
Spinster Homolog 2 () Deficiency Causes Early Onset Progressive Hearing Loss
Progressive hearing loss is common in the human population but we know very little about the molecular mechanisms involved. Mutant mice are useful for investigating these mechanisms and have revealed a wide range of different abnormalities that can all lead to the same outcome: deafness. We report here our findings of a new mouse line with a mutation in the Spns2 gene, affecting the release of a lipid called sphingosine-1-phosphate, which has an important role in several processes in the body. For the first time, we report that this molecular pathway is required for normal hearing through a role in generating a voltage difference that acts like a battery, allowing the sensory hair cells of the cochlea to detect sounds at extremely low levels. Without the normal function of the Spns2 gene and release of sphingosine-1-phosphate locally in the inner ear, the voltage in the cochlea declines, leading to rapid loss of sensitivity to sound and ultimately to complete deafness. The human version of this gene, SPNS2, may be involved in human deafness, and understanding the underlying mechanism presents an opportunity to develop potential treatments for this form of hearing loss.
Published in the journal:
. PLoS Genet 10(10): e32767. doi:10.1371/journal.pgen.1004688
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004688
Summary
Progressive hearing loss is common in the human population but we know very little about the molecular mechanisms involved. Mutant mice are useful for investigating these mechanisms and have revealed a wide range of different abnormalities that can all lead to the same outcome: deafness. We report here our findings of a new mouse line with a mutation in the Spns2 gene, affecting the release of a lipid called sphingosine-1-phosphate, which has an important role in several processes in the body. For the first time, we report that this molecular pathway is required for normal hearing through a role in generating a voltage difference that acts like a battery, allowing the sensory hair cells of the cochlea to detect sounds at extremely low levels. Without the normal function of the Spns2 gene and release of sphingosine-1-phosphate locally in the inner ear, the voltage in the cochlea declines, leading to rapid loss of sensitivity to sound and ultimately to complete deafness. The human version of this gene, SPNS2, may be involved in human deafness, and understanding the underlying mechanism presents an opportunity to develop potential treatments for this form of hearing loss.
Introduction
Spinster homolog 2 (Spns2) is a multi-pass membrane protein belonging to the Spns family. Though the functions of Spns1 and Spns3 are largely unknown, Spns2 is known to act as a sphingosine-1-phosphate (S1P) transporter, based upon previous studies in zebrafish and mouse [1]–[6]. S1P is a vital lipid. It has diverse roles, functioning as a signalling molecule regulating cell growth [7], [8], programmed cell death [9], angiogenesis [10], [11], vascular maturation [12], [13], heart development [14] and immunity [15], [16] by binding specific G-protein-coupled S1P receptors. Red blood cells and endothelial cells are important sources of circulating S1P [17]–[19]. The role of Spns2 in regulating S1P signalling is still elusive.
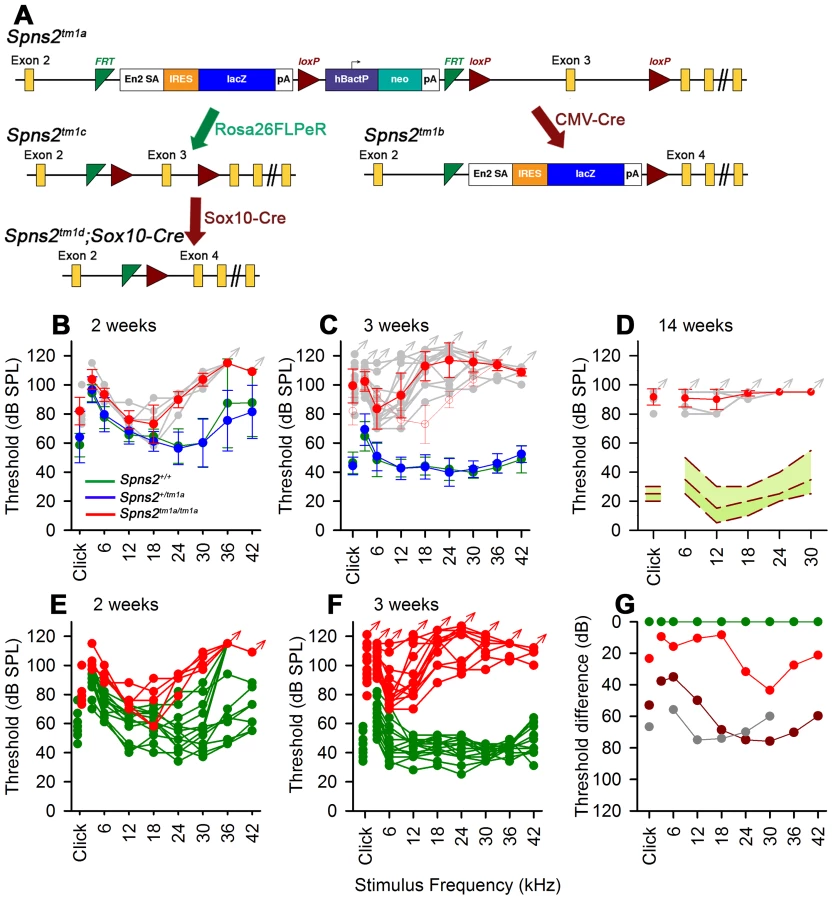
Spns2-deficient mice were initially discovered to be deaf during a large-scale screen of new mouse mutants carried out by the Sanger Institute's Mouse Genetics Project (MGP). The MGP uses the KOMP/EUCOMM resource of over 15,000 genes targeted in embryonic stem (ES) cells and aims to generate new mutants and screen them for a wide range of diseases and traits to reveal the function of 160 mutant genes each year [20]. Screening of hearing using the Auditory Brainstem Response (ABR) is part of the standardised battery of primary phenotypic tests and is carried out at 14 weeks of age [20]. Mutants generated from the KOMP/EUCOMM ES cell resource normally carry LoxP and Frt sites (Fig. 1A) engineered to facilitate further genetic manipulation to generate the conditional allele and then to knock out gene expression selectively [21].
Spns2-deficient mice showed early onset of hearing loss that progressed rapidly to profound deafness. This was associated with declining endocochlear potential (EP), which appeared to be the primary physiological defect. At later stages we observed degeneration of sensory hair cells and decreased expression of several key genes required for normal generation of the EP in the lateral wall of the cochlea, but these appeared to be secondary effects. By producing and analysing different conditional knockouts, we established that Spns2 expression was required locally in the inner ear rather than systemically. Our study suggests a vital role for Spns2 and S1P signalling in hearing.
Results
Spns2 gene targeting and mouse production
The introduction of a cassette with an additional splice acceptor site is predicted to interrupt normal transcription of the Spns2 gene (Fig. 1A) and generate a truncated non-functional transcript encoding the first 146 out of 549 amino acids of the Spns2 protein [4]. The Spns2tm1a/tm1a mice were fertile and can survive to adulthood, but were born at sub-Mendelian ratios (15.9% homozygotes among 747 offspring of heterozygous intercrosses; χ2 test, p<0.001). Quantitative real-time PCR revealed that residual transcript of Spns2 in cochleae, eyes and livers of the homozygous mice was substantially reduced compared to that of the heterozygous and wildtype mice (Fig. 2A). In order to completely inactivate expression of the Spns2 gene, we produced Spns2tm1b/tm1b mice by crossing Spns2tm1a/tm1a with mice ubiquitously expressing Cre recombinase to delete exon 3 in the germline. Spns2tm1b/tm1b mice were also fertile and can survive to adulthood with a birth rate at sub-Mendelian ratios (16.9% homozygotes among 266 offspring of heterozygous intercrosses; χ2 test, p = 0.0044). In other aspects of their phenotype, Spns2tm1b/tm1b mice were broadly similar to Spns2tm1a/tm1a mice (see http://www.sanger.ac.uk/mouseportal/search?query=spns2 for a comparison of the two lines). Spns2tm1a/tm1a mice were the first to be available and were used for most experiments in this study, and may be more relevant to human disease because most disease-causing mutations reduce rather than eliminate gene activity.

Spns2tm1a/tm1a mice have early onset progressive hearing impairment
We found that Spns2tm1a/tm1a mice had profound hearing impairment during screening at 14 weeks old by auditory brainstem response (ABR) measurement (Fig. 1D). Spns2tm1a/tm1a mice displayed no overt signs of abnormal behaviour throughout life up to 12 months old (n = 20 homozygotes, 29 control littermates) suggesting normal vestibular function. ABR measurements were recorded at younger ages to find out the time of onset of hearing loss. Hearing impairment in Spns2tm1a/tm1a mice can be detected as early as 2 weeks of age at high frequencies (Fig. 1B, E, G), and thresholds were raised further at a wide range of frequencies by 3 weeks old (Fig. 1C, F, G). The mutant thresholds recorded at P21 were significantly elevated compared with mutant thresholds at P14, indicating progressive hearing loss (Kruskall-Wallis One-Way Analysis of Variance on ranks, H = 102.857, 17 degrees of freedom, p<0.001). There was no difference in ABR thresholds between the Spns2+/tm1a and Spns2+/+ mice, indicating recessive inheritance. Thresholds continue to improve in control mice from 2 to 3 weeks old as hearing function matures [22].
Spns2 is expressed in the inner ear
Quantitative real-time PCR showed that Spns2 is expressed in the cochlea, both in the lateral wall and organ of Corti, and in eyes and liver in P4 wildtype mice (Fig. 2A). X-gal staining was used to indicate expression domains of Spns2, benefiting from the knockout first design of the allele which includes a LacZ reporter gene (Fig. 1A). At P10, X-gal labeling was detected in the spiral prominence area (Fig. 2C), hair cells (Fig. 2D), Reissner's membrane (Fig. 2B), vessels of inner ear (Fig. 2E) including the spiral modiolar vessels (Fig. 2F), proximal auditory nerve and bony shell of the cochlea (Fig. 2B), and the stria vascularis close to where the Reissner's membrane attaches. There was a similar labelling pattern at P14. The X-gal staining was also detected in the maculae and cristae in the vestibular system (Fig. S1A,B). On the basis of the expression pattern, our further investigation focused on two key components of the inner ear: the organ of Corti, where the pressure wave is transduced into action potentials, and the lateral wall, which maintains the ionic homeostasis of the cochlear endolymph.
Progressive degeneration of organ of Corti
The Spns2tm1a/tm1a mice showed a normal gross morphology of the middle ear and ossicles assessed by dissection and gross inspection, and the cleared inner ears also showed no malformation (Fig. S1C,D). We performed scanning electron microscopy (SEM) of P4, P21, P28 and P56 Spns2tm1a/tm1a mice and littermate controls. The hair cells of Spns2tm1a/tm1a mice appeared normal at P4 (Fig. S1E,F) and at P21 (Fig. 3A,B). There was scattered or patchy degeneration of stereocilia of outer hair cells in the homozygous cochleae at P28 (Fig. 3C,D). Hair cell degeneration became more apparent over time and by P56, only a few outer hair cells remained at the apex with most of them missing in other turns, and inner hair cells showed signs of degeneration such as fused stereocilia (Fig. 3E,F).

We also used transmission electron microscopy to examine the cochlear duct at P28, and observed degeneration of the basal turn organ of Corti and an apparently reduced density of dendrites in Rosenthal's canal in that turn (see later).
The increase in ABR thresholds preceded degeneration of the organ of Corti suggesting that these were secondary changes rather than the primary cause of the hearing impairment.
Decreased endocochlear potential (EP) in Spns2tm1a/tm1a mice
The stria vascularis is responsible for pumping K+ into the endolymph and generation of the endocochlear potential (EP) [23]. The EP starts to develop at around P6 in the mouse reaching adult values around P16 [24] and plays a key role in sound transduction because it provides approximately half of the electrochemical gradient that drives cations from the endolymph, a K+-rich extracellular fluid, into the sensory hair cells through mechanoelectrical transduction channels. The lateral wall of the cochlea is composed of the stria vascularis, spiral prominence and the spiral ligament. A defect in the function of any of these components could interfere with the generation of the EP. Therefore, we measured the EP to evaluate the function of the lateral wall. The EP values of the control mice were normal, around 99 to 120 mV, which matched their normal hearing. Spns2tm1a/tm1a mice had abnormally low EP values of 2 to 41 mV at both P21 and P28 when they were profoundly deaf (Fig. 4). However, at P14 the EP was higher, ranging from 52 to 107 mV, with some measurements within the normal range, corresponding to the partially preserved hearing at that age (Fig. 1C). The EP appeared to develop to near-normal levels and then declined very rapidly between P14 and P21. These data indicate that the cause of hearing loss in Spns2tm1a/tm1a mice is a failure to maintain the normal level of the EP after it develops, suggesting that the primary lesion is more likely to be in the lateral wall than the organ of Corti.

Structural defects of the stria vascularis
In order to understand what causes the reduction in the EP, we investigated the lateral wall, where the EP is produced. Generation of a voltage difference requires efficient separation of different compartments within the cochlear duct with adequate electrical resistance. Therefore we examined the morphology of cell boundaries between adjacent marginal cells and between basal cells in whole-mount samples of the stria vascularis. Filamentous actin was stained by phalloidin to label the cell boundaries at different ages. At P14, both wildtype and homozygous mice showed a distinctive regular hexagonal pattern of the boundaries of marginal cells (Fig. 5A,B). At P28, a subtle change was seen in Spns2tm1a/tm1a mice (Fig. 5C) and it became worse with age with a patchy pattern of different layouts of cell boundaries (Fig. 5D,E). However, the boundaries were always continuous and intact in Spns2tm1a/tm1a mice without any sign of breakdown (Fig. 5C–E). We quantified the marginal cell numbers by using the labelled cell boundaries at P28. The density of marginal cells in Spns2tm1a/tm1a mice was reduced compared with controls (t test, p<0.05), associated with irregular layout of marginal cell boundaries (Fig. 5J). Noticeably, this irregularity of marginal cells was not detected in mutants at P14, a stage when hearing had started to deteriorate, but appeared at later stages. The boundaries between basal cells of the stria vascularis did not show any obvious anomalies in mutants (Fig. S2A,B).

The morphology of strial capillaries was examined as well. Some Spns2tm1a/tm1a mice showed slight dilation (2 out of 5 mice) in patches along the length of the cochlear duct, and apparently increased branching (Fig. 5H) at P14. At P28, these changes were detected in all five tested mice and were more severe than at P14 although were still patchy (Fig. S2C,D). The number of branch points per unit area in homozygotes was significantly more than that in the control mice (Mann-Whitney Rank Sum Test, p<0.05 at both P14 and P28; Fig. 5I).
We analysed the structure of the lateral wall of the cochlea, including the stria vascularis and spiral ligament, using semithin sections and transmission electron microscopy in P28 mice. The position of Reissner's membrane was normal in semi-thin sections of P28 cochleae (Fig. 6A,B), with no evidence of hydrops or collapse. No systematic differences in the appearance of fibrocytes of the spiral ligament were observed (Fig. 6C,D). The inner boundaries of marginal cells of the stria, facing the endolymph, have a typical scallop-shaped surface in wildtype mice with the junctions between adjacent cells raised, but this feature was not seen in the Spns2tm1a/tm1a mice and the luminal surface appeared flat (Fig. 6E,F). Nuclei of marginal and basal cells appeared more rounded in Spns2tm1a/tm1a mice than in wildtypes (Fig. 6E,F). There was also a marked difference in the appearance of endothelial cells and pericytes [25], [26] of strial capillaries, with the nuclei of mutant cells appearing more darkly-stained (Fig. 6G,H). However, this abnormality appeared to be limited to capillaries of the stria vascularis only, and was not seen in the spiral ligament capillaries (Fig. 6I), suggesting a specific effect of Spns2 deficiency on the capillaries of the stria vascularis.

Intermediate cells of the stria vascularis are derived from melanocytes and tend to accumulate pigment during ageing or under stress such as in mice with Pendrin deficiency [27], [28]. The pigmented cells may derive from migratory melanocytes that adopt macrophage-like features during development [27], [29] or may derive from macrophage invasion [28]. At 4 weeks of age, we did not observe any obvious difference in strial pigmentation between Spns2 mutants and control littermates, but by 7 months old the mutant strias appeared more strongly pigmented than controls (Fig. S2E,F). This timecourse, after the onset of raised thresholds, suggests the accumulation of pigment is likely to be a secondary effect, not a cause of cochlear dysfunction.
Normal strial integrity and normal permeability of strial capillaries to BSA-FITC
In view of the abnormal morphology of marginal cell boundaries, we asked whether the diffusion barrier of stria vascularis, for example between adjacent marginal cells, was affected because normal morphology of boundaries at P14 does not necessarily mean normal function. We used biotin as a tracer injected into the endolymphatic and perilymphatic compartments of 6 week old mice to test the barrier permeability of the stria vascularis. There was no evidence of biotin entry into the stria vascularis of Spns2tm1a/tm1a or control mice indicating a normal diffusion barrier of stria vascularis (Fig. 7A,B). As we observed dilated strial capillaries with abnormal endothelial cells and pericytes, we tested their permeability by injecting BSA-FITC into the tail vein. There were no signs of leakage of the tracer to the tissues surrounding the strial capillaries in Spns2tm1a/tm1a mice suggesting that they have normal permeability to BSA-FITC (Fig. 7C,D).

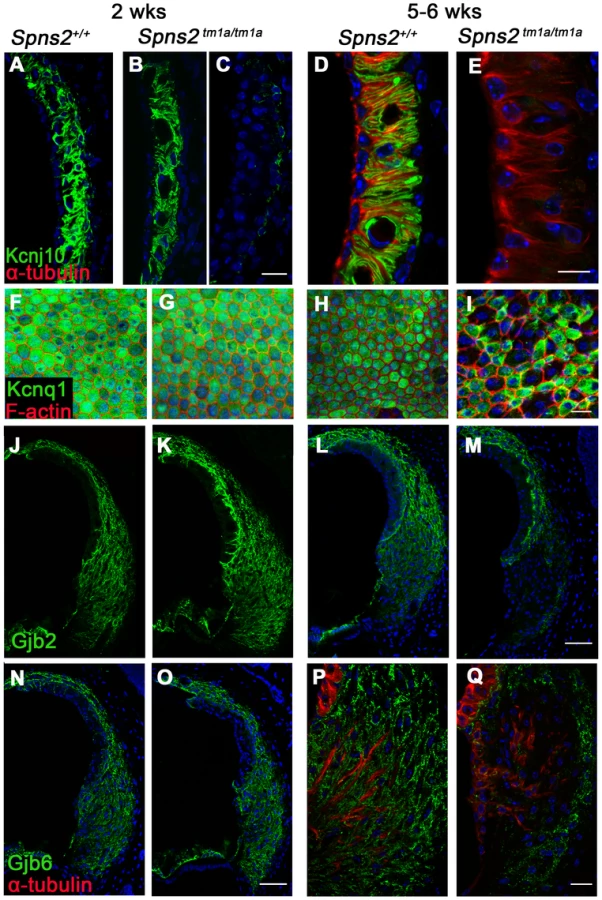
Decreased expression of Kcnj10, Kcnq1, Gjb2 and Gjb6 in lateral wall
To further investigate the reasons underlying the reduced EP and deafness in the Spns2 tm1a/tm1a mice, we analysed expression of some key proteins involved in normal EP formation and maintenance by immunofluorescence labelling, including Kir4.1 (Kcnj10), Kv7.1 (Kcnq1), Cx26 (Gjb2), Cx30 (Gjb6), Na+, K+-ATPase (Atp1a1), NKCC1 (Slc12a2) and ZO-1 (Tjp1). In homozygotes aged P14 (Fig. 8F,G,J,K,N,O), the expression of these proteins appeared normal, apart from the expression of Kcnj10, which appeared normal in most mutants (5/8), with the remaining three mice showing reduced labelling in the basal turn only (Fig. 8A–C). At 5–6 weeks old, we observed similar expression of Na+/K+-ATPase, NKCC1 and ZO-1 in Spns2 tm1a/tm1a mice compared with control mice (Fig. S3A–F). However, the other four proteins showed reduced expression at this age and the expression of Kcnj10 was largely absent in the stria vascularis (Fig. 8D,E). In contrast, there was similar labelling intensity of Kcnj10 in the satellite cells of the spiral ganglion in mutants and control mice (Fig. S3G,H). The expression of Kcnq1 appeared to be evenly distributed on the luminal surface of marginal cells in both wildtype and homozygotes at P14 (Fig. 8F, G), but at 5–6 weeks, labelling of Kcnq1 in the homozygotes was absent in some of the marginal cells that had enlarged boundaries (Fig. 8H,I). There was extensive expression of Gjb2 and Gjb6 in fibrocytes type I, II, and V of the spiral ligament in the wildtype (Fig. 8J,L,N,P) and homozygotes (Fig. 8K,O), but expression appeared greatly reduced behind the spiral prominence area corresponding to the fibrocyte type II region [30] in the Spns2tm1a/tm1a mice (Fig. 8M,Q) aged 5–6 weeks old. As EP started to reduce at P14, while expression of Kcnj10 (in the majority of observed mice), Kcnq1, Gjb2 and Gjb6 appeared normal at this age, these findings suggest that the reduction in expression of these key proteins in adults is secondary to a primary dysfunction of EP generation.
Conditional knock-out of Spns2 suggests a local function
We generated the conditional allele of Spns2 (Spns2tm1c) by crossing the Spns2tm1a allele to a line expressing Flp recombinase to excise the inserted cassette (Fig. 1A). The Spns2tm1c/tm1c mice have the same Spns2 allele as the wildtype except that exon 3 is flanked by two loxP sites. Spns2tm1c/tm1c mice showed normal ABR thresholds and normal morphology of hair cells (Fig. 9A, C). These observations confirmed that the inner ear defects we found in the Spns2tm1a/tm1a mice were due to the insertion of the cassette and its disruption of Spns2 gene function.

We then asked whether the hearing defects of Spns2tm1a/tm1a mice are caused by localised deficiency of Spns2 in the inner ear or are mediated systemically. S1P is known to be released from several other tissues that could affect cochlear function, including various blood cell types and endothelial cells [1], [2]. We generated conditional knockout mice carrying the Spns2tm1d allele in specific tissues by crossing mice carrying the Spns2tm1c allele with mice carrying Cre recombinase under the control of five different promoters: Tie1, Pf4, Lyve1, EpoR and Sox10, driving expression of Cre recombinase in blood vessel endothelial cells, platelets, lymphatic endothelial cells, red blood cells, and the inner ear with surrounding neural crest-derived mesenchyme respectively. Sox10-Cre transgenic mice have been successfully used to express Cre recombinase in the developing inner ear previously [31]. Homozygous Spns2tm1d mutants carrying the Tie1, Pf4, Lyve1 and EpoR Cre alleles all had normal ABR thresholds in young adults (Fig. 10). In contrast, no ABR response was detected in the young adult Spns2tm1d/tm1d;Sox10-Cre mice (Fig. 10). Spns2tm1d/tm1d; Sox10-Cre mice showed a similar pattern of progression of raised thresholds between 2 and 3 weeks old as observed in Spns2tm1a/tm1a mice (Fig. 11). Spns2tm1d/tm1d; Sox10-Cre mice also showed similar inner ear pathological changes as found in Spns2tm1a/tm1a mice, such as degeneration of hair cells (Fig. 9B, D) and irregular arrangement of marginal cell boundaries. Therefore, we propose that Spns2 plays an important role in mammalian hearing through its localised function in the inner ear.


The Spns2tm1b/tm1b mutants, with deletion of exon 3 of Spns2, showed a more severe increase in thresholds than the Spns2tm1a/tm1a and Spns2tm1d/tm1d;Sox10-Cre mice from 2 weeks old, the earliest stage studied, with a lack of detectable response at most frequencies in most mice up to the maximum stimulus intensity used, 95 dB SPL (Fig. 11, middle row).
Eye defects
Spns2tm1a/tm1a mice had other defects detected by the MGP phenotyping pipeline such as low white blood cell count and increased bone mineral density [4], [20], [32]. However, the eye defects were of particular interest because of the relatively common association of retinal defects with deafness, as in Usher syndrome for example, and our finding of Spns2 expression in the eye (Fig. 2A). We assessed the retina for features corresponding to those found in the organ of Corti, and found focal degeneration of the retina (Fig. 12D,E). As focal degeneration can be associated with a Crb1rd8 mutant allele found in some C57BL/6 lines [33], we sequenced this gene and confirmed that the Spns2 retinal phenotype was independent of the rd8 mutation. As the Spns2tm1a/tm1a stria vascularis capillaries showed abnormal morphology, we examined the retinal vasculature in whole mount preparations. Retinal vein morphology also appeared abnormal with some veins appearing thinner in mutants than in controls as well as veins of irregular caliber (Fig. 12F–I). This retinal vascular phenotype was first evident by P10 when the retinal vasculature was still undergoing development, and persisted into adulthood. We therefore undertook branchpoint analysis to quantify any differences between Spns2tm1a/tm1a and Spns2+/tm1a mice. This showed no difference between genotypes at P10 (Fig. 12J). We also analysed the pericyte coverage of the retinal vessels, as a reduction in pericyte coverage is associated with increased vascular permeability. We found no significant difference in pericyte coverage in peripheral vessels, and a small but significantly reduced coverage in central vessels (Fig. S4A). The vitreous and optic nerve appeared normal in the mutants (Fig. 12D,E).

Other eye defects included open eyelids at birth (Fig. S4B) resulting in corneal opacity, vascularization and ulceration (Fig. 12A,B,C). These corneal defects made retinal assessment by ophthalmoscopy impossible in vivo. Histological examination showed corresponding gross morphological defects. Eyes were smaller, the cornea was thickened with vascularisation, the anterior chamber was collapsed, and the lens was small and cataractous (Fig. 12D,E). We observed the anterior eye defects in the Spns2tm1a/tm1a and Spns2tm1b/tm1b mice, but these defects were not seen in Spns2tm1d/tm1d;Sox10-Cre mice or any of the other four conditional lines.
Discussion
Here, we report that Spns2-deficient (Spns2tm1a/tm1a) mice have profound hearing loss and propose an underlying mechanism: a rapid decline in EP paralleling loss of auditory sensitivity and preceding degeneration of hair cells, suggesting that the primary lesion is in the cochlear lateral wall, the site of EP production and maintenance [23], [34], [35]. Reduced EP has been associated with ion transport defects [36]–[39]; defects of tight junctions [40] or gap junctions [41]–[43]; absence of melanocytes [24]; microvascular disease [44], [45]; abnormal spiral ligament development [35]; sphingomyelin metabolic disturbance [46], or the lateral wall can simply be a target in systemic diseases [47]. This suggests complexity underlying the strial/metabolic category of hearing loss described in humans [48]. EP is essential for hair cell function [49]. Degeneration of hair cells secondary to reduced EP has been reported in other mouse mutants [50], [51] and normal EP seems to be important for survival of the hair cells. However, as Spns2 is expressed in hair cells as well as in the lateral wall, we cannot exclude the possibility that disruption of Spns2 function in the organ of Corti also contributed to raised ABR thresholds and hair cell degeneration. Analysis of mice with conditional knockout of Spns2 in hair cells and other cochlear cell types will be useful in dissecting the role of Spns2 further.
Spns2 acts as a transporter of S1P [1]–[6]. S1P may modulate vascular tone [52] and has been shown to regulate the inner ear spiral modiolar artery tone in vitro [53], [54]. S1P-induced vasoconstriction is thought to be important to protect strial capillary beds from high pressure [54]. We found that Spns2 was expressed in blood vessels of the inner ear including spiral modiolar vessels. Any reduction in local S1P level due to Spns2 dysfunction may weaken vasoconstriction and explain the dilation of strial capillaries in Spns2tm1a/tm1a mice. The relationship between capillary size and EP value is not unidirectional; both smaller and larger strial capillaries have been reported in different mouse mutants with low EP [55], [56]. S1P signalling also can affect vascular permeability [57]–[59]. However, we did not see increased permeability of strial capillaries using BSA as a tracer. Recently, Mendoza and colleagues found little difference in lung vascular permeability between Spns2-deficient and control animals [2], similar to our finding in strial capillaries of Spns2tm1a/tm1a mice. BSA is a medium molecular mass tracer (66.4 kDa), so tracers of different sizes and properties such as Evans blue and cadaverine [29] may be useful for further investigation of the strial capillary barrier.
We found decreased expression of several proteins critical for normal EP production at 5–6 weeks of age in Spns2-deficient mice. However, the expression of most of these proteins appeared normal at the time when the EP has already started to decline at P14, suggesting that these are likely to be secondary effects. The morphological changes of marginal cell boundaries and reduction in marginal cell density together with a lack of expression of Kcnq1 in affected cells are also likely to be secondary changes because these features were normal when hearing started to deteriorate at P14 and they did not affect strial permeability. Loss of Kcnq1 expression in marginal cells with expanded luminal surfaces may be a common consequence of strial dysfunction because it has been reported in several different mutants with reduced EP [28], [46]. It has been suggested that these common changes in mutant marginal cells may occur because these cells are relatively sensitive to metabolic stress [60], [61]. The variable decrease in Kcnj10 labelling in the basal turn and slightly dilated strial capillaries in a few mutants at P14 were the earliest abnormalities we have seen, and may correlate with the variable reduction in EP values at the same age. Disrupted expression of Kcnj10 is another common consequence of strial dysfunction [37], [47], [62]. The causal direction between reduced EP and decreased Kcnj10 expression in Spns2-deficient mice needs further investigation.
The most robust labeling for Spns2 expression was consistently in the hair cells and spiral prominence. The function of the spiral prominence is unclear. Two types of voltage-dependent K+ currents are expressed in spiral prominence epithelial cells, which may play a role in the homeostasis of inner ear fluids [63]. Another gene expressed strongly in the spiral prominence is pendrin (Pds), and the Pds mutant mouse also shows reduced EP [64], loss of expression of Kcnj10 [37] and Kcnq1 [28], and increased accumulation of strial pigmentation compared with controls [28]. However, the Pds mutant shows a severe early developmental defect of the inner ear with extensive hydrops [65], which we do not find in Spns2 mutants. This indicates that Spns2-deficient and Pds-deficient mice may have different mechanisms underlying the reduced EP and hearing impairment. Dysfunction of the spiral prominence in Spns2-deficient mice may be the main trigger of reduction of the EP and a series of pathological changes in inner ears.
One later change we saw in the lateral wall was a localised decrease of Gjb2 and Gjb6 expression in the type II fibrocytes of the spiral ligament located adjacent to the spiral prominence. Type II fibrocytes are important for K+ recycling and are considered to mediate K+ translocation between the epithelial cell network of the organ of Corti and the fibrocyte network of the lateral wall, and to facilitate ion flow directed towards the stria vascularis [66]. Intriguingly, this reduction in expression was seen for Gjb2 and Gjb6 only, and no reduction in labelling was found of Atp1a1 and Slc12a2, two other proteins strongly expressed in type II fibrocytes [67], [68]. This may indicate a selective impact of Spns2 deficiency on Gjb2 and Gjb6 expression in nearby cells, or alternatively these two genes may be more sensitive to changes in homeostasis in the cochlear duct than Atp1a1 and Slc12a2.
S1P is a bioactive lipid and acts as a second messenger intracellularly and as a ligand for cell surface G protein-coupled receptors extracellularly [69]. Five different S1P receptors participate in cellular responses based on the cell type and available downstream effectors [70]. S1P signalling has been implicated in maintenance of hair cells via activation of S1P receptor 2 (S1PR2) [71]. S1pr2-null mice are deaf and share some pathological changes with Spns2-deficient mice, such as disorganized cell boundaries of marginal cells, dilated capillaries in the stria vascularis, and degeneration of the organ of Corti [54], [71], [72]. Unlike S1pr2-null or S1pr2/S1pr3 double null mice, no overt vestibular defects were found in Spns2-deficient mice. Thus, we propose that the Spns2-S1P-S1PR2 signalling axis is important for normal hearing function. A similar Spns2-S1P-S1PR2 signalling axis may exist in bones as both S1pr2-deficient [73] and Spns2-deficient [32] mice have strong but brittle bones with high bone mineral density. In contrast, the Spns2-S1P-S1PR1 signalling axis is more important for lymphocyte trafficking [1]–[5].
Systemic disruption of Spns2 function in blood vessel or lymphatic endothelial cells, platelets or red blood cells did not affect hearing, suggesting that systemic loss of Spns2 activity in these tissues does not mediate the hearing loss we see in the Spns2tm1a mutants. However, when we deleted Spns2 locally in the inner ear using the Sox10-Cre recombinase, the resulting mutants were deaf. Sox10 is expressed throughout the otic epithelium from an early stage of development as well as in cranial neural crest-derived cells, so can effectively drive deletion of exon 3 of the Spns2tm1c allele in the entire inner ear [74], [75]. These findings indicate that hearing loss in Spns2tm1a/tm1a mice is due to local loss of Spns2 function in the inner ear.
Defects of the anterior eye were only seen in the Spns2tm1a and Spns2tm1b homozygous mutants. We did not see anterior eye defects in any of the 5 conditional alleles, consistent with normal anterior eye development in another conditional Spns2;Tie2-Cre mutant mouse [1]. The anterior eye phenotype appears to be due to a developmental abnormality resulting in defective eyelid formation and subsequent corneal opacity and vascularisation. Spns2 also plays a role in retinal blood vessels. Our results showed that global Spns2 knockout resulted in a mild phenotype of the retinal vasculature (thin and irregular veins) with decreased pericyte coverage in the central retina which may be related to the widely known role of S1P signalling in angiogenesis [76], [77]. The milder vascular phenotype in the retina than in the cochlea may be due to differences in the requirement for Spns2 in these tissues. We also found focal retinal degeneration in these mutant eyes suggesting a role for Spns2 in the photoreceptor and/or retinal pigment epithelium. Taken together, these findings suggest that SPNS2 is not only a candidate gene for involvement in deafness, but also for deaf-blind syndromes.
In summary, we report here that Spns2-deficient mice displayed rapidly progressive hearing impairment associated with a rapid decline in the EP between P14 and P21. The mechanism by which Spns2 deficiency leads to decreased EP merits further investigation, but it most likely involves local S1P signalling. Following the early drop in the EP, later changes include reduced expression of key proteins involved in cochlear homeostasis and ultimately sensory hair cell loss. Our findings suggest that Spns2 is a promising candidate gene for human deafness. Furthermore, Spns2-deficient mice may serve as a model to learn more about the role of S1P signalling in auditory function and the mechanism underlying at least one form of strial hearing loss.
Materials and Methods
Production and genotyping of Spns2-deficient and conditional knockout mice
Mouse studies were carried out in accordance with UK Home Office regulations and the UK Animals (Scientific Procedures) Act of 1986 (ASPA) under a UK Home Office licence, and the study was approved by the Wellcome Trust Sanger Institute's Ethical Review Committee. Mice were culled using methods approved under this licence to minimize any possibility of suffering. The mice were maintained in individually-ventilated cages at a standard temperature and humidity and in specific pathogen-free conditions. Either sex was used for this study.
The Spns2 mutant allele we used carries a promoter-driven cassette designed to interrupt normal gene transcription but flanked by Frt sites to enable its removal and conversion to a conditional allele with a critical exon surrounded by LoxP sites (a knockout-first design; [21], [78]). The allele is designated Spns2tm1a(KOMP)Wtsi, abbreviated to Spns2tm1a in this study. A schematic of the knockout-first design of Spns2tm1a allele is shown in Fig. 1A. The mutant mice were generated by blastocyst injection of the targeted ES cell using standard techniques [20], [21] and germ line transmission of Spns2tm1a was confirmed by a series of genotyping PCR analyses [79].
The Spns2tm1a colony was maintained on a mixed genetic C57BL/6BrdTyrc-Brd;C57BL/6Dnk;C57BL/6N background. Spns2tm1a/tm1a mice were crossed to HprtTg(CMV-Cre)Brd/Wtsi transgenic mice (on a C57BL/6NTac background) with systemic expression of Cre recombinase to remove the cassette and produce mice carrying the Spns2tm1b allele (Fig. 1A). Mice showing the correct excision were mated to wildtype C57BL/6N mice and offspring carrying the Spns2tm1b allele were mated to breed out the Cre allele and expand the colony. The Spns2tm1c allele was produced by crossing Spns2tm1a/tm1a mice to Gt(ROSA)26Sortm1(FLP1)/Wtsi mice expressing Flp recombinase ubiquitously in which the promoter-driven cassette was excised and exon 3 was retained flanked by LoxP sites (Fig. 1A). All the genotyping PCR primers and product sizes are shown in Table 1. Lack of the rd8 mutant allele was confirmed by conventional sequencing of the Crb1 gene [33].

Spns2tm1c/tm1c mice were mated to Sox10-Cre mice (Tg(Sox10-cre)1Wdr) [31] to delete the floxed exon 3 and to generate a frameshift mutation of Spns2 in the inner ear and craniofacial neural crest-derived tissues [31]. Genotyping was carried out using genomic DNA extracted from pinna tissue, which was mosaic under this conditional knockout strategy, and the conditional Spns2tm1d allele was confirmed by co-presence of Spns2tm1c and Sox10-Cre allele PCR bands. Since S1P is released from different blood cells and endothelial cells, we used mice expressing Tie1-Cre [80], Pf4-Cre [81], Lyve1-Cre [82] and EpoR-Cre [83] to inactivate the Spns2 gene in blood vessel endothelial cells, platelets, lymphatic endothelial cells and red blood cells respectively by crossing with Spns2tm1c/tm1c mice to produce the conditional knockouts.
Real-time PCR
The organ of Corti, lateral wall (stria vascularis and spiral ligament), eyes and livers of postnatal day (P)4 homozygous, heterozygous and wildtype littermate mice were dissected in RNAlater (n = 3 for each genotype). Total RNA was isolated with QIAshredder columns (QIAgen, cat. no. 79654) and the RNAeasy mini kit (QIAgen, cat. no. 74104). RNA was normalized to the same concentration for cDNA synthesis using oligo dT and SuperScrip II (Invitrogen). Real-time PCR was performed in triplicate for each sample using a CFX connect real time PCR machine (BIO-RAD). The Spns2 probe was designed to cover the 3′ untranslated region (Applied Biosystem). Hypoxanthine-guanine phospharibosyltransferase (Hprt) was amplified simultaneously (Applied Biosystem, Mm01318747_g1) as an internal reference. The relative quantity of Spns2 was calculated using the 2−ΔΔCt method [84].
Reporter gene visualisation
X-gal staining can be used to visualise the expression of Spns2 due to the LacZ gene inserted in the cassette of Spns2tm1a allele (Fig. 1A), downstream of the Spns2 promotor. Inner ears of P10 and P14 heterozygous and homozygous mice (at least three mice of each age group) were dissected out and fixed in 4% paraformaldehyde (PFA) for 45 minutes to 2 hrs. These were washed twice with PBS and decalcified in 10% EDTA until soft. After a PBS wash and immersing in 30% sucrose, inner ears were embedded in Agarose type VII (low gelling temperature, Sigma-Aldrich), then mounted using OCT compound ready for cryosectioning at 14 µm. Sections were treated with Solution A (2 mM MgCl2; 0.02% NP-40; 0.01% sodium deoxycholate; PBS) for 15 mins, then incubated with Solution B (Solution A plus 5 mM K3Fe(CN)6; 5 mM K4Fe(CN)6; 1 mg/ml X-Gal in DMSO) over night at 37°C. Sections were rinsed in water then counterstained in Fast Red to label nuclei, mounted and examined.
Auditory Brainstem Response (ABR)
Mice were anaesthetised by ketamine hydrochloride (100 mg/Kg, Ketaset, Fort Dodge Animal Health) and xylazine hydrochloride (10 mg/Kg, Rompun, Bayer Animal Health) and subcutaneous needle electrodes were inserted on the vertex (active), and over the left (reference) and right (ground) bullae. A calibrated sound system was used to deliver free-field click (0.01 ms duration) and tone pip (various frequencies from 6–30 kHz of 5 ms duration, 1 ms rise/fall time) stimuli at a range of intensity levels in 5 dB steps. Averaged responses to 256 stimuli, presented at 42.2 per second, were analysed and thresholds established as the lowest sound intensity giving a visually-detectable ABR response [85]. For P14 and P21 mice, in order to achieve higher sound pressure levels, sound was delivered to the external auditory meatus via a parabolic cone loud speaker attachment for click (0.01 ms duration) and tone pip stimuli (frequencies from 3–42 kHz of 5 ms duration in 3 dB SPL steps). Separate cohorts of P14 and P21 Spns2tm1a mice were used at the standard maximum intensity of 95 dB SPL with free field delivery as shown in Fig. 11 and at the higher sound intensities delivered near field, directly to the external auditory meatus in Fig. 1. The median ABR thresholds recorded in homozygous mutants at P14 and P21 shown in Fig. 1 were compared using the Kruskall-Wallis One-Way Analysis of Variance on Ranks, as thresholds did not show a normal distribution.
Scanning electron microscopy and gross morphology of ears
The temporal bones were isolated. The inner ears were dissected out and fixed by 2.5% glutaraldehyde in 0.1M sodium cacodylate buffer with 3 mM calcium chloride at room temperature for 3 hours. Cochleae were finely dissected in PBS. This was followed by further processing using an osmium-thiocarbohydrazide-osmium (OTOTO) method [86]. The samples were dehydrated in increasing concentrations of ethanol, critical-point dried (Bal-Tec CPD030), mounted and examined under a HITACHI S-4800 scanning electron microscope. At least 3 wildtype, heterozygous and homozygous mice were examined for each age group (P4, P21, P28 and P56). Middle ears were dissected and examined. Inner ears were cleared by a standard glycerol clearing technique and examined for gross structural defects (control, n = 5; homozygotes, n = 5; aged P30–34).
Semi-thin sections and transmission electron microscopy
Inner ears (wildtype, n = 2; heterozygotes, n = 2; homozygotes, n = 4, at P28) were dissected out and gently perfused with 2.5% glutaraldehyde, 1% paraformaldehyde in 0.1M sodium phosphate buffer with 0.8 mM calcium chloride through the round and oval windows and a small hole in the apex then fixed at room temperature for 2 hours. Secondary fixation was in 1% osmium tetroxide in sodium phosphate buffer for 1 hour. After 5 washes in 0.1 M sodium phosphate buffer, inner ears were decalcified in 0.1M EDTA at 4°C until soft. Then the samples were dehydrated through an ethanol series, staining in 2% uranyl acetate at the 30% ethanol stage, embedded in Epon resin mixed 1∶1 and then 3∶1 in propylene oxide for 30 min and infiltrated overnight under vacuum in neat resin. The samples were embedded at 60°C for 24–48 hours. 1 µm sections were cut through the modiolar plane and stained with toluidine blue for light microscope observation. 60 nm sections were cut on a Leica EM UC6 ultramicrotome, stained in 2% uranyl acetate and aqueous lead citrate and imaged on an FEI Spirit Biotwin 120 kV transmission electron microscope using a Tietz F4.15 CCD.
Endocochlear potential measurement
Mice were anaesthetized with 0.01 ml/g body weight of 20% urethane, a tracheal cannula was inserted and the bulla was opened to reveal the cochlea while the body temperature was kept at 37°C by a feedback-controlled heating pad. A small hole was made in the bony wall of the cochlea over the basal turn of scala media, and a micropipette electrode filled with 150 mM potassium chloride was advanced through the hole and through the lateral wall into the scala media. The potential difference between the scala media and a reference silver/silver chloride pellet under the dorsal skin was recorded [24].
Surface preparation of lateral wall and visualization of capillaries
The inner ears were rapidly dissected out and fixed in 4% paraformaldehyde at room temperature for 2 hours. The lateral walls were dissected out in PBS for surface preparation. Filamentous actin was visualized by rhodamine phalloidin (1∶200, Molecular Probe) at room temperature for 2 hours. Strial capillaries were visualized by Isolectin B4 (Vector Laboratories, 1∶50) at 4°C, overnight in PBS with 10% sheep serum). Samples were mounted with Vectashield Mounting medium (Vector, Cat. No: H-1000) and imaged by confocal microscopy (Carl Zeiss, LSM 510 META). The numbers of capillary branch points per field (220×220-µm fields) in the middle turn (40–70% of the distance along the cochlear duct from the base) of the stria vascularis (control, n = 4; homozygotes, n = 5, at P14. control, n = 3; homozygotes, n = 5, at P28) was quantified using image J. Data were presented as a density in a 100×100 µm field and statistics analysis was conducted using Mann-Whitney Rank Sum Test, SigmaPlot v12.0. Surface preparations were also used for analysis of Kcnq1 expression, using overnight incubation at 4°C with goat anti-Kcnq1 polyclonal antibody (Santa Cruz, 1∶200) followed by washing with PBS and incubation with donkey anti-goat secondary antibody (Invitrogen, 1∶500) prior to analysis by confocal microscopy. At least three homozygotes and three controls were used at P14, P28 and 6 months for phalloidin labeling to show marginal cell boundaries, and P14 and 5–6 weeks for Kcnq1 expression. The density of marginal cells was measured in the phalloidin-labelled whole mount preparations by counting the number of cells defined by their labeled boundaries in two areas each 100×100 µm from the middle turn (40–70%) of each cochlea (n = 4 homozygous mutants; 4 littermate controls).
Immunofluorescence labeling
The cochleae were dissected out and fixed in 4% PFA at room temperature for 2 hours. Cryosections were obtained as described above for X-gal staining. We used the following antibodies: rabbit anti-Kcnj10 polyclonal (Alomone labs, 1∶300), rabbit anti-Gjb2 polyclonal (from WH Evans, 1∶300), rabbit anti-Gjb6 polyclonal (Zymed, 1∶400), mouse anti - Na+, K+-ATPase (α1 subunit) monoclonal (Sigma, 1∶300), mouse anti - NKCC1 monoclonal (C. Lytle, Developmental Studies Hybridoma Bank, University of Iowa, 1∶300) and rabbit anti-ZO-1 polyclonal (Zymed, 1∶300). Sections were blocked by incubation with 10% sheep serum (with 0.1% TritonX-100 in PBS) for 40 mins. Sections were incubated with appropriate primary antibodies overnight at 4°C, washed with PBS and incubated with corresponding secondary antibodies at room temperature for 2 hours (donkey anti-rabbit, donkey anti-goat, Invitrogen, 1∶500). After washing with PBS, slides were imaged by confocal microscopy. Three mice of each genotype (Spns2tm1a/tm1a and Spns2+/+) were used for each antibody at P14 and 5–6 weeks old.
Stria vascularis tight junction permeability assessment
The inner ears (wildtype, n = 1; heterozygote, n = 3; homozygote, n = 4 at 6 weeks old) were dissected and round and oval windows were opened in PBS containing 1 mM calcium chloride. A hole was made in the basal turn leading to the scala media. The membranous labyrinth was perfused for 5 minutes with 400 µl Sulfo-NHS-LC-Biotin (Thermo Scientific, 10 mg/ml, in PBS with 1 mM calcium chloride) through the round and oval windows and the hole exposing the endolymphatic compartment. Following a PBS wash, the inner ears were fixed in 4% paraformaldehyde at room temperature for 2 hours, and processed for cryosectioning as described above. The biotin tracer was detected by fluorescein isothiocyanate (FITC)-conjugated streptavidin (Thermo Scientific, 1∶100) incubating at room temperature for 30 min. Samples perfused with PBS alone were used as negative controls.
Strial capillary permeability assessment
Mice (wildtype, n = 2; heterozygote, n = 2; homozygote, n = 2; at 5–6 weeks old) were warmed in a 39°C incubator for 5 minutes and then held in a mouse restrainer so that the tail was accessible and the tail vein visible. A 50 µl aliquot of 5% (w/v) FITC-conjugated bovine serum albumin (BSA-FITC; Sigma cat. no. A9771; size 66.4 kDa), made up in sterile PBS, was injected into the tail vein. The mice were left at room temperature for 45–60 minutes to allow the BSA-FITC to permeate all capillaries and to allow for any vascular extravasation. The mice were sacrificed by CO2 inhalation and the auditory bullae dissected out. Whole cochleae were exposed and fixed by removing a small piece of bone at the apex and gently perfusing 4% PFA through the round and oval windows. Cochleae were then immersed in fixative and left on a rotator for 1.5 hours at room temperature. Whole-mounts of stria vascularis were dissected from fixed cochleae, covered with Vectashield Mounting Media (Vector, Cat. No: H-1000) in glass bottom culture dishes (MatTek Corp.) and imaged using confocal laser-scanning microscopy (Carl Zeiss, LSM 510 META).
Ocular assessment
Mice underwent ophthalmic screening at 15 weeks of age. They were assessed for gross morphological changes to the eye using a slit lamp (Zeiss SL130) and ophthalmoscope (Heine Omega 500). The eye was examined both undilated and dilated (topical tropicamide). Images using the slit lamp were collected using a Leica DFC420 camera. The mice were culled under terminal anaesthesia followed by cervical dislocation and both eyes from 3 male homozygous mutants and 3 wildtype mice were removed and fixed. Pupil-optic nerve sections were processed, stained with hematoxylin and eosin, and standard images were captured under light microscopy for review [87].
For whole mount retinal analysis, heterozygotes and homozygotes were used (n = 3 for each genotype at P10, n = 2 at 8 weeks old) and the eyes removed and fixed in 4% PFA. Retinae were prepared and stained as described [88] using flourescein-conjugated Griffonia simplicifolia Isolectin B4 (Vector Laboratories, UK) to label blood vessels, mouse anti-proteoglycan NG2 (Millipore UK Ltd., UK) to label pericytes, and donkey anti-rabbit secondary Alexa-594 (Molecular Probes, Life Technologies, UK). The homozygote and control mouse retinae were stained in the same well to control for changes in staining efficiency, distinguishing the retinae by different numbers of radial incisions. All tissues were mounted in Vectashield (Vector Laboratories Ltd., Peterborough, UK), imaged by confocal microscopy (Nikon A1R; Nikon Instruments, Inc., Melville, NY), and maximum intensity projections of z-stacks were created using NisElements AR Version 4.0 software (Nikon UK, Kingston Upon Thames, UK). The MetaMorph Angiogenesis Tube Formation application (Molecular Devices, Berkshire, UK) was used for quantification. Confocal images were used to determine the total area covered by vessels and by pericytes to calculate the percentage pericyte coverage of vessels and the total number of capillary branchpoints per unit area. We imaged three areas of each retina: three different regions of the central retina each encompassing an artery and vein, and two images each of peripheral arteries and peripheral veins, a total of five images/retina. Threshold values were kept the same for analysis of samples of the same stage.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. FukuharaS, SimmonsS, KawamuraS, InoueA, OrbaY, et al. (2012) The sphingosine-1-phosphate transporter Spns2 expressed on endothelial cells regulates lymphocyte trafficking in mice. J Clin Invest 122 : 1416–1426.
2. MendozaA, BreartB, Ramos-PerezWD, PittLA, GobertM, et al. (2012) The transporter spns2 is required for secretion of lymph but not plasma sphingosine-1-phosphate. Cell Rep 2 : 1104–1110.
3. HisanoY, KobayashiN, YamaguchiA, NishiT (2012) Mouse SPNS2 functions as a sphingosine-1-phosphate transporter in vascular endothelial cells. PLoS One 7: e38941.
4. NijnikA, ClareS, HaleC, ChenJ, RaisenC, et al. (2012) The role of sphingosine-1-phosphate transporter spns2 in immune system function. J Immunol 189 : 102–111.
5. NagahashiM, KimEY, YamadaA, RamachandranS, AllegoodJC, et al. (2012) Spns2, a transporter of phosphorylated sphingoid bases, regulates their blood and lymph levels and the lymphatic network. FASEB J 27 : 1001–11.
6. KawaharaA, NishiT, HisanoY, FukuiH, YamaguchiA, et al. (2009) The Sphingolipid Transporter Spns2 Functions in Migration of Zebrafish Myocardial Precursors. Science 323 : 524–527.
7. ZhangH, DesaiNN, OliveraA, SekiT, BrookerG, et al. (1991) Sphingosine-1-phosphate, a novel lipid, involved in cellular proliferation. J Cell Biol 114 : 155–167.
8. OliveraA, SpiegelS (1993) Sphingosine-1-phosphate as second messenger in cell proliferation induced by PDGF and FCS mitogens. Nature 365 : 557–560.
9. CuvillierO, PirianovG, KleuserB, VanekPG, CosoOA, et al. (1996) Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature 381 : 800–803.
10. WangF, Van BrocklynJR, HobsonJP, MovafaghS, Zukowska-GrojecZ, et al. (1999) Sphingosine 1-phosphate stimulates cell migration through a G(i)-coupled cell surface receptor. Potential involvement in angiogenesis. J Biol Chem 274 : 35343–35350.
11. LeeMJ, ThangadaS, ClaffeyKP, AncellinN, LiuCH, et al. (1999) Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell 99 : 301–312.
12. LiuY, WadaR, YamashitaT, MiY, DengCX, et al. (2000) Edg-1, the G protein-coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. J Clin Invest 106 : 951–961.
13. GarciaJG, LiuF, VerinAD, BirukovaA, DechertMA, et al. (2001) Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J Clin Invest 108 : 689–701.
14. KuppermanE, AnS, OsborneN, WaldronS, StainierDY (2000) A sphingosine-1-phosphate receptor regulates cell migration during vertebrate heart development. Nature 406 : 192–195.
15. BrinkmannV, DavisMD, HeiseCE, AlbertR, CottensS, et al. (2002) The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J Biol Chem 277 : 21453–21457.
16. MandalaS, HajduR, BergstromJ, QuackenbushE, XieJ, et al. (2002) Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science 296 : 346–349.
17. VenkataramanK, LeeYM, MichaudJ, ThangadaS, AiY, et al. (2008) Vascular endothelium as a contributor of plasma sphingosine 1-phosphate. Circ Res 102 : 669–676.
18. PappuR, SchwabSR, CornelissenI, PereiraJP, RegardJB, et al. (2007) Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science 316 : 295–298.
19. VenkataramanK, ThangadaS, MichaudJ, OoML, AiY, et al. (2006) Extracellular export of sphingosine kinase-1a contributes to the vascular S1P gradient. Biochem J 397 : 461–471.
20. WhiteJK, GerdinAK, KarpNA, RyderE, BuljanM, et al. (2013) Genome-wide Generation and Systematic Phenotyping of Knockout Mice Reveals New Roles for Many Genes. Cell 154 : 452–464.
21. SkarnesWC, RosenB, WestAP, KoutsourakisM, BushellW, et al. (2011) A conditional knockout resource for the genome-wide study of mouse gene function. Nature 474 : 337–342.
22. SteelKP, BockGR (1983) Cochlear dysfunction in the jerker mouse. Behav Neurosci 97 : 381–391.
23. TasakiI, SpyropoulosCS (1959) Stria vascularis as source of endocochlear potential. J Neurophysiol 22 : 149–155.
24. SteelKP, BarkwayC (1989) Another role for melanocytes: their importance for normal stria vascularis development in the mammalian inner ear. Development 107 : 453–463.
25. AlltG, LawrensonJG (2001) Pericytes: cell biology and pathology. Cells Tissues Organs 169 : 1–11.
26. BergersG, SongS (2005) The role of pericytes in blood-vessel formation and maintenance. Neuro Oncol 7 : 452–464.
27. CableJ, SteelKP (1991) Identification of two types of melanocyte within the stria vascularis of the mouse inner ear. Pigment Cell Res 4 : 87–101.
28. JabbaSV, OelkeA, SinghR, MagantiRJ, FlemingS, et al. (2006) Macrophage invasion contributes to degeneration of stria vascularis in Pendred syndrome mouse model. BMC Med 4 : 37.
29. ZhangW, DaiM, FridbergerA, HassanA, DegagneJ, et al. (2012) Perivascular-resident macrophage-like melanocytes in the inner ear are essential for the integrity of the intrastrial fluid-blood barrier. Proc Natl Acad Sci U S A 109 : 10388–10393.
30. SpicerSS, SchulteBA (1991) Differentiation of inner ear fibrocytes according to their ion transport related activity. Hear Res 56 : 53–64.
31. MatsuokaT, AhlbergPE, KessarisN, IannarelliP, DennehyU, et al. (2005) Neural crest origins of the neck and shoulder. Nature 436 : 347–355.
32. BassettJH, GogakosA, WhiteJK, EvansH, JacquesRM, et al. (2012) Rapid-throughput skeletal phenotyping of 100 knockout mice identifies 9 new genes that determine bone strength. PLoS Genet 8: e1002858.
33. MattapallilMJ, WawrousekEF, ChanCC, ZhaoH, RoychoudhuryJ, et al. (2012) The Rd8 mutation of the Crb1 gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Invest Ophthalmol Vis Sci 53 : 2921–2927.
34. SaltAN, MelicharI, ThalmannR (1987) Mechanisms of endocochlear potential generation by stria vascularis. Laryngoscope 97 : 984–991.
35. MinowaO, IkedaK, SugitaniY, OshimaT, NakaiS, et al. (1999) Altered cochlear fibrocytes in a mouse model of DFN3 nonsyndromic deafness. Science 285 : 1408–1411.
36. MarcusDC, WuT, WangemannP, KofujiP (2002) KCNJ10 (Kir4.1) potassium channel knockout abolishes endocochlear potential. Am J Physiol Cell Physiol 282: C403–407.
37. WangemannP, ItzaEM, AlbrechtB, WuT, JabbaSV, et al. (2004) Loss of KCNJ10 protein expression abolishes endocochlear potential and causes deafness in Pendred syndrome mouse model. BMC Med 2 : 30.
38. RickheitG, MaierH, StrenzkeN, AndreescuCE, De ZeeuwCI, et al. (2008) Endocochlear potential depends on Cl - channels: mechanism underlying deafness in Bartter syndrome IV. Embo J 27 : 2907–2917.
39. NorgettEE, GolderZJ, Lorente-CanovasB, InghamN, SteelKP, et al. (2012) Atp6v0a4 knockout mouse is a model of distal renal tubular acidosis with hearing loss, with additional extrarenal phenotype. Proc Natl Acad Sci U S A 109 : 13775–13780.
40. KitajiriS, MiyamotoT, MineharuA, SonodaN, FuruseK, et al. (2004) Compartmentalization established by claudin-11-based tight junctions in stria vascularis is required for hearing through generation of endocochlear potential. J Cell Sci 117 : 5087–5096.
41. Cohen-SalmonM, OttT, MichelV, HardelinJP, PerfettiniI, et al. (2002) Targeted ablation of connexin26 in the inner ear epithelial gap junction network causes hearing impairment and cell death. Curr Biol 12 : 1106–1111.
42. Cohen-SalmonM, RegnaultB, CayetN, CailleD, DemuthK, et al. (2007) Connexin30 deficiency causes instrastrial fluid-blood barrier disruption within the cochlear stria vascularis. Proceedings of the National Academy of Sciences 104 : 6229–6234.
43. TeubnerB, MichelV, PeschJ, LautermannJ, Cohen-SalmonM, et al. (2003) Connexin30 (Gjb6)-deficiency causes severe hearing impairment and lack of endocochlear potential. Hum Mol Genet 12 : 13–21.
44. OhlemillerKK, RiceME, GagnonPM (2008) Strial microvascular pathology and age-associated endocochlear potential decline in NOD congenic mice. Hear Res 244 : 85–97.
45. OhlemillerKK (2009) Mechanisms and genes in human strial presbycusis from animal models. Brain Res 1277 : 70–83.
46. LuMH, TakemotoM, WatanabeK, LuoH, NishimuraM, et al. (2012) Deficiency of sphingomyelin synthase-1 but not sphingomyelin synthase-2 causes hearing impairments in mice. J Physiol 590 : 4029–4044.
47. MustaphaM, FangQ, GongTW, DolanDF, RaphaelY, et al. (2009) Deafness and permanently reduced potassium channel gene expression and function in hypothyroid Pit1dw mutants. J Neurosci 29 : 1212–1223.
48. SchuknechtHF, GacekMR (1993) Cochlear pathology in presbycusis. Ann Otol Rhinol Laryngol 102 : 1–16.
49. SteelKP (1995) Inherited hearing defects in mice. Annu Rev Genet 29 : 675–701.
50. GowA, DaviesC, SouthwoodCM, FrolenkovG, ChrustowskiM, et al. (2004) Deafness in Claudin 11-null mice reveals the critical contribution of basal cell tight junctions to stria vascularis function. J Neurosci 24 : 7051–7062.
51. SteelKP, BarkwayC, BockGR (1987) Strial dysfunction in mice with cochleo-saccular abnormalities. Hear Res 27 : 11–26.
52. LevkauB (2008) Sphingosine-1-phosphate in the regulation of vascular tone: a finely tuned integration system of S1P sources, receptors, and vascular responsiveness. Circ Res 103 : 231–233.
53. SchererEQ, LidingtonD, OestreicherE, ArnoldW, PohlU, et al. (2006) Sphingosine-1-phosphate modulates spiral modiolar artery tone: A potential role in vascular-based inner ear pathologies? Cardiovasc Res 70 : 79–87.
54. KonoM, BelyantsevaIA, SkouraA, FrolenkovGI, StarostMF, et al. (2007) Deafness and stria vascularis defects in S1P2 receptor-null mice. J Biol Chem 282 : 10690–10696.
55. CableJ, SteelKP (1998) Combined cochleo-saccular and neuroepithelial abnormalities in the Varitint-waddler-J (VaJ) mouse. Hear Res 123 : 125–136.
56. OhlemillerKK, LettJM, GagnonPM (2006) Cellular correlates of age-related endocochlear potential reduction in a mouse model. Hear Res 220 : 10–26.
57. CamererE, RegardJB, CornelissenI, SrinivasanY, DuongDN, et al. (2009) Sphingosine-1-phosphate in the plasma compartment regulates basal and inflammation-induced vascular leak in mice. J Clin Invest 119 : 1871–1879.
58. WangL, DudekSM (2009) Regulation of vascular permeability by sphingosine 1-phosphate. Microvasc Res 77 : 39–45.
59. McVerryBJ, PengX, HassounPM, SammaniS, SimonBA, et al. (2004) Sphingosine 1-phosphate reduces vascular leak in murine and canine models of acute lung injury. Am J Respir Crit Care Med 170 : 987–993.
60. SpicerSS, SchulteBA (2005) Pathologic changes of presbycusis begin in secondary processes and spread to primary processes of strial marginal cells. Hear Res 205 : 225–240.
61. OhlemillerKK, RiceME, LettJM, GagnonPM (2009) Absence of strial melanin coincides with age-associated marginal cell loss and endocochlear potential decline. Hear Res 249 : 1–14.
62. ChenJ, NathansJ (2007) Estrogen-related receptor beta/NR3B2 controls epithelial cell fate and endolymph production by the stria vascularis. Dev Cell 13 : 325–337.
63. LeeJH, KimSJ, JungSJ, LimW, KimKW, et al. (2000) Voltage-dependent K(+) currents in spiral prominence epithelial cells of rat cochlea. Hear Res 146 : 7–16.
64. RoyauxIE, BelyantsevaIA, WuT, KacharB, EverettLA, et al. (2003) Localization and functional studies of pendrin in the mouse inner ear provide insight about the etiology of deafness in pendred syndrome. J Assoc Res Otolaryngol 4 : 394–404.
65. EverettLA, BelyantsevaIA, Noben-TrauthK, CantosR, ChenA, et al. (2001) Targeted disruption of mouse Pds provides insight about the inner-ear defects encountered in Pendred syndrome. Hum Mol Genet 10 : 153–161.
66. SpicerSS, SchulteBA (1996) The fine structure of spiral ligament cells relates to ion return to the stria and varies with place-frequency. Hear Res 100 : 80–100.
67. SchulteBA, AdamsJC (1989) Distribution of immunoreactive Na+,K+-ATPase in gerbil cochlea. J Histochem Cytochem 37 : 127–134.
68. CrouchJJ, SakaguchiN, LytleC, SchulteBA (1997) Immunohistochemical localization of the Na-K-Cl co-transporter (NKCC1) in the gerbil inner ear. J Histochem Cytochem 45 : 773–778.
69. MaceykaM, HarikumarKB, MilstienS, SpiegelS (2012) Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol 22 : 50–60.
70. AdadaM, CanalsD, HannunYA, ObeidLM (2013) Sphingosine-1-phosphate receptor 2. FEBS J
71. HerrDR, GrilletN, SchwanderM, RiveraR, MullerU, et al. (2007) Sphingosine 1-phosphate (S1P) signaling is required for maintenance of hair cells mainly via activation of S1P2. J Neurosci 27 : 1474–1478.
72. MacLennanAJ, BennerSJ, AndringaA, ChavesAH, RosingJL, et al. (2006) The S1P2 sphingosine 1-phosphate receptor is essential for auditory and vestibular function. Hear Res 220 : 38–48.
73. IshiiM, KikutaJ, ShimazuY, Meier-SchellersheimM, GermainRN (2010) Chemorepulsion by blood S1P regulates osteoclast precursor mobilization and bone remodeling in vivo. J Exp Med 207 : 2793–2798.
74. BreuskinI, BodsonM, ThelenN, ThiryM, BorgsL, et al. (2009) Sox10 promotes the survival of cochlear progenitors during the establishment of the organ of Corti. Dev Biol 335 : 327–339.
75. WakaokaT, MotohashiT, HayashiH, KuzeB, AokiM, et al. (2013) Tracing Sox10-expressing cells elucidates the dynamic development of the mouse inner ear. Hear Res 302 : 17–25.
76. JungB, ObinataH, GalvaniS, MendelsonK, DingBS, et al. (2012) Flow-regulated endothelial S1P receptor-1 signaling sustains vascular development. Dev Cell 23 : 600–610.
77. MendelsonK, ZygmuntT, Torres-VazquezJ, EvansT, HlaT (2013) Sphingosine 1-phosphate receptor signaling regulates proper embryonic vascular patterning. J Biol Chem 288 : 2143–2156.
78. TestaG, SchaftJ, van der HoevenF, GlaserS, AnastassiadisK, et al. (2004) A reliable lacZ expression reporter cassette for multipurpose, knockout-first alleles. Genesis 38 : 151–158.
79. RyderE, GleesonD, SethiD, VyasS, MiklejewskaE, et al. (2013) Molecular Characterization of Mutant Mouse Strains Generated from the EUCOMM/KOMP-CSD ES Cell Resource. Mamm Genome 24 : 286–294.
80. GustafssonE, BrakebuschC, HietanenK, FasslerR (2001) Tie-1-directed expression of Cre recombinase in endothelial cells of embryoid bodies and transgenic mice. J Cell Sci 114 : 671–676.
81. TiedtR, SchomberT, Hao-ShenH, SkodaRC (2007) Pf4-Cre transgenic mice allow the generation of lineage-restricted gene knockouts for studying megakaryocyte and platelet function in vivo. Blood 109 : 1503–1506.
82. PhamTH, BalukP, XuY, GrigorovaI, BankovichAJ, et al. (2010) Lymphatic endothelial cell sphingosine kinase activity is required for lymphocyte egress and lymphatic patterning. J Exp Med 207 : 17–27.
83. HeinrichAC, PelandaR, KlingmullerU (2004) A mouse model for visualization and conditional mutations in the erythroid lineage. Blood 104 : 659–666.
84. LivakKJ, SchmittgenTD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25 : 402–408.
85. InghamNJ, PearsonS, SteelKP (2011) Using the auditory brainstem response (ABR) to determine sensitivity of hearing in mutant mice. Current Protocols in Mouse Biology 1 : 279–287.
86. Hunter-DuvarIM (1978) A technique for preparation of cochlear specimens for assessment with the scanning electron microscope. Acta Otolaryngol Suppl 351 : 3–23.
87. MahajanVB, SkeieJM, AssefniaAH, MahajanM, TsangSH (2011) Mouse eye enucleation for remote high-throughput phenotyping. J Vis Exp 2011: pii: 3184.
88. WestH, RichardsonWD, FruttigerM (2005) Stabilization of the retinal vascular network by reciprocal feedback between blood vessels and astrocytes. Development 132 : 1855–1862.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 10
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- The Master Activator of IncA/C Conjugative Plasmids Stimulates Genomic Islands and Multidrug Resistance Dissemination
- A Splice Mutation in the Gene Causes High Glycogen Content and Low Meat Quality in Pig Skeletal Muscle
- Keratin 76 Is Required for Tight Junction Function and Maintenance of the Skin Barrier
- A Role for Taiman in Insect Metamorphosis
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy