
The C-Terminal Domain of the Bacterial SSB Protein Acts as a DNA Maintenance Hub at Active Chromosome Replication Forks
We have investigated in vivo the role of the carboxy-terminal domain of the Bacillus subtilis Single-Stranded DNA Binding protein (SSBCter) as a recruitment platform at active chromosomal forks for many proteins of the genome maintenance machineries. We probed this SSBCter interactome using GFP fusions and by Tap-tag and biochemical analysis. It includes at least 12 proteins. The interactome was previously shown to include PriA, RecG, and RecQ and extended in this study by addition of DnaE, SbcC, RarA, RecJ, RecO, XseA, Ung, YpbB, and YrrC. Targeting of YpbB to active forks appears to depend on RecS, a RecQ paralogue, with which it forms a stable complex. Most of these SSB partners are conserved in bacteria, while others, such as the essential DNA polymerase DnaE, YrrC, and the YpbB/RecS complex, appear to be specific to B. subtilis. SSBCter deletion has a moderate impact on B. subtilis cell growth. However, it markedly affects the efficiency of repair of damaged genomic DNA and arrested replication forks. ssbΔCter mutant cells appear deficient in RecA loading on ssDNA, explaining their inefficiency in triggering the SOS response upon exposure to genotoxic agents. Together, our findings show that the bacterial SSBCter acts as a DNA maintenance hub at active chromosomal forks that secures their propagation along the genome.
Published in the journal:
. PLoS Genet 6(12): e32767. doi:10.1371/journal.pgen.1001238
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001238
Summary
We have investigated in vivo the role of the carboxy-terminal domain of the Bacillus subtilis Single-Stranded DNA Binding protein (SSBCter) as a recruitment platform at active chromosomal forks for many proteins of the genome maintenance machineries. We probed this SSBCter interactome using GFP fusions and by Tap-tag and biochemical analysis. It includes at least 12 proteins. The interactome was previously shown to include PriA, RecG, and RecQ and extended in this study by addition of DnaE, SbcC, RarA, RecJ, RecO, XseA, Ung, YpbB, and YrrC. Targeting of YpbB to active forks appears to depend on RecS, a RecQ paralogue, with which it forms a stable complex. Most of these SSB partners are conserved in bacteria, while others, such as the essential DNA polymerase DnaE, YrrC, and the YpbB/RecS complex, appear to be specific to B. subtilis. SSBCter deletion has a moderate impact on B. subtilis cell growth. However, it markedly affects the efficiency of repair of damaged genomic DNA and arrested replication forks. ssbΔCter mutant cells appear deficient in RecA loading on ssDNA, explaining their inefficiency in triggering the SOS response upon exposure to genotoxic agents. Together, our findings show that the bacterial SSBCter acts as a DNA maintenance hub at active chromosomal forks that secures their propagation along the genome.
Introduction
Maintaining genome integrity is a permanent challenge for all organisms, particularly during genome duplication, when accidental replication fork arrests expose the genome to damage. Numerous mechanisms have evolved to counteract the deleterious consequences of fork arrest (reviewed in [1], [2]). The multiplicity of these fork repair mechanisms reflects the need to respond appropriately to a variety of damaged fork structures. A key question is therefore how these multiple rescue pathways are appropriately and efficiently triggered and coordinated in the cell.
Bacteria can manage chromosomal replication fork arrest without necessarily interrupting other key cell cycle events. Their genome is generally composed of one circular DNA molecule (of several Mbp) replicated by a single pair of divergent forks fired at a fixed origin, oriC. Thus, effective repair of accidentally arrested replication forks is vital to bacteria. In addition to a requirement for removal and repair of the damage originally responsible for a particular replication fork arrest, the cell possesses the machinery necessary for re-assembling the replication machinery (the replisome) at these rescued forks [3]. An emerging model is that components of the replisome determine the recruitment of accessory proteins at the forks to assist their progression. One of these is DnaN, a dimeric protein that forms a ring around double-stranded DNA (dsDNA) and clamps the replicative DNA polymerase [4], and also interacts with several proteins involved in DNA replication and repair (reviewed in [5]). Another protein of the replisome, the Single-Stranded DNA Binding protein (SSB), is also known to interact with accessory proteins at the fork. The primary role of SSB at the fork is to facilitate the activities of replisomal enzymes by preventing the formation of ssDNA secondary structures (for a review, see [6]). SSB is composed of two domains: an N-terminal ssDNA binding domain and a C-terminal domain, SSBCter, enriched in glycine and acidic amino-acids. A short hexapeptide motif with a consensus signature D-D-D-I/L-P-F emerges from the end of the protein [7]. The SSBCter is dispensable for SSB tetramerisation and interaction with ssDNA [8], [9] but permits interaction with many proteins of the DNA recombination, repair and replication machineries. The E. coli SSB (EcSSB) interactome is currently estimated to include 14 proteins (reviewed in [6]).
Many of the SSB partners are involved in distinct replication fork repair pathways. Thus, SSB might be responsible for coordinating recruitment of these repair proteins at active replication forks. As judged by the analysis of SSB localization in B. subtilis and E. coli [10]–[12], active forks are the subcellular sites where SSB accumulates in replicating cells grown without genotoxic stress. We previously provided strong support for the idea that SSB acts as a protein recruitment platform at active replication forks by localizing in living B. subtilis cells three conserved DNA helicases as GFP fusions. These were PriA, the primary restart protein, which directs replisome re-assembly on branched DNA originating from arrested forks [13]–[15], and RecG and RecQ, two recombination proteins involved in the maintenance of the genome and of chromosome forks [16], [17]. These three proteins accumulate at chromosomal forks in an SSBCter-dependent manner, a discrete localization that does not depend on accidental fork arrest [8]. In addition, we have characterized a B. subtilis PriA mutant unable to interact with SSB, which was no longer targeted to active chromosomal forks and did not support replication restart unless overproduced. This underlines the direct benefit of pre-recruitment and targeting of PriA by SSB on active chromosomal forks: in anticipation of a requirement of PriA repair action, which can occur at any stage of genome replication [8]. Thus, an hypothesis raised by such a preparatory mode of PriA action at replication forks would also apply to the other SSB protein partners.
In this study, we have further defined the B. subtilis SSBCter interactome at active chromosomal forks. First, using cytological and biochemical approaches we have extended the number of B. subtilis SSB partner proteins targeted at forks to twelve, including RarA, SbcC and XseA, which are also present in E. coli but not previously known to interact with EcSSB. Among the other proteins identified were key effectors of the RecFOR loading machinery for RecA. In addition, 3 others, including the DNA polymerase DnaE, appear to be specific to the B. subtilis SSB interactome. Paradoxically, although DnaE is one of the two essential B. subtilis DNA polymerases, this interaction is not essential since we have been able to delete SSBCter while retaining cell viability [8]. In parallel to this screening, we have undertaken detailed analysis of the multiple defects caused by the deletion of the SSBCter in vivo. Based on these results we propose an integrated model for replication fork rescue in which SSB coordinates the multiple processes potentially involved in a cascade-like manner. In an initial response to replication fork blockage, the system would first attempt to repair the damage and restart the stalled fork by the coordinated action of proteins present at the fork prior to its blockage. Failure to circumvent the blockage in this way would lead to a second set of responses, in particular the de novo loading of RecA on ssDNA at arrested forks by specific SSB-associated proteins. This would facilitate fork remodeling by homologous recombination (reviewed in [2]). Failure at this step would then lead to a more robust response by induction of the SOS system to provide increased levels of repair proteins to repair damaged forks.
Results
Defining the B. subtilis SSBCter interactome at chromosome replication forks
A remarkable feature of the B. subtilis SSB protein is that deletion of its C-terminal end is not lethal to the cell, in sharp contrast to that of E. coli [9]. This enabled the demonstration that PriA, RecG and RecQ proteins are targeted to active chromosome replication forks in B. subtilis in a manner which depends on the C-terminal region of SSB [8]. To identify additional proteins targeted to active chromosomal forks in the same way, we have extended these studies by using two B. subtilis ssb alleles truncated for the last 35 (ssbΔ35) or 6 (ssbΔ6) codons.
Candidate proteins were chosen according different criteria. Some were E. coli homologues already shown or proposed to interact physically with the EcSSBCter (reviewed in [6]). Others were selected because of their sequence or functional homology with known partners of B. subtilis SSB. A third group included those already known to be present at active chromosomal forks. The final group comprised proteins selectively purified with B. subtilis SSB using the Tap-tag procedure [18] and identified by mass spectrometry.
All candidates were screened for localization at active replication forks as GFP fusions, expressed ectopically from the amyE locus, in SSBCter deletion strains and in the isogenic wild type strain (ssb3+). Most candidates were also screened for physical interaction with SSB as purified proteins in vitro or by Tap-tag analysis with the use of the SPA motif fused to their C-terminal end at their original genetic locus. The combination of these three approaches identified 9 additional proteins which together represent an extended view of the SSBCter interactome targeted to active B. subtilis chromosomal forks. The results of this screening are compiled in Table 1 and described in the following sections.

RecS, a paralogue of RecQ in B. subtilis, interacts with SSB in association with YpbB
B. subtilis and closely related bacteria encode two RecQ homologues [19]. The first, initially annotated as YocI, was renamed RecQ since it is highly homologous to the single RecQ protein generally encoded in bacterial genomes (including E. coli). B. subtilis RecQ co-localizes with active replication forks in an SSBCter-dependent manner and Tap-tag analysis of RecQ provided further evidence for its ability to interact unaided with SSB [8]. The second RecQ homologue, RecS (also annotated as YpbC), is smaller. The RecQ family is typified by 8 helicase motifs. In both RecQ and RecS, they are located towards the N-terminal (Nter) region and are followed by a more divergent C-terminal (Cter) region. In EcRecQ, the latter region carries the site of interaction with SSB [20], [21].
As shown in Figure 1A, Tap-tag analysis of RecS revealed a prominent interaction with SSB and a protein of unknown function, YpbB, encoded immediately upstream of recS. The stop codon of ypbB overlaps the start codon of recS, suggesting translational coupling of the two proteins (Figure 1B). In the few bacterial species encoding a ypbB homologue, this invariably appears upstream of a recS homologue in a common operon (see Figure S1). In the Tap-tag experiments, RecS and YpbB appear in almost equimolar amounts after purification, as does the co-captured SSB (Figure 1A). In addition, the cellular concentrations of RecS and RecQ appear similar (between 150 and 300 copies per cell) as judged by western-blotting of total protein extracts of cells expressing the RecS-SPA or RecQ-SPA fusions and probed with anti-Flag antibodies against the SPA motif (not shown). To test whether RecS and/or YpbB are targeted to chromosomal replication forks, we first constructed N-ter GFP fusions of each gene individually at amyE. Both GFP-RecS and GFP-YpbB fusion proteins appeared largely dispersed throughout the cell (Figure 1C) although some cells were found to exhibit tiny foci on their nucleoid (Table S1). In view of their tandem genetic configuration, it seemed possible that both might be required for correct targeting. We therefore inserted a construction (GFP-ypbB/recS) including both genes at the amyE locus, to retain potential translational coupling. As shown in Figure 1D, all ssb3+ cells carrying this construct exhibit a regular GFP focus pattern identical to that observed previously with GFP-recQ ([8]; Table S1). The simplest explanation for this, as implied by the Tap-tag analysis (Figure 1A), is that YpbB and RecS assemble into a single complex able to interact with SSB, resulting in its targeting to active chromosome replication forks. Biochemical evidence for physical interaction between YpbB and RecS comes from our attempts to purify them from E. coli (described in Text S1): YpbB could not be prepared as a soluble protein alone but only as a stable complex with RecS. Furthermore, the YpbB/RecS complex, but not RecS alone, interacts physically with SSB (Figure S2). Finally, no localization of GFP-YpbB was observed in ssbΔ35 and ssbΔ6 cells (Figure 1D and Table S1).

Altogether, these results show that YpbB is targeted to active chromosomal forks in an SSBCter-dependent manner. They also indicate that the GFP-YpbB foci depend on RecS, with which YpbB forms a stable complex. Reciprocally, RecS could also be present at forks via its association with YpbB. The detection of RecS in the Tap-tag of SSB argues for this is the case (see below).
PcrA and DinG DNA helicases do not belong to the interactome of SSB
To test whether localization at active forks is a property shared by other DNA helicases known to act in repair of arrested replication forks, we analyzed PcrA [22]. A functional GFP-PcrA fusion did not form foci in growing cells but appeared to localize non-specifically to the nucleoid (Figure S3A). In addition, SSB was not detected among the proteins that co-purified with PcrA in Tap-tag experiments (Figure S3B). The Tap-tag is nevertheless validated by the recovery of 2 known partners of PcrA with the functional PcrA-SPA fusion: RNA polymerase [23], and RecA [24], [25]. Furthermore, we did not detect an interaction between purified SSB and PcrA (active as a DNA helicase) in a specific SSB pull-down assay (Figure S4C) detailed below. A lack of discrete targeting in the cell was also observed for the widespread DinG DNA helicase (Figure S3A), recently demonstrated in E. coli to function in concert with Rep or UvrD (the functional equivalents of PcrA; [26], [27]) in resolving accidents caused by collision between the replication and transcription machineries. In addition, SSB was not detected in the Tap tag of DinG (Figure S3B). Most of the proteins co-purified with DinG were ribosomal proteins, indicative for a putative role of DinG in translation and/or ribosome biogenesis in B. subtilis.
Thus, anchorage to active chromosomal forks visualizable by focus formation would not be a hallmark of all effectors of DNA replication rescue. We could not exclude, however, that specific interactions of PcrA and DinG with one component of the fork might occur without being strong or cumulative enough to generate a detectable focus.
The essential DNA polymerase DnaE accumulates at active chromosomal DNA replication forks in an SSBCter–dependent manner
We previously showed that DnaX, a homologue of the E. coli Holopolymerase III (EcPolIII) τ subunit, still formed foci in ssbΔ35 dividing cells [8]. This is also true for other components of EcPolIII conserved in B. subtilis, i.e. HolA, HolB and DnaN, as well as for the replicative DNA helicase, DnaC (Table S1). We could not test the primase, DnaG, since neither N - nor C-terminal GFP fusions gave rise to discrete foci in wild-type B. subtilis cells [10]. B. subtilis encodes two DNA polymerases essential for genome duplication, PolC and DnaE [28]. Both are homologous to the single essential E. coli DNA polymerase, EcDnaE. GFP fusions to PolC and DnaE were both shown to localize at active chromosomal forks [28], [29]. Remarkably, and in sharp contrast with PolC-GFP, we found that DnaE-GFP did not form foci in ssbΔ35 cells (Figure 2A and Table S1) nor in ssbΔ6 cells (Table S1). This suggests that DnaE accumulates at active chromosomal forks via a physical interaction with SSB whereas PolC does not. Tap-tag analysis of DnaE was not informative, since only the DnaE-SPA prey was recovered (not shown). We therefore explored the potential interaction between DnaE and SSB in vitro with purified recombinant proteins. We used a pull-down assay based on magnetic beads coated with ssDNA fully bound by purified SSB or SSBΔ6 (or SSBΔ35, which behaved as SSBΔ6; Figure S5) to test specific interaction between a protein and the SSBCter. This assay was validated with RecQ and RecG (see Figure S4A and S4B; Figure S5). Purified DnaE was found to interact with the SSBCter in this assay and poorly with SSBΔ6 and SSBΔ35 (Figure 2B and Figure S5), supporting the notion that the interaction between DnaE and SSB is direct and accounts for the accumulation of DnaE-GFP at active chromosomal forks.

Extending the B. subtilis SSB interactome at active forks
We next examined the localization of other proteins known or expected to co-localize with B. subtilis chromosomal forks but not essential for their propagation. These included SbcC, a subunit of the heterodimeric SbcCD nuclease that acts specifically on ssDNA palindromic structures [30]; YabA, a negative regulator of initiation of DNA replication at oriC in B. subtilis [31]; RarA, which, in E. coli, is required for RecA loading on arrested chromosomal forks [32]; and RecO and RecJ, which are also involved with RecA loading at arrested forks in concert with RecF, RecR and RecQ.
Among these, only YabA was found to localize in ssbΔ35 cells (Figure 2C, Table S1). Since YabA localizes at forks in a DnaA - and DnaN-dependent manner [33], this result implies that DnaN and DnaA act in ssbΔ35 cells as in wild-type cells.
GFP-SbcC was previously shown to co-localize with B. subtilis replication forks [10]. Here we find that this localization is dependent on the C-terminal domain of SSB (Figure 2C and Table S1).
The three others candidate proteins, i.e. RarA, RecO, and RecJ, which did not localize in ssbΔ35 cells in contrast to wild type ssb3+ cells (Figure 2C and Table S1), were found to interact physically with SSB. This was demonstrated, by Tap-tag analysis in the case of RecJ (Figure 2D), by pull-down and gel filtration assays for RarA (Figure 2E and Figure S6) and by pull-down assays for RecO (Figure 2F). The precise targeting of the functional GFP-RecO fusion to active forks (and not to ssDNA gaps that could be formed elsewhere on the genome) was confirmed by its co-localization with the replisome protein DnaX (Figure S7).
These results imply that although RecA is not normally present at active forks [34], these are equipped with key components of the RecFOR machinery (i.e. RecO, RecJ, RecQ and RarA) permitting recruitment of RecA at replication forks upon accidental arrest [34].
Tap-tag analysis of B. subtilis SSB
To identify a more complete repertoire of SSB partners, we used SSB as a prey in Tap-tag analysis. However, in contrast to EcSSB [35], we were unable to construct an SSB-SPA fusion at the ssb locus suggesting that capping SSBCter with the SPA motif inactivates SSB and leads to cell lethality. We therefore inserted the ssb-SPA construct under the Pxyl promoter at amyE to generate mixed SSB complexes composed of both SSB-SPA and wild-type SSB subunits. Their selective capture, via the SSB-SPA component should permit the co-capture of protein partners interacting with the uncapped wild-type SSB subunits. Ectopic expression of SSB-SPA had no observable negative effect on cell growth (not shown). As shown in Figure 3, wild type SSB and SSB-SPA subunits were recovered in equal amounts from cells grown with (lane 3) or without (lane 1) D-xylose induction of SSB-SPA expression. As expected, the total yield of the hetero-tetrameric SSB/SSB-SPA complexes was higher with than without D-xylose.

Many proteins were observed to co-purify with SSB/SSB-SPA complexes (Figure 3). These included those reproducibly recovered in other Tap-tag experiments performed with other B. subtilis proteins (e.g. GyrA and many ribosomal proteins) and were not considered further. The others appeared to be specific partners of SSB. They were not observed in control experiments where the ectopic ssb-SPA was replaced with a wild type ssb allele (lane 2). In addition, their levels were increased when SSB-SPA expression was induced (compare lanes 1 and 3). Many of these proteins (e.g. RecJ, RecQ, RecG, and RarA) were identified in the experiments described above as direct partners of B. subtilis SSB. Very few peptides of RecS were unambiguously detected by mass spectrometry. As reported above, RecS alone could not stably interact with SSB in vitro but could do so in a complex with YpbB (Figure S2). YpbB was not detected in the Tap tag of SSB, possibly because the number of YpbB molecules recovered was below the level of detection by mass spectrometry, RecS being very close to this limit. The Ung protein was identified in the Tap tag of SSB. Ung was identified previously as a partner of EcSSB [36]. However, some known SSB partners, such as RecO, PriA, DnaE, SbcC and YpbB, were absent. This is probably due to differences in affinity between SSB and each of its partners and to variation in their natural cellular levels since some are known to be present in very low amounts (for instance, ∼50 copies of PriA per cell; [15]). The same dual explanation may also account for the differential yield of some SSB partners recovered in the experiment e.g. RecJ which is by far the most abundant protein co-purified with the SSB/SSB-SPA complex (Figure 3).
We also identified several new candidate partners. These included XseA, the large subunit of ExoVII, and YrrC, a protein of unknown function conserved in gram positive bacteria and predicted to be a helicase/nuclease by sequence analysis. GFP fusions to XseA and YrrC were also both found to form foci on the nucleoid of ssb3+ cells but not in ssbΔ35 cells (Table S1).
This screening identified a repertoire of 12 proteins belonging to the B. subtilis SSB interactome escorting active chromosomal forks. Together, these proteins fulfill a large variety of functions concerned with DNA processing. As a result, the SSBCter emerges as a central hub of DNA processing functions at chromosomal replication forks, where SSB naturally concentrates in the cell.
Growth and cellular defects of B. subtilis ssbΔ35 mutant cells
While deletion of the SSBCter is not lethal, ssbΔ35 cells show viability defects. They exhibit a ∼5-10 fold lower plating efficiency during growth in rich medium, i.e. under fast growing conditions (Figure 4A), as well as in minimal medium (not shown), and smaller colonies on solid medium (Figure 5A). The reduced viability was also directly inferred by observation of exponentially growing cells (in rich medium) under the microscope where up to 15% of ssbΔ35 cells show various kinds of cellular and/or nucleoid morphological defects (i.e. distribution, shape, length, segregation; see Figure S8). Similar observations were made with the ssbΔ6 strain (Figure S9A and S9B and not shown).


The SSBCter is crucial for rescuing the damaged genome
The SSBCter interactome includes many proteins involved in maintaining genome integrity. The importance of the SSBCter might therefore be expected to be more pronounced under growth conditions that are stressful for the genome. Indeed, ssbΔ35 and ssbΔ6 cells are nearly as sensitive to UV irradiation as recA− cells (Figure 4B and Figure S9C). Similarly, both mutants are also more sensitive to Mitomycin C (MMC) than the ssb3+ strain (Figure S9D).
To investigate further the intracellular role of the SSBCter, we examined the effect of complementation of the defects of ssbΔ35 cells by ectopic expression of wild-type SSB at amyE in a controlled manner from the Pxyl promoter. Upon induction with D-xylose production of SSB from the Pxyl promoter was ∼10% that of the natural SSB level (Figure 4C; compare lane 2 with lane 6). This low concentration was, however, sufficient to fully suppress the plating defect of the ssbΔ35 strain (not shown). It also fully restored UV resistance at doses up to 10 J/m2 (Figure 4D). Above this dose, the cells exhibited sensitivity intermediate between that of ssb3+ and ssbΔ35 cells indicating that the intracellular concentration of SSBCter is determinant for an optimal response to DNA damage.
We next investigated whether SOS, a well known cellular response to DNA damaging agents, could be triggered in ssbΔ35 cells. In B. subtilis, the SOS system is regulated by RecA-induced auto-cleavage of the LexA repressor. We used a PlexA:lacZ construct as a reporter of SOS activity and another DNA damaging agent, MMC, as an inducer [37]. The MMC-induced SOS response was dramatically reduced in ssbΔ35 cells compared to ssb3+ cells (Figure 4E). These results therefore underline a pivotal role for the SSBCter in triggering the SOS response.
SSBCter deletion mutants are temperature-sensitive
Another notable defect of ssbΔ35 strains is their temperature-sensitive growth, as measured by plating assay (Figure 5A). This lethality is fully corrected by SSB expression induced from the ectopic Pxyl:ssb (not shown). Since some SSB partners are independently important for cell viability, we tested whether the temperature sensitivity of ssbΔ35 cells could be corrected by increasing individual expression of these partners placed under the Pxyl promoter from the amyE locus. Overexpression of DnaE or PriA, the two most important components of the SSB interactome for cell viability, did not alleviate the temperature sensitivity of ssbΔ35 cells (not shown). Unexpectedly, however, induced expression of RecO (as a functional RecO-SPA fusion) did so (Figure 5B). In contrast, the plating defect characteristic of the ssbΔ35 strain observed at permissive temperature is not corrected by RecO overexpression (Figure 5B).
RecO is a recombination mediator protein that, with RecR and RecF, directs loading of RecA onto ssDNA coated by SSB [38]. We therefore tested whether suppression of ssbΔ35 temperature sensitivity by overexpression of RecO was dependent on RecA. We introduced the recA::tet allele into the ssbΔ35 and ssb3+ strains carrying the Pxyl:recO-SPA cassette. Inactivation of recA in the ssb3+ strain provoked weak temperature sensitivity (Figure 5C). Disruption of recA prevented RecO suppression of ssbΔ35 temperature sensitivity (Figure 5C). This implies that RecA loading on ssDNA is needed for this suppression. The formation of a RecA-ssDNA nucleofilament is the pre-synaptic intermediate of homologous DNA recombination and the inducing signal of SOS, which we have shown above to be defective in MMC-treated ssbΔ35 cells (Figure 4E). However, individual overexpression of RecO neither restored SOS induction by MMC in this mutant, nor suppressed its sensitivity to MMC (not shown).
Thus, the suppression is not solely due to RecO action, but also relies on RecA, leading to the proposal that it proceeds through the RecO-dependent loading of RecA on ssDNA.
The SSBCter is needed for supporting genomic DNA replication
The previous experiments demonstrated that SSBCter was required for repair of lesions throughout the genome. They did not address the question of whether the SSBCter specifically assists chromosomal fork progression. To specifically stress the replication fork, we used two B. subtilis strains bearing temperature sensitive alleles, dnaN5 and dnaX51, whose products are exclusively associated with the replisome [39] and analyzed how deletion of the SSBCter affects their viability. The ssbΔ35 allele, genetically linked to the erythromycin resistance marker (EryR), was introduced by transformation at low temperature into strains carrying the replication mutations. An isogenic EryR-linked ssb3+ allele was also used as a control. Viable EryR clones were obtained upon transformation at 30°C of the dnaN5 and dnaX51 strains with the ssbΔ35 and ssb3+ alleles. The ssbΔ35 dnaN5 and ssbΔ35 dnaX51 recombinants exhibited the characteristic plating and growth defects of the ssbΔ35 strain (Figure 6). Interestingly, they were both significantly more temperature sensitive for growth than their ssb3+ counterparts (Figure 6A–6C). Thus the SSBCter is crucial for growth of ssb3+ dnaN5 and ssb3+ dnaX51 cells at semi-permissive temperatures. In addition, we also found that ssbΔ35 cells were markedly more sensitive than ssb3+ cells to DNA replication stresses induced either by hydroxyurea, which diminishes the dNTP pools, or by HPUra, an antibiotic that specifically inactivates the essential DNA polymerase, PolC (not shown).

The exact nature of the defects provoked by a dysfunction of the replisomes made with the mutated DnaN or DnaX proteins is not known. These defects could be either fork arrest or lesions left by continuing forks. These results provide evidence that the SSBCter is crucial for ensuring the proper duplication of a genome damaged by stresses that specifically impair the replisome.
Discussion
In this study, we have extended the number of known B. subtilis proteins involved in the SSBCter interactome and targeted to active chromosomal DNA replication forks. It includes at least 9 additional members (Table 1), in addition to the PriA, RecG and RecQ DNA helicases [8]. Collectively these constitute a multipurpose DNA processing toolbox able to unwind, replicate or cleave DNA and to promote homologous DNA recombination. Replication forks can consequently marshal a large repertoire of enzymes for their rescue or for preventing their accidental arrest. This is revealed by an increased sensitivity to a variety of replication stresses of B. subtilis cells carrying an ssb allele truncated for its C-terminal domain, SSBCter (Figure 6). Thus, in addition to assisting the activities of the enzymes of the replisome by polymerizing along ssDNA via its Nter domain, SSB provides constant support for fork progression via its Cter domain by mediating multiple DNA transactions. It does so by concentrating a specific subset of proteins of the DNA recombination, repair and replication machineries at active forks.
The SSBCter acts as a general hub of DNA maintenance proteins
SSB is not the only source of accessory proteins at the replication fork. DnaN and the replicative helicase also act as anchors for distinct replication accessory proteins (for reviews, see [5], [40]). While DnaN and the replicative helicase are expected to be confined to replication forks, the spectrum of SSB activity on the genome could be larger since its localisation is primarily determined by availability of ssDNA. Assembly of the SSB interactome at a precise site on the genome is nevertheless expected to be qualitatively and quantitatively modulated by the length of the ssDNA available for SSB polymerisation. Indeed, the local concentration of SSBCter will increase with the length of the SSB-ssDNA nucleofilament. With an average length of 1 kb for single strand DNA on the lagging strand template at an active bacterial DNA replication fork and a binding mode of SSB of ∼65 nts per tetramer, a minimum of ∼60 copies of SSBCter may be present at each fork. This would generate a filament capable of attracting many molecules of the different SSB interactome members. Consequently, chromosomal DNA replication forks constitute permanent subcellular sites for assembling the SSB interactome in dividing cells. In addition, all DNA processes that generate stretches of ssDNA accessible to SSB tetramers are expected to produce such centers for the SSBCter interactome anywhere on the genome (as in the case of the repair of DNA double-strand breaks). Thus, the DNA toolbox associated with the SSBCter should not be considered as exclusively devoted to the progression of replication forks. In contrast to DnaN and the replicative helicase, the SSBCter may therefore be a general determinant for the maintenance of genome integrity.
The SSBCter interactome is specific
Comparison of the SSBCter interactome of E. coli (compiled in [6]) and that determined here for B. subtilis provides several important general conclusions concerning SSBCter function.
Homologues of prominent EcSSB partners such as PriA, RecQ, RecG, RecJ, RecO and Ung which are conserved in B. subtilis (and generally widespread in bacteria) have also been demonstrated to interact with B. subtilis SSB. This points to a strong selective pressure in maintaining such a conserved and abundant SSBCter interactome. In this study, we have identified additional B. subtilis SSB partners (i.e. RarA, SbcC and XseA) also widely conserved in bacteria (including E. coli) but not yet identified as part of the EcSSB interactome. Conversely, we have not yet identified other known conserved EcSSB partners, such as DnaG primase and Topoisomerase III [41], [42], in the SSB interactome of B. subtilis. Further experiments will be required to determine whether these proteins are indeed members of the SSB interactome.
B. subtilis SSB partners that are not widely conserved in bacteria have also been identified, e.g. YrrC (a putative helicase/nuclease encoded in the genomes of gram positive bacteria also annotated as RecD) and the YpbB/RecS complex (see Figure S1). Conversely, EcSSB partners, such as the χ subunit of the EcHolopolymerase III [43], [44], have no equivalent in B. subtilis. Thus, the interactome of SSB also includes some specific proteins representative of subgroups of bacteria. This reveals particular needs in genome metabolism, suggesting that not all bacteria require these functions and/or have evolved distinct alternative strategies to execute identical functions.
Another specific part of the B. subtilis SSBCter interactome is the replisomal DNA polymerase DnaE [28]. Neither its homologue PolC, nor any other known proteins of the B. subtilis replisome, depend on the SSBCter for their targeting to active chromosomal forks (Table S1). DnaE remains essential for viability of ssbΔ35 cells (not shown). Interestingly, it has recently been shown that the essential role of DnaE in the replisome is to elongate a short DNA stretch on the RNA primers synthesized by the DnaG primase before a hand-off to the bona fide replicative polymerase, PolC [45]. Thus, DnaE has presumably evolved distinct interactions with the replisome to functionally link the activities of DnaG and PolC. However, these interactions are not strong enough to produce detectable fluorescent foci with the DnaE-GFP fusion at active forks in ssbΔ35 cells. In line with this reasoning, the DnaE interaction with the SSBCter might serve an additional role. E. coli encodes a single DNA polymerase of the DnaE family, which is not part of the EcSSB interactome [6]. In contrast, the EcDNA polymerase II (EcPolII) has been found to interact with the EcSSB [46]. EcPolII is not essential for the cell, is involved in distinct pathways of replication re-activation and belongs to the E. coli SOS system [47]. Remarkably, B. subtilis dnaE also belongs to the SOS regulon [48]. This raises the possibility that B. subtilis DnaE might also be involved in certain fork maintenance pathways, as demonstrated for many members of the SSB interactome.
The SSBCter is needed for RecA loading
A central piece of the SSB interactome is RecO, which acts with RecR and RecF to direct the loading of RecA on SSB-coated ssDNA [49]. Temperature sensitivity of ssbΔ35 cells can be suppressed by RecO overexpression, and in a RecA-dependent manner. This shows that ssDNA accessible to RecA is generated in ssbΔ35 cells at high temperature, and that RecA then mediates cell rescue. It also reveals a RecO dysfunction in the ssbΔ35 strain, which can be compensated by increasing its cellular concentration. This parallels the results obtained previously with a PriA mutant unable to interact with SSB, whose inefficiency in directing replication restart was compensated by its overexpression [8]. Thus, a consequence of deleting SSBCter is a reduction in the activities of certain of its partners, resulting from loss of SSBCter-assisted and targeted recruitment to their sites of action on the genome.
Importantly, RecO overexpression does not suppress the growth defect of ssbΔ35 cells observed at permissive temperature. This points to the importance of the other SSB partners for sustaining optimal growth of wild-type cells. The growth defect of ssbΔ35 cells could not be corrected by the overexpression of DnaE or of PriA alone; these are the two proteins of the SSB interactome known to be essential for growth (in rich medium in the case of PriA; [15]). Thus, it is possible that more than one SSB partner must be overexpressed to circumvent this defect, if this stems solely from the loss of their SSB-assisted targeting in the cell. Clearly, more work is needed to understand the growth defect caused by the deletion of the SSBCter.
Another marked defect of ssbΔ35 cells at permissive temperature is their inefficiency in inducing the SOS response upon treatment by MMC. This reflects a failure to generate the RecA-ssDNA filament which would normally act as a triggering signal. The RecFOR apparatus is needed for the MMC-mediated SOS induction in B. subtilis [50]. However, RecO overexpression in MMC-treated ssbΔ35 cells did not lead to SOS induction (not shown). Conversely to what is observed at non permissive temperature, this strongly indicates that other members of the SSBCter interactome are needed for generating and/or stabilizing the ssDNA template for the loading of RecA upon MMC treatment. Obvious candidates are the RecQ and RecJ proteins, a helicase/exonuclease couple known to generate the ssDNA from damaged DNA or inactivated replication forks, onto which the RecFOR machinery mediates RecA delivery (reviewed in [2]). The SOS response defect in ssbΔ35 cells has an important bearing on the results of a recent study on RecA localization in B. subtilis cells. GFP-RecA focus formation on the genome provoked by DNA damaging agents (including MMC) was shown to depend on replisome activity although RecA does not appear to be pre-recruited at active forks [34]. Our results suggest a mechanism to explain this conditional RecA localization. We propose that the active fork itself has the potential to load RecA directly onto ssDNA already available or produced de novo via the SSBCter.
Defining the role of the SSBCter in the rescue of arrested forks
Together, these results support a model in which the SSBCter interactome associated with active forks provides a series of solutions for promoting their restart upon accidental blockage (Figure 7), as well as for dealing with errors left behind the passage of the fork. A key step is replisome assembly on the branched DNA backbone of the fork. The PriA protein and its interaction with the SSBCter are central to this event [8]. This could be the only response necessary if arrest is due to replisome dismantling. In more complex situations, other actions aim at protecting and/or clearing the fork, via individual or concerted actions of the many members of SSBCter interactome. DNA repair is obviously crucial. This could be handled either immediately by the proteins already present, or delayed via RecA loading that could then act in two ways. RecA may reconstruct the fork by homologous recombination, and may induce the SOS response to provide more effectors for DNA repair. Amongst the new effectors coming into play are the error-prone DNA polymerases. Interestingly, it has been shown that the recruitment of E. coli PolV at the 3′end of a DNA gap flanked by RecA filaments is increased by an interaction with the EcSSBCter [51]. A distinct class of repair pathways not drawn in the model of Figure 7, are those acting on lesions caused by the replisome but not accompanied by fork arrest. ssDNA gaps are prominent examples of such lesions. In such cases, ssDNA gaps are expected to remain coated by several copies of SSB still interacting with or attracting the proteins that will promote repair.
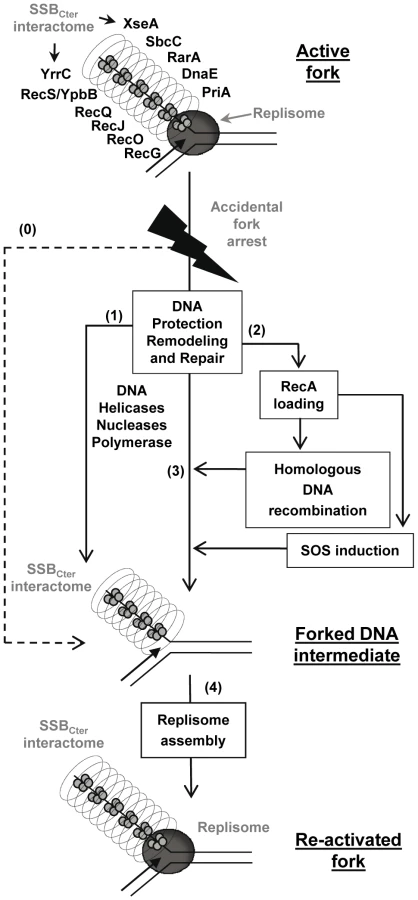
In conclusion, the SSBCter emerges as a general maintenance pivot of bacterial genome integrity. Long stretches of ssDNA are intimately associated with the functioning of active bacterial forks. These form primary targets of SSB in living cells and, consequently, of its interactome. One consequence is that replisomes of chromosomal forks are escorted throughout their progression along the bacterial genome (generally for more than 2 Mbp per fork, as in the case of E. coli and B. subtilis model bacteria). In addition and in a reciprocal way, the forks behave as vehicles for many DNA repair proteins, providing also a convenient way to scan DNA integrity during genome duplication.
Materials and Methods
Bacterial strains and plasmids
B. subtilis strains used in this study, all based on the 168 or L1430 derivatives, are listed in Table S2 along with the strategies used for their construction. They were propagated in LB medium supplemented, unless otherwise indicated, with appropriate antibiotics (erythromycin, 0.6 µg/ml; spectinomycin, 60 µg/ml; chloramphenicol, 5 µg/ml; tetracycline, 15 µg/ml, phleomycin, 2 µg/ml). ssb3+, ssbΔ35, ssbΔ6 and all strains carrying a gene tagged with the SPA motif at its locus were maintained with IPTG (1 mM). Expression of a gene under the Pxyl promoter was achieved by adding 0.2% of D-xylose to the medium. All new chromosomal structures were verified by PCR using appropriate pairs of primers. In case of insertions at the amyE locus, these were also verified by the loss of amylase activity on starch containing media plates.
E. coli strains used were MiT898 [15] for plasmid constructions and ER2566 from NEB, Rosetta (DE3 pLys) or BL21-Gold (DE3) from Novagen for protein expression and purification.
All plasmids used in this study are listed in Table S3. Details of their construction are presented in Text S1.
Microscopy and analysis of the localization patterns
Microscopy analyses were done as described previously [8]. Cells, grown at 30°C until mid-exponential growth phase in LB medium supplemented with appropriate antibiotics and 0.2% D-xylose, were examined with a Leica DMRA2 microscope equipped with a ×100 magnification oil-immersion objective and a COOLSNAP HQ camera (Roper Scientific, USA). Images were captured and processed with METAMORPH V7.5r5.
Tap-tag of SPA fusions in B. subtilis
Except for SSB, the SPA purification tag [52] was joined to the 3′ end of each gene candidate at its original locus. Tandem affininity purifications were performed as previously described [8] with slight modifications, which are detailed in Text S1.
Purification of the proteins produced in E. coli
Purification procedures of all the proteins produced in E. coli used in this study are described in Text S1.
Pull-down assays on magnetic ssDNA beads coated by SSB proteins
25 µl of Dynabeads M-280 streptavidin per assay (Invitrogen) were incubated 15 min at 4°C in 20 mM Tris-HCl pH 7.5, 2 M NaCl with 50 pmol of a 65-mer oligonucleotide 5′-CGTCGTTTTACAACGTCGTGACTGGGAAAACCCTGGCGTTACCCAACTTAATCGCCTTGCAGCA-3′ biotinilated (with biotin TEG; Genecust). Beads were washed with 200 µl of the same buffer, resuspended in 200 µl of buffer B (20 mM Tris-HCl pH 7.5; 200 mM NaCl) supplemented with 80 pmol of purified SSB or SSBΔ6 and incubated at 20°C under agitation (800 rpm) in a 96 wells plate in a Thermomixer (Eppendorf). Beads were washed in 200 µl of buffer B and resuspended in 200 µl of the same buffer supplemented with various quantities of purified DnaE, PcrA, RecO, RecG, RecQ, or RarA proteins as indicated in the figures. After 30 min of incubation at 800 rpm and 20°C, beads were washed in 200 µl of buffer B, drained and resuspended in 10 µl of SDS-PAGE loading buffer. The proteins were separated on 14% SDS-PAGE and revealed by Coomassie blue staining.
Cell survival assays
Spot assays were used to measure the viability of B. subtilis strains used in this study. O/N cultures, incubated at 30°C or 37°C, as indicated in the figure legend, were diluted in fresh LB medium at the same temperature supplemented as indicated in the figure legend with erythromycin, IPTG and with or without D-xylose. At mid-log phase (A650nm≈0.3), 10 µl of 10-fold dilutions (100 to 10−5 in Figure 5 and 10−1 to 10−6 in Figure 6) were spotted on LB agar plates containing the same antibiotics and inducers as those used in the liquid culture. Plates were then incubated O/N at different temperatures (as indicated in the figure legends). In UV resistance assays, plates were exposed to UV irradiation at the indicated doses prior to O/N incubation at 37°C. Colonies were counted after 24 or 48 hours of growth (depending on their growth rate and/or the incubation temperature). Cell survival was expressed as the ratios of the CFU (Colony Forming Units) of UV-treated to untreated cells or of CFU obtained at the tested temperature to CFU obtained at 37°C (Figure 5) or 30°C (Figure 6) for each strain.
SOS response assays
O/N cultures of strains containing the PlexA:lacZ cassette at amyE were propagated at 37°C in LB medium supplemented with erythromycin, spectinomycin and IPTG. Cells in exponential phase (A650nm≈0.03), obtained by inoculating O/N cultures in fresh LB medium supplemented with erythromycin and IPTG, were treated or not with 40 ng/ml MMC to induce or not the SOS response respectively. Sample of ∼0.5 ml per unit of A650nm were taken from cultures every 30 min and treated as described previously [53] for the determination of β-galactosidase activity, expressed in nmol of ONP produced per minute and per mg of protein.
Western blot analysis
Whole protein extracts and western blot analysis were done as previously described [8] with slight modifications as reported in Text S1.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. MichelB
GromponeG
FloresMJ
BidnenkoV
2004 Multiple pathways process stalled replication forks. Proc Natl Acad Sci U S A 101 12783 12788
2. MichelB
BoubakriH
BaharogluZ
LeMassonM
LestiniR
2007 Recombination proteins and rescue of arrested replication forks. DNA Repair (Amst) 6 967 980
3. HellerRC
MariansKJ
2006 Replisome assembly and the direct restart of stalled replication forks. Nat Rev Mol Cell Biol 7 932 943
4. KongXP
OnrustR
O'DonnellM
KuriyanJ
1992 Three-dimensional structure of the beta subunit of E. coli DNA polymerase III holoenzyme: a sliding DNA clamp. Cell 69 425 437
5. Lopez de SaroF
2009 Regulation of interactions with sliding clamps during DNA replication and repair. Curr Genomics 10 206 215
6. SheredaRD
KozlovAG
LohmanTM
CoxMM
KeckJL
2008 SSB as an organizer/mobilizer of genome maintenance complexes. Crit Rev Biochem Mol Biol 43 289 318
7. LuD
KeckJL
2008 Structural basis of Escherichia coli single-stranded DNA-binding protein stimulation of exonuclease I. Proc Natl Acad Sci U S A 105 9169 9174
8. LecointeF
SerenaC
VeltenM
CostesA
McGovernS
2007 Anticipating chromosomal replication fork arrest: SSB targets repair DNA helicases to active forks. Embo J 26 4239 4251
9. CurthU
GenschelJ
UrbankeC
GreipelJ
1996 In vitro and in vivo function of the C-terminus of Escherichia coli single-stranded DNA binding protein. Nucleic Acids Res 24 2706 2711
10. MeileJC
WuLJ
EhrlichSD
ErringtonJ
NoirotP
2006 Systematic localisation of proteins fused to the green fluorescent protein in Bacillus subtilis: identification of new proteins at the DNA replication factory. Proteomics 6 2135 2146
11. PossozC
FilipeSR
GraingeI
SherrattDJ
2006 Tracking of controlled Escherichia coli replication fork stalling and restart at repressor-bound DNA in vivo. Embo J 25 2596 2604
12. Reyes-LamotheR
PossozC
DanilovaO
SherrattDJ
2008 Independent positioning and action of Escherichia coli replisomes in live cells. Cell 133 90 102
13. LiuJ
MariansKJ
1999 PriA-directed assembly of a primosome on D loop DNA. J Biol Chem 274 25033 25041
14. BruandC
FaracheM
McGovernS
EhrlichSD
PolardP
2001 DnaB, DnaD and DnaI proteins are components of the Bacillus subtilis replication restart primosome. Mol Microbiol 42 245 255
15. PolardP
MarsinS
McGovernS
VeltenM
WigleyDB
2002 Restart of DNA replication in Gram-positive bacteria: functional characterisation of the Bacillus subtilis PriA initiator. Nucleic Acids Res 30 1593 1605
16. RudolphCJ
UptonAL
HarrisL
LloydRG
2009 Pathological replication in cells lacking RecG DNA translocase. Mol Microbiol 73 352 366
17. ZhangJ
MahdiAA
BriggsGS
LloydRG
2010 Promoting and avoiding recombination: contrasting activities of the Escherichia coli RuvABC Holliday junction resolvase and RecG DNA translocase. Genetics 185 23 37
18. RigautG
ShevchenkoA
RutzB
WilmM
MannM
1999 A generic protein purification method for protein complex characterization and proteome exploration. Nat Biotechnol 17 1030 1032
19. SanchezH
KidaneD
Castillo CozarM
GraumannPL
AlonsoJC
2006 Recruitment of Bacillus subtilis RecN to DNA double-strand breaks in the absence of DNA end processing. J Bacteriol 188 353 360
20. SheredaRD
BernsteinDA
KeckJL
2007 A central role for SSB in Escherichia coli RecQ DNA helicase function. J Biol Chem 282 19247 19258
21. SheredaRD
ReiterNJ
ButcherSE
KeckJL
2009 Identification of the SSB binding site on E. coli RecQ reveals a conserved surface for binding SSB's C terminus. J Mol Biol 386 612 625
22. PetitMA
DervynE
RoseM
EntianKD
McGovernS
1998 PcrA is an essential DNA helicase of Bacillus subtilis fulfilling functions both in repair and rolling-circle replication. Mol Microbiol 29 261 273
23. NoirotP
Noirot-GrosMF
2004 Protein interaction networks in bacteria. Curr Opin Microbiol 7 505 512
24. VeauteX
DelmasS
SelvaM
JeussetJ
Le CamE
2005 UvrD helicase, unlike Rep helicase, dismantles RecA nucleoprotein filaments in Escherichia coli. Embo J 24 180 189
25. AnandSP
ZhengH
BiancoPR
LeubaSH
KhanSA
2007 DNA helicase activity of PcrA is not required for the displacement of RecA protein from DNA or inhibition of RecA-mediated strand exchange. J Bacteriol 189 4502 4509
26. BoubakriH
de SeptenvilleAL
VigueraE
MichelB
2010 The helicases DinG, Rep and UvrD cooperate to promote replication across transcription units in vivo. Embo J 29 145 157
27. PetitMA
EhrlichD
2002 Essential bacterial helicases that counteract the toxicity of recombination proteins. Embo J 21 3137 3147
28. DervynE
SuskiC
DanielR
BruandC
ChapuisJ
2001 Two essential DNA polymerases at the bacterial replication fork. Science 294 1716 1719
29. LemonKP
GrossmanAD
1998 Localization of bacterial DNA polymerase: evidence for a factory model of replication. Science 282 1516 1519
30. ConnellyJC
KirkhamLA
LeachDR
1998 The SbcCD nuclease of Escherichia coli is a structural maintenance of chromosomes (SMC) family protein that cleaves hairpin DNA. Proc Natl Acad Sci U S A 95 7969 7974
31. Noirot-GrosMF
DervynE
WuLJ
MerveletP
ErringtonJ
2002 An expanded view of bacterial DNA replication. Proc Natl Acad Sci U S A 99 8342 8347
32. LestiniR
MichelB
2007 UvrD controls the access of recombination proteins to blocked replication forks. Embo J 26 3804 3814
33. Noirot-GrosMF
VeltenM
YoshimuraM
McGovernS
MorimotoT
2006 Functional dissection of YabA, a negative regulator of DNA replication initiation in Bacillus subtilis. Proc Natl Acad Sci U S A 103 2368 2373
34. SimmonsLA
GrossmanAD
WalkerGC
2007 Replication is required for the RecA localization response to DNA damage in Bacillus subtilis. Proc Natl Acad Sci U S A 104 1360 1365
35. ButlandG
Peregrin-AlvarezJM
LiJ
YangW
YangX
2005 Interaction network containing conserved and essential protein complexes in Escherichia coli. Nature 433 531 537
36. HandaP
AcharyaN
VarshneyU
2001 Chimeras between single-stranded DNA-binding proteins from Escherichia coli and Mycobacterium tuberculosis reveal that their C-terminal domains interact with uracil DNA glycosylases. J Biol Chem 276 16992 16997
37. DuigouS
EhrlichSD
NoirotP
Noirot-GrosMF
2004 Distinctive genetic features exhibited by the Y-family DNA polymerases in Bacillus subtilis. Mol Microbiol 54 439 451
38. HobbsMD
SakaiA
CoxMM
2007 SSB protein limits RecOR binding onto single-stranded DNA. J Biol Chem 282 11058 11067
39. MauelC
KaramataD
1984 Prophage induction in thermosensitive DNA mutants of Bacillus subtilis. Mol Gen Genet 194 451 456
40. KaguniJM
2006 DnaA: controlling the initiation of bacterial DNA replication and more. Annu Rev Microbiol 60 351 375
41. YuzhakovA
KelmanZ
O'DonnellM
1999 Trading places on DNA–a three-point switch underlies primer handoff from primase to the replicative DNA polymerase. Cell 96 153 163
42. SuskiC
MariansKJ
2008 Resolution of converging replication forks by RecQ and topoisomerase III. Mol Cell 30 779 789
43. GloverBP
McHenryCS
1998 The chi psi subunits of DNA polymerase III holoenzyme bind to single-stranded DNA-binding protein (SSB) and facilitate replication of an SSB-coated template. J Biol Chem 273 23476 23484
44. KelmanZ
YuzhakovA
AndjelkovicJ
O'DonnellM
1998 Devoted to the lagging strand-the subunit of DNA polymerase III holoenzyme contacts SSB to promote processive elongation and sliding clamp assembly. Embo J 17 2436 2449
45. SandersGM
DallmannHG
McHenryCS
2010 Reconstitution of the B. subtilis replisome with 13 proteins including two distinct replicases. Mol Cell 37 273 281
46. MolineuxIJ
GefterML
1974 Properties of the Escherichia coli in DNA binding (unwinding) protein: interaction with DNA polymerase and DNA. Proc Natl Acad Sci U S A 71 3858 3862
47. BonnerCA
RandallSK
RayssiguierC
RadmanM
EritjaR
1988 Purification and characterization of an inducible Escherichia coli DNA polymerase capable of insertion and bypass at abasic lesions in DNA. J Biol Chem 263 18946 18952
48. Le ChatelierE
BecherelOJ
d'AlenconE
CanceillD
EhrlichSD
2004 Involvement of DnaE, the second replicative DNA polymerase from Bacillus subtilis, in DNA mutagenesis. J Biol Chem 279 1757 1767
49. WebbBL
CoxMM
InmanRB
1997 Recombinational DNA repair: the RecF and RecR proteins limit the extension of RecA filaments beyond single-strand DNA gaps. Cell 91 347 356
50. FernandezS
AyoraS
AlonsoJC
2000 Bacillus subtilis homologous recombination: genes and products. Res Microbiol 151 481 486
51. AradG
HendelA
UrbankeC
CurthU
LivnehZ
2008 Single-stranded DNA-binding protein recruits DNA polymerase V to primer termini on RecA-coated DNA. J Biol Chem 283 8274 8282
52. ZeghoufM
LiJ
ButlandG
BorkowskaA
CanadienV
2004 Sequential Peptide Affinity (SPA) system for the identification of mammalian and bacterial protein complexes. J Proteome Res 3 463 468
53. MsadekT
KunstF
HennerD
KlierA
RapoportG
1990 Signal transduction pathway controlling synthesis of a class of degradative enzymes in Bacillus subtilis: expression of the regulatory genes and analysis of mutations in degS and degU. J Bacteriol 172 824 834
54. DavidsenT
BeckE
GanapathyA
MontgomeryR
ZafarN
2010 The comprehensive microbial resource. Nucleic Acids Res 38 D340 D345
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 12
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Functional Comparison of Innate Immune Signaling Pathways in Primates
- Expression of Linear and Novel Circular Forms of an -Associated Non-Coding RNA Correlates with Atherosclerosis Risk
- Genome-Wide Interrogation of Mammalian Stem Cell Fate Determinants by Nested Chromosome Deletions
- Histone H2A C-Terminus Regulates Chromatin Dynamics, Remodeling, and Histone H1 Binding
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy