
An AGEF-1/Arf GTPase/AP-1 Ensemble Antagonizes LET-23 EGFR Basolateral Localization and Signaling during Vulva Induction
In the nematode, Caenorhabditis elegans, an evolutionarily conserved Epidermal Growth Factor Receptor (EGFR) signaling pathway is required to induce three epithelial cells to initiate a program of vulva development. EGFR on the basolateral membrane is essential to engage and transmit this signal. Here we demonstrate that AGEF-1 and the AP-1 clathrin adaptor complex function with two Arf GTPases to regulate EGFR localization and signaling. In humans, EGFR also localizes to the basolateral membrane of epithelial cells, and excessive EGFR signaling is a major driver of cancer. In C. elegans, we show that loss of AGEF-1 results in an increase in basolateral EGFR localization in the vulva precursor cells, and in sensitized genetic backgrounds, a corresponding increase in vulva induction. While the human AGEF-1 proteins, BIG1 and BIG2, have not been previously implicated in EGFR signaling and cancer, mutations in BIG2 are causal of periventricular heterotopia, a condition whereby neurons fail to migrate to the cerebral cortex during brain development. As migrating neurons require polarized protein localization, BIG2 and AGEF-1 may have similar functions in these polarized cell types.
Published in the journal:
. PLoS Genet 10(10): e32767. doi:10.1371/journal.pgen.1004728
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004728
Summary
In the nematode, Caenorhabditis elegans, an evolutionarily conserved Epidermal Growth Factor Receptor (EGFR) signaling pathway is required to induce three epithelial cells to initiate a program of vulva development. EGFR on the basolateral membrane is essential to engage and transmit this signal. Here we demonstrate that AGEF-1 and the AP-1 clathrin adaptor complex function with two Arf GTPases to regulate EGFR localization and signaling. In humans, EGFR also localizes to the basolateral membrane of epithelial cells, and excessive EGFR signaling is a major driver of cancer. In C. elegans, we show that loss of AGEF-1 results in an increase in basolateral EGFR localization in the vulva precursor cells, and in sensitized genetic backgrounds, a corresponding increase in vulva induction. While the human AGEF-1 proteins, BIG1 and BIG2, have not been previously implicated in EGFR signaling and cancer, mutations in BIG2 are causal of periventricular heterotopia, a condition whereby neurons fail to migrate to the cerebral cortex during brain development. As migrating neurons require polarized protein localization, BIG2 and AGEF-1 may have similar functions in these polarized cell types.
Introduction
C. elegans vulval cell induction requires a highly conserved Epidermal Growth Factor Receptor (EGFR)/Ras GTPase/Mitogen Activated Protein Kinase (MAPK) signaling pathway providing an in vivo model in which to study signaling in a polarized epithelia [1], [2]. During larval development, an equivalence group of six vulval precursor cells (VPCs), P3.p-P8.p, have the potential to be induced to generate the vulva. The anchor cell in the overlying gonad secretes the LIN-3 EGF-like ligand, inducing the closest VPC, P6.p, to adopt the primary vulval fate, and a combination of graded LIN-3 EGF signal and lateral signaling by LIN-12 Notch specifies the neighboring VPCs, P5.p and P7.p, to adopt the secondary vulval fate. Together P5.p-P7.p generate the 22 nuclei of the mature vulva, eight cells from the primary cell and seven from each of the secondary cells. The remaining VPCs, P3.p, P4.p, and P8.p, divide once and fuse with the surrounding hypodermal syncytium (50% of the time P3.p fuses prior to dividing) and thus adopt a tertiary non-vulval fate. Inhibition of LET-23 EGFR signaling causes a Vulvaless (Vul) phenotype in which less than the normal three VPCs are induced. Conversely, increased LET-23 EGFR signaling causes a Multivulva (Muv) phenotype in which greater than three VPCs are induced.
LET-23 EGFR localizes to both the apical and basolateral membranes of the VPCs, though, it is the basolateral localization that is thought to engage LIN-3 EGF and induce vulva induction [3], [4], [5]. A tripartite complex of proteins, LIN-2 Cask, LIN-7 Veli, and LIN-10 Mint (LIN-2/7/10), interacts with the C-terminal tail of LET-23 EGFR and is required for its basolateral localization [3], [4]. Mutations in any component of the complex, or the let-23(sy1) mutation, which deletes the last six amino acids of LET-23 EGFR that are required for its interaction with LIN-7, result in LET-23 EGFR localizing only to the apical membrane and a strong Vul phenotype [3], [4], [6], [7], [8]. The Vul phenotype of lin-2/7/10 mutants or the let-23(sy1) mutant are easily suppressed to a wild-type or even a Muv phenotype by loss of negative regulators of LET-23 EGFR signaling such as sli-1 Cbl, gap-1 RasGAP, rab-7 GTPase, and unc-101 AP-1μ [5], [9], [10], [11], [12]. Thus far, no suppressors of the lin-2/7/10 mutant Vul phenotype have been shown to restore LET-23 EGFR to the basolateral membrane.
UNC-101 and APM-1 are two μ1 subunits for the AP-1 adaptor protein complex, which function redundantly to antagonize vulva cell induction [12], [13]. In mammals, AP-1 localizes to the trans-Golgi network (TGN) and endosomes, promotes formation of clathrin-coated vesicles, and is involved in regulated secretion from the TGN. [14], [15], [16]. In epithelial cells, AP-1 sorts cargo, including EGFR, to the basolateral membrane, which would be inconsistent with AP-1 antagonizing signaling [17], [18]. The small GTPase, Arf1, recruits AP-1 to the TGN and thus facilitates the formation of clathrin-coated vesicles [15], [19], [20]. BIG1 and BIG2 are Sec7 domain containing guanine nucleotide exchange factors (GEFs) for class I Arf GTPases [21], [22], and are required for recruitment of AP-1 to the TGN and endosomes [23], [24]. To date, neither Arf1 nor the BIG1/2 GEFs have been implicated in EGFR/Ras/MAPK signaling.
Here we identify C. elegans AGEF-1, a homolog of yeast Sec7p and the mammalian BIG1 and BIG2 Arf GEFs, as negatively regulating EGFR/Ras/MAPK-mediated vulva induction. We show that AGEF-1 regulates protein secretion in multiple tissues, regulates polarized localization of the SID-2 transmembrane protein in the intestine, and regulates the size of late endosomes/lysosomes with the AP-1 complex in the macrophage/scavenger cell-like coelomocytes. Genetic epistasis places AGEF-1 upstream or in parallel to LET-23 EGFR. We find that the ARF-1.2 and ARF-3 GTPases also negatively regulate LET-23 EGFR signaling. Moreover, our genetics are consistent with AGEF-1 BIG1/2, ARF-1.2 Arf1 and UNC-101 AP-1μ1 functioning together in preventing ectopic vulva induction. It has been 20 years since UNC-101 was identified as a negative regulator of LET-23 EGFR signaling, however its mechanism of action has remained an enigma [12]. Contrary to the role of AP-1 in basolateral sorting in mammalian cells, we demonstrate that AGEF-1 BIG1/2 and UNC-101 AP-1μ1 antagonize the basolateral membrane localization of LET-23 EGFR in the VPCs. Thus, the AGEF-1/Arf GTPase/AP-1 ensemble antagonizes LET-23 EGFR-mediated vulva induction via regulation of LET-23 EGFR membrane localization.
Results
Identification of agef-1(vh4) as a suppressor of the lin-2(e1309) Vul phenotype
We previously reported that rab-7(ok511), a maternal effect embryonic lethal mutant, strongly suppresses the lin-2(e1309) Vul phenotype [11]. To identify new candidate regulators of LET-23 EGFR trafficking and signaling we conducted a clonal screen for essential suppressors of lin-2(e1309) (see Materials and Methods). In this screen we identified vh4 as a strong suppressor of the lin-2(e1309) Vul phenotype (Figure 1A–D; Table 1, lines 1–4). The vh4 mutation can suppress the 100% Vul phenotype of lin-2(e1309) to 20% Vul, and 30% Muv. In a lin-2(+) background, however, vh4 mutant animals have 100% wild-type vulva induction. Consistent with a potential role in vesicular trafficking, the coelomocytes (macrophage-like scavenger cells) of vh4 mutants accumulate abnormally large vesicular structures (Figure 1E, F). Additionally, vh4 mutants have a dumpy body morphology, uncoordinated movement and ∼50 percent embryonic lethality (Figure 1G, H).


To determine the molecular identity of vh4, we used a single nucleotide polymorphism (SNP) mapping strategy [25]. Genome-wide mapping located vh4 to the right arm of chromosome I, and interval mapping placed vh4 in a 2.75 map unit region between SNPs haw14137 and pkP1071 at positions 20.65 and 23.4 map units, respectively (Figure 2A). We further refined the genomic interval by complementation with chromosomal deficiencies dxDf2 and eDf3. vh4 failed to complement the large deficiency dxDf2, but complemented the small eDf3, indicating that vh4 lies in a 0.9 map unit region (20.65–21.51) containing 27 genes. We found that one obvious candidate, vps-28, was an RNAi suppressor of the lin-2 Vul phenotype [11]. However, vh4 complemented the vps-28(tm3767) deletion allele and no lesion in the vps-28 coding sequence of vh4 animals was detected by DNA sequencing suggesting that vh4 is not an allele of vps-28. Whole genome sequencing revealed a homozygous G to A transition at position 3082 in exon 11 of the agef-1 gene (AAA TTT TTG GAA AAG GGA GAA CTT CCG AAT TTC CGA TTT) that corresponds to a glutamate to lysine substitution in a conserved region of the predicted AGEF-1 protein (Figure 2B). Consistent with vh4 being a mutation in agef-1, we find that agef-1(RNAi) suppresses the severity of the lin-2 Vul phenotype (Table 1, lines 3 and 7) and agef-1(vh4) mutant oocytes have defects in CAV-1 body formation as previously seen with agef-1(RNAi) (Figure S1J–M) [26]. Finally, agef-1(vh4) fails to complement two deletion alleles, agef-1(ok1736) and agef-1(tm1693), resulting in a strong embryonic lethal phenotype. These data indicate that vh4 is a hypomorphic allele of agef-1.

The coelomocytes of agef-1(vh4) mutants have enlarged late endosomes/lysosomes
To determine the identity of the large vesicles in agef-1(vh4) coelomocytes, we used GFP tagged endosomal and Golgi proteins. Since the vesicles are presumably in flux, we measured the diameter of the largest GFP-positive vesicle per coelomocyte. We found a modest, but significant increase in the size of vesicles positive for the early endosomal 2×FYVE::GFP and the pan-endosomal RME-8::GFP markers in agef-1(vh4) animals as compared to wild-type (Figure 3A–C and Figure S2A–C). However, the large vesicles in agef-1(vh4) coelomocytes visible by DIC optics correspond to LMP-1::GFP, a marker for late endosomes/lysosomes (Figure 3D–F). This finding corroborates a concurrent study identifying large LMP-1 positive vesicles in the coelomocytes of agef-1(RNAi) animals [27]. LMP-1 is a transmembrane protein, whose mammalian homolog, Lamp1, can transit from Golgi via the plasma membrane and endosomes to the lysosome [28]. Therefore, we assessed the morphology of the Golgi in agef-1(vh4) mutants using a mannosidase II::GFP marker. While there might be a slight increase in the size of the Golgi mini-stacks, they were distinct from the large LMP-1::GFP positive vesicles seen by DIC (Figure S2D–F). Of note, the mannosidase II::GFP strands that appear to interconnect the Golgi mini-stacks in wild-type (30/37 coelomocytes) were largely absent in agef-1(vh4) mutants (6/42 coelomocytes are interconnected). These data show that agef-1(vh4) disrupts endosome and Golgi morphology and possibly trafficking.

The body wall muscle cells and intestinal cells of agef-1(vh4) mutants have defects in protein secretion
To test if agef-1(vh4) coelomocytes have an endocytosis defect we analyzed the internalization of a signal secreted GFP (ssGFP) that is expressed in body wall muscle cells, secreted into the pseudocoelom, and endocytosed by the coelomocytes [29]. We found less ssGFP in the coelomocytes of agef-1(vh4) animals as compared to wild-type (Figure 4A–E). However, we did not detect a significant accumulation of ssGFP in the pseudocoelom of agef-1(vh4) mutants as would be expected for an endocytosis defect. Rather there was a clear accumulation of ssGFP in the body wall muscle cells of agef-1(vh4) animals as compared to wild-type (Figure 4F–J). While this does not rule out a potential endocytosis defect in agef-1(vh4) coelomocytes, it does indicate that agef-1(vh4) mutants have a secretion defect in the body wall muscle cells.

We also analyzed a Yolk::GFP fusion (YP170::GFP) that is secreted from the intestine, and internalized by maturing oocytes [30]. We did not detect a difference in the uptake of YP170::GFP by oocytes (Figure S1A–D), however YP170::GFP levels in the intestine were higher in agef-1(vh4) animals than in wild-type (Figure S1E–I). An independent study also found impaired secretion of yolk in agef-1(RNAi) animals [31]. Thus, agef-1(vh4) mutants have impaired protein secretion from both body wall muscle and intestinal cells.
AGEF-1 antagonizes signaling upstream or in parallel to LET-23 EGFR
To understand the role of AGEF-1 in the LET-23 EGFR/LET-60 Ras signaling pathway, we made double mutants with agef-1 and several mutations in core components of the pathway. A gain of function mutation in let-60 ras (n1046) causes a Muv phenotype that can be enhanced by loss of a negative regulator of the pathway [11], [32], [33], [34]. agef-1(vh4) significantly enhances the Muv phenotype of let-60(n1046), consistent with AGEF-1 being a negative regulator of signaling (Table 1, lines 10–11). We performed epistasis analysis to determine at which step of the pathway AGEF-1 functions. We found that agef-1(vh4) strongly suppresses the Vul phenotype of the let-23(sy1) mutant (Table 1, lines 12–13). The sy1 allele truncates the last six amino acids of LET-23 EGFR that are required for its interaction with the LIN-2/7/10 complex, and thus behaves identical to mutations in components of this complex [3], [8]. However, agef-1(vh4) fails to suppress the Vul phenotype of the let-23(sy97) allele that results in a more severe truncation of LET-23 EGFR that blocks signaling to the LET-60 Ras (Table 1, lines 14–15) [7]. We next tested if agef-1(vh4) can suppress the Vul phenotype of lin-3(e1417), a strong hypomorphic allele of lin-3 EGF [35]. We found that agef-1(vh4) partially suppressed the lin-3(e1417) Vul phenotype (Table 1, lines 16–17). These data are consistent with AGEF-1 antagonizing signaling upstream or in parallel to LET-23 EGFR.
SLI-1 Cbl, a putative E3-ubiquitin ligase, and UNC-101 AP-1μ are negative regulators of LET-23 EGFR signaling that also function at the level of LET-23 EGFR [9], [12], [36]. Like agef-1(vh4), mutations in sli-1 Cbl and unc-101 AP-1μ do not cause a vulval phenotype alone, but double mutants cause a synergistic Muv phenotype. Therefore, we tested if agef-1(vh4) is Muv in combination with strong loss-of-function alleles of sli-1 Cbl and unc-101 AP-1μ. We found that agef-1(vh4); sli-1(sy143) animals are strongly Muv, suggesting that AGEF-1 might function in parallel to SLI-1 Cbl (Table 1, lines 18–19). We were unable to identify unc-101(sy108) agef-1(vh4) double mutants segregating from unc-101(sy108) agef-1(vh4)/unc-101(sy108) mothers suggesting that they are zygotic lethal. Thus, we fed L1 larvae RNAi and found that both unc-101(RNAi) agef-1(vh4) and unc-101(sy108) agef-1(RNAi) animals have a strong Muv phenotype (Table 1, lines 20–22). Since there are two AP-1µ genes, unc-101 and apm-1, that are functionally redundant, we cannot conclude whether AGEF-1 functions in parallel to UNC-101, or whether they function together; we favor the later, see below. However, the strong genetic interactions of agef-1(vh4) and mutations in sli-1 and unc-101 further support AGEF-1 functioning at the level of LET-23 EGFR to negatively regulate signaling.
ARF-1.2 and ARF-3 antagonize LET-23 EGFR signaling
The identification of a putative Arf GEF as a negative regulator of LET-23 EGFR signaling suggests that one or more of the four C. elegans Arf GTPases might also regulate LET-23 EGFR signaling. The mammalian Arf GTPases have been placed in three classes based on homology [37]. To gain a better understanding of the relationship of the C. elegans and human Arf GTPases we undertook a phylogenetic analysis (Figure 2C). From this we conclude that C. elegans ARF-1.2 is homologous to Class I Arfs; ARF-3 is related to both Class I and II Arfs, but clusters with the Class II; ARF-6 is a homolog of the Class III Arf, whereas ARF-1.1 appears to be a Caenorhabiditis specific Arf GTPase that is distinct from the Arf-like Arl GTPases (Figure 2C). We used RNAi and deletion mutants to test each arf gene for suppression of the lin-2(e1309) Vul phenotype. RNAi of either arf-1.2 or arf-3 partially suppressed of the lin-2(e1309) Vul phenotype (Table 2, lines 1–3). The arf-1.2(ok796) deletion mutant was a much more potent suppressor of the lin-2(e1309) Vul phenotype consistent with RNAi being less effective in the VPCs [11; this study] (Table 2, lines 4–5). The arf-3(tm1877) deletion is zygotic lethal and did not permit analysis. However, arf-3(RNAi) into arf-1.2(ok796); lin-2(e1309) animals led to an even stronger suppression of the Vul phenotype comparable to that of agef-1(vh4); lin-2(e1309) (Table 2, line 8). Neither the arf-6(tm1447) nor the arf-1.1(ok1840) deletions were able to suppress the lin-2(e1309) Vul phenotype (Table 2, lines 9–12). These data suggest that ARF-1.2 and ARF-3 function in a partly redundant manner, possibly with AGEF-1, to antagonize LET-23 EGFR signaling.

ARF-1.2 and AGEF-1 antagonize LET-23 EGFR signaling in the VPCs
To test if arf-1.2 was required in the VPCs we generated two transgenic extrachromosomal arrays, vhEx7 and vhEx8, expressing ARF-1.2::GFP under the control of the VPC-specific promoter, lin-31 [38]. Both transgenic lines were able to strongly rescue arf-1.2(ok796) suppression of the lin-2(e1309) Vul phenotype (Table 2, lines 6–7). Thus, ARF-1.2 functions in the VPCs to negatively regulate LET-23 EGFR signaling. We next tested whether VPC-specific overexpression of ARF-1.2::GFP can revert the suppression of Vul phenotype in agef-1(vh4); lin-2(e1309). Both lines, vhEx7 and vhEx8, led to a more severe Vul phenotype when expressed in agef-1(vh4); lin-2(e1309) animals (Table 1, lines 5–6). This suggests that AGEF-1 functions in the VPCs through ARF-1.2 to antagonize LET-23 EGFR signaling.
AGEF-1, ARF-1.2, and UNC-101 AP-1μ cooperate to repress ectopic vulva induction
Having observed a strong Muv phenotype in unc-101(RNAi) agef-1(vh4) and unc-101(sy108) agef-1(RNAi) doubles (Table 1, lines 21–22), we hypothesized that arf-1.2(ok796) would have similar interactions. Indeed, both unc-101(sy108); arf-1.2(ok796) and agef-1(vh4); arf-1.2(ok796) animals have a strong Muv phenotype (Table 1, lines 24–25). Given that agef-1(vh4) is a weak hypomorphic allele and ARF-1.2 and UNC-101 AP-1μ are each functionally redundant with ARF-3 and APM-1 AP-1μ, respectively; these data are consistent with AGEF-1, ARF-1.2 and UNC-101 AP-1μ functioning together to negatively regulate LET-23 EGFR signaling.
AGEF-1 and UNC-101 have shared phenotypes in coelomocytes and the intestine
If AGEF-1, the ARF GTPases and the AP-1 complex function together, we expect that they will have shared phenotypes. We tested whether the ARFs and the AP-1 complex regulate the size of vesicles in coelomocytes as does AGEF-1. While unc-101(sy108) mutants do not have large vesicles, further depletion of the AP-1 complex by RNAi of apm-1 AP-1μ or apg-1 AP-1γ in the unc-101(sy108) background resulted in enlarged LMP-1::GFP vesicles in the coelomocytes (Figure 3G–I). Consistent with previous studies, we found no evidence for arf-1.2 or arf-3 in regulating the size of vesicles in the coelomocytes [27], nor do deletions in arf-1.1 or arf-6. The complement of ARF GTPases that function with AGEF-1 and AP-1 in coelomocytes remains to be determined.
The AP-1 complex has recently been shown to restrict both apical and basolateral membrane protein localization in the C. elegans intestine [39], [40]. Similarly, we found that the apically localized SID-2 transmembrane protein [41], was mislocalized to the cytoplasm and basolateral membranes in agef-1(vh4) mutants (Figure S3A–D), suggesting that AGEF-1 and AP-1 might function together to regulate polarized localization of membrane proteins in the intestine.
AGEF-1 and UNC-101 antagonize LET-23 EGFR basolateral localization
The role of AGEF-1 in restricting SID-2::GFP on the apical membrane suggests that AGEF-1, the ARF GTPases and the AP-1 complex might restrict LET-23 EGFR to the apical membrane in the VPCs. To test this hypothesis we made use of two transgenic strains expressing a LET-23 EGFR GFP fusion (zhIs035 and zhIs038) that mimic the localization of endogenous LET-23 EGFR as seen by antibody staining [5]. In wild-type animals, LET-23::GFP localizes to both the apical and basolateral membranes of P6.p and in the lin-2(e1309) animals LET-23::GFP localizes strictly to the apical membrane (Figure 5A, C and Figure S4A, C). At the Pn.px stage, some basolateral, or lateral only localization is seen in lin-2(e1309) animals. Despite the lack of basolateral localization at the Pn.p stage, we find that the LET-23::GFP transgenes fully rescue the lin-2(e1309) Vul phenotype (Table 1, lines 8 and 9), suggesting that the levels of LET-23 EGFR at the basolateral membrane required for VPC induction are below the level of detection. Similarly, the gaIs27 LET-23::GFP transgene, that is only detectable by immunostaining with anti-GFP antibody, suppressed the lin-2(e1309) egg-laying defective phenotype [11].
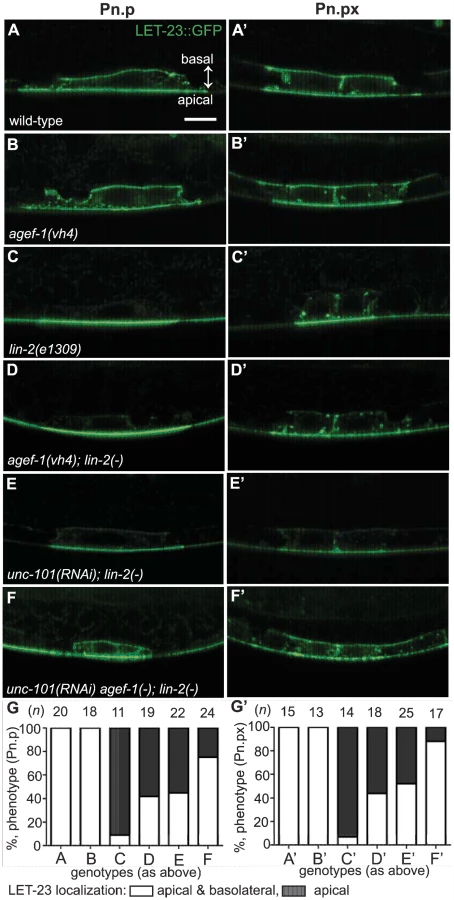
To determine if AGEF-1 regulates LET-23 EGFR localization we compared the ratio of basolateral versus apical localization of LET-23::GFP in the P6.p cell of wild-type and agef-1(vh4) animals (Table 3). In wild-type, the average basal/apical intensity of LET-23::GFP in P6.p was 0.49 for zhIs035 and 0.65 for zhIs038. In agef-1(vh4) animals, the average basal/apical intensity of LET-23::GFP in the P6.p cell is 0.79 for zhIs035 and 0.93 for zhIs038 reflecting a decrease in apical intensity and an increase in basolateral intensity. We also found LET-23::GFP is present on the basolateral membrane of the intestinal cells in agef-1(vh4) animals whereas we did not see this in wild-type by confocal microscopy (Figure S3E–H). Therefore, AGEF-1 represses basolateral localization of LET-23::GFP in the VPCs and intestinal cells.

We next tested if agef-1(vh4) could restore the basolateral localization of LET-23::GFP in lin-2(1309) animals. We found that ∼40% of agef-1(vh4); lin-2(e1309) animals with zhIs035 have weak basolateral membrane localization of LET-23::GFP in P6.p compared to 9% in lin-2(e1309) animals (Figure 5C–D, G). Similarly, at the P6.px stage, we see an increase in the number of animals with basolateral localization of LET-23::GFP in agef-1(vh4); lin-2(e1309) as compared to lin-2(e1309) single mutants (Figure 5C′–D′, G′). No basolateral LET-23::GFP was seen with agef-1(vh4); lin-2(e1309) animals with the lower expressing zhIs038 (Figure S4C′–D′, F′). Since agef-1(vh4) is a weak hypomorphic mutation, we tested if knocking down the AP-1 complex via unc-101(RNAi) can further restore basolateral localization of LET-23 EGFR in agef-1(vh4); lin-2(e1309) mutants. We found that unc-101(RNAi) agef-1(vh4); lin-2(e1309) animals with either zhIs035 or zhIs038 had an increase in basolateral membrane localization of LET-23::GFP in both the intensity and the number of animals (Figure 5F–G′ and Figure S4E, F). unc-101(RNAi); lin-2(e1309) animals only showed mild restoration of LET-23::GFP using the zhIs035 transgene (Figure 5E, E′, G and G′). The restoration of LET-23 EGFR on the basolateral membrane in agef-1(vh4); lin-2(e1309), unc-101(RNAi); lin-2(e1309) and unc-101(RNAi) agef-1(vh4); lin-2(e1309) animals suggests that AGEF-1 and UNC-101 AP-1μ negatively regulate LET-23 EGFR signaling by limiting basolateral membrane localization.
Discussion
Regulators of LET-23 EGFR trafficking are likely required for viability, as is the case for the RAB-7 GTPase [11]. In a screen for essential negative regulators of LET-23 EGFR-mediated vulva induction we identified a hypomorphic allele in the agef-1 gene. AGEF-1 is the C. elegans homolog of the yeast Sec7p and human BIG1 and BIG2 Arf GEFs, which function with class I Arf GTPases and the AP-1 complex to regulate cargo sorting and trafficking from the TGN [42]. We demonstrate that AGEF-1 regulates protein secretion, polarized protein localization, and late endosome/lysosome morphology. We show that AGEF-1 antagonizes signaling in the VPCs, upstream or in parallel to LET-23 EGFR, and that the class I/II Arf GTPases, ARF-1.2 and ARF-3, also negatively regulate signaling. Our genetic and phenotypic data are consistent with AGEF-1, the ARF-1.2 and ARF-3 GTPases, and the AP-1 complex together preventing ectopic vulva induction. The AGEF-1/Arf GTPase/AP-1 ensemble antagonizes the basolateral membrane localization of LET-23 EGFR in the VPCs; and hence, LET-23 EGFR-mediated vulva induction.
The clonal screen for suppressors of the lin-2(e1309) Vul phenotype was initially aimed at identifying maternal effect lethal mutants, like rab-7(ok511). Instead, we identified two strong suppressors of lin-2(e1309) with partial embryonic lethal phenotypes, agef-1(vh4) and vh22 (J. Meng, O.S. and C.E.R., unpublished data); which we currently believe function independently of each other and rab-7. The agef-1 deletion alleles are zygotic lethal and RNAi in the VPCs with agef-1, arf-1.2 and rab-7 has proven less effective than their corresponding genetic mutations [11; this study]. Therefore, the agef-1(vh4) mutation, being a recessive partial loss-of-function allele, provides a unique tool to study the function of agef-1, particularly in tissues refractory to RNAi such as the VPCs and neurons.
The agef-1(vh4) lesion changes a conserved negatively charged Glutamic Acid in the HDS2 domain to a positively charged Lysine. Collectively, the HDS2, HDS3, and HDS4 domains of yeast Sec7p have been shown to have an autoinhibitory function [43]. However, the specific function of the HDS2 domain is not known. Given the recessive nature of the agef-1(vh4) allele, it suggests that the HDS2 domain has a positive role in promoting AGEF-1 function.
Consistent with yeast Sec7p and human BIG1/BIG2 functioning in the secretory pathway, we found that agef-1(vh4) animals had defects in secretion of ssGFP from body wall muscles and Yolk::GFP from the intestine. Similar yolk secretion defects were recently reported for agef-1(RNAi) [31]. We found that agef-1(vh4) coelomocytes accumulated enlarged LMP-1::GFP positive late endosomes/lysosomes. Independently, Tang et al. [27] found that agef-1(RNAi) also caused enlargement of LMP-1::GFP vesicles and proposed a role for AGEF-1 in late endosome to lysosome trafficking, however, they did not find a defect in lysosome acidification or protein degradation. We do not know the reason for the enlarged late endosomes/lysosomes, but it could reflect a defect in retrograde transport from late endosomes to the Golgi as has been shown for knockdown of BIG1 and BIG2 or the AP-1 complex in mammalian cells [23]. Consistent with this idea, we found that knockdown of the AP-1 complex also induced enlarged LMP-1::GFP vesicles. Tang et al. [27] also reported that ssGFP accumulated in the pseudocoelom suggesting an uptake defect in the coelomocytes. We found that agef-1(vh4) mutants accumulated ssGFP in the body wall muscles rather than the pseudocoelom; thus the reduced ssGFP in coelomocytes could be explained by reduced secretion from the body wall muscles. However, we cannot rule out an uptake defect in the coelomocytes as well. Perhaps these discrepancies reflect a difference in reducing the levels of agef-1 by RNAi versus the vh4 missense mutation.
Our genetic analysis with agef-1(vh4) indicate that AGEF-1 is a potent negative regulator of LET-23 EGFR-mediated vulva induction. Similar to other negative regulators, agef-1 enhanced the Muv phenotype of the gain-of-function Ras mutant, let-60(n1046), and was a potent suppressor the Vul phenotypes of lin-2(e1309) and let-23(sy1) mutations, restoring vulva induction and even inducing a Muv phenotype. However, agef-1(vh4) failed to suppress a strong let-23(sy97) allele similar to sli-1 and unc-101 mutations and consistent with a role for AGEF-1 upstream or in parallel to LET-23 EGFR. In accordance with AGEF-1 being an Arf GEF, we found that ARF-1.2 and ARF-3, Class I/II Arf GTPases, also negatively regulate LET-23 EGFR signaling. The arf-1.2(ok796) deletion allele was a less potent suppressor of the lin-2(e1309) Vul phenotype as compared to the agef-1(vh4) mutant. However, arf-3(RNAi) in arf-1.2(ok796); lin-2(e1309) doubles showed suppression comparable to that in agef-1(vh4); lin-2(e1309) mutants. Therefore, ARF-1.2 and ARF-3 appear to function in a partly redundant manner during vulva development. Furthermore, expression of an ARF-1.2::GFP fusion in the VPCs rescued the suppressed Vul phenotype of both arf-1.2(ok796); lin-2(e1309) and agef-1(vh4); lin-2(e1309) animals indicating that ARF-1.2 antagonizes signaling in the VPCs likely downstream of AGEF-1.
In mammalian cells, the BIG1/BIG2 proteins and Arf1 recruit the AP-1 adaptor protein complex to the TGN and endosomes [15], [19], [20]. Both of the C. elegans AP-1μ subunits, unc-101 and apm-1, negatively regulate LET-23 EGFR mediated vulva development [12], [13]. In fact, apm-1(RNAi) unc-101(sy108) animals had a Muv phenotype, indicating that UNC-101 and APM-1 are functionally redundant during vulva induction, thus revealing a role for the AP-1 complex in inhibiting ectopic vulva induction [13]. Our findings that various double-mutant combinations between agef-1(vh4), arf-1.2(ok796) and unc-101(sy108) AP-1μ result in a synergistic Muv phenotype are consistent with AGEF-1, the Arfs and AP-1 functioning together to inhibit ectopic vulva induction. However, we cannot conclude whether they function in parallel pathways or in a common pathway due to the fact that agef-1(vh4) is not a null allele and the unc-101(sy108) and arf-1.2(ok796) mutations, while severe loss-of-function or null alleles, function in a partly redundant manner with apm-1 and arf-3, respectively. We favor a model whereby AGEF-1, the Arfs, and AP-1 function in a common pathway since this is most consistent with data from yeast and mammals, and that loss of AGEF-1 and components of the AP-1 complex have similar phenotypes in coelomocytes and the intestine. While synergistic genetic interactions are typically more indicative of genes in parallel pathways, we interpret that no single mutation in the AGEF-1/Arf/AP-1 pathway is sufficient to increase LET-23 EGFR signaling above a threshold necessary for ectopic induction. It is only when the activity of the AGEF-1/Arf/AP-1 pathway is further compromised by two mutations that LET-23 EGFR signaling increases above a threshold to induce a synergistic Muv phenotype. It is important to note that the AGEF-1/Arf/AP-1 pathway is essential, and only animals that survive to the fourth larval stage can be scored for vulva induction phenotypes. Thus, LET-23 EGFR signaling and localization phenotypes would likely be more severe if we were able to assess true null mutations in the VPCs only.
In polarized epithelial cells, the AP-1 complex mediates sorting and polarized distribution of transmembrane proteins, including EGFR, and thus the AGEF-1/Arf GTPase/AP-1 ensemble could regulate signaling via LET-23 EGFR localization. In the P6.p cell, we showed that the localization of LET-23 EGFR is altered in agef-1(vh4) animals using two transgenic lines (zhIs035 and zhIs038) expressing LET-23::GFP [5]. In wild-type animals, LET-23::GFP is present on both the apical and basolateral domains, however the average levels of LET-23:GFP on the apical membrane are double (zhIs035) or close to double (zhIs038) that on the basolateral membrane (Figure 6A). In agef-1(vh4) animals there was a redistribution of LET-23::GFP from apical to basolateral membrane bringing the average intensities closer to equal, suggesting that AGEF-1 either promotes apical localization or antagonizes basolateral localization of the receptor (Figure 6A, B). In the lin-2(e1309) background, LET-23::GFP is apical only (Figure 6C). In the more highly expressed line, zhIs035, we see some lateral only or faint basolateral in the P6.p descendants, P6.pa and P6.pp of lin-2(e1309) larvae. In the zhIs035 line, agef-1(vh4) partially restores LET-23::GFP on the basolateral membrane in lin-2(e1309) larvae. RNAi of unc-101 also partially restores basolateral localization and enhances the effect of agef-1(vh4) such that we see increased levels of LET-23::GFP, with both lines, in lin-2(e1309) larvae (Figure 6D). Therefore, AGEF-1 and UNC-101 AP-1μ cooperate to antagonize LET-23 EGFR basolateral localization and thus provide a mechanism by which these genes/proteins antagonize LET-23 EGFR signaling. Despite the lack of basolateral localization of LET-23::GFP in lin-2 mutant animals, the two LET-23::GFP transgenes used in this study rescued the lin-2(e1309) Vul phenotype, suggesting that the levels of receptor required for VPC induction are below detection. Therefore, the modest amount of LET-23::GFP restored to the basolateral membrane in agef-1(vh4); lin-2(e1309) or unc-101(RNAi); lin-2(e1309) could be more than sufficient to explain the strong restoration of VPC induction in these double mutants.

Our findings that an AGEF-1/Arf GTPase/AP-1 ensemble antagonizes the basolateral localization of LET-23 EGFR is contradictory to the established role of the mammalian AP-1A and AP-1B complexes in sorting transmembrane proteins to the basolateral membrane through the specific binding of basolateral sorting motifs in the cytoplasmic tail [18]. In fact, the AP-1B complex promotes the basolateral localization of EGFR in MDCK cells [17]. LET-23 EGFR does have several putative AP-1 sorting motifs, and thus could be a direct target for AP-1 regulation, but this would imply that AP-1 is impeding basolateral localization. A precedent for AP-1 having an antagonistic role in protein sorting or secretion has been found with the yeast Chs3p and Fus1p proteins, which rely on the exomer for secretion [44], [45]. In the absence of exomer, Chs3p and Fus1p are retained internally in an AP-1 dependent manner [45], [46], [47]. An analogous situation whereby the LIN-2/7/10 complex sorts/maintains LET-23 EGFR localization on the basolateral membrane and the AGEF-1/Arf/AP-1 pathway plays an antagonistic role could exist.
Recent studies in C. elegans and mice have shown that both basolateral and apical membrane cargos are mislocalized in the absence of the AP-1 complex [39], [40], [48], suggesting that the AP-1 complex is required to maintain the polarity of the epithelial cells [reviewed in 18]. Similarly, we find that agef-1(vh4) mutants mislocalized the SID-2 protein to the basolateral membranes, which is strictly apical in wild-type animals. Therefore, AGEF-1 might function with AP-1 to maintain polarity in the intestinal epithelia and by extension the AGEF-1/Arf GTPase/AP-1 ensemble could indirectly regulate LET-23 EGFR localization via maintenance of VPC polarity.
In summary, an AGEF-1/Arf GTPase/AP-1 ensemble functions opposite the LIN-2/7/10 complex to regulate apical versus basolateral localization of LET-23 EGFR in the VPCs, thus explaining how it negatively regulates LET-23 EGFR-mediated vulva induction. We don't yet know whether the AGEF-1/Arf GTPase/AP-1 ensemble directly regulates LET-23 EGFR sorting and localization or whether it is indirect via maintenance of VPC polarity. Further studies will be required to sort out the mechanisms by which the AGEF-1/ARF GTPase/AP-1 ensemble regulates LET-23 EGFR localization.
Materials and Methods
Strains and alleles
General methods for the handling and culturing of C. elegans were as previously described [49]. C. elegans Bristol strain N2 is the wild-type parent for all the strains used in this study; E. coli stain HB101 was used as a food source. The Hawaiian strain CB4856 was used for SNP mapping. All experiments were performed at 20°C. Information on the genes and alleles used in this work can be found on WormBase (www.wormbase.org) and are available through Caenorhabditis Genetics Center (www.cbs.umn.edu/cgc) unless otherwise noted in the strain list (Table S1).
lin-2(e1309) suppressor screen
lin-2(e1309) L4 hermaphrodites were mutagenized as previously described [49]. F1 progeny (m/+; lin-2) were transferred to individual plates. Due to the strong Vul phenotype of lin-2(e1309) animals, the self-progeny hatch internally [6]. F2 progeny were screened at the adult stage in order to identify plates that had a large number of eggs and egg-layers, additional preference was given to plates that had Muv, embryonic lethality, or dumpy phenotypes, similar to the rab-7(ok511) mutant. Progeny of a total of 2430 F1 animals (4860 haploid genomes) were screened and two lin-2 (e1309) suppressor mutants that are dumpy and partly embryonic lethal were identified, vh4 and vh22.
Genetic mapping and cloning of agef-1(vh4)
Single nucleotide polymorphism (SNP) mapping was used to place vh4 to the right arm of chromosome I [25]. Chromosome mapping showed linkage of vh4 to SNPs at 13 (F58D5), 14 (T06G6) and 26 (Y105E8B) map units (m.u.). Interval mapping using two sets of recombinants, 141 animals in total, was conducted using the following SNPs: pkP1133 at 17.4 m.u (A/T Bristol/CB4856, RFLP DraI); pkP1134 at 18.95 m.u. (T/C Bristol/CB4856, RFLP AflIII); haw14061 at 19.51 m.u. (T/C Bristol/CB4856, sequencing); haw14137 at 20.65 m.u. (T/A Bristol/CB4856, sequencing); haw14164 at 21.04 m.u. (C/T Bristol/CB4856, sequencing); CE1-248 at 21.97 m.u. (T/A Bristol/CB4856, sequencing); CE1-220 at 23.16 m.u. (A/G Bristol/CB4856, sequencing); pkP1071 at 23.4 m.u. (C/T Bristol/CB4856, RFLP EcoRI). In the course of interval mapping the following predicted sequencing SNPs were confirmed: haw14061 and haw14137 as T/C and T/A Bristol/CB4856, respectively. Genomic DNA from vh4 and vh22 was isolated and submitted to Genome Quebec for Illumina sequencing. Within the defined map region, the agef-1 gene was the only gene carrying a non-synonymous mutation unique to the vh4 strain.
RNA interference
RNAi feeding was performed essentially as previously described [50] using the unc-101 (I-6G20), agef-1 (I-6L22), arf-1.2 (III-3A13), and arf-3 (IV-4E13) clones from Ahringer RNAi library (Geneservice, Cambridge, United Kingdom). Clones were verified by DNA sequencing. To avoid embryonic and larval lethal phenotypes, synchronized L1 larvae were placed on RNAi plates and scored for vulva induction when the animals reached L4 stage 36–48 hours later.
Microscopy and phenotype analysis
General methods for live animal imaging using Nomarski differential interference contrast (DIC) microscopy were as previously described [51]. Animals were analyzed on an Axio Zeiss A1 Imager compound microscope (Zeiss, Oberkochen, Germany) and images were captured using an Axio Cam MRm camera and AxioVision software (Zeiss, Oberkochen, Germany). Muv and Vul phenotypes were scored by counting the numbers of vulval and non-vulval descendants of P3.p–P8.p in L4 stage larvae as described previously [11]. Fisher's exact test (www.graphpad.com/quickcalcs) was used for statistical analysis of the vulval phenotypes. Comparison of GFP intensities wild-type and agef-1(vh4) was performed using identical exposure times for conditions being compared. Fiji image processing tool was used to measure intensities in raw images; any adjustments to contrast/brightness were for presentation purposes and were performed after analysis [52]. Tissue/organ of interest was outlined using free hand selection tool followed by measurement of the average pixel intensity. Images selected for figures are representative of the mean value for average pixel intensity for the group. Statistical analysis and graphing was done using Prism 5 (GraphPad Software, Inc., La Jolla, CA).
Confocal analysis was performed using a Zeiss LSM-510 Meta laser scanning microscope with 63× oil immersion lens (Zeiss, Oberkochen, Germany) in a single-track mode using a 488 nm excitation for GFP. Images were captured using ZEN 2009 Image software (Zeiss, Oberkochen, Germany). Animals at the L4 larval stage were selected for visualization of endocytic/secretory compartments in the coelomocytes. Images selected for figures are representative of the mean value for the largest vesicle diameter for the group. Statistical analysis and graphing was done using Prism 5 (GraphPad Software, Inc., La Jolla, CA). Confocal analysis of zhIs038 transgene-carrying animals was performed at early L3 larval stage using the Zeiss LSM-510 Meta laser scanning microscope. Confocal analysis of zhIs035 was performed using the Zeiss Axio Observer Z1 LSM-780 laser scanning microscope with 63× oil immersion lens (Zeiss, Oberkochen, Germany) in a single-track mode using an Argon multiline laser with 488 nm excitation for GFP. Images were captured using ZEN 2010 Image software (Zeiss, Oberkochen, Germany). The apical and basal LET-23::GFP intensities were measured using Fiji by drawing a line through the center of the nucleus in the DIC channel and transferring the selection into the GFP channel to prevent bias.
Plasmid and transgenic construction
arf-1.2 was amplified by PCR from wild-type cDNA using the primers 5′-CATAAGAATAGTCGACATGGGAAACGTGTTCGGCAGC-3′ (forward) and 5′-GATTCTGATTACCGGTTCAGATCTATTCTTGAGCT-3′ (reverse) containing SalI and AgeI cut sites, respectively. The PCR product was cloned into pEGFP-N1 plasmid using SalI 639 and AgeI 666 sites. arf-1.2::GFP was digested using SalI and NotI and subcloned into the p255 lin-31 promoter plasmid. Transgenic animals were generated by DNA microinjection [53] of the Plin-31::ARF-1.2::GFP plasmid and a marker plasmid Pttx-3::GFP at a concentration of 50 ng/µl of each into N2 animals using maxiprep quality DNA. Two of three lines were used for this study, vhEx7 and vhEx8. Rescue of agef-1(vh4) and arf-1.2(ok796) mediated suppression of the lin-2(e1309) Vul phenotype was scored in animals expressing ARF-1.2::GFP in the VPCs.
Phylogenetic analysis
Analysis of the Arf GTPases was performed using MAFFT version 7 multiple alignment program for amino acid or nucleotide sequences online (http://mafft.cbrc.jp) [54]. Input sequences were human NP_001649.1 (Arf1), NP_001650.1 (Arf3), NP_001651.1 (Arf4), NP_001653.1 (Arf5), AAV38671.1 (Arf6), NP_001168.1 (Arl1) and C. elegans NP_501242.1 (ARF-1.1), NP_498235.1 (ARF-1.2), NP_501336.1 (ARF-3), NP_503011.1 (ARF-6), NP_495816.1 (ARL-1). Phylogenetic tree was constructed and visualized using Archaeopteryx [55], [56].
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. SternbergPW (2005) Vulval development. WormBook 1–28.
2. SundaramMV (2013) Canonical RTK-Ras-ERK signaling and related alternative pathways. WormBook 1–38.
3. KaechSM, WhitfieldCW, KimSK (1998) The LIN-2/LIN-7/LIN-10 complex mediates basolateral membrane localization of the C. elegans EGF receptor LET-23 in vulval epithelial cells. Cell 94 : 761–771.
4. WhitfieldCW, BenardC, BarnesT, HekimiS, KimSK (1999) Basolateral localization of the Caenorhabditis elegans epidermal growth factor receptor in epithelial cells by the PDZ protein LIN-10. Mol Biol Cell 10 : 2087–2100.
5. HaagA, GutierrezP, BuhlerA, WalserM, YangQ, et al. (2014) An In Vivo EGF Receptor Localization Screen in C. elegans Identifies the Ezrin Homolog ERM-1 as a Temporal Regulator of Signaling. PLoS Genet 10: e1004341.
6. FergusonEL, HorvitzHR (1985) Identification and characterization of 22 genes that affect the vulval cell lineages of the nematode Caenorhabditis elegans. Genetics 110 : 17–72.
7. AroianRV, SternbergPW (1991) Multiple functions of let-23, a Caenorhabditis elegans receptor tyrosine kinase gene required for vulval induction. Genetics 128 : 251–267.
8. AroianRV, LesaGM, SternbergPW (1994) Mutations in the Caenorhabditis elegans let-23 EGFR-like gene define elements important for cell-type specificity and function. EMBO J 13 : 360–366.
9. JongewardGD, ClandininTR, SternbergPW (1995) sli-1, a negative regulator of let-23-mediated signaling in C. elegans. Genetics 139 : 1553–1566.
10. HajnalA, WhitfieldCW, KimSK (1997) Inhibition of Caenorhabditis elegans vulval induction by gap-1 and by let-23 receptor tyrosine kinase. Genes Dev 11 : 2715–2728.
11. SkorobogataO, RocheleauCE (2012) RAB-7 antagonizes LET-23 EGFR signaling during vulva development in Caenorhabditis elegans. PLoS One 7: e36489.
12. LeeJ, JongewardGD, SternbergPW (1994) unc-101, a gene required for many aspects of Caenorhabditis elegans development and behavior, encodes a clathrin-associated protein. Genes Dev 8 : 60–73.
13. ShimJ, SternbergPW, LeeJ (2000) Distinct and redundant functions of mu1 medium chains of the AP-1 clathrin-associated protein complex in the nematode Caenorhabditis elegans. Mol Biol Cell 11 : 2743–2756.
14. GonzalezA, Rodriguez-BoulanE (2009) Clathrin and AP1B: key roles in basolateral trafficking through trans-endosomal routes. FEBS Lett 583 : 3784–3795.
15. RobinsonMS (2004) Adaptable adaptors for coated vesicles. Trends Cell Biol 14 : 167–174.
16. BraulkeT, BonifacinoJS (2009) Sorting of lysosomal proteins. Biochim Biophys Acta 1793 : 605–614.
17. RyanS, VergheseS, CianciolaNL, CottonCU, CarlinCR (2010) Autosomal Recessive Polycystic Kidney Disease Epithelial Cell Model Reveals Multiple Basolateral Epidermal Growth Factor Receptor Sorting Pathways. Molecular Biology of the Cell 21 : 2732–2745.
18. BonifacinoJS (2014) Adaptor proteins involved in polarized sorting. J Cell Biol 204 : 7–17.
19. StamnesMA, RothmanJE (1993) The binding of AP-1 clathrin adaptor particles to Golgi membranes requires ADP-ribosylation factor, a small GTP-binding protein. Cell 73 : 999–1005.
20. TraubLM, OstromJA, KornfeldS (1993) Biochemical dissection of AP-1 recruitment onto Golgi membranes. J Cell Biol 123 : 561–573.
21. MorinagaN, TsaiSC, MossJ, VaughanM (1996) Isolation of a brefeldin A-inhibited guanine nucleotide-exchange protein for ADP ribosylation factor (ARF) 1 and ARF3 that contains a Sec7-like domain. Proc Natl Acad Sci U S A 93 : 12856–12860.
22. TogawaA, MorinagaN, OgasawaraM, MossJ, VaughanM (1999) Purification and cloning of a brefeldin A-inhibited guanine nucleotide-exchange protein for ADP-ribosylation factors. J Biol Chem 274 : 12308–12315.
23. IshizakiR, ShinHW, MitsuhashiH, NakayamaK (2008) Redundant roles of BIG2 and BIG1, guanine-nucleotide exchange factors for ADP-ribosylation factors in membrane traffic between the trans-Golgi network and endosomes. Mol Biol Cell 19 : 2650–2660.
24. ManoleaF, ClaudeA, ChunJ, RosasJ, MelanconP (2008) Distinct functions for Arf guanine nucleotide exchange factors at the Golgi complex: GBF1 and BIGs are required for assembly and maintenance of the Golgi stack and trans-Golgi network, respectively. Mol Biol Cell 19 : 523–535.
25. DavisMW, HammarlundM, HarrachT, HullettP, OlsenS, et al. (2005) Rapid single nucleotide polymorphism mapping in C. elegans. BMC Genomics 6 : 118.
26. SatoK, SatoM, AudhyaA, OegemaK, SchweinsbergP, et al. (2006) Dynamic regulation of caveolin-1 trafficking in the germ line and embryo of Caenorhabditis elegans. Mol Biol Cell 17 : 3085–3094.
27. TangL, FaresH, ZhaoX, DuW, LiuBF (2012) Different endocytic functions of AGEF-1 in C. elegans coelomocytes. Biochim Biophys Acta 1820 : 829–840.
28. SaftigP, KlumpermanJ (2009) Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat Rev Mol Cell Biol 10 : 623–635.
29. FaresH, GreenwaldI (2001) Regulation of endocytosis by CUP-5, the Caenorhabditis elegans mucolipin-1 homolog. Nat Genet 28 : 64–68.
30. GrantB, HirshD (1999) Receptor-mediated endocytosis in the Caenorhabditis elegans oocyte. Mol Biol Cell 10 : 4311–4326.
31. AckemaKB, SauderU, SolingerJA, SpangA (2013) The ArfGEF GBF-1 Is Required for ER Structure, Secretion and Endocytic Transport in C. elegans. PLoS One 8: e67076.
32. BersetTA, HoierEF, HajnalA (2005) The C. elegans homolog of the mammalian tumor suppressor Dep-1/Scc1 inhibits EGFR signaling to regulate binary cell fate decisions. Genes Dev 19 : 1328–1340.
33. HopperNA, LeeJ, SternbergPW (2000) ARK-1 inhibits EGFR signaling in C. elegans. Mol Cell 6 : 65–75.
34. KritikouEA, MilsteinS, VidalainPO, LettreG, BoganE, et al. (2006) C. elegans GLA-3 is a novel component of the MAP kinase MPK-1 signaling pathway required for germ cell survival. Genes Dev 20 : 2279–2292.
35. HwangBJ, SternbergPW (2004) A cell-specific enhancer that specifies lin-3 expression in the C. elegans anchor cell for vulval development. Development 131 : 143–151.
36. YoonCH, LeeJ, JongewardGD, SternbergPW (1995) Similarity of sli-1, a regulator of vulval development in C. elegans, to the mammalian proto-oncogene c-cbl. Science 269 : 1102–1105.
37. KahnRA, CherfilsJ, EliasM, LoveringRC, MunroS, et al. (2006) Nomenclature for the human Arf family of GTP-binding proteins: ARF, ARL, and SAR proteins. J Cell Biol 172 : 645–650.
38. TanPB, LacknerMR, KimSK (1998) MAP kinase signaling specificity mediated by the LIN-1 Ets/LIN-31 WH transcription factor complex during C. elegans vulval induction. Cell 93 : 569–580.
39. Shafaq-ZadahM, BrocardL, SolariF, MichauxG (2012) AP-1 is required for the maintenance of apico-basal polarity in the C. elegans intestine. Development 139 : 2061–2070.
40. ZhangH, KimA, AbrahamN, KhanLA, HallDH, et al. (2012) Clathrin and AP-1 regulate apical polarity and lumen formation during C. elegans tubulogenesis. Development 139 : 2071–2083.
41. WinstonWM, SutherlinM, WrightAJ, FeinbergEH, HunterCP (2007) Caenorhabditis elegans SID-2 is required for environmental RNA interference. Proc Natl Acad Sci U S A 104 : 10565–10570.
42. CasanovaJE (2007) Regulation of Arf activation: the Sec7 family of guanine nucleotide exchange factors. Traffic 8 : 1476–1485.
43. RichardsonBC, McDonoldCM, FrommeJC (2012) The Sec7 Arf-GEF is recruited to the trans-Golgi network by positive feedback. Dev Cell 22 : 799–810.
44. WangCW, HamamotoS, OrciL, SchekmanR (2006) Exomer: A coat complex for transport of select membrane proteins from the trans-Golgi network to the plasma membrane in yeast. J Cell Biol 174 : 973–983.
45. BarfieldRM, FrommeJC, SchekmanR (2009) The exomer coat complex transports Fus1p to the plasma membrane via a novel plasma membrane sorting signal in yeast. Mol Biol Cell 20 : 4985–4996.
46. ValdiviaRH, BaggottD, ChuangJS, SchekmanRW (2002) The yeast clathrin adaptor protein complex 1 is required for the efficient retention of a subset of late Golgi membrane proteins. Dev Cell 2 : 283–294.
47. StarrTL, PagantS, WangCW, SchekmanR (2012) Sorting signals that mediate traffic of chitin synthase III between the TGN/endosomes and to the plasma membrane in yeast. PLoS One 7: e46386.
48. HaseK, NakatsuF, OhmaeM, SugiharaK, ShiodaN, et al. (2013) AP-1B-mediated protein sorting regulates polarity and proliferation of intestinal epithelial cells in mice. Gastroenterology 145 : 625–635.
49. BrennerS (1974) The genetics of Caenorhabditis elegans. Genetics 77 : 71–94.
50. KamathRS, Martinez-CamposM, ZipperlenP, FraserAG, AhringerJ (2001) Effectiveness of specific RNA-mediated interference through ingested double-stranded RNA in Caenorhabditis elegans. Genome Biol 2: RESEARCH0002.
51. SulstonJE, HorvitzHR (1977) Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev Biol 56 : 110–156.
52. SchindelinJ, Arganda-CarrerasI, FriseE, KaynigV, LongairM, et al. (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9 : 676–682.
53. BerkowitzLA, KnightAL, CaldwellGA, CaldwellKA (2008) Generation of stable transgenic C. elegans using microinjection. J Vis Exp 18 doi:10.3791/833
54. KatohK, MisawaK, KumaK, MiyataT (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30 : 3059–3066.
55. ZmasekCM, EddySR (2001) ATV: display and manipulation of annotated phylogenetic trees. Bioinformatics 17 : 383–384.
56. HanMV, ZmasekCM (2009) phyloXML: XML for evolutionary biology and comparative genomics. BMC Bioinformatics 10 : 356.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 10
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- The Master Activator of IncA/C Conjugative Plasmids Stimulates Genomic Islands and Multidrug Resistance Dissemination
- A Splice Mutation in the Gene Causes High Glycogen Content and Low Meat Quality in Pig Skeletal Muscle
- Keratin 76 Is Required for Tight Junction Function and Maintenance of the Skin Barrier
- A Role for Taiman in Insect Metamorphosis
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy