
Expression of Linear and Novel Circular Forms of an -Associated Non-Coding RNA Correlates with Atherosclerosis Risk
Human genome-wide association studies have linked single nucleotide polymorphisms (SNPs) on chromosome 9p21.3 near the INK4/ARF (CDKN2a/b) locus with susceptibility to atherosclerotic vascular disease (ASVD). Although this locus encodes three well-characterized tumor suppressors, p16INK4a, p15INK4b, and ARF, the SNPs most strongly associated with ASVD are ∼120 kb from the nearest coding gene within a long non-coding RNA (ncRNA) known as ANRIL (CDKN2BAS). While individuals homozygous for the atherosclerotic risk allele show decreased expression of ANRIL and the coding INK4/ARF transcripts, the mechanism by which such distant genetic variants influence INK4/ARF expression is unknown. Here, using rapid amplification of cDNA ends (RACE) and analysis of next-generation RNA sequencing datasets, we determined the structure and abundance of multiple ANRIL species. Each of these species was present at very low copy numbers in primary and cultured cells; however, only the expression of ANRIL isoforms containing exons proximal to the INK4/ARF locus correlated with the ASVD risk alleles. Surprisingly, RACE also identified transcripts containing non-colinear ANRIL exonic sequences, whose expression also correlated with genotype and INK4/ARF expression. These non-polyadenylated RNAs resisted RNAse R digestion and could be PCR amplified using outward-facing primers, suggesting they represent circular RNA structures that could arise from by-products of mRNA splicing. Next-generation DNA sequencing and splice prediction algorithms identified polymorphisms within the ASVD risk interval that may regulate ANRIL splicing and circular ANRIL (cANRIL) production. These results identify novel circular RNA products emanating from the ANRIL locus and suggest causal variants at 9p21.3 regulate INK4/ARF expression and ASVD risk by modulating ANRIL expression and/or structure.
Published in the journal:
. PLoS Genet 6(12): e32767. doi:10.1371/journal.pgen.1001233
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001233
Summary
Human genome-wide association studies have linked single nucleotide polymorphisms (SNPs) on chromosome 9p21.3 near the INK4/ARF (CDKN2a/b) locus with susceptibility to atherosclerotic vascular disease (ASVD). Although this locus encodes three well-characterized tumor suppressors, p16INK4a, p15INK4b, and ARF, the SNPs most strongly associated with ASVD are ∼120 kb from the nearest coding gene within a long non-coding RNA (ncRNA) known as ANRIL (CDKN2BAS). While individuals homozygous for the atherosclerotic risk allele show decreased expression of ANRIL and the coding INK4/ARF transcripts, the mechanism by which such distant genetic variants influence INK4/ARF expression is unknown. Here, using rapid amplification of cDNA ends (RACE) and analysis of next-generation RNA sequencing datasets, we determined the structure and abundance of multiple ANRIL species. Each of these species was present at very low copy numbers in primary and cultured cells; however, only the expression of ANRIL isoforms containing exons proximal to the INK4/ARF locus correlated with the ASVD risk alleles. Surprisingly, RACE also identified transcripts containing non-colinear ANRIL exonic sequences, whose expression also correlated with genotype and INK4/ARF expression. These non-polyadenylated RNAs resisted RNAse R digestion and could be PCR amplified using outward-facing primers, suggesting they represent circular RNA structures that could arise from by-products of mRNA splicing. Next-generation DNA sequencing and splice prediction algorithms identified polymorphisms within the ASVD risk interval that may regulate ANRIL splicing and circular ANRIL (cANRIL) production. These results identify novel circular RNA products emanating from the ANRIL locus and suggest causal variants at 9p21.3 regulate INK4/ARF expression and ASVD risk by modulating ANRIL expression and/or structure.
Introduction
Atherosclerotic vascular disease (ASVD) is a leading cause of human mortality worldwide [1]. While there are well-recognized risk factors for ASVD such as tobacco use, obesity and hyperlipidemia, the identification of common genetic variants associated with the disease has proven difficult despite strong evidence that susceptibility is heritable. Recently, multiple unbiased genome-wide wide association studies (GWAS) have linked single nucleotide polymorphisms (SNPs) on chromosome 9p21 to ASVD and other related conditions (i.e. coronary artery disease, stroke, myocardial infarction and aortic aneurysm) [2]–[12]. These associations have been replicated in multiple independent studies and are not associated with “classical” ASVD risk factors such as hypertension, obesity, tobacco use or lipid levels. While causal variants within 9p21.3 have yet to be identified, the risk associated SNPs cluster together within a 53 kb region roughly 100 kb centromeric to the INK4/ARF (CDKN2a/b) tumor suppressor locus ([13] and Figure S1). Congenic mapping using mice differentially susceptible to ASVD has identified syntenic susceptibility alleles near the murine Ink4/Arf locus [14], suggesting that this risk interval is conserved in mammals.
The INK4/ARF locus encodes three archetypal tumor suppressor genes: p16INK4a, p15INK4b and ARF (p14ARF in humans, p19ARF in mice), as well as a long non-coding RNA called Antisense Non-coding RNA in the INK4 Locus (ANRIL or CDKN2BAS). Other than a role for ARF in development of the optic vasculature, all of the INK4/ARF proteins are thought to be largely dispensable for normal mammalian development, but play important roles in restraining aberrant proliferation associated with cancer and other disease states (reviewed in: [15]). In purified T-cells from healthy individuals, we have shown a pronounced effect of ASVD-associated SNPs at 9p21 on INK4/ARF expression, with those harboring the risk alleles demonstrating reduced levels of p15INK4b, p16INK4a, ARF and ANRIL [16]. Decreased expression of such anti-proliferative molecules could promote pathologic monocytic or vascular proliferation, thus accelerating ASVD development (reviewed in [17], [18]). For example, mice lacking p16INK4a exhibit increased vascular hyperplasia following intra-arterial injury [19] and ARF deficiency has been implicated in atherosclerotic plaque formation [20]. Additionally, TGF-β signaling, which induces the expression of p16INK4a and p15INK4b, is anti-atherogenic in some settings [21]–[23]. Most recently, excess proliferation of hematopoietic progenitor cells, which is in part controlled by p16INK4a expression during aging [24], has been associated with atherosclerosis in a murine model [25]. Moreover, targeted deletion of a region syntenic to the ASVD risk interval in mice resulted in severely attenuated expression of p15INK4b and p16INK4a [26]. Although these results suggest that the ASVD-associated 9p21 SNPs control INK4/ARF expression, and that decreased expression of the INK4/ARF tumor suppressors may promote ASVD, it is not known how polymorphisms located ∼120 kb away from the locus might influence INK4/ARF expression.
ANRIL was first uncovered in a genetic analysis of familial melanoma patients with neural system tumors [27]. Based upon EST assembly, ANRIL has 19 exons with no identified open reading frame [27] (Figure S2). Although cloning a full-length version of the predicted transcript has proven difficult, a growing number of alternatively spliced ANRIL transcripts have recently been reported in the literature [28], [29]. Many of these reports suggest that multiple ANRIL isoforms can be expressed in a single cell type. For example, two ANRIL variants have been reported in testes, five in HUVECs and three in lung [27], [28]. Further confounding the study of ANRIL, the majority of predicted exons are <100 nucleotides in length and many consist entirely of repetitive LINE, SINE and Alu elements (i.e. exons 7, 8, 12, 14 and 16) [30]. Without a firm understanding of ANRIL structure, deciphering the biological function of this non-coding RNA has become increasingly complicated.
More than a decade ago, elegant genetic work by Van Lohuizen, DePinho and colleagues demonstrated that the Ink4/Arf locus is potently repressed by Polycolmb group (PcG) complexes [31]. Such repression appears critical for the persistence and proliferation of somatic stem cells and other self-renewing tissues such as pancreatic beta-cells [32], [33]. Until recently little progress was made in understanding the biochemical basis for PcG-mediated INK4/ARF silencing, however two independent groups have since shown that the PcG complexes, PRC-1 and PRC-2, localize to the INK4/ARF locus and repress its activity through the establishment of repressive chromatin modifications such as H3K27 trimethylation [34]–[36]. Prior work has also shown that long, non-coding RNAs such as Xist, Kcnq1ot1 and HOTAIR can repress genes in cis - or trans- through interaction with PcG complexes [37]–[40]. Moreover, dysregulation of HOTAIR has been recently implicated in breast cancer progression, suggesting that long, non-coding RNAs play an important role in human disease [41]. Based on this evidence, we and others postulated that ANRIL could play a similar role in PcG-mediated repression of the INK4/ARF locus [16].
Against this background of prior work, we sought to better determine ANRIL structure and expression in relation to ASVD-SNP genotype and INK4/ARF expression. Toward that end, we performed comprehensive DNA and RNA analyses of ANRIL using RACE and next-generation sequencing in primary and transformed cell lines. We showed that the expression of ANRIL isoforms containing exons 1–2 or 4–6 correlated with ASVD-SNP genotype, however those containing exons 18–19 did not. Surprisingly, we also uncovered circular ANRIL (cANRIL) RNA species whose expression correlated with ASVD-SNP genotype. These new data suggest that the causal variant(s) within 9p21 influence ASVD susceptibility through regulation of ANRIL expression and splicing, leading to differential PcG recruitment and INK4/ARF repression.
Results
ANRIL transcription produces multiple rare, non-coding RNA species
Multiple ANRIL isoforms have been proposed based upon the assembly of ESTs and the sequencing of cDNA libraries (See Figure S2). To determine which of these isoforms predominates in vivo, we performed RNA ligase mediated (RLM)-RACE in cell lines and primary human peripheral blood T-lymphocytes (PBTL). In addition to using the RLM procedure to maximize the detection of mRNA transcripts, we employed a high-fidelity Taq polymerase capable of amplifying complex DNAs such as those containing SINE, LINE and Alu elements. Primers for 3′ and 5′ RACE were designed within exons 1, 2, 4, 6, 9, 13, 16 and 18 of the originally reported transcript, NR_003529 [27], but only primers in exons 4 and 6 selectively amplified ANRIL sequences (data not shown). These amplicons were cloned and sequenced to verify the resulting DNA sequences. Using exon 4 and 6 primers, we identified multiple ANRIL variants, including novel splice isoforms that were not previously reported (Figure 1A, novel exons shown in blue). We also detected several transcripts with a peculiar, non-colinear exon sequence, most notably in the HeLa and primary PBTL populations (shown in red in Figure 1A). Given that no particular ANRIL isoform appeared to predominate from our RACE data, we exploited the observation that ANRIL RNAs containing exon 15 frequently maintained the canonical exonic structure (e.g. 15–16–17–18–19). We termed these exons as “distal” because they are located at the 3′ end of ANRIL, and those prior to exon 15 as “proximal”.

To examine the absolute abundance of transcripts containing the “proximal” or “distal” exons in a variety of cell types, we developed and validated quantitative Taqman primer-probe sets spanning ANRIL exons 1–2, 4–6 and 18–19 (See Materials and Methods and Figure 1B). We also quantified the expression of other INK4/ARF transcripts (e.g. p15INK4b, p16INK4a and ARF) since we previously observed a correlation between the expression of ANRIL4-6 and p16INK4a in PBTLs (r2 = 0.32, p<0.0001; [42]). Importantly, none of these Taqman strategies amplify ANRIL exons containing repetitive sequences. Moreover, the products from these assays were gel-purified, cloned and sequenced to verify their identities. Equal amounts of RNA from a panel of 27 primary and transformed cell lines were subjected to reverse transcription using a combination of random hexamer and oligo dT primers. Deletion or methylation of the 9p21 tumor suppressors is a common event in cancer and was observed in 59% of our cell line panel (Figure S4). Even after exclusion of these lines from our analyses, all three ANRIL species (exon 1–2, 4–6 and 18–19) were rare: based on an estimate of 15 pg of total RNA per cell, expression of ANRIL species ranged between 0.01 to 1 copy per cell (Figure 1B). For comparison, INK4/ARF expression is considered quite low in primary cells [43], [44], yet p16INK4a and p15INK4b were more abundant than any ANRIL species in non-transformed Hs68, HUVEC and IMR90 cell lines (on average 155.9±18.9 - and 14.5±5.5-fold, respectively; See Figure S4). These data indicate that ANRIL species are readily detectable but non-abundant in a wide variety of cell types.
Central ANRIL exons are weakly detected in cell line and RNA-seq analyses
To further characterize 9p21 transcription in a manner unbiased by PCR primer choice, we analyzed publically available, next-generation RNA sequencing (RNA-seq) datasets of oligo dT-primed and reverse transcribed RNA from primary human brain or HeLa cells. ([45]; Genbank Short Read Archive Study ID: SRP002274 and ENCODE RNA-seq replicates, respectively). These datasets were solely chosen based upon the high-depth of sequencing provided (>368 million and >110 million reads, respectively), allowing for the study of non-abundant ncRNAs. In accord with the TaqMan-based analyses, reads mapping to ANRIL were rare in both the human brain and HeLa datasets (Figure 2). In fact, the peak height of reads mapping to p16INK4a was >40 times higher than that of any ANRIL exon, even though brain expresses low levels of p16INK4a [44], [46]. The level of ANRIL expression detected in both datasets (maximal peak height of 12) was comparable to that of other long, non-coding RNAs including HOTAIR and Kcnq1ot1 (maximal peak heights of 10 and 18, respectively).

Intriguingly, by both TaqMan (Figure 1B) and RNA-seq (Figure 2B), we observed a disparity in the number of molecules of each ANRIL exon detected, with those containing the ‘central’ ANRIL (4–12) exons being the least abundant when averaged across the entire locus. The most prevalent ANRIL peaks localized to the 5′ and 3′ ends of the transcript (i.e. exons 1–3 and 13–19). The relative excess of the more distal exons might be a consequence of 9p21 deletion, as we observed splicing between MTAP, a gene 100 kb telomeric to the INK4/ARF locus, and the 3′ end of ANRIL in RNA-seq datasets from cell lines with INK4/ARF loss (i.e. SUM102 and MCF7, data not shown). Although such MTAP-ANRIL fusions have been previously described [47], using a highly sensitive Taqman strategy we only detected these fusions in cell lines with 9p21 deletion, and not in any primary cells or lines with two copies of an intact INK4/ARF locus (data not shown). Therefore, while exon deletions and/or MTAP splicing explain the relative excess transcription of distal ANRIL exons in some cancer cell lines harboring 9p21 deletion, these mechanisms do not explain the decreased transcription of exons 4–12 versus exons 1–3, in any cell line. Likewise, somatic deletions and MTAP splicing do not explain the uniform decrease of the central exons compared to the proximal and distal exons in cultures of primary cells (Figure S4). Together, these RNA-seq and TaqMan analyses indicate that ANRIL is a rare, multi-variant RNA species in which transcripts containing exons 1–3 or 13–19 predominate over those containing exons 4–12. Based on these findings as well as the observation of non-colinear RNA species by RACE (Figure 1A), we hypothesized that the reduced level of the central exons (4–12) in mature, polyadenylated RNA may be the result of alternative splicing events in which the central exons of ANRIL are frequently skipped.
Select ANRIL transcripts containing internal exons are circular
We observed non-colinear RNA species in RACE analysis of several cell lines and primary cells (Figure 1A). We considered the possibility that this represented alterations of the germline DNA sequence (e.g. duplications or transversions) but found no evidence for this using BreakDancer [48] to analyze next-generation DNA sequencing from 10 individuals. Given that cryptic germline DNA alterations did not appear to explain these non-colinear forms, we considered that non-canonical RNA splicing events such as trans-splicing or re-splicing of RNA lariats might occur as part of ANRIL processing. In the latter case, the splice sites of certain consecutive exons are not recognized by the splicing machinery, resulting in the inclusion of exonic sequences within the RNA lariat [49]. Internal splicing of this exon-containing lariat structure may then occur, leading to the production of exon-only RNA circles in which non-consecutive exon junctions are generated by splicing across the branch site (e.g. exon 13–14–4–5).
To determine the structure of such non-colinear ANRIL RNAs, we designed and validated a Taqman strategy using outward facing primers to detect transcripts containing the non-colinear exon 14-5 junction (Figure 3A). This detection scheme was chosen based upon preliminary PCR studies wherein amplification of the 14-5 splice junction predominated over that of the exon 14-4 junction. As lariat or circular RNAs would not be polyadenylated, we assayed the amount of each ANRIL species detected in RNA samples reverse transcribed with random hexamers (HEX), oligo dT (dT) or both (Figure 3B). As would be expected for polyadenylated RNAs, conversion of ANRIL1-2, ANRIL18-19, p16INK4a and p15INK4b into cDNA was efficiently accomplished with either oligo dT or HEX primers alone. Conversely, ANRIL4-6 and 14-5 were effectively primed with HEX but not oligo dT, confirming that these transcripts were not polyadenylated.
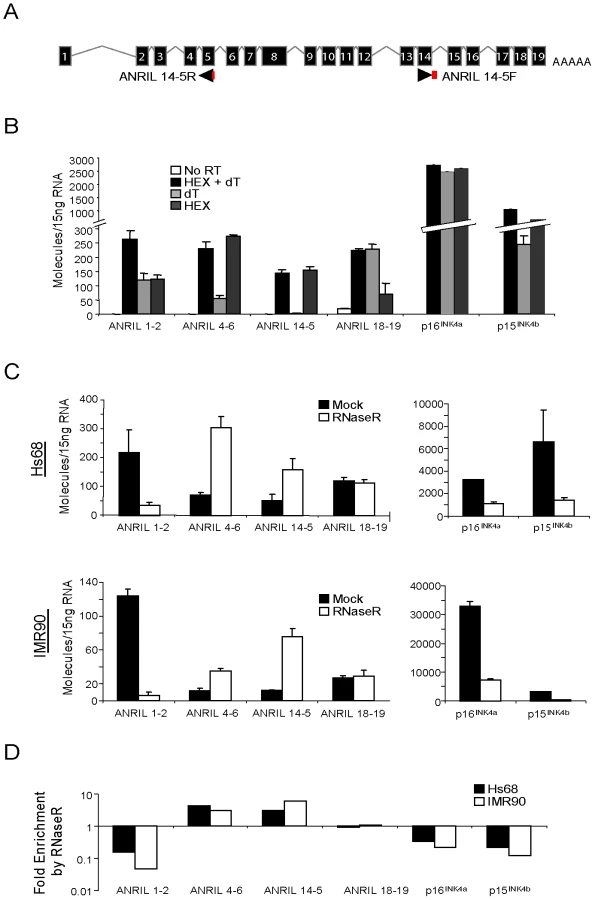
To further examine the structure of these non-canonical transcripts, we determined transcript sensitivity to the RNA exonuclease, RNAse R. RNAse R specifically digests both structured and non-structured linear RNAs, but spares RNA circles and lariats [50]. Using equal amounts of RNA from normal and immortalized human fibroblasts (IMR90 and Hs68, respectively) treated with or without RNAse R, we generated cDNA and conducted TaqMan analysis for ANRIL expression. As expected for linear species, RNAse R treatment caused a marked reduction in the number of p16INK4a and p15INK4b transcripts detected (4.5 - and 8.4-fold decrease, respectively), demonstrating that these coding transcripts are predominantly linear (Figure 3C and 3D). In contrast, we observed a 6-fold enrichment of ANRIL14-5 molecules with RNAse R treatment, confirming their circular nature. We were surprised to find that ANRIL4-6 expression also exhibited RNAse R-dependent enrichment in both cell lines. ANRIL1-2 levels decreased consistent with a predominantly linear species, and ANRIL18-19 demonstrated an intermediate behavior consistent with a mix of linear and circular forms (Figure 3C and 3D). Together, these data provide evidence that ANRIL4-6 and 14-5 are predominantly contained within non-polyadenylated, circular (or lariat) ANRIL (cANRIL) transcripts.
Circular ANRIL species are observed in a wide variety of cell types
Based upon analysis of non-sequential cDNA and EST sequences, stable RNA circles have been hypothesized to represent <1% of the total transciptome [51]. To determine the ubiquity of cANRIL RNAs, we employed the Taqman-based strategy shown in Figure 3A. Using this method, we observed amplification of ANRIL14-5 in 16 of 20 ANRIL expressing primary cultures and cell lines (Figure S5). The finding of cANRIL in diverse cell types is perhaps surprising given the low abundance of ANRIL transcripts and that RNA lariats are usually unstable intermediates which undergo rapid degradation. These data suggest that cANRIL is a stable, naturally occurring, circular RNA species produced in most INK4/ARF expressing cells.
To better define the structure of cANRIL RNAs, we used outward-facing primers located in five ANRIL exons to amplify cDNA produced using a mix of random hexamers and oligo dT (Figure 4A). In Hs68 and IM90 cells, PCR products specific to 9p21 were detected using primers sets targeting exons 4 and 6. These products were more efficiently amplified using cDNAs from RNase R-treated samples, suggesting that they arose from circular RNAs. In contrast, PCR products specific to chromosome 9 were not detected using primers internal to exons 1 and 18, and only detected in one instance with primers internal to exon 16, suggesting these exons were not commonly included in stable, circular ANRIL species. To determine the contents of the circular transcripts containing ANRIL exons 4 and 6, we cloned and sequenced the observed PCR products. The resulting sequences predominantly included ANRIL exons 4 to14. Several exons were never observed in the looped structures (i.e. exons 1, 2, 3, 8, 9, 11, and 12) (Figure 4B and 4C). Novel sequences were also discovered within the products, most notably, regions intronic to the previously described ANRIL transcript (i.e. parts of intron 3). These data explain the paucity of expression of ANRIL exons 4 to 14 as determined by Taqman and RNA-seq (Figure 1B and Figure 2B), suggesting that ANRIL processing may involve exon skipping events which preferentially incorporate the central exons into lariats which may then be internally spliced to form cANRIL.

Expression of cANRIL and proximal ANRIL species correlate with INK4/ARF expression and ASVD–associated SNP genotype
We have previously shown [42] a correlation between expression of the coding INK4/ARF transcripts and ANRIL4-6. We now recognize that the latter is largely contained in circular RNA species (Figure 3B and 3C). Next, we investigated correlations between other, newly identified ANRIL isoforms and the coding INK4/ARF transcripts (Figure 5A and Figure S6). We performed Taqman-based gene expression analyses in 106 primary human peripheral blood T-lymphocyte (PBTL) samples from two previously published cohorts [16], [42]. Remarkably, cANRIL expression (ANRIL14-5 and ANRIL4-6) was observed in all PBTL samples. Expression of ANRIL18-19 did not correlate with any other ANRIL or INK4/ARF transcript (Figure 5A). In contrast, a strong correlation was observed between the expression of ANRIL4-6 and all of the INK4/ARF tumor suppressors (p<1*10−15 for all pair-wise comparisons). Significant associations (p<0.05) were also observed between ANRIL1-2 and ARF, and between ANRIL14-5 and p16INK4a.

We have also demonstrated an effect of a replicated SNP associated with atherosclerosis (rs10757278) on the expression of p15INK4b, p16INK4a, ARF and ANRIL4-6 in PBTLs [16]. We therefore examined whether expression of other ANRIL isoforms correlated with ASVD SNP genotype (Figure 5B). While no significant correlation between ANRIL18-19 with SNP genotype was noted, expression of ANRIL1-2 and ANRIL14-5 showed significantly decreased expression in individuals harboring the risk (G) allele (p<0.0001 and p<0.04, respectively). In aggregate, these data demonstrate that expression of both circular and linear transcripts containing the proximal but not distal ANRIL exons correlates with ASVD-SNP genotype and expression of the coding INK4/ARF transcripts.
Deep sequencing of the ASVD risk interval reveals ANRIL exon 15 variants predicted to influence cANRIL production
We performed next-generation DNA sequencing of the ASVD risk interval to identify polymorphisms that might influence ANRIL expression or splicing. To enhance detection of rare variants, we pooled DNA from 5 individuals of Asian and European descent who were homozygous for either the A or G allele of rs10757278. To increase the chance that the analyzed DNAs would harbor causal variants that influence INK4/ARF expression, we selected individuals based on their age-adjusted expression of p16INK4a (i.e. AA donors with higher than average expression, GG donors with lower than average expression [16]). We performed sequence capture using an Illumina tiling array on the AA vs. GG pooled samples. The tiling array was designed to bind all non-repetitive human chromosome 9 regions from 22,054,888 to 22,134,171 bp, a region chosen as it contains the previously identified ASVD and type 2 diabetes mellitus risk intervals (Figure 6A). The 9p21 enriched DNA was sequenced using both Roche 454 (400 bp reads) and Illumina GAII (35 bp reads) technologies. The resulting sequences were aligned to the UCSC reference genome (hg18) using three separate alignment algorithms: gsMapper, BWA and SOAP [52], [53]. As shown in Figure 6B, SOAP identified the highest number of polymorphisms within each pooled sample; however, many of these were not recapitulated using other mapping techniques. Thus, for further analysis we focused our efforts on polymorphisms which were identified by at least two mapping algorithms. Employing these criteria, we discovered 101 SNPs that differed from the reference genome within our samples: 11 in the AA pool, 64 in the GG pool, and 26 in both (Figure 6C). As expected, more differences from the reference genome were identified in the GG sample, as the reference genome harbors the A allele, and most SNPs in the captured region are in moderate to strong linkage disequilibrium with rs10757278 (Figure S1). Therefore, using next generation sequencing of captured DNA from 10 informative individuals biased to harbor causal variants, the chosen sequencing approach found 75 (11+64) SNPs that differed between the two pooled samples.
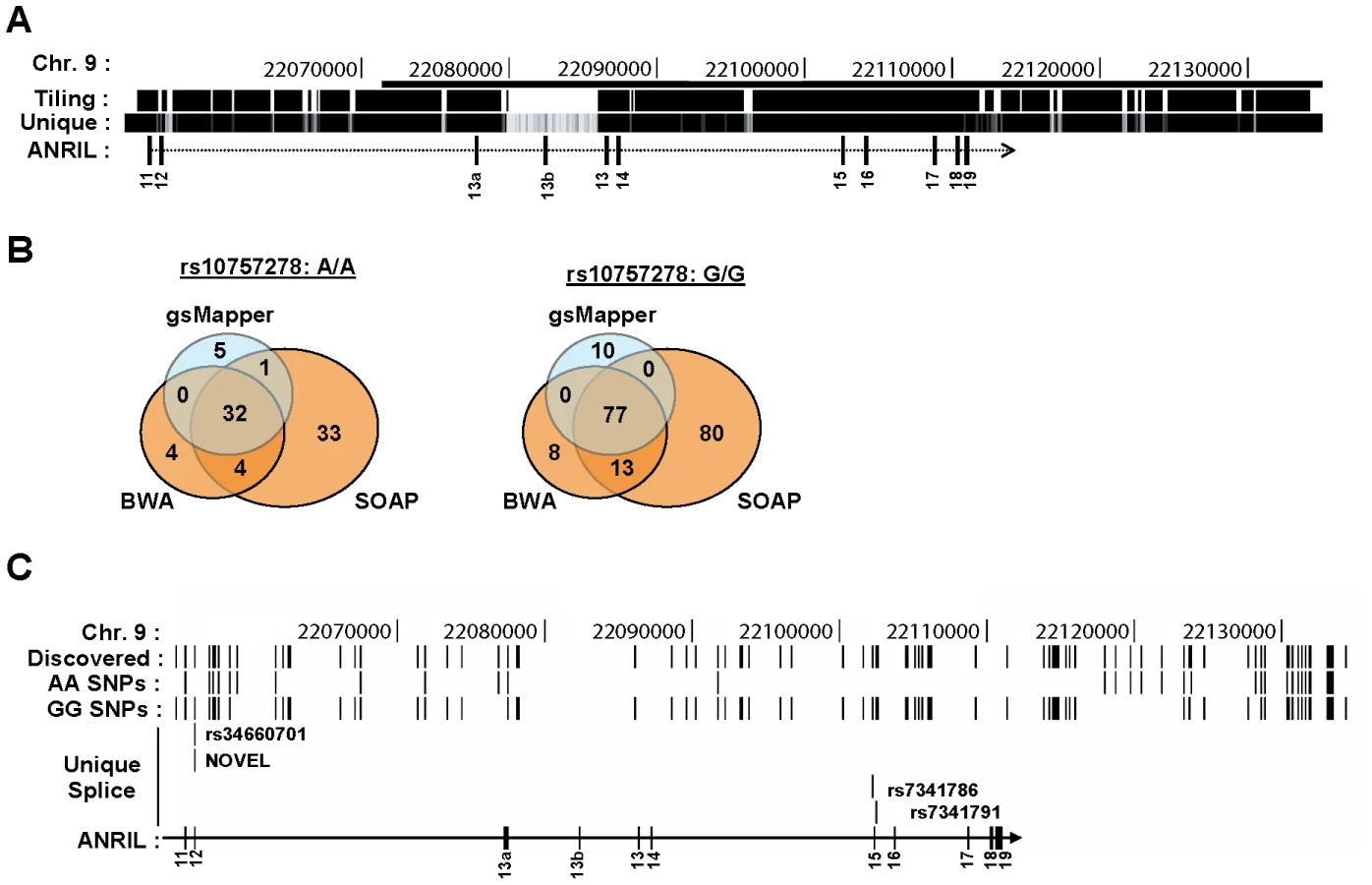
Given the finding that cANRIL expression correlates with rs10757278 genotype, we sought to determine if any of the identified SNPs might influence ANRIL splicing. For this analysis, we restricted the SNP list to include only those within 200 bp of an ANRIL intron-exon boundary, as genetic alterations in this region have the highest potential to influence RNA splicing [54]. Of the 75 SNPs present in only one of the two pooled samples, four were within 200 bp of an ANRIL intron-exon boundary (Figure 6C, Table S1). Using previously described prediction algorithms [54], [55], we determined the likelihood of these variants near intron-exon boundaries to change cis-regulatory splice elements including exon splicing enhancers (ESEs) and silencers (ESSs) as well as intronic splicing enhancers (ISEs) and silencers (ISSs). A score of -1 indicates that the minor allele destroys one cis-element and +1 indicates that the minor allele creates one cis-element (Table S1). ESEs and ISEs favor exon recognition by the spliceosome when they occur in exons or introns, respectively. However, ISSs and ESSs promote exon skipping irrespective of position [56]. A pair of SNPs (rs34660702 and NOVEL) 10 bp apart near the start of exon 12 were predicted to alter ANRIL splicing, but the effects of these SNPs worked in opposite directions, and both exhibited low frequencies in the CEU population (Figure 6C and Table S1). More provocatively, we also observed two SNPs (rs7341786 and rs7341791) in strong linkage disequalibrium with rs10757278 and which bracketed exon 15. These SNPs demonstrated high minor allele (CC and GG, respectively) frequencies and with the major allele of both SNPs enhancing the “exon-ness” of exon 15. These results indentify common, linked SNPs whose major allele is likely to inhibit skipping of exon 15 and thereby promote the production of cANRIL species ending in exon 14 in individuals homozygous for the A (protective) allele of rs10757278.
Due to the small number of pooled individuals for sequencing (n = 5/group), we also sought to identify additional, rare polymorphisms with the potential to modulate ANRIL splicing. We analyzed all SNPs reported in the HapMap database in the chromosome 9 region 22,054,888 to 22,134,171 within 200 bp of ANRIL intron-exon boundaries for their potential to alter RNA splicing in cis-. Using this method, we identified 31 additional SNPs in HapMap that modified ESE, ESS, ISE or ISS sequences (Table S1). Notably, the majority of these variants were rare and almost never reported in the CEU population [57]. Therefore, among all of the SNPs examined from the ASVD risk interval, rs7341786 and rs7341791 appear most likely to influence ANRIL splicing. These SNPs are also in very strong linkage disequilibrium with the ASVD-associated SNP, rs10757278 (r2>0.96, based upon the nearest database SNPs in CEU), and therefore would be expected to correlate with ANRIL and INK4/ARF expression. However, it is important to note that other classes of germline variants (e.g. complex indels or alterations in repetitive elements) would not have been identified by our sequencing strategy, and therefore we are unable to exclude a role for other such variants in ANRIL expression and/or splicing.
Discussion
Motivated by the role of other long, non-coding RNAs in PcG repression, we investigated whether ANRIL transcription and/or structure was SNP-dependent. In primary and cultured cell lines, we used RACE and RNA-seq to identify novel ANRIL variants (Figure 1 and Figure 2). Intriguingly, the central ANRIL exons were underrepresented in these data (Figure 1B and Figure 2B). This observation, along with non-colinear products detected by RACE, led to the unexpected discovery of multiple circular RNAs emanating from the ANRIL locus. Sequencing of these circular species showed non-sequential linkages between various ANRIL exons, especially those from the central portion of the transcript (e.g. exons 4-14) (Figure 4B and 4C). Expression of both circular and linear ANRIL isoforms proximal to the INK4/ARF locus strongly correlated with INK4/ARF transcription and the ASVD risk genotype at rs10757278 (Figure 5 and Figure S6). In contrast, distal ANRIL variants containing exons 18 and 19 were expressed in a genotype-independent manner and did not correlate with the levels of any INK4/ARF transcript. Using next-generation sequencing to genotype captured DNA from the ASVD risk interval, we identified a common pair of linked SNPs near exon 15 predicted to influence ANRIL splicing (Figure 6 and Table S1). Together, these findings suggest that common polymorphisms in the ASVD risk interval could modulate ANRIL transcription and/or splicing, thereby influencing PcG-mediated INK4/ARF repression and atherosclerosis susceptibility.
ANRIL encodes multiple, non-abundant linear and circular species
Multiple ANRIL isoforms have been reported in the literature and EST databases, with some exhibiting differential expression patterns and SNP associations (Figure S2 and [28], [29]). Using RACE, RNA-seq and sensitive quantitative real-time PCR techniques, we now show that no single ANRIL species predominates in vivo, and that splicing to MTAP does not occur in cells with an intact 9p21 locus. Moreover, our analyses identified new ANRIL exons and variants, uncovering a novel group of circular ANRIL (cANRIL) species (Figure 4). Using independent approaches, we found that all ANRIL exons were expressed at very low levels, orders of magnitude lower than even the relatively rare INK4/ARF tumor suppressors, p15INK4b and p16INK4a (Figure 1B, Figure 2B, and Figure S4). This low level of expression is comparable to what we observed for other regulatory non-coding RNAs (i.e. HOTAIR and Kcnq1ot1) associated with PcG-mediated repression.
The discovery of non-colinear ANRIL species whose expression correlated with INK4/ARF transcription suggested that alternative splicing events might modify ANRIL structure leading to changes in PcG-mediated INK4/ARF repression. There are two major mechanisms by which non-colinear RNAs are thought to arise: trans-splicing and exon skipping [58]. During trans-splicing, a Y-branched RNA structure is formed that is sensitive to RNase R digestion [50]. In contrast, exon skipping events generate large lariat structures, which can then undergo cis-splicing to create RNase R-resistant circular RNAs. To confirm the circular nature of the ANRIL species, we showed that transcripts containing ANRIL4-6 and 14-5 were resistant to RNase R degradation, were not polyadenylated and could be PCR amplified using sets of outward facing primers (Figure 3 and Figure 4). Therefore, cANRIL species appear to result from exon skipping events occurring during RNA splicing.
INK4/ARF regulation and ASVD genotype
Based on these observations, we propose a model suggesting that common polymorphisms in 9p21.3 modifiy INK4/ARF gene expression through cis-regulation of ANRIL transcription and/or splicing (Figure 7). In turn, such changes in ANRIL levels or structure influence the ability of these ncRNAs to repress the INK4/ARF locus. Specifically, changes in PcG-mediated repression of p15INK4b, p16INK4a and/or ARF occur, altering potentially atherogenic cellular proliferation and ASVD risk as previously suggested [16]–[20], [26].

Prior studies provide evidence for this model. For example, we and others have described an effect of ASVD-genotype on ANRIL transcription [16], [30], [59], [60]. Moreover, meticulous allele-specific expression analyses by Cunnington et al. suggested that the cis-effects of 9p21 SNPs were stronger for ANRIL than for the coding INK4/ARF transcripts (20% vs. <8%)[60]. In contrast, the effect of these SNPs on expression of the coding INK4/ARF transcripts predominantly occurred in trans-. Such findings are consistent with our model whereby causal variants directly influence ANRIL structure in cis-, thereby controlling repression of the INK4/ARF locus, possibly in trans - (Figure 7). Likewise, recent work has supported a role for ncRNAs in INK4/ARF repression. Knockdown of the RNA helicase, Mov10, led to INK4/ARF deregulation, suggesting a role for RNA metabolism in regulation of the locus [61]. Supporting these data, Yap et al. recently demonstrated that the 5′ end of ANRIL contains stem-loop structures capable of binding CBX7, a member of the PRC-1 complex [62]. Disruption of this interaction led to premature senescence marked by increased INK4/ARF expression. Therefore, strong evidence supports the notion that ANRIL expression correlates with ASVD-SNP genotype and that PcG-mediated repression of the INK4/ARF locus is modulated by this expression.
The outstanding question of this model is the biochemical mechanism whereby causal variants at 9p21.3 located more than 100 kb distant from the proximal exons of ANRIL (Figure S1) modulate ANRIL expression and/or structure. Our data are consistent with a “transcriptional model” wherein distal cis-regulatory elements within the ASVD risk interval directly influence ANRIL transcription. Jarinova et al. have identified an enhancer sequence within ANRIL intron 17, which in heterologous reporter assays, enhanced transcription in a genotype-specific manner [30]. Consistent with this view, we also observed a significant effect of ASVD-SNP genotype on the expression of the linear ANRIL1-2 transcript (Figure 5B). As such, the correlation of cANRIL expression with ASVD-SNP genotype would presumably reflect passive production of these circular species in the setting of increased or decreased in total ANRIL transcription. However, this model would not easily explain the positive correlation we observed between the expression of coding INK4/ARF transcripts and proximal ANRIL exons (Figure 5A and Figure S6), as the 5′end of ANRIL is reported to foster INK4/ARF repression [62].
Alternatively, we believe our present data are more consistent with a “splicing model” in which causal variants in the ASVD risk interval influence ANRIL splicing by regulating exon skipping. Provocatively, the ASVD risk interval includes exon 15 where the termination of most exon skipping events that produce cANRIL occur (Figure 4B and 4C). Using next-generation sequencing of this region in individuals biased to harbor causal variants, we identified exon 15 SNPs (rs7341786 and rs7341791) which are in strong linkage disequilibrium with the ASVD-associated SNP rs10757278 (r2>0.96; Table S1). These polymorphisms have high minor allele frequencies in our sample and the CEU population and are predicted to alter ANRIL splicing (Figure 6, Table S1). In particular, the major allele (‘A’) of rs7341786, detected only in individuals homozygous for the A-allele at rs10757278, is predicted to increase the strength of exon 15 as a splice acceptor, which would promote the expression of spliced RNA circles ending in exon 14, as observed (Figure 4B and 4C).
For the splicing model to explain the observed positive correlation between the expression of ANRIL and the coding INK4/ARF transcripts, distinct species of ANRIL would need to differ in their ability to repress the INK4/ARF locus. Supporting this possibility, PcG-mediated repression of the murine Kcnq1 locus is dependent upon the length of the long, ncRNA, Kcnq1ot1 [63], [64]. Specifically, longer Kcnq1ot1 transcripts are associated with increased PcG recruitment and Kcnq1 silencing potential. Therefore, shorter ANRIL variants (i.e. those lacking exons 4–16) generated by exon skipping events may also be less efficient at repressing the INK4/ARF locus. Important predictions of the splicing model are that cANRIL expression (reflecting ANRIL splicing) should correlate with INK4/ARF expression (which it does, Figure 5A and Figure S6), that individuals homozygous for the ‘A’ allele of rs7341786 (and rs10757278) should exhibit increased production of cANRIL species containing exon 14 but not exon 15 (which they do, see Figure 4B and 4C) and that these individuals with increased propensity for splicing should exhibit de-repressed INK4/ARF expression (which they do, [16]).
A caveat to the splicing model is that the identified exon 15 SNPs need not be the true causal variant(s) influencing ANRIL splicing. It is possible that other polymorphisms not detected by our sequencing strategy could regulate ANRIL splicing. In particular, the sequencing strategy employed would not find differences between the pooled samples in repetitive regions, such as the large LINE element in the 12th intron of ANRIL (Figure 6A). ANRIL harbors several LINE and SINE elements, and such repetitive motifs have been reported to modulate RNA splicing in other systems [65], [66]. Therefore, while the exon 15 SNPs appear to be prime candidates to regulate ANRIL splicing, a variety of other classes of polymorphisms could also influence splicing and would not have been observed by the chosen sequencing approach.
Importantly, the splicing and transcriptional models are not mutually exclusive. A single causal variant may influence both processes or there may be multiple causal variants that influence either process within the ASVD risk interval. Finally, while we believe the correlation of cANRIL expression and ASVD-SNP genotype is most likely explained by an effect of common 9p21 polymorphisms on transcription and/or splicing, a third possibility also exists. While circular RNA byproducts of exon skipping have generally been regarded as inconsequential, circular RNAs with catalytic activities (e.g. group I and some group II introns) are well described in bacteria, lower eukaryotes, plants [67]. In addition, some viroids and the hepatitis delta satellite virus have circular RNA genomes [68], [69]. Although we are not aware of any endogenously produced circular RNA with discrete function in mammals, clearly circular RNAs species can possess independent functions in non-mammalian species, and we remain open to the possibility that cANRIL itself can directly participate in INK4/ARF regulation.
In summary, this work links ASVD-genotype to ANRIL structure and INK4/ARF regulation, providing evidence for what we believe is a first association between endogenous circular RNA expression and a mammalian phenotype (ASVD). Even were cANRIL expression merely a marker of exon skipping with no specific biologic function, we believe its expression may be a useful marker of ANRIL isoform selection, which our findings suggest is of pathogenic relevance to ASVD susceptibility. We believe this work has implications beyond ASVD, as altered ANRIL splicing could influence INK4/ARF expression, explaining the association of other nearby 9p21 SNPs with a variety of non-ASVD phenotypes in humans including longevity, type II diabetes, endometriosis and several tumors types [70]–[77].
Materials and Methods
Ethics statement
Research involving human subjects was approved by the University of North Carolina Institutional Review Board and all participants provided informed, written consent.
Cell lines and culture conditions
WM266-4, UACC 257, A2058, A375, SUM-149, RPMI-8322 and telomerized Hs68 cells were obtained and grown as previously described [78]–[80]. MDA-MB-468, MDA-MB-436, MDA-MB-231, MCF7, BT-474, BT-549, T-47D, COLO 205, T84, LoVo, LS 174T, SW480, LS1034, HeLa, HUVEC, IMR90, Ramos, Raji, Jurkat and U-87 cells were originally obtained from ATCC and cultured as suggested.
3′ and 5′ rapid amplification of cDNA ends (RACE)
CD3 positive T-cells were isolated from human peripheral blood samples as previously described [42]. RNA was generated from proliferating cell lines and isolated human T-cells using the RNAeasy system (Qiagen Inc., Valencia, CA). 3′ and 5′ RACE was performed as described in the Firstchoice RLM-RACE manual (Ambion Inc., Austin, TX). This procedure is optimized for the detection of rare transcripts and provides additional steps to improve the specificity of mRNA amplification. Gene-specific primers were designed within ANRIL exons 4 and 6 as shown in Figure 1A and Table S2; RACE primers for other ANRIL exons tested did not amplify chromosome 9 specific products. All PCR reactions were conducted using SuperTaq-Plus (Ambion) in a Bio-Rad DNA Engine thermocycler. SuperTaq-Plus is a high fidelity, long range polymerase with the capability to amplify complex DNAs such as repetitive SINE, LINE and Alu elements. Cycling conditions for 5′RACE were: 94°C 3 min, 34×[94°C 30 s, 60°C 30 s, 68°C 3 min], 68°C 5 min (inner reaction) and 94°C 3 min, 34×[94°C 30 s, 62°C 30 s, 68°C 3 min], 68°C 5 min (outer reaction). Cycling conditions for 3′RACE were: 94°C 3 min, 34× [94°C 30 s, 57 or 60°C 30 s, 68°C 3 min] (outer reaction) and 94°C 3 min, 34×[94°C 30 s, 60°C 30 s, 68°C 3 min] (inner reaction). Cloning of the resulting PCR products was conducted using the TOPO-Blunt cloning kit (Invitrogen). Sequencing of the resulting clones was conducted using both M13F and M13R primers.
Quantitative real-time PCR (qRT-PCR)
RNA was isolated using the Qiagen RNAeasy Kit and subjected to reverse transcription using the ImPromII reverse transcription system (Promega, Madison, WI). RNA samples from breast and colorectal cell lines were kindly provided by Drs. N. Mitin and J. Yeh (UNC). Taqman primer and probe sets for the detection of ANRIL exons 1–2, 4–6, 14–5, and 18–19 as well as those for p15INK4b, p16INK4a, ARF and 18S rRNA are described in Table S2. The ANRIL primer sets were designed to span at least one intron and were shown to have high specificity with linear amplification efficiencies between 88 and 94% (Figure S3). Final primer and probe concentrations were 900 and 250 nM, respectively. Products from the ANRIL 1–2, 4–6, 14–5 and 18–19 qRT-PCR reactions were cloned separately into the pBluntII-TOPO vector (Invitrogen) and verified. Real-time PCR was carried out in triplicate on an ABI 7900HT thermocyler.
For relative expression studies, differentials were first calculated between each sample and the average 18S cycle threshold (Ct) for the entire experiment. Transcript Ct values were normalized using the equation: Expression = Lower limit of detection (37 or 40) - (Ct target – Ct 18S differential) and plotted on a log2 scale. The relative expression of p16INK4a, p15INK4b, ARF and ANRIL4-6 in PBTLs has been previously reported [42]; in this work, these same samples were reanalyzed for ANRIL1-2, ANRIL14-5 and ANRIL18-19 expression as shown in Figure 5 and Figure S6. In Figure 5 and Figure S6, data was corrected to account for batch effects between the pilot and verification datasets previously described [42]. To determine the absolute number of transcripts present, a standard curve of five dilutions was generated for each experimental plate using known amounts of linearized plasmid containing the target sequence. The number of molecules detected in each sample was calculated using the equation: #Molecules = 10∧((Ct – y-intercept)/slope). Primer efficiencies were calculated using the equation: Efficiency = (10∧(-1/slope))-1. Multiple comparison plots and statistical analysis appearing in Figure 5A and Figure S6 was performed using the gpairs function of the R YaleToolkit library.
Analysis of RNA-seq data
A total of 368,846,235 reads generated on the Illumina platform (study SRP002274) were downloaded from the NCBI Short Read Archive. The reads were first screened for unique 20mers deriving from chromosome 9∶21,700,000–22,300,000 using the UCSC genome bowser Duke uniqueness mapability table. The resulting reads were mapped using the TopHat spliced aligner (PMID: 19289445) to the reference human genome (hg18). The resulting coverage plot was imported into the UCSC genome browser for display. Also analyzed were two independent CalTech ENCODE mRNA-seq datasets (http://genome.ucsc.edu/cgi-bin/hgTrackUi?g=wgEncodeCaltechRnaSeq) from HeLa cells. These datasets were chosen because they lacked deletion of 9p21 and showed the highest level of ANRIL expression of any publically available RNA-seq data we analyzed. For Figure 2B, the peak number of reads at a given base (SRP002274 data) or peak reads per kilobase mapping (CalTech HeLa data) were quantified and scaled relative to the average exonic coverage.
RNase R digestion
Total RNA was isolated from proliferating Hs68 and IMR90 cells using a Qiagen RNAeasy kit. Equal amounts of RNA (43–50 µg, depending on the experiment) were incubated with or without 40 U of Rnase R (RNR07250, Epicentre Biotechnologies) for 2.5 hours at 37°C. The resulting RNA was purified using the RNAeasy column and quantified. Equal amounts of RNA were then subjected to reverse transcription using the ImPromII reverse transcription system and a mixture of random hexamer and oligo dT primers (Promega, Madison, WI). Transcripts were quantified using Taqman-based real-time PCR.
Loop PCR
PCR primers pointing in opposite directions on ANRIL exons 1, 4, 6, 13, 16 and 18 were designed using Primer3 software and analyzed for hairpins using Netprimer (Premier Biosoft and http://frodo.wi.mit.edu/primer3/) (Table S2). PCR reactions were conducted using cDNA representing 15 ng of mock or Rnase R-treated RNA. Reactions were performed using Apex Hot Start Taq DNA polymerase and Buffer 2 (Genesee Scientific) in a Bio-Rad DNA Engine thermocycler. The cycling conditions were as follows: 95°C 15 min, 40×[94°C 30 s, 59°C 30 s, 72°C 1 min], 72°C 2 min. The resulting PCR products were cloned into the TOPO-Blunt cloning kit (Invitrogen) and sequenced using M13F and M13R primers.
Sequence capture and next-generation sequencing
Genomic DNA was generated from T-cells of healthy human volunteers of known rs10757278 genotype, age and p16INK4a expression status using the Qiagen Peripheral Blood DNA isolation kit. For sequence capture, 21 ug of DNA was pooled from five individuals homozygous for the G-allele of rs10757278 and another five individuals homozygous for the A-allele. Samples were sent to NimbleGen for sequence capture using a tiled array spanning human chromosome 9 (22,054,888-22,134,171). The resulting amplified DNA fragments were analyzed at the UNC Genome Analysis Facility using both Illumina GAII and Roche 454 technology. For sequencing on the Illumina platform, DNA was randomly sheered and appropriate adapters ligated. Resulting sequences were aligned to the entire human genome (hg18) using MAQ, SOAP, and gsMapper software [52], [53]. MAQ was run with default settings and output was translated into BAM format. SOAP alignment was performed allowing up to 10 gap bases and 2 mismatches and also translated into BAM format. Mapping with gsMapper was performed with default settings. SNP calls from MAQ and SOAP were generated using the pileup function of the SAMtools library [81]. Calls were culled to include only those SNPs appearing in ≥20 percent of reads.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. World Health Organization W 2009 Cardiovascular diseases Fact Sheet
2. BirosE
CooperM
PalmerLJ
WalkerPJ
NormanPE
2010 Association of an allele on chromosome 9 and abdominal aortic aneurysm. Atherosclerosis
3. BownMJ
BraundPS
ThompsonJ
LondonNJ
SamaniNJ
2008 Association between the coronary artery disease risk locus on chromosome 9p21.3 and abdominal aortic aneurysm. Circ Cardiovasc Genet 1 39 42
4. ThompsonAR
GolledgeJ
CooperJA
HafezH
NormanPE
2009 Sequence variant on 9p21 is associated with the presence of abdominal aortic aneurysm disease but does not have an impact on aneurysmal expansion. Eur J Hum Genet 17 391 394
5. HelgadottirA
ThorleifssonG
ManolescuA
GretarsdottirS
BlondalT
2007 A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science 316 1491 1493
6. HelgadottirA
ThorleifssonG
MagnussonKP
GretarsdottirS
SteinthorsdottirV
2008 The same sequence variant on 9p21 associates with myocardial infarction, abdominal aortic aneurysm and intracranial aneurysm. Nat Genet 40 217 224
7. McPhersonR
PertsemlidisA
KavaslarN
StewartA
RobertsR
2007 A common allele on chromosome 9 associated with coronary heart disease. Science 316 1488 1491
8. MatarinM
BrownWM
SingletonA
HardyJA
MeschiaJF
2008 Whole genome analyses suggest ischemic stroke and heart disease share an association with polymorphisms on chromosome 9p21. Stroke 39 1586 1589
9. GschwendtnerA
BevanS
ColeJW
PlourdeA
MatarinM
2009 Sequence variants on chromosome 9p21.3 confer risk for atherosclerotic stroke. Ann Neurol 65 531 539
10. SmithJG
MelanderO
LovkvistH
HedbladB
EngstromG
2009 Common genetic variants on chromosome 9p21 confers risk of ischemic stroke: a large-scale genetic association study. Circ Cardiovasc Genet 2 159 164
11. CluettC
McDermottMM
GuralnikJ
FerrucciL
BandinelliS
2009 The 9p21 myocardial infarction risk allele increases risk of peripheral artery disease in older people. Circ Cardiovasc Genet 2 347 353
12. YeS
WilleitJ
KronenbergF
XuQ
KiechlS
2008 Association of genetic variation on chromosome 9p21 with susceptibility and progression of atherosclerosis: a population-based, prospective study. J Am Coll Cardiol 52 378 384
13. BroadbentHM
PedenJF
LorkowskiS
GoelA
OngenH
2008 Susceptibility to coronary artery disease and diabetes is encoded by distinct, tightly linked SNPs in the ANRIL locus on chromosome 9p. Hum Mol Genet 17 806 814
14. SeidelmannSB
KuoC
PleskacN
MolinaJ
SayersS
2008 Athsq1 is an atherosclerosis modifier locus with dramatic effects on lesion area and prominent accumulation of versican. Arterioscler Thromb Vasc Biol 28 2180 2186
15. KimWY
SharplessNE
2006 The regulation of INK4/ARF in cancer and aging. Cell 127 265 275
16. LiuY
SanoffHK
ChoH
BurdCE
TorriceC
2009 INK4/ARF transcript expression is associated with chromosome 9p21 variants linked to atherosclerosis. PLoS One 4 e5027
17. WesselyR
2010 Atherosclerosis and cell cycle: put the brakes on! Critical role for cyclin-dependent kinase inhibitors. J Am Coll Cardiol 55 2269 2271
18. BoehmM
NabelEG
2003 The cell cycle and cardiovascular diseases. Prog Cell Cycle Res 5 19 30
19. GizardF
AmantC
BarbierO
BellostaS
RobillardR
2005 PPAR alpha inhibits vascular smooth muscle cell proliferation underlying intimal hyperplasia by inducing the tumor suppressor p16INK4a. J Clin Invest 115 3228 3238
20. Gonzalez-NavarroH
Abu NabahYN
VinueA
Andres-ManzanoMJ
ColladoM
2010 p19(ARF) deficiency reduces macrophage and vascular smooth muscle cell apoptosis and aggravates atherosclerosis. J Am Coll Cardiol 55 2258 2268
21. KalininaN
AgrotisA
AntropovaY
IlyinskayaO
SmirnovV
2004 Smad expression in human atherosclerotic lesions: evidence for impaired TGF-beta/Smad signaling in smooth muscle cells of fibrofatty lesions. Arterioscler Thromb Vasc Biol 24 1391 1396
22. ReynisdottirI
PolyakK
IavaroneA
MassagueJ
1995 Kip/Cip and Ink4 Cdk inhibitors cooperate to induce cell cycle arrest in response to TGF-beta. Genes Dev 9 1831 1845
23. GraingerDJ
2004 Transforming growth factor beta and atherosclerosis: so far, so good for the protective cytokine hypothesis. Arterioscler Thromb Vasc Biol 24 399 404
24. JanzenV
ForkertR
FlemingHE
SaitoY
WaringMT
2006 Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature 443 421 426
25. Yvan-CharvetL
PaglerT
GautierEL
AvagyanS
SiryRL
2010 ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science 328 1689 1693
26. ViselA
ZhuY
MayD
AfzalV
GongE
2010 Targeted deletion of the 9p21 non-coding coronary artery disease risk interval in mice. Nature 464 409 412
27. PasmantE
LaurendeauI
HeronD
VidaudM
VidaudD
2007 Characterization of a germ-line deletion, including the entire INK4/ARF locus, in a melanoma-neural system tumor family: identification of ANRIL, an antisense noncoding RNA whose expression coclusters with ARF. Cancer Res 67 3963 3969
28. FolkersenL
KyriakouT
GoelA
PedenJ
MalarstigA
2009 Relationship between CAD risk genotype in the chromosome 9p21 locus and gene expression. Identification of eight new ANRIL splice variants. PLoS One 4 e7677
29. KyriakouT
PalA
PedenJ
GreenF
GloynA
2009 ANRIL, The non-coding RNA present in the chromosome 9 CAD associated locus, has multiple splice variants and a potential regulatory role in CDKN2B expression. Atherosclerosis 207 e3
30. JarinovaO
StewartAF
RobertsR
WellsG
LauP
2009 Functional analysis of the chromosome 9p21.3 coronary artery disease risk locus. Arterioscler Thromb Vasc Biol 29 1671 1677
31. JacobsJJ
KieboomK
MarinoS
DePinhoRA
van LohuizenM
1999 The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 397 164 168
32. DhawanS
TschenSI
BhushanA
2009 Bmi-1 regulates the Ink4a/Arf locus to control pancreatic beta-cell proliferation. Genes Dev 23 906 911
33. ChenH
GuX
SuIH
BottinoR
ContrerasJL
2009 Polycomb protein Ezh2 regulates pancreatic beta-cell Ink4a/Arf expression and regeneration in diabetes mellitus. Genes Dev 23 975 985
34. KotakeY
CaoR
ViatourP
SageJ
ZhangY
2007 pRB family proteins are required for H3K27 trimethylation and Polycomb repression complexes binding to and silencing p16INK4alpha tumor suppressor gene. Genes Dev 21 49 54
35. BrackenAP
Kleine-KohlbrecherD
DietrichN
PasiniD
GargiuloG
2007 The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev 21 525 530
36. GilJ
PetersG
2006 Regulation of the INK4b-ARF-INK4a tumour suppressor locus: all for one or one for all. Nat Rev Mol Cell Biol 7 667 677
37. TerranovaR
YokobayashiS
StadlerMB
OtteAP
van LohuizenM
2008 Polycomb group proteins Ezh2 and Rnf2 direct genomic contraction and imprinted repression in early mouse embryos. Dev Cell 15 668 679
38. RinnJL
KerteszM
WangJK
SquazzoSL
XuX
2007 Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129 1311 1323
39. ZhaoJ
SunBK
ErwinJA
SongJJ
LeeJT
2008 Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 322 750 756
40. PandeyRR
MondalT
MohammadF
EnrothS
RedrupL
2008 Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol Cell 32 232 246
41. GuptaRA
ShahN
WangKC
KimJ
HorlingsHM
2010 Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464 1071 1076
42. LiuY
SanoffHK
ChoH
BurdCE
TorriceC
2009 Expression of p16(INK4a) in peripheral blood T-cells is a biomarker of human aging. Aging Cell 8 439 448
43. HaraE
SmithR
ParryD
TaharaH
StoneS
1996 Regulation of p16CDKN2 expression and its implications for cell immortalization and senescence. Mol Cell Biol 16 859 867
44. KrishnamurthyJ
TorriceC
RamseyMR
KovalevGI
Al-RegaieyK
2004 Ink4a/Arf expression is a biomarker of aging. J Clin Invest 114 1299 1307
45. AuKF
JiangH
LinL
XingY
WongWH
2010 Detection of splice junctions from paired-end RNA-seq data by SpliceMap. Nucleic Acids Res
46. NielsenGP
Stemmer-RachamimovAO
ShawJ
RoyJE
KohJ
1999 Immunohistochemical survey of p16INK4A expression in normal human adult and infant tissues. Lab Invest 79 1137 1143
47. SchmidM
SenM
RosenbachMD
CarreraCJ
FriedmanH
2000 A methylthioadenosine phosphorylase (MTAP) fusion transcript identifies a new gene on chromosome 9p21 that is frequently deleted in cancer. Oncogene 19 5747 5754
48. ChenK
WallisJW
McLellanMD
LarsonDE
KalickiJM
2009 BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods 6 677 681
49. ZaphiropoulosPG
1996 Circular RNAs from transcripts of the rat cytochrome P450 2C24 gene: correlation with exon skipping. Proc Natl Acad Sci U S A 93 6536 6541
50. SuzukiH
ZuoY
WangJ
ZhangMQ
MalhotraA
2006 Characterization of RNase R-digested cellular RNA source that consists of lariat and circular RNAs from pre-mRNA splicing. Nucleic Acids Res 34 e63
51. DixonRJ
EperonIC
HallL
SamaniNJ
2005 A genome-wide survey demonstrates widespread non-linear mRNA in expressed sequences from multiple species. Nucleic Acids Res 33 5904 5913
52. LiH
DurbinR
2009 Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25 1754 1760
53. LiR
LiY
KristiansenK
WangJ
2008 SOAP: short oligonucleotide alignment program. Bioinformatics 24 713 714
54. WangZ
RolishME
YeoG
TungV
MawsonM
2004 Systematic identification and analysis of exonic splicing silencers. Cell 119 831 845
55. FairbrotherWG
YehRF
SharpPA
BurgeCB
2002 Predictive identification of exonic splicing enhancers in human genes. Science 297 1007 1013
56. WangZ
BurgeCB
2008 Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. Rna 14 802 813
57. FrazerKA
BallingerDG
CoxDR
HindsDA
StuveLL
2007 A second generation human haplotype map of over 3.1 million SNPs. Nature 449 851 861
58. NigroJM
ChoKR
FearonER
KernSE
RuppertJM
1991 Scrambled exons. Cell 64 607 613
59. HoldtLM
BeutnerF
ScholzM
GielenS
GabelG
2010 ANRIL expression is associated with atherosclerosis risk at chromosome 9p21. Arterioscler Thromb Vasc Biol 30 620 627
60. CunningtonMS
Santibanez KorefM
MayosiBM
BurnJ
KeavneyB
2010 Chromosome 9p21 SNPs Associated with Multiple Disease Phenotypes Correlate with ANRIL Expression. PLoS Genet 6 e1000899
61. Messaoudi-AubertSE
NichollsJ
MaertensGN
BrookesS
BernsteinE
2010 Role for the MOV10 RNA helicase in polycomb-mediated repression of the INK4a tumor suppressor. Nat Struct Mol Biol 17 862 868
62. YapKL
LiS
Munoz-CabelloAM
RaguzS
ZengL
2010 Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol Cell 38 662 674
63. Mancini-DinardoD
SteeleSJ
LevorseJM
IngramRS
TilghmanSM
2006 Elongation of the Kcnq1ot1 transcript is required for genomic imprinting of neighboring genes. Genes Dev 20 1268 1282
64. KanduriC
ThakurN
PandeyRR
2006 The length of the transcript encoded from the Kcnq1ot1 antisense promoter determines the degree of silencing. Embo J 25 2096 2106
65. DixonRJ
EperonIC
SamaniNJ
2007 Complementary intron sequence motifs associated with human exon repetition: a role for intragenic, inter-transcript interactions in gene expression. Bioinformatics 23 150 155
66. CocquerelleC
DaubersiesP
MajerusMA
KerckaertJP
BailleulB
1992 Splicing with inverted order of exons occurs proximal to large introns. Embo J 11 1095 1098
67. BonenL
VogelJ
2001 The ins and outs of group II introns. Trends Genet 17 322 331
68. KosA
DijkemaR
ArnbergAC
van der MeidePH
SchellekensH
1986 The hepatitis delta (delta) virus possesses a circular RNA. Nature 323 558 560
69. TsagrisEM
Martinez de AlbaAE
GozmanovaM
KalantidisK
2008 Viroids. Cell Microbiol 10 2168 2179
70. ZegginiE
WeedonMN
LindgrenCM
FraylingTM
ElliottKS
2007 Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science 316 1336 1341
71. ScottLJ
MohlkeKL
BonnycastleLL
WillerCJ
LiY
2007 A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science 316 1341 1345
72. SheteS
HoskingFJ
RobertsonLB
DobbinsSE
SansonM
2009 Genome-wide association study identifies five susceptibility loci for glioma. Nat Genet 41 899 904
73. WrenschM
JenkinsRB
ChangJS
YehRF
XiaoY
2009 Variants in the CDKN2B and RTEL1 regions are associated with high-grade glioma susceptibility. Nat Genet 41 905 908
74. UnoS
ZembutsuH
HirasawaA
TakahashiA
KuboM
2010 A genome-wide association study identifies genetic variants in the CDKN2BAS locus associated with endometriosis in Japanese. Nat Genet 42 707 710
75. BeiJX
LiY
JiaWH
FengBJ
ZhouG
2010 A genome-wide association study of nasopharyngeal carcinoma identifies three new susceptibility loci. Nat Genet 42 599 603
76. EmanueleE
FontanaJM
MinorettiP
GeroldiD
2010 Preliminary evidence of a genetic association between chromosome 9p21.3 and human longevity. Rejuvenation Res 13 23 26
77. SebastianiP
SolovieffN
PucaA
HartleySW
MelistaE
2010 Genetic Signatures of Exceptional Longevity in Humans. Science
78. HankerAB
MoritaS
RepaskyGA
RossDT
SeitzRS
2008 Tools to study the function of the Ras-related, estrogen-regulated growth inhibitor in breast cancer. Methods Enzymol 439 53 72
79. BrookesS
RoweJ
RuasM
LlanosS
ClarkPA
2002 INK4a-deficient human diploid fibroblasts are resistant to RAS-induced senescence. Embo J 21 2936 2945
80. ShieldsJM
ThomasNE
CreggerM
BergerAJ
LeslieM
2007 Lack of extracellular signal-regulated kinase mitogen-activated protein kinase signaling shows a new type of melanoma. Cancer Res 67 1502 1512
81. LiH
HandsakerB
WysokerA
FennellT
RuanJ
2009 The Sequence Alignment/Map format and SAMtools. Bioinformatics 25 2078 2079
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 12
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Functional Comparison of Innate Immune Signaling Pathways in Primates
- Expression of Linear and Novel Circular Forms of an -Associated Non-Coding RNA Correlates with Atherosclerosis Risk
- Genome-Wide Interrogation of Mammalian Stem Cell Fate Determinants by Nested Chromosome Deletions
- Histone H2A C-Terminus Regulates Chromatin Dynamics, Remodeling, and Histone H1 Binding
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy