 85% overlap in probes (303/358; P = 2.01×10−168; Figure 4A) or 87.2% overlap in genes (285/327; P = 1.05×10−191) when we filter out probes in each predicted module that have an expression value = <1 (based on a log2 scale). The same approach was used to predict repressed targets of Neurog3. When we filter probes with an expression value >−1, 67% of probes overlap (220/328, P-value = 2.60×10−85; Figure S6A) or 73% genes overlap (192/263; P = 7.02×10−102). Fisher's exact test was used to determine statistical significance with P<0.05 against the total number of probes or genes. Next, we performed functional enrichment analysis on each list of Genomica predicted targets through DAVID using default settings and compared these results to targets obtained by gene expression profiling of Neurog3-null cells (FDR<0.05).
The analysis was integrated through GenomeSpace (http://www.genomespace.org/). Venn diagrams were obtained with the Venny program [89] (http://bioinfogp.cnb.csic.es/tools/venny/index.html). Fisher's exact analysis was performed using the following website: http://www.langsrud.com/fisher.htm
Statistical analyses
Each variable was analyzed using the two-tailed Student's t test. For all analyses, a P value of less than 0.05 was considered significant. Results are given as mean +/ − SEM.
Over-expression of Neurog3
A mouse duct cell line (mPAC) was infected with adenoviruses expressing the mouse Neurog3 gene and the red fluorescent protein (RFP) from separate CMV promoters. The control sample included mPAC cells that were infected with Adenoviruses expressing RFP. We verified that RFP did not have an effect on the expression of Neurog3 downstream genes in non-treated mPAC cells. Cells were cultured with the virus for 1 day and then media was changed. Cells were harvested after 3–4 days. Each experimental condition was performed in triplicate.
Immunohistology
For measurement of endocrine-cell mass, a minimum of 12 pancreas sections spanning the entire pancreas were assessed for at least 3 different mice per genotype. The total cross-sectional area of hormone+ cells was summed and normalized to total pancreatic area using Image-Pro Plus analysis software (Media Cybernetics). Statistical analysis was performed using a two-tailed Student's t-test. For staining Runx1t1 and Etv1-LacZ expression, E15 and 2-month old mouse pancreata were dissected and fixed with 4% paraformaldehyde overnight at 4°C, and cryo-embedded. Sections were permeabilized with 1% Triton-X-100 for 1 hr before blocking with 2%BSA, 1% DMSO in PBS. We used the following primary antibodies: Goat anti-Runx1t1 (1∶200, Santa Cruz, C-20), Rabbit anti-LacZ (1∶500, Invitrogen), and Rat anti-E-cadherin (1∶400, Invitrogen). Secondary antibodies were from Jackson ImmunoResearch and Molecular Probes. Samples were mounted with Vectashield containing DAPI (Vector Laboratories). Microscopic images were obtained using a Leica SP2 AOBS confocal laser-scanning microscope.
Supporting Information
Zdroje
1. PradoCL, Pugh-BernardAE, ElghaziL, Sosa-PinedaB, SusselL (2004) Ghrelin cells replace insulin-producing beta cells in two mouse models of pancreas development. Proceedings of the National Academy of Sciences of the United States of America 101 : 2924–2929.
2. LynnFC, SmithSB, WilsonME, YangKY, NekrepN, et al. (2007) Sox9 coordinates a transcriptional network in pancreatic progenitor cells. Proceedings of the National Academy of Sciences of the United States of America 104 : 10500–10505.
3. SeymourPA, FreudeKK, TranMN, MayesEE, JensenJ, et al. (2007) SOX9 is required for maintenance of the pancreatic progenitor cell pool. Proceedings of the National Academy of Sciences of the United States of America 104 : 1865–1870.
4. JonssonJ, CarlssonL, EdlundT, EdlundH (1994) Insulin-promoter-factor 1 is required for pancreas development in mice. Nature 371 : 606–609.
5. KrappA, KnoflerM, LedermannB, BurkiK, BerneyC, et al. (1998) The bHLH protein PTF1-p48 is essential for the formation of the exocrine and the correct spatial organization of the endocrine pancreas. Genes & development 12 : 3752–3763.
6. KoppJL, DuboisCL, SchafferAE, HaoE, ShihHP, et al. (2011) Sox9+ ductal cells are multipotent progenitors throughout development but do not produce new endocrine cells in the normal or injured adult pancreas. Development 138 : 653–665.
7. BenitezCM, GoodyerWR, KimSK (2012) Deconstructing pancreas developmental biology. Cold Spring Harbor perspectives in biology 4 doi: 10.1101/cshperspect.a012401
8. ShihHP, WangA, SanderM (2013) Pancreas Organogenesis: From Lineage Determination to Morphogenesis. Annual review of cell and developmental biology
9. ArdaHE, BenitezCM, KimSK (2013) Gene regulatory networks governing pancreas development. Developmental cell 25 : 5–13.
10. NovershternN, SubramanianA, LawtonLN, MakRH, HainingWN, et al. (2011) Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell 144 : 296–309.
11. McKinney-FreemanS, CahanP, LiH, LacadieSA, HuangHT, et al. (2012) The transcriptional landscape of hematopoietic stem cell ontogeny. Cell stem cell 11 : 701–714.
12. ScearceLM, BrestelliJE, McWeeneySK, LeeCS, MazzarelliJ, et al. (2002) Functional genomics of the endocrine pancreas: the pancreas clone set and PancChip, new resources for diabetes research. Diabetes 51 : 1997–2004.
13. GuG, WellsJM, DombkowskiD, PrefferF, AronowB, et al. (2004) Global expression analysis of gene regulatory pathways during endocrine pancreatic development. Development 131 : 165–179.
14. HoffmanBG, ZavagliaB, WitzscheJ, Ruiz de AlgaraT, BeachM, et al. (2008) Identification of transcripts with enriched expression in the developing and adult pancreas. Genome biology 9: R99.
15. van ArensbergenJ, Garcia-HurtadoJ, MoranI, MaestroMA, XuX, et al. (2010) Derepression of Polycomb targets during pancreatic organogenesis allows insulin-producing beta-cells to adopt a neural gene activity program. Genome Res 20 : 722–732.
16. SugiyamaT, RodriguezRT, McLeanGW, KimSK (2007) Conserved markers of fetal pancreatic epithelium permit prospective isolation of islet progenitor cells by FACS. Proceedings of the National Academy of Sciences of the United States of America 104 : 175–180.
17. SugiyamaT, BenitezCM, GhodasaraA, LiuL, McLeanGW, et al. (2013) Reconstituting pancreas development from purified progenitor cells reveals genes essential for islet differentiation. Proceedings of the National Academy of Sciences of the United States of America 110 : 12691–12696.
18. SeymourPA, SanderM (2011) Historical perspective: beginnings of the beta-cell: current perspectives in beta-cell development. Diabetes 60 : 364–376.
19. SuAI, WiltshireT, BatalovS, LappH, ChingKA, et al. (2004) A gene atlas of the mouse and human protein-encoding transcriptomes. Proceedings of the National Academy of Sciences of the United States of America 101 : 6062–6067.
20. SegalE, FriedmanN, KollerD, RegevA (2004) A module map showing conditional activity of expression modules in cancer. Nature genetics 36 : 1090–1098.
21. BenitezCM, GoodyerWR, KimSK (2012) Deconstructing pancreas developmental biology. Cold Spring Harb Perspect Biol 4.
22. ZhouQ, BrownJ, KanarekA, RajagopalJ, MeltonDA (2008) In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature 455 : 627–632.
23. LeeJ, SugiyamaT, LiuY, WangJ, GuX, et al. (2013) Expansion and conversion of human pancreatic ductal cells into insulin-secreting endocrine cells. Elife 2: e00940.
24. PagliucaFW, MeltonDA (2013) How to make a functional beta-cell. Development 140 : 2472–2483.
25. MezzaT, KulkarniRN (2014) The regulation of pre - and post-maturational plasticity of mammalian islet cell mass. Diabetologia 57 : 1291–1303.
26. ReinertRB, CaiQ, HongJY, PlankJL, AamodtK, et al. (2014) Vascular endothelial growth factor coordinates islet innervation via vascular scaffolding. Development 141 : 1480–1491.
27. CleaverO, DorY (2012) Vascular instruction of pancreas development. Development 139 : 2833–2843.
28. GeorgiaS, HinaultC, KawamoriD, HuJ, MeyerJ, et al. (2010) Cyclin D2 is essential for the compensatory beta-cell hyperplastic response to insulin resistance in rodents. Diabetes 59 : 987–996.
29. TetaM, LongSY, WartschowLM, RankinMM, KushnerJA (2005) Very slow turnover of beta-cells in aged adult mice. Diabetes 54 : 2557–2567.
30. GuC, SteinGH, PanN, GoebbelsS, HornbergH, et al. (2010) Pancreatic beta cells require NeuroD to achieve and maintain functional maturity. Cell Metab 11 : 298–310.
31. GoodyerWR, GuX, LiuY, BottinoR, CrabtreeGR, et al. (2012) Neonatal beta cell development in mice and humans is regulated by calcineurin/NFAT. Developmental cell 23 : 21–34.
32. McKnightKD, WangP, KimSK (2010) Deconstructing pancreas development to reconstruct human islets from pluripotent stem cells. Cell Stem Cell 6 : 300–308.
33. De FrancoE, Shaw-SmithC, FlanaganSE, ShepherdMH, HattersleyAT, et al. (2013) GATA6 mutations cause a broad phenotypic spectrum of diabetes from pancreatic agenesis to adult-onset diabetes without exocrine insufficiency. Diabetes 62 : 993–997.
34. SegalE, ShapiraM, RegevA, Pe'erD, BotsteinD, et al. (2003) Module networks: identifying regulatory modules and their condition-specific regulators from gene expression data. Nature genetics 34 : 166–176.
35. NovershternN, RegevA, FriedmanN (2011) Physical Module Networks: an integrative approach for reconstructing transcription regulation. Bioinformatics 27: i177–185.
36. KanamoriM, KonnoH, OsatoN, KawaiJ, HayashizakiY, et al. (2004) A genome-wide and nonredundant mouse transcription factor database. Biochem Biophys Res Commun 322 : 787–793.
37. RavasiT, SuzukiH, CannistraciCV, KatayamaS, BajicVB, et al. (2010) An atlas of combinatorial transcriptional regulation in mouse and man. Cell 140 : 744–752.
38. ZhangHM, ChenH, LiuW, LiuH, GongJ, et al. (2012) AnimalTFDB: a comprehensive animal transcription factor database. Nucleic Acids Res 40: D144–149.
39. JoshiA, De SmetR, MarchalK, Van de PeerY, MichoelT (2009) Module networks revisited: computational assessment and prioritization of model predictions. Bioinformatics 25 : 490–496.
40. SubramanianA, TamayoP, MoothaVK, MukherjeeS, EbertBL, et al. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America 102 : 15545–15550.
41. ImamuraM, MaedaS (2011) Genetics of type 2 diabetes: the GWAS era and future perspectives [Review]. Endocr J 58 : 723–739.
42. MorrisAP, VoightBF, TeslovichTM, FerreiraT, SegreAV, et al. (2012) Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet 44 : 981–990.
43. DimasAS, LagouV, BarkerA, KnowlesJW, MagiR, et al. (2014) Impact of type 2 diabetes susceptibility variants on quantitative glycemic traits reveals mechanistic heterogeneity. Diabetes 63 : 2158–2171.
44. GradwohlG, DierichA, LeMeurM, GuillemotF (2000) neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proceedings of the National Academy of Sciences of the United States of America 97 : 1607–1611.
45. LeeCS, PerreaultN, BrestelliJE, KaestnerKH (2002) Neurogenin 3 is essential for the proper specification of gastric enteroendocrine cells and the maintenance of gastric epithelial cell identity. Genes & development 16 : 1488–1497.
46. WangS, JensenJN, SeymourPA, HsuW, DorY, et al. (2009) Sustained Neurog3 expression in hormone-expressing islet cells is required for endocrine maturation and function. Proceedings of the National Academy of Sciences of the United States of America 106 : 9715–9720.
47. JuhlK, SarkarSA, WongR, JensenJ, HuttonJC (2008) Mouse pancreatic endocrine cell transcriptome defined in the embryonic Ngn3-null mouse. Diabetes 57 : 2755–2761.
48. WhiteP, MayCL, LamounierRN, BrestelliJE, KaestnerKH (2008) Defining pancreatic endocrine precursors and their descendants. Diabetes 57 : 654–668.
49. SoyerJ, FlasseL, RaffelsbergerW, BeucherA, OrvainC, et al. (2010) Rfx6 is an Ngn3-dependent winged helix transcription factor required for pancreatic islet cell development. Development 137 : 203–212.
50. ArberS, LadleDR, LinJH, FrankE, JessellTM (2000) ETS gene Er81 controls the formation of functional connections between group Ia sensory afferents and motor neurons. Cell 101 : 485–498.
51. CalabiF, PannellR, PavloskaG (2001) Gene targeting reveals a crucial role for MTG8 in the gut. Molecular and cellular biology 21 : 5658–5666.
52. HerronBJ, LuW, RaoC, LiuS, PetersH, et al. (2002) Efficient generation and mapping of recessive developmental mutations using ENU mutagenesis. Nature genetics 30 : 185–189.
53. SankaranVG, XuJ, RagoczyT, IppolitoGC, WalkleyCR, et al. (2009) Developmental and species-divergent globin switching are driven by BCL11A. Nature 460 : 1093–1097.
54. KarsunkyH, ZengH, SchmidtT, ZevnikB, KlugeR, et al. (2002) Inflammatory reactions and severe neutropenia in mice lacking the transcriptional repressor Gfi1. Nature genetics 30 : 295–300.
55. HockH, HamblenMJ, RookeHM, TraverD, BronsonRT, et al. (2003) Intrinsic requirement for zinc finger transcription factor Gfi-1 in neutrophil differentiation. Immunity 18 : 109–120.
56. PinheiroI, MargueronR, ShukeirN, EisoldM, FritzschC, et al. (2012) Prdm3 and Prdm16 are H3K9me1 methyltransferases required for mammalian heterochromatin integrity. Cell 150 : 948–960.
57. CollombatP, Hecksher-SorensenJ, KrullJ, BergerJ, RiedelD, et al. (2007) Embryonic endocrine pancreas and mature beta cells acquire alpha and PP cell phenotypes upon Arx misexpression. J Clin Invest 117 : 961–970.
58. MiyoshiH, KozuT, ShimizuK, EnomotoK, MasekiN, et al. (1993) The t(8;21) translocation in acute myeloid leukemia results in production of an AML1-MTG8 fusion transcript. The EMBO journal 12 : 2715–2721.
59. KimYR, KimMS, LeeSH, YooNJ (2011) Mutational analysis of RUNX1T1 gene in acute leukemias, breast and lung carcinomas. Leukemia research 35: e157–158.
60. ChiangC, AyyanathanK (2013) Snail/Gfi-1 (SNAG) family zinc finger proteins in transcription regulation, chromatin dynamics, cell signaling, development, and disease. Cytokine Growth Factor Rev 24 : 123–131.
61. BramswigNC, EverettLJ, SchugJ, DorrellC, LiuC, et al. (2013) Epigenomic plasticity enables human pancreatic alpha to beta cell reprogramming. The Journal of clinical investigation 123 : 1275–1284.
62. ThorelF, NepoteV, AvrilI, KohnoK, DesgrazR, et al. (2010) Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature 464 : 1149–1154.
63. CourtneyM, GjernesE, DruelleN, RavaudC, VieiraA, et al. (2013) The inactivation of Arx in pancreatic alpha-cells triggers their neogenesis and conversion into functional beta-like cells. PLoS Genet 9: e1003934.
64. DhawanS, GeorgiaS, TschenSI, FanG, BhushanA (2011) Pancreatic beta cell identity is maintained by DNA methylation-mediated repression of Arx. Developmental cell 20 : 419–429.
65. HebrokM (2012) Generating beta cells from stem cells-the story so far. Cold Spring Harbor perspectives in medicine 2: a007674.
66. PeirisH, RaghupathiR, JessupCF, ZaninMP, MohanasundaramD, et al. (2012) Increased expression of the glucose-responsive gene, RCAN1, causes hypoinsulinemia, beta-cell dysfunction, and diabetes. Endocrinology 153 : 5212–5221.
67. Al-HasaniK, PfeiferA, CourtneyM, Ben-OthmanN, GjernesE, et al. (2013) Adult Duct-Lining Cells Can Reprogram into beta-like Cells Able to Counter Repeated Cycles of Toxin-Induced Diabetes. Developmental cell 26 : 86–100.
68. EllardS, Lango AllenH, De FrancoE, FlanaganSE, HysenajG, et al. (2013) Improved genetic testing for monogenic diabetes using targeted next-generation sequencing. Diabetologia 56 : 1958–1963.
69. LiuP, KellerJR, OrtizM, TessarolloL, RachelRA, et al. (2003) Bcl11a is essential for normal lymphoid development. Nature immunology 4 : 525–532.
70. SatterwhiteE, SonokiT, WillisTG, HarderL, NowakR, et al. (2001) The BCL11 gene family: involvement of BCL11A in lymphoid malignancies. Blood 98 : 3413–3420.
71. XuJ, PengC, SankaranVG, ShaoZ, EsrickEB, et al. (2011) Correction of sickle cell disease in adult mice by interference with fetal hemoglobin silencing. Science 334 : 993–996.
72. SealeP, BjorkB, YangW, KajimuraS, ChinS, et al. (2008) PRDM16 controls a brown fat/skeletal muscle switch. Nature 454 : 961–967.
73. BjorkBC, Turbe-DoanA, PrysakM, HerronBJ, BeierDR (2010) Prdm16 is required for normal palatogenesis in mice. Human molecular genetics 19 : 774–789.
74. MorishitaK (2007) Leukemogenesis of the EVI1/MEL1 gene family. International journal of hematology 85 : 279–286.
75. ChuikovS, LeviBP, SmithML, MorrisonSJ (2010) Prdm16 promotes stem cell maintenance in multiple tissues, partly by regulating oxidative stress. Nature cell biology 12 : 999–1006.
76. OhS, ShinS, JanknechtR (2012) ETV1, 4 and 5: an oncogenic subfamily of ETS transcription factors. Biochimica et biophysica acta 1826 : 1–12.
77. AmannJM, ChylaBJ, EllisTC, MartinezA, MooreAC, et al. (2005) Mtgr1 is a transcriptional corepressor that is required for maintenance of the secretory cell lineage in the small intestine. Molecular and cellular biology 25 : 9576–9585.
78. BjerknesM, ChengH (2010) Cell Lineage metastability in Gfi1-deficient mouse intestinal epithelium. Dev Biol 345 : 49–63.
79. DorrellC, SchugJ, LinCF, CanadayPS, FoxAJ, et al. (2011) Transcriptomes of the major human pancreatic cell types. Diabetologia 54 : 2832–2844.
80. Munoz-BravoJL, Hidalgo-FigueroaM, PascualA, Lopez-BarneoJ, Leal-CerroA, et al. (2013) GDNF is required for neural colonization of the pancreas. Development 140 : 3669–3679.
81. BuenrostroJD, GiresiPG, ZabaLC, ChangHY, GreenleafWJ (2013) Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10 : 1213–1218.
82. SeymourPA, FreudeKK, DuboisCL, ShihHP, PatelNA, et al. (2008) A dosage-dependent requirement for Sox9 in pancreatic endocrine cell formation. Dev Biol 323 : 19–30.
83. HaraM, WangX, KawamuraT, BindokasVP, DizonRF, et al. (2003) Transgenic mice with green fluorescent protein-labeled pancreatic beta -cells. American journal of physiology Endocrinology and metabolism 284: E177–183.
84. ReimannF, HabibAM, TolhurstG, ParkerHE, RogersGJ, et al. (2008) Glucose sensing in L cells: a primary cell study. Cell metabolism 8 : 532–539.
85. BlumB, HrvatinSS, SchuetzC, BonalC, RezaniaA, et al. (2012) Functional beta-cell maturation is marked by an increased glucose threshold and by expression of urocortin 3. Nature biotechnology 30 : 261–264.
86. de HoonMJ, ImotoS, NolanJ, MiyanoS (2004) Open source clustering software. Bioinformatics 20 : 1453–1454.
87. EdgarR, DomrachevM, LashAE (2002) Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30 : 207–210.
88. MurphyR, EllardS, HattersleyAT (2008) Clinical implications of a molecular genetic classification of monogenic beta-cell diabetes. Nat Clin Pract Endocrinol Metab 4 : 200–213.
89. Oliveros JC (2007) VENNY: An interactive tool for comparing lists with Venn Diagrams. BioinfoGP, CNB-CSIC.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 10
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- The Master Activator of IncA/C Conjugative Plasmids Stimulates Genomic Islands and Multidrug Resistance Dissemination
- A Splice Mutation in the Gene Causes High Glycogen Content and Low Meat Quality in Pig Skeletal Muscle
- Keratin 76 Is Required for Tight Junction Function and Maintenance of the Skin Barrier
- A Role for Taiman in Insect Metamorphosis
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy