
Role of CTCF Protein in Regulating Locus Transcription
Fragile X syndrome (FXS), the leading cause of inherited intellectual disability, is caused by epigenetic silencing of the FMR1 gene, through expansion and methylation of a CGG triplet repeat (methylated full mutation). An antisense transcript (FMR1-AS1), starting from both promoter and intron 2 of the FMR1 gene, was demonstrated in transcriptionally active alleles, but not in silent FXS alleles. Moreover, a DNA methylation boundary, which is lost in FXS, was recently identified upstream of the FMR1 gene. Several nuclear proteins bind to this region, like the insulator protein CTCF. Here we demonstrate for the first time that rare unmethylated full mutation (UFM) alleles present the same boundary described in wild type (WT) alleles and that CTCF binds to this region, as well as to the FMR1 gene promoter, exon 1 and intron 2 binding sites. Contrariwise, DNA methylation prevents CTCF binding to FXS alleles. Drug-induced CpGs demethylation does not restore this binding. CTCF knock-down experiments clearly established that CTCF does not act as insulator at the active FMR1 locus, despite the presence of a CGG expansion. CTCF depletion induces heterochromatinic histone configuration of the FMR1 locus and results in reduction of FMR1 transcription, which however is not accompanied by spreading of DNA methylation towards the FMR1 promoter. CTCF depletion is also associated with FMR1-AS1 mRNA reduction. Antisense RNA, like sense transcript, is upregulated in UFM and absent in FXS cells and its splicing is correlated to that of the FMR1-mRNA. We conclude that CTCF has a complex role in regulating FMR1 expression, probably through the organization of chromatin loops between sense/antisense transcriptional regulatory regions, as suggested by bioinformatics analysis.
Published in the journal:
. PLoS Genet 9(7): e32767. doi:10.1371/journal.pgen.1003601
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003601
Summary
Fragile X syndrome (FXS), the leading cause of inherited intellectual disability, is caused by epigenetic silencing of the FMR1 gene, through expansion and methylation of a CGG triplet repeat (methylated full mutation). An antisense transcript (FMR1-AS1), starting from both promoter and intron 2 of the FMR1 gene, was demonstrated in transcriptionally active alleles, but not in silent FXS alleles. Moreover, a DNA methylation boundary, which is lost in FXS, was recently identified upstream of the FMR1 gene. Several nuclear proteins bind to this region, like the insulator protein CTCF. Here we demonstrate for the first time that rare unmethylated full mutation (UFM) alleles present the same boundary described in wild type (WT) alleles and that CTCF binds to this region, as well as to the FMR1 gene promoter, exon 1 and intron 2 binding sites. Contrariwise, DNA methylation prevents CTCF binding to FXS alleles. Drug-induced CpGs demethylation does not restore this binding. CTCF knock-down experiments clearly established that CTCF does not act as insulator at the active FMR1 locus, despite the presence of a CGG expansion. CTCF depletion induces heterochromatinic histone configuration of the FMR1 locus and results in reduction of FMR1 transcription, which however is not accompanied by spreading of DNA methylation towards the FMR1 promoter. CTCF depletion is also associated with FMR1-AS1 mRNA reduction. Antisense RNA, like sense transcript, is upregulated in UFM and absent in FXS cells and its splicing is correlated to that of the FMR1-mRNA. We conclude that CTCF has a complex role in regulating FMR1 expression, probably through the organization of chromatin loops between sense/antisense transcriptional regulatory regions, as suggested by bioinformatics analysis.
Introduction
Fragile X syndrome (FXS, OMIM #300624), the most studied and best known FRAXopathy, is the leading cause of inherited intellectual disability (ID) [1]. FXS is caused by the expansion beyond 200 repeats (full mutation) and subsequent methylation of the polymorphic CGG sequence within the 5′ untranslated region (5′ UTR) of the FMR1 gene, an X-linked gene which contains a CpG island in its promoter [2]. The methylation of cytosines of both the expanded CGGs and of the neighboring CpGs, as well as other heterochromatic histone modifications, cause the transcriptional silencing of the FMR1 gene and the lack of the FMRP protein [3], [4]. FMRP is an RNA-binding protein, which inhibits the translation of messenger RNAs (mRNAs), especially within post-synaptic vesicles of the dendritic spines. Its absence impairs synaptic plasticity, which is thought to be the cause of ID [5]. Previous reports described rare individuals of normal intelligence, carrying a transcriptionally active unmethylated full mutation (UFM) [6]–[8]. Cell lines derived from these individuals might reflect the status of FXS cells before epigenetic silencing, that is thought to occur at about 11 weeks of gestation [9]. Indeed, the epigenetic characterization of their FMR1 locus showed histone H3 and H4 hyperacetylation, lysine 4 of histone 3 (H3-K4) methylation, lysine 9 of histone 3 (H3-K9) hypomethylation, lysine 27 of histone 3 (H3-K27) dimethylation and lack of DNA methylation [7], [8]. This epigenetic status is compatible with an euchromatic conformation of the FMR1 locus, allowing transcription. A similar epigenetic status can be induced by treatment of FXS cells with the DNA demethylating agent 5-aza-2-deoxycytidine (5-azadC), which also causes histone changes (hyperacetylation, H3-K4 methylation), the latter actually preceding DNA demethylation [4], [10], [11]. In accordance with these results, silencing of FMR1 in human embryonic stem cells seems to begin from histone modifications prior to DNA methylation [12].
In FXS cell lines DNA methylation extends further to approximately 1 kb upstream the CGG repeat sequence [13]. In wild-type (WT) alleles a zone of transition between methylated and unmethylated sequences was described around 650 to 800 nucleotides upstream the CGG repeat, with CpGs being unmethylated all the way down to the FMR1 promoter. This methylation boundary (MB) appears to be lost in completely methylated FXS alleles. The boundary is also conserved in the mouse genome, even if human and mouse are only 46.7% identical in the 5′ region upstream the FMR1 gene [13].
Methylation boundary regions are characterized by the presence of binding sites for various nuclear proteins including CTCF (CCCTC-binding factor), the first insulator protein found in mammals [14]. CTCF is a widely expressed nuclear protein, which binds different DNA target sequences through its 11 zinc-finger domains [15], [16]. It was first discovered as a negative transcriptional regulator, interacting with various sequences in the promoter of the chicken, mouse and human C-MYC oncogene [17], [18]. Subsequent studies recognized its involvement in several functions, including transcriptional activation or repression, X chromosome inactivation, genomic imprinting, methylation-dependent chromatin insulation and higher-order chromatin organization through the establishment of DNA loops [19]–[22]. CTCF has been implicated in the organization of both the structure of the chromosomal fiber within each individual chromosome and of the chromosome territories within the cell nucleus. Many CTCF binding sites reside within promoters, as well as in inter - and intra-genic regions [23]. The relationship between CTCF binding patterns and DNA methylation is currently unknown. Pre-existing methylation can antagonize CTCF binding in vitro [24]–[26]. A recent study of overall methylation status showed that 98% of CTCF sites were unmethylated in at least one of the 13 cell types tested, confirming an inverse relationship between DNA methylation and CTCF occupancy [27]. Despite that, it is still unclear whether demethylation facilitates subsequent CTCF binding and whether bound CTCF maintains the corresponding domain in an unmethylated status.
An important regulatory role of CTCF was described in expanded triplet diseases. Specific binding sites for this protein were recognized flanking the CTG triplet at the DM1 locus of myotonic dystrophy [28]. Recent evidence suggests that both CTCF binding and CpG methylation may contribute to CTG repeats instability [29], [30]. In a transgenic mouse model for spinocerebellar ataxia type 7 (SCA7), CTCF regulates ataxin-7 gene expression and is required for SCAANT1 (SCA7 antisense noncoding transcript 1) expression. Loss of SCAANT1 de-represses ataxin-7 sense transcription in a cis-dependent manner and is accompanied by chromatin remodeling [31]. In Friedreich ataxia (FRDA), caused by expansion of a GAA repeat sequence in intron 1 of the FXN gene, CTCF depletion was observed in the 5′ UTR of the mutant alleles. This depletion is associated with high levels of the transcript antisense of FXN (FAST-1), supporting the hypothesis of an epigenetic silencing of the corresponding “sense” gene [32].
Four CTCF binding sites have been identified within the FMR1 locus, suggesting a role of this protein in the regulation of the gene [33]. In the same report, an antisense transcript of the FMR1 gene (FMR1-AS1) spanning the expanded CGG repeat was identified in normal and premutated alleles, but not in FXS alleles. The authors suggested a possible pathogenic role of FMR1-AS1 in FXS and also in the fragile X tremor-ataxia syndrome (FXTAS) associated with premutated alleles. However, they did not study the presence of the antisense transcript in UFM cells.
In this paper we investigate the role of CTCF in transcriptional regulation of the FMR1 gene and in chromatin organization of the corresponding locus including the methylation boundary region, in different cell lines derived from normal (WT), FXS and UFM individuals, respectively. Through molecular and bioinformatics approaches we demonstrate that CTCF does not preserve the methylation boundary of the FMR1 locus, but is required for its proper transcription. Significant results were obtained from the further characterization of the rare UFM cell lines by mapping the methylation boundary region and by measuring the FMR1 antisense transcript.
Results
Identification of methylation boundary and FMR1-AS1 in UFM cell lines
The extended region upstream the CGG repeats described by Naumann et al. [2009] [13] was analyzed in three classes of cell lines (WT, FXS and UFM), both lymphoblasts and fibroblasts. Bisulfite sequencing of the methylation boundary in WT cell lines confirmed the results already reported [13], with a DNA methylation boundary located at CpG pairs 70–71 in lymphoblastoid cells (Figure S1A) and 73–74 in fibroblasts (Figure S1B), respectively. As expected, no boundary was present in FXS cells. Despite the presence of the CGG expansion, the transcriptionally active UFM cell lines retained the methylation boundary as in WT cells, both in lymphoblasts and in fibroblasts (Figure S1A and B).
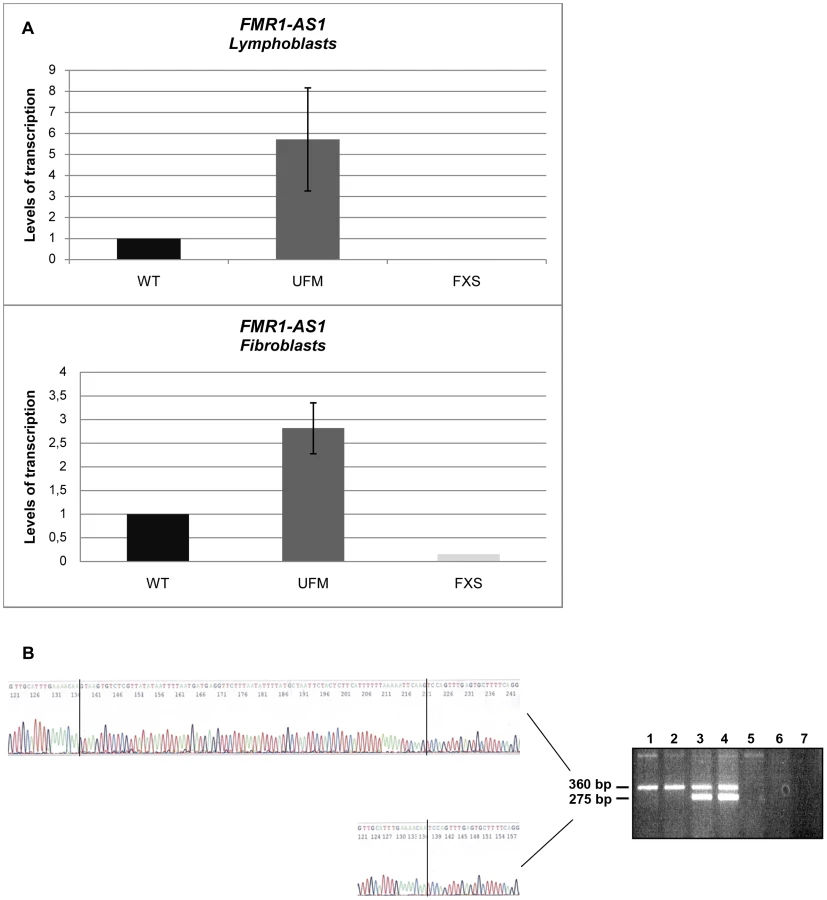
We went on to quantify FMR1-AS1 transcript levels and observed higher levels of transcription in UFM lymphoblasts (around 6-fold higher) and fibroblasts (around 3-fold higher) compared to WT, while no antisense transcript was detected in FXS cells, as expected [33] (Figure 1A). These results showed that the antisense transcript follows the same expression pattern as the sense RNA [8]. Amplification and sequencing analysis of FMR1-AS1 cDNA in WT and UFM cells confirmed the presence of the splicing corresponding to the intron 1 of the sense transcript (Figure 1B), despite the recognition of a non-canonical AC-CT splice site in the antisense mRNA. Moreover, UFM cells presented a second isoform of antisense transcript, which retained the non-canonical splicing in intron 2, like in premutation alleles [33] (Figure 1B). Based on FMR1-AS1 data, we may hypothesize a co-regulation mechanism for sense and antisense transcription at the FMR1 locus.
CTCF binding to FMR1 locus is not restored after DNA demethylation
CTCF binding sites on the FMR1 gene were previously reported [33]. We now include one additional site obtained from the database available online at http://insulatordb.uthsc.edu/ [34], designated MR (methylated region) site, located at −5557 bp upstream the FMR1 transcription start site. A schematic outline of all CTCF binding sites within the FMR1 locus included in our study is represented in Figure 2. We first studied the three CTCF binding sites in the promoter and near exon1, flanking the CGG repeat sequence, and in the intron 2 region, near one of the transcription starting site of FMR1-AS1 in UFM cell lines. ChIP assay results demonstrated the binding of CTCF to these three sites in UFM fibroblasts and lymphoblasts (Figure 3A–C). The level of binding in UFM was significantly higher compared to FXS cells, both fibroblasts and lymphoblasts, in all sites analyzed. In promoter and exon 1 regions lymphoblasts showed similar CTCF binding levels in UFM and WT (Figure 3A), while in WT fibroblasts CTCF binding levels were significantly higher (p<0.05) compared to UFM (Figure 3B).


In WT cells, we confirmed CTCF binding to the MB site between CpG pairs 66–69. As expected, no CTCF binding was found in FXS fibroblasts, given the complete methylation of this region. Instead, UFM fibroblasts showed binding levels similar to those of WT cells (Figure 3D), demonstrating that CTCF binding is strictly related with the unmethylated status of FMR1 locus. The MR binding site at −5557 bp corresponds to CpG 98, which is fully methylated in all cell lines under investigation. Expectedly, we did not detect CTCF binding in any of them, both fibroblasts and lymphoblasts (data not shown).
We speculated that after DNA demethylation CTCF might rebind to its sites on the FMR1 locus in FXS cells. Our previous studies demonstrated that treatment of FXS lymphoblastoid cells with the demethylating agent 5-azadC induces FMR1 transcriptional reactivation, consequent to demethylation of the 52 CpGs of the promoter [10], [11]. After a 7 day-treatment with 5-azadC of a FXS lymphoblastoid line, we did not observe any significant change in cell viability. We obtained a 25% transcriptional reactivation of FMR1 and a related eight-fold increase of FMR1-AS1 transcript (data not shown). However, as indicated in Figure 4, 5-azadC treatment did not restore CTCF binding to the reactivated FMR1 gene in exon 1, promoter and boundary region (MB site).

CTCF involvement in FMR1 transcriptional regulation
After demonstrating that CTCF binds to the FMR1 regulatory region in transcriptionally active cells, we went on to investigate whether CTCF protein had a regulatory function in FMR1 gene transcription.
We transfected synthetic siRNAs specific for CTCF transcript into WT and UFM fibroblasts to reduce CTCF mRNA and to verify the effect of this reduction on FMR1 transcription. In each knock-down experiment CTCF mRNA depletion was confirmed by quantitative RT-PCR, in comparison with GAPDH mRNA levels, used as control (data not shown). The CTCF reduction was also confirmed on protein levels both in WT and UFM cells (Figure 5A). The residual CTCF transcription was around 15–20% in both fibroblast lines (Figure 5B). On the other hand, the effect on FMR1 transcription was variable. In about two thirds of all knock-down experiments performed on both cell lines, no modification in FMR1 transcription was observed, while in the remaining third we observed a near 50% reduction of FMR1 transcription, as exemplified in Figure 5B. Interestingly, the FMR1 mRNA decrease was accompanied by a similar reduction of the FMR1-AS1 transcription in both cell lines (Figure 5B). We also found that CTCF knock-down coupled with FMR1 reduction resulted in lower levels of CTCF bound to the FMR1 sites in the promoter and exon 1 of WT cells (Figure 6). In those CTCF knock-down experiments in which FMR1-mRNA remained unmodified, ChIP assay demonstrated no variation in CTCF binding at the promoter and exon 1 in WT as well as in UFM cells (Table 1).



The next step was to establish whether overexpression of CTCF transcript could affect the transcription of FMR1. This was accomplished by transfecting a plasmid containing the variant 1 of human CTCF open reading frame into WT, UFM and FXS fibroblasts. The levels of overexpression ranged from 40 to 180 folds compared to the untreated controls, as confirmed by qRT-PCR (Figure 7A). Even in presence of the highest CTCF overexpression, the level of FMR1 transcript remained substantially unmodified in all cell lines analyzed (Figure 7B).

CTCF contributes to maintain the euchromatic status of the FMR1 locus
To understand the molecular events underlying the variable results of CTCF knock-down experiments, we investigated the DNA methylation status and the chromatin organization of the FMR1 locus after CTCF depletion coupled with FMR1 reduction in WT and in UFM fibroblasts. Surprisingly, when we analyzed the methylation of promoter CpGs by bisulfite sequencing, all 52 CpGs were found unmethylated, as in the untreated controls. We extended our observation to the upstream region, observing that the methylation boundary persisted after CTCF depletion and FMR1 transcript reduction (Figure 8A and B). Therefore, CTCF knock-down did not induce the spreading of methylation from the boundary to the FMR1 promoter region, even in presence of a CGG expansion (Figure 8B).

On the other hand, FMR1 transcriptional reduction was found to correlate with histone epigenetic changes. In fact, in those experiments in which CTCF knock-down did not correlate with FMR1 reduction, no variation of epigenetic marks (i.e. methylation of H3-K4 and H3-K9) was observed in the promoter and exon 1 of WT fibroblasts (Table 1). Instead, in those experiments in which CTCF knock-down correlated with FMR1 transcript reduction, we observed a decreased methylation of H3-K4 in both regions analyzed (promoter and exon 1) and increased methylation of H3-K9 in the promoter region, compared to the untreated WT cells (Figure 9). These changes are representative of a more heterochromatic configuration of the locus, correlating with the reduction of FMR1 transcription.

Computational prediction of chromatin loops inside the FMR1 locus
Our data support a mechanism of transcriptional co-regulation between FMR1 sense and antisense, supporting a different role for CTCF protein rather than that of insulator. Based on the variability of FMR1 transcription after CTCF knock-down, we shifted our focus on the role of this protein as chromatin organizer particularly in the loops formation. In order to explore the possibility that CTCF bound to its sites near the FMR1 gene transcription start site (TSS) shapes regulatory chromatin loops, we performed a statistical and computational analysis of DNA structural properties of known regulatory loops determined by 5C experiments [35], compared to those of control genomic regions, and trained a machine learning algorithm to discriminate between real and control DNA loops (Text S1 and Figure S2).
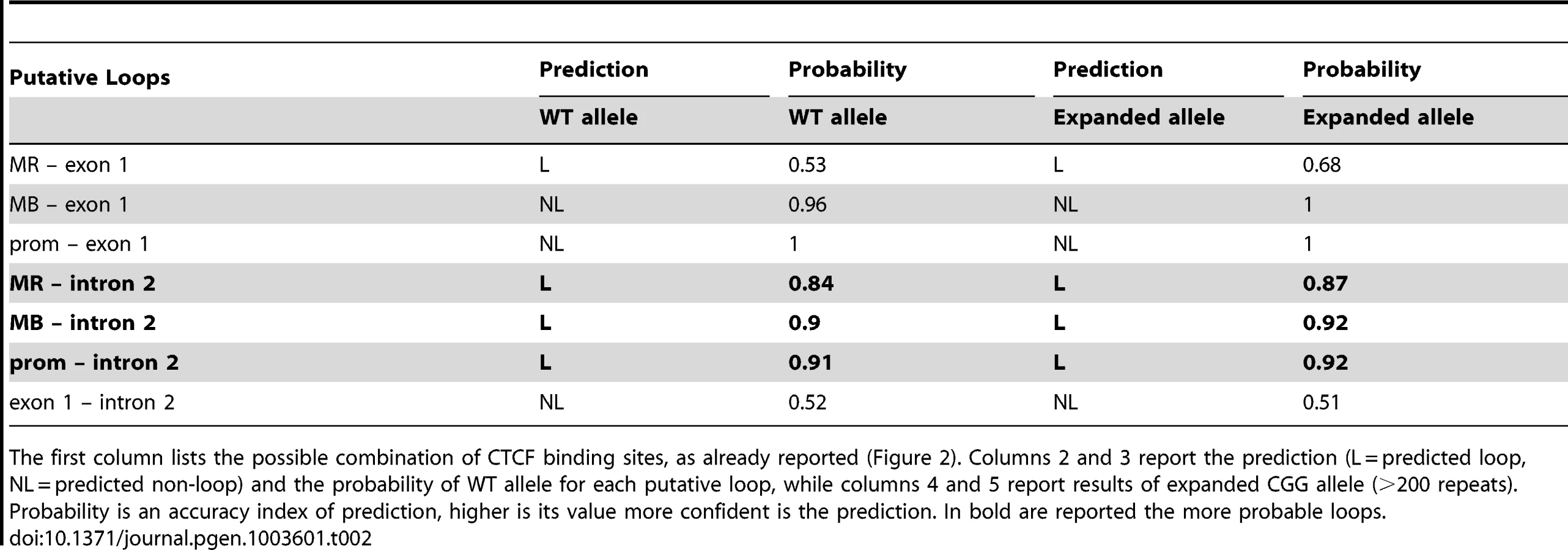
All putative CTCF-mediated loops in the proximity of the FMR1 gene TSS were tested in silico, pairing the CTCF binding sites illustrated in Figure 2. We simulated the CGG expansion by adding 200 CGG triplets to the 5′UTR of the FMR1 gene. The results of this predictions are reported in Table 2. All loops involving the intron 2 binding site, in which a FMR1-AS1 transcriptional start site was identified, were predicted with high confidence both in WT and in the expanded allele. The in silico analysis excluded loops formation between exon 1 and all the other CTCF binding sites.
Discussion
Emerging evidence underlines the dynamic status of the chromatin, previously thought to be static, showing that a given region may be condensed (heterochromatin) and decondensed (euchromatin), according to the cell needs for transcriptional activity of that region. The discovery of proteins capable of establishing physical, as well as functional connections among distant genomic regions, even among different chromosomes, adds complexity to an already intricate network of gene-gene interactions. CTCF can be considered a leading candidate mediating these complex interactions [14]. In fact, it plays different roles in a gene-specific and context-specific manner depending on the possibility of creating homodimers and heterodimers with other proteins, such as cohesin, RNA Polymerase II and Parp1 [36]–[38]. CTCF was the first protein to be identified with a role of insulator, involved in the maintenance of the methylation boundaries in mammals [21]. Recently a methylation boundary region, which seems to prevent methylation to spread downstream, was reported in WT cell lines approximately 1 kb upstream the FMR1 gene promoter, but not in FXS cells [13]. Other regions with this function were described in the myotonic dystrophy gene DMPK, in the ICR (Imprinted Control Region) of IGF2 and in the neighboring BLU and RASSF1A loci of the 3p21.3 gene cluster region [30], [24], [39]. Triplet repeat expansion disorders often undergo transcriptional regulation by the CTCF protein, suggesting a role of CTCF also in FMR1 gene transcriptional regulation.
Binding sites for CTCF in the FMR1 locus were already identified [33], and now confirmed by our study, particularly in the promoter, exon 1 and intron 2, in which is located one of the transcriptional start site of the FMR1-AS1. We firstly showed that these three sites are bound to CTCF in UFM cells, both lymphoblasts and fibroblasts, and the binding level is quite similar to WT cells. These latter cell lines showed differences in CTCF binding in the two cell types analyzed (lymphoblasts and fibroblasts) and these variations should be related to differences between primary fibroblasts and Epstein-Barr-transformed and clonal lymphoblasts, as previously observed for other chromatin marks [4], [8], [27].
A CTCF binding site located in the FMR1 methylation boundary was already described [33]. We now demonstrate for the first time the existence of the methylation boundary in UFM cells, supporting the hypothesis of a regulatory role played by the boundary region in preventing gene silencing. Interestingly, the CTCF binding site located in this border region, between CpG pairs 66 and 69 in WT cells, was also observed in UFM cell lines, but not in FXS cells, as expected given the CpGs methylation status of the latter.
We then tried to restore CTCF binding to the FMR1 gene in FXS cell lines by inducing DNA demethylation with 5-azadC. DNA demethylation resulted in FMR1 transcription reactivation as expected, while CTCF binding to its specific sites on promoter, exon 1 and boundary region was not restored. This result might be explained by failure of drug-induced DNA demethylation to reverse all modifications that occur during gene silencing. As observed on p16 and MLH1 gene, 5-azadC treatment did not completely restore normal histone code and post-translational modifications of DNA binding proteins to reestablish long-term expression [40], [41]. We previously observed that transcriptionally reactivated FXS cell lines restored epigenetic changes consistent with an euchromatic status, without fully reaching the euchromatic configuration typical of normal control cell lines [4]. We also demonstrated that 5-azadC-induced demethylation is partial and transient. After 4 weeks from 5-azadC withdrawal, the FMR1 promoter resumed its methylated status [11]. Therefore it can be inferred that CTCF binding, even if it occurred after 5-azadC demethylation, would not by itself sufficient to maintain the unmethylated status of the FMR1 gene.
These data seemed to suggest a functional role of the CTCF protein in regulating FMR1 gene transcription. To investigate this potential role, we induced both silencing and overexpression of CTCF transcript. In those experiments in which siRNA-mediated CTCF knock-down did not correlate with FMR1 transcript reduction, epigenetic marks (CTCF binding, H3-K4/H3-K9 methylation) were unmodified in promoter and exon 1 regions. On the other hand, the level of CTCF protein still bound to the gene was found reduced in CTCF knock-down experiments coupled with FMR1 mRNA reduction. Moreover, FMR1 decreased expression correlated with increased levels of heterochromatinic marks, such as H3-K4 demethylation and H3-K9 hypermethylation in the 5′ UTR of the gene. Interestingly, these epigenetic changes, known to favor heterochromatinic configuration, were not followed by the spreading of DNA methylation from the boundary region towards the FMR1 promoter, not only in WT alleles, but also in UFM alleles, suggesting that a CGG expansion is not by itself sufficient to induce methylation, even in absence of CTCF. This latter result implies that CTCF does not work as an insulator at the FMR1 locus. Therefore, other still unknown proteins must act as barrier elements in this specific region, as already hypothesized [13]. There are a number of boundaries that may function in a CTCF-independent manner through the binding of proteins known to act as transcriptional regulators, such as USF1 [42], YY1 and EVI1, or through non-coding RNAs [43]. Particularly, USF1 is one of the major transcription factors that bind the FMR1 promoter region. Its binding is partially inhibited by DNA methylation and it might be a hypothetical candidate as insulator for the FMR1 gene [44].
Interesting results came from the FMR1 antisense transcript characterization, particularly in UFM cell lines, both before and after CTCF transcriptional silencing. The FMR1 antisense RNA is transcribed starting from the second intron of the gene in WT and premutated alleles [33]. We detected, for the first time, FMR1-AS1 RNA in UFM cell lines and also showed that the levels of this antisense transcript were higher in UFM cells, compared to normal controls, similar to what happens with the sense transcript [7]. The antisense transcript splices a 9.7 kb intron corresponding to the FMR1 intron 1, that uses the complementary splice donor and acceptor to FMR1, representing a non-consensus CT to AC splice site. Moreover we observed in UFM cells the same splicing variant of the FMR1-AS1 previously described as premutation-specific alternative splicing in intron 2 that also uses a non-consensus CT-AC splice site [33]. Furthermore, after CTCF depletion the reduction of FMR1 mRNA was always coupled with the decrease of FMR1-AS1 transcript. These data indicated a co-regulation of transcription and splicing mechanisms at the FMR1 locus in transcriptional active alleles. On the other hand, CTCF knock-down did not have always the same effect: in only one third of all the experiments we observed a diminished transcription of both sense and antisense FMR1. These results suggested a partial and/or indirect role of CTCF in regulating FMR1 expression and led us to hypothesize that the sites located within the FMR1 locus may form chromatin loops mediated by CTCF homodimers capable of bringing in close proximity molecular machineries for transcription, splicing and epigenetic modifications. The formation of these loops would be partially affected by CTCF knockdown but not by CTCF overexpression, i.e. additional CTCF protein would not affect loop formation [45], [46]. Loss of CTCF-mediated chromosomal organization through disruption of this loop could exert a negative effect on FMR1 transcription. On the other hand, it would seem that other factors, yet to be identified, could activate self-preserving mechanisms that maintain FMR1 transcription unchanged despite the absence of the loop, as observed in a fraction of our experiments. Indeed, how chromatin configurations may influence gene expression still remains unclear. The “loop” hypothesis was supported by antisense transcription data, as well as by CTCF depletion/overexpression experiments. The presence of a CTCF binding site in FMR1 intron 2, near one of the transcription starting sites of FMR1-AS1, previously observed by Ladd et al. [33], was confirmed in our cell lines by ChIP assays. Our hypothesis was that this CTCF site is involved in the chromatin looping together with one of the 5′-UTR sites within the active FMR1 gene both in normal and in the expanded alleles, such as UFM. This loop may not form after 5-azadC-induced demethylation, which cannot reestablish native epigenetic modifications. In fact, as previously observed, 5-azadC effect is only transient [11]. The region surrounding the FMR1 promoter (approximately 50 kb) was previously studied through 3C technique, which demonstrated reduced interaction frequencies [47]. This work did not take into account the behavior of the chromatin region surrounding the active FMR1 gene with CGG expansion, such as in premutation and UFM cells. The 3C technique is only capable of detecting chromatin loop interactions greater than 10 kb and for this reason a chromatin loop formation in our region of interest cannot be excluded. We investigated the possibility of looping between CTCF sites using an in silico analysis of DNA structural characteristics of experimentally validated DNA regulatory loops. For this purpose, we elaborated a new predictor system that showed good performances in discriminating between real loops and control genomic regions. This predictor (SVM) confirmed that putative loops can form involving the CTCF binding site in intron 2, both in WT and in expanded alleles.
The bioinformatics approach takes into account parameters concerning the nucleotide sequence but not molecular and epigenetic characteristics, such as DNA methylation. In silico data should be interpreted considering the biological context in which the FMR1 gene is located. Therefore, loop formation in FXS alleles was excluded by the existence of DNA methylation of the entire region upstream the FMR1 promoter, that prevents CTCF from binding its sites. The formation of loops between intron 2 and MR sites could also be excluded because the MR site is located in a region that is extensive methylated in WT and in expanded alleles. Our in silico results affirmed that a chromatin loop mediated by CTCF homodimers can exist between intron 2 and the methylation boundary region or promoter in normal and UFM alleles. These bioinformatics data will deserve further experimental validations.
In conclusion our results delineate a role for CTCF as transcriptional regulator of FMR1 expression through chromatin organization. CTCF was firstly described as the only known insulator [48], but we show that it does not act as an insulator on the methylation boundary upstream the FMR1 gene. A role of CTCF in genome and locus organization acting to secure long-range intra - and inter-chromosomal interactions was abundantly described [22]. Our results define an indirect role for CTCF in modulating bidirectional transcription through FMR1 locus chromatin organization and loop formation. Indeed, reduction of FMR1 sense and antisense transcription after CTCF depletion underscores the importance of the CTCF-mediated loop complex. This study will be help in further clarifying the processes by which cell type specific patterns of gene expression can be established and maintained.
Materials and Methods
Cell lines and pharmacological treatments
Lymphoblastoid cell lines were established by Epstein–Barr virus transformation from peripheral blood lymphocytes of FXS, UFM and normal control (WT) males. The FXS cell lines employed in these experiments were E3 and S1, with 250 and 450 CGGs, respectively; the UFM cell line (MA) contains 265–430 CGGs [8]; two different WT cell lines obtained from normal control males. Lymphoblasts were grown in RPMI1640 medium (Sigma Aldrich) supplemented with 20% fetal bovine serum, 2.5% L-glutamine and 1% penicillin/streptomycin at 37°C with 5% CO2.
Primary fibroblast cultures were obtained from skin biopsies derived from the UFM individual (MA). We have also employed one FXS line (GM04026) and three WT lines (GM05381, GM03349 and GM07492), provided by the Coriell Institute (Camden, USA). Fibroblasts were grown in BIO-AMF2 complete medium (Biological Industries).
FXS lymphoblasts were treated with the demethylating agent 5-azadC (Sigma-Aldrich), as previously described [10]. Cells were seeded at 7×105 cells/ml and 5-azadC was added daily at 1 µM (final concentration) for 7 days. At the end of the treatment, cells were harvested to measure viability with the propidium iodide method (Nucleocounter, Sartorius/Stedim) and to perform RNA and DNA extraction.
Transfection experiments
Knock-down of CTCF transcripts was carried out in UFM and in all three WT fibroblast lines with synthetic siRNAs (Dharmacon, USA). Complete sequences of the siRNAs are listed in Table S1. Negative control to check the efficiency of CTCF depletion was performed using scramble siRNA (IDT). In accordance with the protocol of the manufacturer, 40 nM of siRNA were transfected by Lipofectamine RNAiMAX (Invitrogen, USA) and cultures were harvested after 72 hours.
The human open reading frame of CTCF was transfected into the cells through the expression plasmid pCMV6-Entry (C-terminal Myc - and DDK-tagged) (Origene). 100 ng of plasmid DNA was transfected in fibroblasts with Lipofectamine 2000 (Invitrogen, USA) and cells were collected after 48 h, according to manufacturer's instructions, and after 120 h to asses if a longer overexpression could affect FMR1 transcription.
Western blotting analysis
Proteins extracted from untreated and siRNA-treated WT and UFM fibroblasts were resuspended in Laemli buffer, boiled, separated on 8% polyacrylamide gel electrophoresis, transferred to Hybond-ECL membrane (GE Healthcare), immunostained and visualized after film exposure using the ECL Western Blotting Kit (GE Healthcare), according to the manufacturer. Primary antibodies were used at the following concentrations: 1∶1000 anti-CTCF rabbit policlonal antibody (Millipore) and 1∶10000 anti-GAPDH mouse antibody (Sigma-Aldrich).
Methylation analysis
Genomic DNA was isolated from siRNA-treated and untreated fibroblasts both WT and UFM by DNeasy Blood & Tissue kit (Qiagen) The DNA concentration was checked both by absorbance measurements at 260 and 280 nm and on agarose gel. Bisulfite DNA transformation was performed as previously described [11]. Each transformed DNA was amplified in 7 independent PCR reactions, then pooled and recovered from the agarose gel with the StrataPrep DNA Gel extraction kit (Stratagene). The purified PCR products were cloned with the StrataClone PCR cloning kit (Stratagene), according to the manufacturer's instructions. After bacterial plating and overnight incubation at 37°C, white colonies were picked and plasmid DNA was extracted. After a pre-screening of the clones with PCR using specific plasmid primers (M13 forward and reverse), amplification products were sequenced in both directions with BigDye Terminator v3.1 Cycle Sequencing kit (Applied Biosystems) on a 3130 Genetic Analyzer (Applied Biosystems). The modified primers are those described by Naumann et al. [13].
Quantitative RT-PCR analysis
Total RNA was extracted by TRIzol (Invitrogen, USA). RNA concentration and purity were checked on agarose gel and by UV spectrophotometer. RNA samples were treated with TURBO DNA-free DNase (Ambion) to remove contaminating DNA. Afterwards, 1 µg of total RNA was retro-transcribed into cDNA by MoMLV-RT (Invitrogen, USA) using random hexamers. For a relative quantification of each transcript, the following pre-developed TaqMan assays (Applied Biosystems) were used: CTCF (Hs00902008_m1), GAPDH (402869), FMR1 (Hs00233632_m1). For FMR1-AS1, custom-made assay was designed (ASFMR1F 5′-CCTCTGCCAACTCAGTGCTATTAG-3′; ASFMR1R 5′-CATGACCTAGTCTGGGGTGGAG-3′; ASFMR1Probe 5′-(FAM)-TGGAATCATCTCCCC-(TAMRA)-3′(Applied Biosystems), according to Ladd et al. [33]. The real-time RT-PCR was performed on a ABI7900HT (Applied Biosystems). The cycle parameters were: 2 minutes at 50°C and 10 minutes at 95°C, followed by 40 cycles with 15 seconds at 95°C (denaturation) and 1 minute at 60°C (annealing/extension).
Strand-specific RT-PCR
To analyze the FMR1-AS1 transcript, cDNA was generated using specific primers, with a linker (LK) sequence: 5′-CGACTGGAGCACGAGGACACTGA-3′attached to the 5′ end. Primers were those employed by Ladd et al. [33]. cDNA was produced using Superscript III (Invitrogen), according to the manufacturer instruction's. PCR were performed using the LK primer (as forward) and antisense specific reverse primers. The amplicons were sequenced on an 3130 Genetic Analyzer (Applied Biosystems).
Chromatin Immunoprecipitation (ChIP) and quantification of IP-DNA
ChIP assay was performed according to the manufacturer (Upstate Biotechnology, USA). After 10 minutes at 37°C with 1% formaldehyde, cells were seeded and washed with 1× PBS and Protease Inhibitor Cocktail (Sigma-Aldrich). To obtain 200–1000 bp DNA fragments, cell pellets were sonicated. Histone methylation analysis was performed using two different antibodies against dimethyl lysine 9 (H3-K9, 07–441, Upstate Biotechnology) and dimethyl lysine 4 (H3-K4, 07–030, Upstate Biotechnology) on histone 3. Binding of CTCF protein was assayed using the specific antibody (07-729, Millipore). In each ChIP assay antibody against rabbit IgG (1862244, Thermo Scientific) was employed and also no template control was included. Immunoprecipitated DNA (IP-DNA) was extracted by standard procedure (phenol/chloroform/isoamilic alcohol 25∶24∶1) and then quantified by real-time PCR (ABI7900HT, Applied Biosystems) using fluorescent probe and primers specific for both FMR1 and HPRT.
Primers and probes employed for PCR analysis are listed in Table S2. Standard curves for the three FMR1 and for the single HPRT amplicon were constructed with five different DNA dilutions of known concentration (X axis = log[X]) and the corresponding Ct values (Y axis). The unknown amount of methylated histone and CTCF-binding IP-DNA of FMR1 and HPRT (X axis = log[X]) was calculated from Ct values, through the standard curve plot. Normalized FMR1 levels were estimated dividing the amount of FMR1 IP-DNA by the amount of HPRT IP-DNA.
Statistical analysis
All variables were analyzed by means of descriptive statistics (mean, median, standard deviation and standard error of mean). Data were analyzed with non-parametric statistical Kruskal-Wallis test and with K sample test. The level of significance was set at p≤0.05. Data analysis was performed using STATA Intercooled v. 9.2 software (Stata Co.; College Station, Lakewag, TX, USA).
Computational structural analysis and prediction of CTCF-mediated DNA loops
In order to analyze the structural characteristics of CTCF-mediated DNA loops, a bioinformatics approach was developed and is detailed in the Text S1. Briefly, a machine learning method was trained to recognize known chromatin loops from control genomic regions, and then used to test putative regulative loops in the proximity of FMR1 transcription start site.
Supplementary Data are available online: Supplementary Figures S1, S2, Supplementary Tables S1, S2, Supplementary Text S1 and Supplementary References S1 [34], [35],[49]–[58].
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. PirozziF, TabolacciE, NeriG (2011) The FRAXopathies: Definition, overview, and update. Am J Med Genet part A 8 : 1803–1816.
2. VerkerkAJ, PierettiM, SutcliffeJS, FuYH, KuhlDP, et al. (1991) Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65 : 905–914.
3. PierettiM, ZhangFP, FuYH, WarrenST, OostraBA, et al. (1991) Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 66 : 817–822.
4. TabolacciE, PietrobonoR, MoscatoU, OostraBA, ChiurazziP, et al. (2005) Differential epigenetic modifications in the FMR1 gene of the fragile X syndrome after reactivating pharmacological treatments. Eur J Hum Genet 13 : 641–648.
5. ZalfaF, GiorgiM, PrimeranoB, MoroA, Di PentaA, et al. (2003) The fragile X syndrome protein FMRP associates with BC1 RNA and regulates the translation of specific mRNAs at synapses. Cell 112 : 317–327.
6. SmeetsHJ, SmitsAP, VerheijCE, TheelenJP, WillemsenR, et al. (1995) Normal phenotype in two brothers with a full FMR1 mutation. Hum Mol Genet 11 : 331–334.
7. PietrobonoR, TabolacciE, ZalfaF, ZitoI, TerraccianoA, et al. (2005) Molecular dissection of the events leading to inactivation of the FMR1 gene. Hum Mol Genet 14 : 267–277.
8. TabolacciE, MoscatoU, ZalfaF, BagniC, ChiurazziP, et al. (2008) Epigenetic analysis reveals a euchromatic configuration in the FMR1 unmethylated full mutations. Eur J Hum Genet 16 : 1487–1498.
9. WillemsenR, BontekoeCJ, SeverijnenLA, OostraBA (2002) Timing of the absence of FMR1 expression in full mutation chorionic villi. Hum Genet 110 : 601–605.
10. ChiurazziP, PomponiMG, WillemsenR, OostraBA, NeriG (1998) In vitro reactivation of the FMR1 gene involved in fragile X syndrome. Hum Mol Genet 7 : 109–113.
11. PietrobonoR, PomponiMG, TabolacciE, OostraB, ChiurazziP, et al. (2002) Quantitative analysis of DNA demethylation and transcriptional reactivation of the FMR1 gene in fragile X cells treated with 5-azadeoxycytidine. Nucleic Acids Res 30 : 3278–3285.
12. EigesR, UrbachA, MalcovM, FrumkinT, SchwartzT, et al. (2007) Developmental study of fragile X syndrome using human embryonic stem cells derived from preimplantation genetically diagnosed embryos. Cell Stem Cell 1 : 568–577.
13. NaumannA, HochsteinN, WeberS, FanningE, DoerflerW (2009) A distinct DNA-methylation boundary in the 5′-upstream sequence of the FMR1 promoter binds nuclear proteins and is lost in fragile X syndrome. Am J Hum Genet 85 : 606–616.
14. PhillipsJE, CorcesVG (2009) CTCF: master weaver of the genome. Cell 137 : 1194–1211.
15. KlenovaEM, NicolasRH, PatersonHF, CarneAF, HeathCM, et al. (1993) CTCF, a conserved nuclear factor required for optimal transcriptional activity of the chicken c-myc gene, is an 11-Zn-finger protein differentially expressed in multiple forms. Mol Cell Biol 13 : 7612–7624.
16. OhlssonR, RenkawitzR, LobanenkovV (2001) CTCF is a uniquely versatile transcription regulator linked to epigenetics and disease. Trends Genet 17 : 520–527.
17. LobanenkovVV, NicolasRH, AdlerVV, PatersonH, KlenovaEM, et al. (1990) A novel sequence specific DNA binding protein which interacts with three regularly spaced direct repeats of the CCCTC-motif in the 5′-flanking sequence of the chicken c-myc gene. Oncogene 5 : 1743–1753.
18. FilippovaGN, FagerlieS, KlenovaEM, MyersC, DehnerY, et al. (1996) An exceptionally conserved transcriptional repressor, CTCF, employs different combinations of zinc fingers to bind diverged promoter sequences of avian and mammalian c-myc oncogenes. Mol Cell Biol 16 : 2802–2813.
19. WallaceJA, FelsenfeldG (2007) We gather together: insulators and genome organization. Curr Opin Genet Dev 17 : 400–407.
20. FilippovaGN (2008) Genetics and epigenetics of the multifunctional protein CTCF. Curr Top Dev Biol 80 : 337–360.
21. OhlssonR, LobanenkovV, KlenovaE (2010) Does CTCF mediate between nuclear organization and gene expression? Bioessays 32 : 37–50.
22. HandokoL, XuH, LiG, NganCY, ChewE, et al. (2011) CTCF-mediated functional chromatin interactome in pluripotent cells. Nat Genet 43 : 630–638.
23. BottaM, HaiderS, LeungIX, LioP, MozziconacciJ (2010) Intra - and inter-chromosomal interactions correlate with CTCF binding genome wide. Mol Syst Biol 6 : 426–4431.
24. BellAC, FelsenfeldG (2000) Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 405 : 482–485.
25. KanduriC, PantV, LoukinovD, PugachevaE, QiCF, et al. (2000) Functional association of CTCF with the insulator upstream of the H19 gene is parent of origin-specific and methylation-sensitive. Curr Biol 10 : 853–856.
26. HarkAT, SchoenherrCJ, KatzDJ, IngramRS, LevorseJM, et al. (2000) CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature 405 : 486–489.
27. WangH, MauranoMT, QuH, VarleyKE, GertzJ, et al. (2012) Widespread plasticity in CTCF occupancy linked to DNA methylation. Genome Res 22 : 1680–1688.
28. ChoDH, ThienesCP, MahoneySE, AnalauE, FilippovaGN, et al. (2005) Antisense transcription and heterochromatin at the DM1 CTG repeats are constrained by CTCF. Mol Cell 20 : 483–489.
29. ClearyJD, ToméS, López CastelA, PanigrahiGB, FoiryL, et al. (2010) Tissue - and age-specific DNA replication patterns at the CTG/CAG-expanded human myotonic dystrophy type 1 locus. Nat Struct Mol Biol 17 : 1079–1087.
30. López CastelA, NakamoriM, ToméS, ChitayatD, GourdonG, et al. (2011) Expanded CTG repeat demarcates a boundary for abnormal CpG methylation in myotonic dystrophy patient tissues. Hum Mol Genet 20 : 1–15.
31. SopherBL, LaddPD, PinedaVV, LibbyRT, SunkinSM, et al. (2011) CTCF regulates ataxin-7 expression through promotion of a convergently transcribed, antisense noncoding RNA. Neuron 70 : 1071–1084.
32. De BiaseI, ChutakeYK, RindlerPM, BidichandaniSI (2009) Epigenetic silencing in Friedreich ataxia is associated with depletion of CTCF (CCCTC-binding factor) and antisense transcription. PLoS One 4: e7914.
33. LaddPD, SmithLE, RabaiaNA, MooreJM, GeorgesSA, et al. (2007) An antisense transcript spanning the CGG repeat region of FMR1 is upregulated in premutation carriers but silenced in full mutation individuals. Hum Mol Genet 16 : 3174–3187.
34. BaoL, ZhouM, CuiY (2008) CTCFBSDB: a CTCF-binding site database for characterization of vertebrate genomic insulators. Nucleic Acids Res 36(Database issue): D83–87.
35. DostieJ, DekkerJ (2007) Mapping networks of physical interactions between genomic elements using 5C technology. Nat Protoc 2 : 988–1002.
36. WendtKS, YoshidaK, ItohT, BandoM, KochB, et al. (2008) Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature 451 : 796–801.
37. ChernukhinI, ShamsuddinS, KangSY, BergströmR, KwonYW, et al. (2007) CTCF interacts with and recruits the largest subunit of RNA polymerase II to CTCF target sites genome-wide. Mol Cell Biol 27 : 1631–1648.
38. ZampieriM, GuastafierroT, CalabreseR, CiccaroneF, BacaliniMG, et al. (2012) ADP-ribose polymers localized on Ctcf-Parp1-Dnmt1 complex prevent methylation of Ctcf target sites. Biochem J 441 : 645–652.
39. ChangJW, HsuHS, NiHJ, ChuangCT, HsiungCH, et al. (2010) Distinct epigenetic domains separated by a CTCF bound insulator between the tandem genes, BLU and RASSF1A. PLoS One 5: e12847.
40. WitcherM, EmersonBM (2009) Epigenetic silencing of the p16(INK4a) tumor suppressor is associated with loss of CTCF binding and a chromatin boundary. Mol Cell 34 : 271–284.
41. McGarveyKM, FahrnerJA, GreeneE, MartensJ, JenuweinT, et al. (2006) Silenced tumor suppressor genes reactivated by DNA demethylation do not return to a fully euchromatic chromatin state. Cancer Res 66 : 3541–3549.
42. HuangS, LiX, YusufzaiTM, QiuY, FelsenfeldG (2007) USF1 recruits histone modification complexes and is critical for maintenance of a chromatin barrier. Mol Cell Biol 27 : 7991–8002.
43. WangJ, LunyakVV, JordanIK (2012) Genome-wide prediction and analysis of human chromatin boundary elements. Nucleic Acids Res 40 : 511–529.
44. KumariD, UsdinK (2001) Interaction of the transcription factors USF1, USF2, and alpha -Pal/Nrf-1 with the FMR1 promoter. Implications for Fragile X mental retardation syndrome. J Biol Chem 276 : 4357–4364.
45. TaftRJ, HawkinsPG, MattickJS, MorrisKV (2011) The relationship between transcription initiation RNAs and CCCTC-binding factor (CTCF) localization. Epigenetics Chromatin 4 : 1–13.
46. ZhangH, NiuB, HuJF, GeS, WangH, et al. (2011) Interruption of intrachromosomal looping by CCCTC binding factor decoy proteins abrogates genomic imprinting of human insulin-like growth factor II. J Cell Biol 193 : 475–487.
47. GheldofN, TabuchiTM, DekkerJ (2006) The active FMR1 promoter is associated with a large domain of altered chromatin conformation with embedded local histone modifications. Proc Natl Acad Sci USA 103 : 12463–12468.
48. BellAC, WestAG, FelsenfeldG (1999) The protein CTCF is required for the enhancer blocking activity of vertebrate insulators. Cell 98 : 387–396.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 7
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Bacterial Adaptation through Loss of Function
- SLC26A4 Targeted to the Endolymphatic Sac Rescues Hearing and Balance in Mutant Mice
- The Cohesion Protein SOLO Associates with SMC1 and Is Required for Synapsis, Recombination, Homolog Bias and Cohesion and Pairing of Centromeres in Drosophila Meiosis
- Gene × Physical Activity Interactions in Obesity: Combined Analysis of 111,421 Individuals of European Ancestry
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy