
Keratin 76 Is Required for Tight Junction Function and Maintenance of the Skin Barrier
The generation of knockout mice is a central approach to studying gene function. We have examined the consequences of the germ line inactivation of Keratin 76 in mice and in doing so we reveal a previously undescribed mechanism by which keratin intermediate filaments regulate cellular interactions and tissue homeostasis. Our study supports an emerging body of evidence which challenges the classical view of the keratin intermediate filaments as simple structural proteins, highlighting Krt76 as a gene whose function is indispensable for barrier function and skin wound repair as a result of its novel interaction with tight junction complexes. This study identifies a previously unknown and critical link between intermediate filaments and tight junctions where intermediate filament dysfunction influences skin disease.
Published in the journal:
. PLoS Genet 10(10): e32767. doi:10.1371/journal.pgen.1004706
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004706
Summary
The generation of knockout mice is a central approach to studying gene function. We have examined the consequences of the germ line inactivation of Keratin 76 in mice and in doing so we reveal a previously undescribed mechanism by which keratin intermediate filaments regulate cellular interactions and tissue homeostasis. Our study supports an emerging body of evidence which challenges the classical view of the keratin intermediate filaments as simple structural proteins, highlighting Krt76 as a gene whose function is indispensable for barrier function and skin wound repair as a result of its novel interaction with tight junction complexes. This study identifies a previously unknown and critical link between intermediate filaments and tight junctions where intermediate filament dysfunction influences skin disease.
Introduction
The epidermis provides a stable and selectively permeable barrier essential to terrestrial life. Together with microfilaments and microtubules, intermediate filaments (IFs) make up the major components of the epidermal cytoskeleton. Keratins are the largest subgroup of the IF proteins and comprise the major structural proteins in epithelial cells [1]. Keratins are composed of a central, filament forming, alpha-helical rod domain of ∼310 amino acids that is flanked by non-helical head and tail domains [1], [2], [3], [4], [5]. They act as a flexible scaffold enabling cells to resist physical stress. Consequently, defects in IFs can lead to cell fragility and are linked to a wide array of genodermatoses and cancers [5], [6]. The classical view that keratins simply provide a structural scaffold has been challenged by recent studies demonstrating their increasingly specialised and diverse functions [7]. These include protection from apoptosis [8], [9] and injury [10], regulation of epithelial polarity [11], [12] and influence on cell size and protein translation[10], [13], [14], [15].
The functional integration of cytoskeletal elements and cellular junctions is critical for the establishment and maintenance of the epidermal barrier. Tight junctions (TJ) form a seal between cells which make up the layers of the epidermis [16]. This barrier is selectively permeable, allowing passage of small molecules, but restricting water loss, and allowing for antigen sampling by immune cells [16], [17], [18]. TJs are composed of scaffolding and adhesion molecules including claudins, junctional adhesion molecules and occludins. Defective tight junction organization has been linked to compromised barrier function [17] and the development of various dermopathies including psoriasis [19], [20]. The TJs are thought to interact with the IF network by binding of a number of integral or associated TJ proteins that complex to to F-actin [21] but their associations, if any, with the keratin IF network are unclear.
In this report we have studied the effects of Krt76 disruption in mice and demonstrate that the KRT76 protein is essential for postnatal survival beyond ∼3 months of age. Loss of KRT76 leads to the acquisition and infection of skin wounds which fail to properly resolve over time. This phenotype correlates with observations showing that the gene is up-regulated during normal wound healing and is required for this process. At a mechanistic level we show that loss of KRT76 is associated with defective tight junction function through the mislocalization of Claudin1 (CLDN1), an integral TJ component which we show binds to KRT76. These findings identify a critical new relationship between the IF network and TJs which we propose is essential for epidermal homeostasis.
Results
Loss of Krt76 causes gross epidermal defects and results in lethality
As part of the Wellcome Trust Sanger Institute (WTSI) Mouse Genetics Programme [22], we screened the skin of the mutant mouse strains generated. This skin screen is discussed in an accompanying article in this issue of PLoS Genetics [23]. From this screen we identified significant cutaneous defects in mice homozygous for the gene trap “knockout first” [24] allele of Keratin 76 (Krt76tm1a(KOMP)Wtsi hereafter Krt76tm1a) (Figure 1A) [23]. These animals have a splice acceptor-LacZ reporter integrated upstream of floxed exon 2 that allows gene expression to be traced whilst disrupting gene function. Quality control of this mutant allele and correct genome positioning has been confirmed by long range PCR (http://www.sanger.ac.uk/mouseportal/search?query=krt76). Krt76 expression has previously been reported in the palatal and gingival epithelium [25]. By utilising the integrated LacZ reporter in our Krt76tm1a/+ model we confirmed expression at these locations but also detected previously unreported expression in the vagina and the eyelid (Figure 1B).
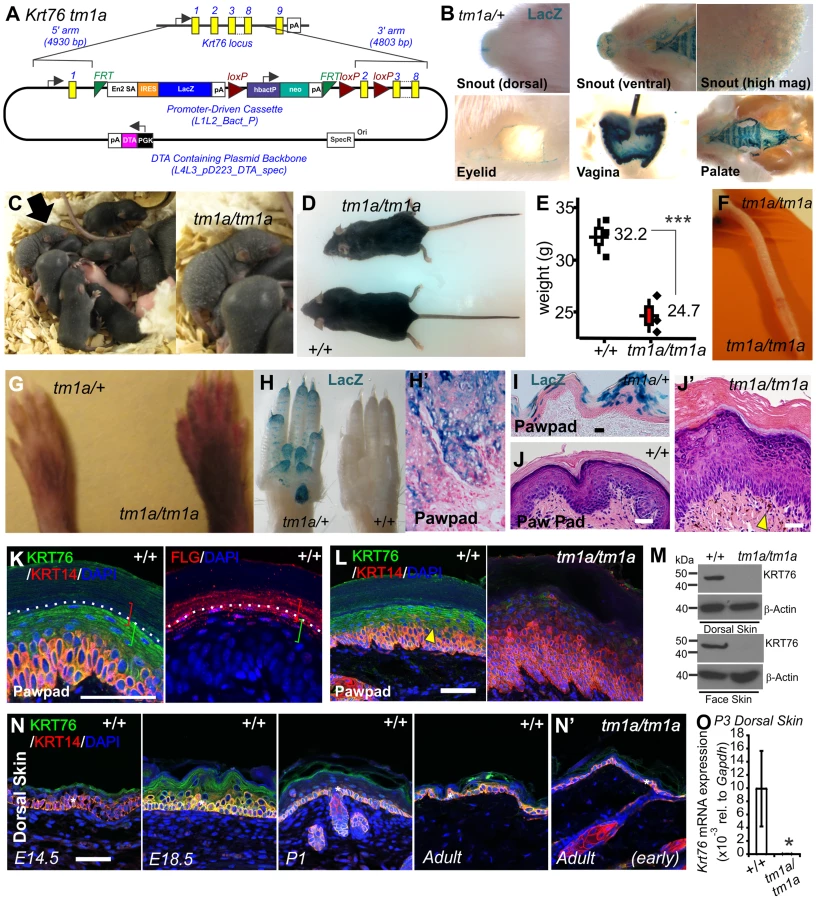
Krt76tm1a mice were then further back-crossed onto a C57BL/6 genetic background and bred back to homozygosity to determine the full consequences of Krt76 disruption. Krt76tm1a/tm1a neonates exhibit flaky skin (Figure 1C, arrow-insert), although these defects diminish somewhat with the emergence of hair follicles. After weaning, mutant mice are distinguished by their unkempt, dull coats and smaller body size (Figure 1D, E) and by scaling of skin of the tail (Figure 1F) [26]. Krt76tm1a/tm1a mice also show abnormal paw pad hyperpigmentation (Figure 1G) which corresponds with Krt76 expression as reported by LacZ, which is observed throughout the stratified epidermal layers and in the exocrine glands (Figure 1H, H′, I).
Haematoxylin and Eosin (H&E) staining of wild type (WT) and Krt76tm1a/tm1a paw pads revealed an overall epidermal thickening, with reduced granular layer compaction and an increased cornified layer (Figure 1J, J′). Pigment was also observed in the dermis (Figure 1J′ see arrowhead). To further explore KRT76 expression, we performed immuno-staining on paw pad skin using antibodies raised against human KRT76 epitopes that are predicted to be disrupted in tm1a animals. Specific KRT76 expression was highest in the granular cell layer where it overlapped with Filaggrin (FLG) in serial sections (Figure 1K). This site of expression correlates with β-galactosidase activity detected via the integrated reporter gene (Figure 1I). Importantly granular layer staining was absent from Krt76tm1a/tm1a animals (Figure 1L) and Western blots from epidermal extracts of the mid-dorsum and face epidermis confirmed that the tm1a allele results in complete loss of KRT76 protein (Figure 1M). We did detect low levels of immuno-staining in basal keratinocytes in Krt76tm1a/tm1a skin which was slightly reduced compared to wild type mice when imaging was performed using the same confocal settings. Given the unequivocal Western results, one interpretation is that this basal signal is a combination of non-specific cross reactivity and low levels of bona fide expression at this location. However, we cannot exclude the possibility that this change instead relates to alterations in expression of the cross-reacting species that might occur as a consequence of loss of KRT76. Further studies, perhaps using different antibodies, would be required to confirm this.
To examine a developmental role for the gene we profiled protein expression during embryonic and postnatal skin development, showing increasing levels of protein associated with the differentiation of the skin during late embryonic development, followed by a subsequent reduction in expression levels after birth (Figure 1N). Importantly though, low levels of KRT76 were still detectable in the spinous and granular cell layers in intact adult dorsal skin (Figure 1N). As expected, KRT76 protein was absent in Krt76 tm1a/tm1a animals by both immunofluorescence (compare Figure 1N with Figure 1N′) and qRT-PCR (Figure 1O), further validating this allele as a bona fide knockout model. This profile also suggests KRT76 may have a role in the later steps of keratinocyte differentiation, an observation which correlates with the flaky skin phenotypes observed in Krt76tm1a/tm1a neonates (Figure 1C).
Krt76 is associated with barrier maturation wound healing
As they age, Krt76tm1a/tm1a mice develop spontaneous wounds that fail to heal, especially on the dorsal skin around sites of active grooming (Figure 2A, see arrow head). Histological examination showed no obvious phenotypic change in young Krt76tm1a/tm1a mice prior to significant wound acquisition (which we refer to as the “early” phenotype), but large scabs, immune dermal infiltrates, extreme IFE thickening (Figure 2B) and hyperpigmentation in the dermis and epidermis develop over time (arrrowheads, Figure 2B). Phospho-histone H3 staining demonstrated a hyperproliferative response in these mice (Figure 2C, D). The morbidity associated with loss of KRT76 is such that animals rarely survive beyond 12 weeks of age. To assess whether cutaneous bacterial infection of these spontaneous wounds may exacerbate morbidity, we treated Krt76tm1a/tm1a mice with a broad spectrum antibiotic (Baytril) and observed a considerable improvement in lifespan (median survival = 70 days versus 32 (p<0.04)) (Figure 2E).

The wounding phenotypes associated with Krt76tm1a/tm1a mice led us to examine whether KRT76 was directly involved in the healing of induced wounds. As a first step in addressing this question we sampled dorsal skin from Krt76tm1a/+ and WT mice 1, 3, 5, 7, and 10 days after wounding by punch biopsy to examine gene expression. Immunofluorescence staining for KRT76 showed an up-regulation of KRT76 protein in the healing wound 5 days after injury (Figure 2F). This was confirmed by LacZ staining in Krt76tm1a/+ wound sections (Figure 2G). Expression profiling by qRT-PCR in WT mice confirmed Krt76 mRNA upregulation in response to wounding, with a profile slightly delayed in comparison to the “classical” wounding keratins Krt6b and Krt16 (Figure 2H). Similar punch biopsy experiments in the dorsal epidermis in Krt76tm1a/tm1a mice resulted in a significant impairment in wound closure at day 3 and day 5, correlating with the peak of Krt76 expression in the wound (Figure 2I). These observations indicate that that KRT76 is normally upregulated in response to skin damage and is required to facilitate wound healing during the latter phases of this process.
Biochemical analysis of Krt76 disrupted skin
We next examined whether the skin of Krt76tm1a/tm1a mice underwent a normal program of differentiation. The basal keratin marker, Keratin 14 (KRT14) and the hair follicle expression of Keratin 6 (KRT6) [27] were normal in “early” phenotype Krt76tm1a/tm1a dorsal skin but both expanded in the interfollicular epidermis (IFE) of “late” phenotype Krt76tm1a/tm1a indicative of a wounding response (Figure 3A). Likewise, the psoriasis and wounding associated factor, Fatty acid binding protein 5 (FABP5) [28], [29], showed normal weak suprabasal IFE expression in WT and “early” phenotype Krt76tm1a/tm1a mice which increased dramatically when wounds developed in “late” phenotype Krt76tm1a/tm1a mice (Figure 3B). Keratin 10 (KRT10), a marker of the stratum spinosum, and Filaggrin (FLG), a marker of the stratum granulosum, were again normal in early phenotype Krt76tm1a/tm1a dorsal skin but expanded upon wounding in late phenotype mice (Figure 3C, D). We also surveyed lipid profiles of the cornified envelope with Nile Red, demonstrating that the deposition of extracellular lipid lamellae were unaffected in mutant animals (Figure 3E). The terminal products of epidermal differentiation, the corneocytes, also appeared to form normally, albeit with a small but significant reduction in surface area which we propose derives from hypercellularity in the epidermis (Figure 3F). While the overall differentiation of keratinocytes in late phenotype Krt76tm1a/tm1a dorsal skin was mostly normal, the hyperplasia, immune infiltrate and IFE expression of KRT6 and FABP5, were reminiscent of the hyperproliferative skin disorder, psoriasis [30], [31], [32]. We also observed enlargement of sebaceous glands shown histologically in Figure 2B and further indicated by sebocyte markers, FABP5 and FASN [33], [34] (Figure 3B, G). Hyperpigmentation was also analysed using an MELAN-A (MEL-A) antibody which revealed dermal melanocytes were abnormally increased in density in the dermis and pigment increased in late phenotype Krt76tm1a/tm1a epidermis (Figure 3H). Their location concurred with the increased incidence of pigment detected in H&E sections [35] (Fig. 2B, upper arrowhead).

The progressive deterioration of the skin in these mice led us to examine whether the barrier function and integrity of the skin was compromised as a result of loss of KRT76 function. Dorsal skin from 3 week old mice (without overt wounding) were subjected to toluidine blue dye exclusion tests and no dye penetrance was observed indicating an intact outside to inside barrier (Figure 3I). Unlike other models of intermediate filament dysfunction, we observed no evidence of cell fragility and intraepidermal cell breakages by histology. This was confirmed using tape stripping assays, which showed no increased susceptibility to dye uptake (Figure S1A) and similar yields of corneocytes in tape stripping assays (Figure S1B).
Histological and biochemical analysis of conditional Krt76 knockout skin
To further confirm that the phenotypes we observed were representative of a null allele, and to confirm the phenotype we observed was driven by gene deletion in the epidermis and not in another organ, we generated a conditional KRT76 allele by crossing these mice with a flippase expressing line to remove the LacZ and NeoR cassettes; thereby generating a Krt76tm1c allele (Figure 4A). Mice homozygous for Krt76tm1c were functionally and phenotypically wild type. This allele was then manipulated to achieve gene deletion by crossing to Cre-driver strains (Figure 4A and Protocol S1). Global gene inactivation using CMV-Cre recapitulated the gene trap phenotype, resulting in early postnatal lethality. Temporally controlled Krt76 deletion specifically in the epidermis was achieved by topical application of 4-hydroxytamoxifen (4OHT) to the dorsal skin of 8 weeks old Krt76tm1c/tm1c mice carrying a K14-CreER transgene. These animals (Krt76tm1d/tm1d) showed regions of IFE hyperplasia and wounding after 3 weeks of treatment (Figure 4B) which was consistent with KRT76 deletion in these areas (Figure 4C, see granular layer absence indicated by arrowhead). As with Krt76tm1a/tm1a mice, hyperproliferation was increased in conditional knockouts (Figure 4D) as well as up-regulation of KRT14, KRT6 and FABP5 IFE expression (Figure 4E, F). Both KRT10 and FLG cell layers appeared to differentiate in normal sequence and showed wound related expansion (Figure 4G, H). The sebaceous glands were again enlarged, as shown by both FABP5 and FASN staining (Figure 4F, I) and like genetrap Krt76tm1a/tm1a mice, an increase in Melanin-A reactivity was seen (Figure 4J). Taken together these experiments confirm that the phenotypes we observe in these mice are due to epidermal specific knockout of KRT76.

Krt76 mutant mice show barrier function defects
Hyper-proliferation, induction of wounding keratins, unresolved wounds, and follicular dysmorphology are phenotypes associated with a loss of barrier function. Neonatal barrier function in dorsal skin was examined using a transepidermal water loss (TEWL) assay and identified a significant defect in the cutaneous barrier in Krt76tm1a/tm1a pups compared to their control littermates (Figure 5A). Importantly, this dorsal skin defect (at P3) was apparent before obvious skin wound lesions develop. As our previous phenotypic characterisation indicated this barrier function breakdown was unlikely to be linked to overt defects in cell stability, epidermal stratification, lipid deposition or terminal differentiation we examined tight junctions (TJ). Loss of TJ functionality can result in a compromised epidermal barrier independent of defects in lipid deposition or keratinocyte differentiation [16], [17]. Furthermore, alterations in TJ proteins are an early event in psoriasis [20], a disease with phenotypes that parallel some of those evident in Krt76tm1a/tm1a and Krt76tm1d/tm1d mice. To investigate TJ integrity we subcutaneously injected P3 mouse paw pads with membrane impermeable Sulfo-NHS-Biotin and tracked its diffusion using streptavidin immunohistochemistry. In WT epidermis, the diffusion of this high molecular weight compound was restricted before the interface of the granular and cornified layers, defined by FLG expression (Figure 5B), but in Krt76tm1a/tm1a littermates the tracer was detected within the cornified layer (Figure 5B, see arrowhead). Co-staining with a cell surface marker (CLDN1) showed regions of distal dye exclusion in wild type animals (Figure 5C, see region defined by arrowheads), which were absent in mutant mice, further indicating that TJ function in these animals was disrupted (Figure 5C). The ultrastructure of TJ's in P3 paw pad was grossly normal (e.g. kissing points) and their number and position were comparable to their WT and heterozygote littermates (Figure S1C). Desmosomes also appeared normal (Figure S1C).

Krt76 stabilises Claudin1 at tight junctions
In assessing the diffusion of the biotin tracer in paw pad skin we noted that CLDN1 exhibited broader margins at the cell periphery and acquired a partial (albeit weak) nuclear localization (Figure 5C). This altered distribution was also observed and quantified in samples stained with CLDN1, DAPI and E-cadherin (Figure 5D, E, F), which confirmed the inward shift and partial nuclear localisation. While CLDN1 is typically a cytoplasmic protein, nuclear redistribution of CLDN1 has been previously reported [36]. Dorsal skin from young animals taken prior to the development of wounding phenotypes also exhibits mislocalisation of membranous CLDN1 (Figure 5G) and this was further exacerbated when wounds formed (Figure 5H), although CLDN1 in the nucleus was not evident at this anatomical site (Figure 5I). Mislocalisation was also confirmed in Krt76tm1d/tm1d samples (Figure 5J, K). No difference in total CLDN1 protein levels were observed in mutant skin relative to ECAD (Figure 5I) nor was there a difference in Cldn1 mRNA expression (Figure S1D). This data collectively suggests KRT76 is required to correctly position CLDN1. Analysis of other TJ components ZO-1 and OCLN confirmed that the mislocalisation was specific to CLDN1 (Figure S2 and S3). In conclusion, our observations using several different experimental approaches indicate that KRT76 is required for normal TJ composition and in particular, the correct membrane localization of CLDN1.
KRT76 interacts with Claudin1
Given that KRT76 is required for normal CLDN1 localization we next assessed a possible physical association between the proteins. Although KRT76 antibodies proved unsuitable for co-immunoprecipitation experiments, we were able to express the tail domain of the protein and conjugate this to nickel magnetic beads. Paw pad lysates were then applied to the beads and interacting proteins eluted. Using this approach we were able to identify a specific interaction between the tail domain of KRT76 and endogenous CLDN1 (23 kDa) and a second higher molecular weight species (∼50 kDa) which may represent previously reported CLDN1 dimers [37], [38]. No such interactions were observed with the HIS-tag control protein (Figure 6A). These bands were absent from samples containing bound HIS-tail domain protein not incubated with paw skin extracts. This assay thereby shows that KRT76 can physically complex with CLDN1 although we cannot determine if this interaction is direct or indirect. The available reagents meant that performing the reverse reaction (pull-down on CLDN1) was impossible in the mouse, so we instead employed the human A549 adenocarcinomic alveolar basal epithelial cell line which we determined to endogenously express both proteins (Figure 6B). Using these cells we were able to co-immunoprecipitate KRT76 with CLDN1. Furthermore, ZO-1 (another TJ component) did not form part of this interaction complex, indicating the interaction between KRT76 and CLDN1 is specific amongst TJ components (Figure 6B). CLDN1 and KRT76 were also observed to co-localise in punctate structures within the cytoplasm of A549 cells (Figure 6C, see arrowheads). These interaction and co-localization assays confirm a physical complex between CLDN1 and KRT76 which we propose is important for mediating the barrier dysfunction and wound healing phenotypes of the KRT76 knockout mouse.

Discussion
The keratins are classically regarded as structural proteins whose role is to form the fabric of the cytoskeleton and to stabilise epithelial cells. However, this somewhat simplistic view has increasingly been challenged by the description of their specialised and dynamic functions in a number of cellular and developmental contexts. The keratins are the most diverse class of intermediate filament proteins and in many cases their functions are poorly defined. In this study we describe the characterisation of KRT76, one of the least understood of the protein family, delineating its essential role in the maintenance of the integrity of the skin. Under resting conditions, Krt76 is expressed at its highest levels in the paw, oral epithelium and vagina, localising to the granular layer. It is also expressed in the dorsal epithelium, particularly during the late stages of embryonic development. Wounding induces Krt76 expression, although the profile of this induction is distinct from other wounding keratins like Krt6 and Krt16.
To examine the functional relevance of this expression and its role in epidermal homeostasis we inactivated the gene in mice globally and in a skin specific manner. Loss of Krt76 results in the rapid appearance of extensive non-healing wounds (especially at sites of active grooming), and the subsequent infection of these lesions contributes significantly to morbidity and mortality in the mice. Unlike other knockout models of structural keratins we failed to observe cytolysis and/or blistering in the skin. Instead we observed a relatively unperturbed program of keratinocyte differentiation although this gives way to a phenotype of hyperproliferation as the phenotype of the animals worsens. What triggers this change remains to be determined, however the frequency of wounds around active grooming sites suggests that KRT76 deletion may impair the ability of the skin to recover from physical insults normally experienced in the life of the mouse. This theory is supported by the demonstration that induced wounds in the skin of these mice, administered prior to the accumulation of significant cutaneous damage, failed to heal normally.
As well as the progressive wounding phenotype observed in these mice, we also noted cellular changes which were consistent with defects in the barrier function of the epidermis. This was confirmed using trans-epidermal water loss assays in neonatal animals. We were unable to establish a role for defective keratinocyte stability or termination in driving this defect, nor was lipid transport affected in the mice to any appreciable level. Instead, we observed the specific mislocalisation of the TJ component CLDN1, even in newborn mice and in animals without overt or severe cutaneous defects. Indeed previous reports have shown that even significant hyperproliferation induced by two step carcinogenesis treatments is unable to elicit similar changes [39]. Although TJs appeared normal at an ultra-structural level, their reduced capacity to limit the movement of molecules between differentiating keratinocytes in our mice suggests that they were functioning abnormally. Importantly mislocalisation was not observed for other structural elements of the TJ. It is therefore notable that the phenotype of the Krt76 KO mice is strikingly similar to animals carrying homozygous mutations in Cldn1 [17]. In both cases, barrier function defects are detectable by biotin tracer and TEWL assays (but not by dye exclusion), and both have apparently normal formation of TJ structures as assessed by EM. Overall, the phenotypes of Krt76 null mice are somewhat milder than their CLDN1 counterparts, suggesting that despite loss of KRT76, some CLDN1 can still contribute to partial TJ function. By studying both skin extracts and cell lines endogenously expressing both CLDN1 and KRT76 we were able to demonstrate a physical association between these proteins, mediated by the tail domain of the latter. At present we do not know whether this interaction is direct, or whether the proteins exist in a larger complex. In either case, the loss of KRT76 is clearly required for normal tight junction function and for CLDN1 localisation. Although links between the tight junction and the cytoskeleton have been described for actin, this is the first report detailing an interaction with the keratin intermediate filaments.
In summary, we believe that the KRT76 protein represents a new and essential protein required for maintaining epidermal integrity. Its expression during fetal development and during wound healing suggests it is required to establish and/or stabilise the development of TJs in differentiating keratinocytes, specifically through mediating the correct localisation off CLDN1 to these structures. Deletion of the protein leads to defects in TJ function that are at least in part associated with the development of progressively worsening wounds. Whether this severe later phenotype, which ultimately leads to the death of the animals, reflects a separate, non-TJ, role for the protein in wound repair is unclear. Mislocalization of CLDN1 is a feature of a number of cutaneous diseases such as psoriasis [40], and in a number of cancers [36], [41], [42]. KRT76 depletion has also been linked with human oral carcinomas and premaligant epidermal changes [43]. It will therefore be interesting to determine the extent to which this new cytoskeletal-TJ interface between CLDN1 and KRT76 interaction plays a role in the development or progression of these diseases.
Materials and Methods
Ethics statement
Animal models were maintained under the auspices of ethics applications to Monash University and subject to the conditions of the Australian Bureau of Animal Welfare.
Transgenic mice
Krt76tm1a/tm1a mice were generated in the Mouse Genetics Programme at the Wellcome Trust Sanger Institute [24]. Animals were bred and maintained on a mixed background of C57BL/6JTyrcBrd; C57BL/6N. The Krt76tm1a/tm1a characterisation data presented is available at www.mousephenotype.org. Targeting vector information is available at http://www.mousephenotype.org/martsearch_ikmc_project/martsearch/ikmc_project/38047. Flip recombinase (Flipper) mice [44], K14-CreER mice [45] and CMV-Cre mice [46] have been described previously. 1.5 mg of 4-hydrotamoxifen (H6278, Sigma-Aldrich) was applied to a shaved region of lower back skin in 100 µl of acetone every second day for 21 days before mice were harvested for analysis.
Genotyping transgenic mice
The PCR conditions were set for amplification of small PCR fragments only. Details of primer sequences, reaction composition and cycling profile are provided in Protocol S1.
Histological preparation and staining
Staining for LacZ expression was performed as previously described [47] on frozen sections and counterstained with Nile Red. Immunofluorescence experiments were performed after citrate based antigen retrieval. Primary antibodies were ZO-1 (Invitrogen cat# 339100), Occludin (BD Transduction cat# 611090), Claudin-1 (ABCAM cat# ab15098), cytokeratin14 - LLOO2 (ABCAM cat # ab7800), keratin10 (Covance PRB-159P), keratin6 (Covance cat # PRB-169P), Ecadherin (Life Technologies, 13-1900), phospho-histone H3 (Cell Signalling, #9708), PCNA (Santa Cruz Biotechnology sc-9857), CLDN1 (Santa Cruz Biotechnology, sc-81796), Keratin 76 (Sigma-Aldrich HPA019696) and Keratin 76 (Sigma-Aldrich HPA019656), Filaggrin (FLG - Covance PRB-417P), FASN (Santa Cruz Biotechnology, sc-48357), and Melan-A (MEL-A, Santa Cruz Biotechnology, sc-20032). All secondary antibodies were AlexaFluor conjugated (Invitrogen).
Imaging and analysis
Sections were imaged using Lecia SP5 5 Channel, Olympus FV500 confocal microscopes or Aperio slide scanners. Bright field images of wound healing experiments were taken with Olympus dotslide brightfield microscope. Images for CLE assays were acquired with Olympus CKX41 and exported to FIJI software [48] for cell analysis.
Wound healing experiments
Mice (age-matched males; 6 weeks) were isofluorane-anaesthetized and 2 full-thickness excisional wounds were made with a 5 mm biopsy punch (Livingstone International). Wound tissue was harvested with an 8 mm biopsy punch.
qRT-PCR
One µg of DNase (Ambion) treated RNA was used for cDNA synthesis (SuperScriptVILO). Multiplex quantitative PCR was performed using Taqman probes for Gapdh (VIC-primer limited labelled, cat# 4448484) and Krt76 (FAM labelled, Cat#4351372) with TaqMan Fast Advanced Master Mix Protocol (PN 4444605B). Gene specific primers were designed and used in conjunction with SYBR Green PCR Master Mix (Applied Biosystems) for the detection and quantification of Claudin1 (5′-ATTTCAGGTCTGGCGACATT-3′ fwd, 5′-ACACCTCCCAGAAGGCAGAG-3 - rev) , Krt6b (5′-CAGACCCCAGATACCCTGGC-3′ fwd, 5′-GAGCAGAGATGGCATCATGTGAGCAACAGG-3′ rev) , Krt16 (5′ - AACAGCCTAGAAGAGACCAAAGGC-3′ fwd, 5′-GGTAGGGGAGACAGATGGGGAATGCGC-3′ rev) mRNA as compared to Gapdh (5′ - CTGCACCACCAACTGCTTAG-3′ fwd,5′ - GTCTTCTGGGTGGCAGTGAT-3′ rev).
Protein fractionation
All fractionation experiments were performed on dorsal epidermis of P3 animals. Pups were euthanized (Pentobarbital) and skin was removed as previously described [49]. Skins were floated on 2.3 U/mL Dispase (Life Technologies) in PBS overnight at 4°C. The epidermis was separated from the dermis and protein fractionated using a Qproteome Cell Compartment Kit (Qiagen). Western blots for E-cadherin (Life Technologies) and total Histone H3 (Cell Signaling) were performed on the nuclear and membrane fractions. Image Quant software was used to calculate densitometry and quantify protein levels. Claudin-1 levels in the membrane fraction were normalized to E-cadherin for each sample.
Biotin tracer assays
TJ permeability assays were undertaken as previously described [17], [50]. Briefly, a solution of 10 mg/ml EZ-Link Sulfo-NHS-LC-Biotin (Pierce) in PBS containing 1 mM CaCl2 was injected into the paw pads of P3 pups. Paw pads were incubated at room temperature for 30 minutes prior to frozen sectioning and IHC with conjugated Streptavidin Alexafluor 594 (Life Technologies, S-11227).
Ultrastructural analysis
Tissue was fixed in Karnovsky's fixative (2% paraformaldehyde, 2.5% glutaraldehyde in 0.1 M Cacodylate buffer) for 2 hours. Then washed in 3×10 min changes of 0.1 M Cacodylate buffer. Post-fixation was with 2% osmium tetroxide in 0.1 M Cacodylate buffer followed by dehydration through a graded series of alcohols, two acetone rinses and embedding in Spurrs resin. 80 nm sections were cut with a diamond knife (Diatome, Switzerland) on an Ultracut-S ultramicrotome (Leica, Mannheim, Germany) and contrasted with uranyl acetate and lead citrate. Images were captured with a Megaview II cooled CCD camera (Soft Imaging Solutions, Olympus, Australia) in a JEOL 1011 transmission electron microscope.
HIS-tag Pull-down from Mammalian Lysates
Recombinant HIS-tagged proteins were produced by IPTG induction (0.4 mM) of T7 Express lysY/Iq Competent E.coli (New England Biolabs C3013I) transformed with HIS-tag expressing control vector, pET-30a+ (Novagen) or HIS-tagged KRT76 domains in pDEST17 gateway backbone (Life Technologies), grown for 6–8 hours at 37°C in low salt LB, supplemented with 100 µg/ml ampicillin or 50 µg/mL kanamycin (as required). Recombinant protein was purified using 0.1 ml per 1 ml of culture of PopCulture lysis reagent (Novagen), 1 µl per mL of culture of 40 U/ml of Lysonase bioprocessing reagent (Novagen), protease inhibitors (Sigma P8849), and His-Mag beads (Novagen) according to manufacturer's protocols. Bound recombinant HIS and HIS-KRT76 protein were washed and stored at 4°C as a 1∶2 resin slurry in Tris-saline pH 7.4 containing protease inhibitors. Paw pad skin of adult was collected in RIPA lysis buffer and incubated with HIS or HIS-KRT76 overnight at 4°C. HisMag bead-bound HIS and HIS-KRT76 + lysates were then washed four times in Tris-saline pH 7.4 including 1% Triton X-100 and immunoblotted for mCLDN1 (Santa Cruz Biotechnology, SC-81796) and the HIS tag (Sigma-Aldrich, clone HIS-1).
A549 analysis and CO-IP
A549 cells (ATCC CCL-185) were cultured in low-glucose DMEM including 10% FCS, Penicillin Streptomycin and L-glutamine. For CO-IP, cells at confluence were scraped and lysed in 1% Triton X-100 in 1xTBS with Roche complete protease inhibitor tablet, extracted for 2 hrs at 4°C then supernatant collected. The supernatant was applied to binding columns prepared using the Pierce Crosslink IP Kit and CO-IP performed as per manufacturers protocol. Bound fractions were washed 3 times in lysis buffer before elution and standard WB analysis. For Immunofluorescence, 2×105 cells were seeded on Collagen type 1 coated glass coverslips in 6 well plate format and processed as previously described [51].
Dye exclusion
E18.5 embryos or 3 week old dorsal skin were collected and transferred through a Methanol gradient with emersion for 1 minute each: 25% methanol in water, 50% methanol in water, 75% methanol in water, 100% methanol, 75% methanol in water, 50% methanol in water, 25%methanol in water and equilibrated in PBS. All reagents were chilled. Tissue was then exposed to 0.1% Toluidine Blue solution in water for 2 minutes and destained in 1xPBS pH7.4. For tape stripping, clipped dorsal skins were first tape stripped twelve times with adhesive tape before tissue collection.
Cornified envelope assay
Analysis of the size of corneocytes in the cornified lipid envelope (CLE) assay was performed as previously published [52].
Statistical analysis
Statistical analysis was performed using unpaired students test, values of p<0.05 were deemed significant. A minimum of 3 mice were analysed per condition unless otherwise stated. In graphs, error bars represent Standard Error of the Mean (S.E.M).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. FuchsE, WeberK (1994) Intermediate filaments: structure, dynamics, function, and disease. Annu Rev Biochem 63 : 345–382.
2. GeislerN, WeberK (1982) The amino acid sequence of chicken muscle desmin provides a common structural model for intermediate filament proteins. EMBO J 1 : 1649–1656.
3. LaneEB, McLeanWH (2004) Keratins and skin disorders. J Pathol 204 : 355–366.
4. ParryDA, StrelkovSV, BurkhardP, AebiU, HerrmannH (2007) Towards a molecular description of intermediate filament structure and assembly. Exp Cell Res 313 : 2204–2216.
5. FuchsE, ClevelandDW (1998) A structural scaffolding of intermediate filaments in health and disease. Science 279 : 514–519.
6. OsbornM (1983) Intermediate filaments as histologic markers: an overview. J Invest Dermatol 81 : 104s–109s.
7. SimpsonCL, PatelDM, GreenKJ (2011) Deconstructing the skin: cytoarchitectural determinants of epidermal morphogenesis. Nat Rev Mol Cell Biol 12 : 565–580.
8. OmaryMB, KuNO, StrnadP, HanadaS (2009) Toward unraveling the complexity of simple epithelial keratins in human disease. J Clin Invest 119 : 1794–1805.
9. CoulombePA, OmaryMB (2002) 'Hard' and 'soft' principles defining the structure, function and regulation of keratin intermediate filaments. Curr Opin Cell Biol 14 : 110–122.
10. KuNO, ZhouX, ToivolaDM, OmaryMB (1999) The cytoskeleton of digestive epithelia in health and disease. Am J Physiol 277: G1108–1137.
11. OrioloAS, WaldFA, RamsauerVP, SalasPJ (2007) Intermediate filaments: a role in epithelial polarity. Exp Cell Res 313 : 2255–2264.
12. ToivolaDM, TaoGZ, HabtezionA, LiaoJ, OmaryMB (2005) Cellular integrity plus: organelle-related and protein-targeting functions of intermediate filaments. Trends Cell Biol 15 : 608–617.
13. KimS, WongP, CoulombePA (2006) A keratin cytoskeletal protein regulates protein synthesis and epithelial cell growth. Nature 441 : 362–365.
14. XuJ, MarzettiE, SeoAY, KimJS, ProllaTA, et al. (2010) The emerging role of iron dyshomeostasis in the mitochondrial decay of aging. Mech Ageing Dev 131 : 487–493.
15. KimS, CoulombePA (2010) Emerging role for the cytoskeleton as an organizer and regulator of translation. Nat Rev Mol Cell Biol 11 : 75–81.
16. NiessenCM (2007) Tight junctions/adherens junctions: basic structure and function. J Invest Dermatol 127 : 2525–2532.
17. FuruseM, HataM, FuruseK, YoshidaY, HaratakeA, et al. (2002) Claudin-based tight junctions are crucial for the mammalian epidermal barrier: a lesson from claudin-1-deficient mice. J Cell Biol 156 : 1099–1111.
18. KirschnerN, BohnerC, RachowS, BrandnerJM (2010) Tight junctions: is there a role in dermatology? Arch Dermatol Res 302 : 483–493.
19. De BenedettoA, RafaelsNM, McGirtLY, IvanovAI, GeorasSN, et al. (2011) Tight junction defects in patients with atopic dermatitis. J Allergy Clin Immunol 127 : 773–786–e771–777.
20. KirschnerN, PoetzlC, von den DrieschP, WladykowskiE, MollI, et al. (2009) Alteration of tight junction proteins is an early event in psoriasis: putative involvement of proinflammatory cytokines. Am J Pathol 175 : 1095–1106.
21. FanningAS, JamesonBJ, JesaitisLA, AndersonJM (1998) The tight junction protein ZO-1 establishes a link between the transmembrane protein occludin and the actin cytoskeleton. J Biol Chem 273 : 29745–29753.
22. WhiteJK, GerdinAK, KarpNA, RyderE, BuljanM, et al. (2013) Genome-wide generation and systematic phenotyping of knockout mice reveals new roles for many genes. Cell 154 : 452–464.
23. DiTommaso T, Jones L, Cottle DL, Program. TWMG, Gerdin A-K, et al. (2014) Identification of genes important for cutaneous function revealed by a large scale reverse genetic screen in the mouse. PLoS Gen, DOI: 101371/journalpgen1004705
24. SkarnesWC, RosenB, WestAP, KoutsourakisM, BushellW, et al. (2011) A conditional knockout resource for the genome-wide study of mouse gene function. Nature 474 : 337–342.
25. CollinC, OuhayounJP, GrundC, FrankeWW (1992) Suprabasal marker proteins distinguishing keratinizing squamous epithelia: cytokeratin 2 polypeptides of oral masticatory epithelium and epidermis are different. Differentiation 51 : 137–148.
26. Liakath-AliK, VancollieVE, HeathE, SmedleyDP, EstabelJ, et al. (2014) Novel skin phenotypes revealed by a genome-wide mouse reverse genetic screen. Nat Commun 5 : 3540.
27. PaladiniRD, TakahashiK, BravoNS, CoulombePA (1996) Onset of re-epithelialization after skin injury correlates with a reorganization of keratin filaments in wound edge keratinocytes: defining a potential role for keratin 16. J Cell Biol 132 : 381–397.
28. MadsenP, RasmussenHH, LeffersH, HonoreB, CelisJE (1992) Molecular cloning and expression of a novel keratinocyte protein (psoriasis-associated fatty acid-binding protein [PA-FABP]) that is highly up-regulated in psoriatic skin and that shares similarity to fatty acid-binding proteins. J Invest Dermatol 99 : 299–305.
29. OgawaE, OwadaY, IkawaS, AdachiY, EgawaT, et al. (2011) Epidermal FABP (FABP5) regulates keratinocyte differentiation by 13(S)-HODE-mediated activation of the NF-kappaB signaling pathway. J Invest Dermatol 131 : 604–612.
30. BaranW, SzepietowskiJC, Szybejko-MachajG (2005) Expression of p53 protein in psoriasis. Acta dermatovenerologica Alpina, Panonica, et Adriatica 14 : 79–83.
31. de RieMA, GoedkoopAY, BosJD (2004) Overview of psoriasis. Dermatologic therapy 17 : 341–349.
32. ThewesM, StadlerR, KorgeB, MischkeD (1991) Normal psoriatic epidermis expression of hyperproliferation-associated keratins. Archives of dermatological research 283 : 465–471.
33. BertaMA, BakerCM, CottleDL, WattFM (2010) Dose and context dependent effects of Myc on epidermal stem cell proliferation and differentiation. EMBO Mol Med 2 : 16–25.
34. CottleDL, KretzschmarK, SchweigerPJ, QuistSR, GollnickHP, et al. (2013) c-MYC-Induced Sebaceous Gland Differentiation Is Controlled by an Androgen Receptor/p53 Axis. Cell reports 3 : 427–441.
35. LinJY, FisherDE (2007) Melanocyte biology and skin pigmentation. Nature 445 : 843–850.
36. FrenchAD, FioriJL, CamilliTC, LeotlelaPD, O'ConnellMP, et al. (2009) PKC and PKA phosphorylation affect the subcellular localization of claudin-1 in melanoma cells. Int J Med Sci 6 : 93–101.
37. SjoA, MagnussonKE, PetersonKH (2010) Protein kinase C activation has distinct effects on the localization, phosphorylation and detergent solubility of the claudin protein family in tight and leaky epithelial cells. J Membr Biol 236 : 181–189.
38. MrsnyRJ, BrownGT, Gerner-SmidtK, BuretAG, MeddingsJB, et al. (2008) A key claudin extracellular loop domain is critical for epithelial barrier integrity. Am J Pathol 172 : 905–915.
39. ArabzadehA, TroyTC, TurksenK (2007) Changes in the distribution pattern of Claudin tight junction proteins during the progression of mouse skin tumorigenesis. BMC cancer 7 : 196.
40. WatsonRE, PoddarR, WalkerJM, McGuillI, HoareLM, et al. (2007) Altered claudin expression is a feature of chronic plaque psoriasis. J Pathol 212 : 450–458.
41. DhawanP, SinghAB, DeaneNG, NoY, ShiouSR, et al. (2005) Claudin-1 regulates cellular transformation and metastatic behavior in colon cancer. J Clin Invest 115 : 1765–1776.
42. HoughCD, Sherman-BaustCA, PizerES, MontzFJ, ImDD, et al. (2000) Large-scale serial analysis of gene expression reveals genes differentially expressed in ovarian cancer. Cancer Res 60 : 6281–6287.
43. AmbatipudiS, BhosalePG, HeathE, PandeyM, KumarG, et al. (2013) Downregulation of keratin 76 expression during oral carcinogenesis of human, hamster and mouse. PloS one 8: e70688.
44. FarleyFW, SorianoP, SteffenLS, DymeckiSM (2000) Widespread recombinase expression using FLPeR (flipper) mice. Genesis 28 : 106–110.
45. VasioukhinV, DegensteinL, WiseB, FuchsE (1999) The magical touch: genome targeting in epidermal stem cells induced by tamoxifen application to mouse skin. Proc Natl Acad Sci U S A 96 : 8551–8556.
46. SchwenkF, BaronU, RajewskyK (1995) A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res 23 : 5080–5081.
47. Adams N, Gale N (2006) High Resolution Gene Expression Analysis in Mice Using Genetically Inserted Reporter Genes. In: Pease S, Lois C, editors. Mammalian and Avian Transgenesis — New Approaches: Springer Berlin Heidelberg. pp. 131–172.
48. SchindelinJ, Arganda-CarrerasI, FriseE, KaynigV, LongairM, et al. (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9 : 676–682.
49. LichtiU, AndersJ, YuspaSH (2008) Isolation and short-term culture of primary keratinocytes, hair follicle populations and dermal cells from newborn mice and keratinocytes from adult mice for in vitro analysis and for grafting to immunodeficient mice. Nat Protoc 3 : 799–810.
50. ChenY, MerzdorfC, PaulDL, GoodenoughDA (1997) COOH terminus of occludin is required for tight junction barrier function in early Xenopus embryos. J Cell Biol 138 : 891–899.
51. WiradjajaF, CottleDL, JonesL, SmythI (2013) Regulation of PDGFC signalling and extracellular matrix composition by FREM1 in mice. Dis Model Mech 6 : 1426–1433.
52. SmythI, HackingDF, HiltonAA, MukhamedovaN, MeiklePJ, et al. (2008) A mouse model of harlequin ichthyosis delineates a key role for Abca12 in lipid homeostasis. PLoS Genet 4: e1000192.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 10
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- The Master Activator of IncA/C Conjugative Plasmids Stimulates Genomic Islands and Multidrug Resistance Dissemination
- A Splice Mutation in the Gene Causes High Glycogen Content and Low Meat Quality in Pig Skeletal Muscle
- Keratin 76 Is Required for Tight Junction Function and Maintenance of the Skin Barrier
- A Role for Taiman in Insect Metamorphosis
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy