
Season of Conception in Rural Gambia Affects DNA Methylation at Putative Human Metastable Epialleles
Throughout most of the mammalian genome, genetically regulated developmental programming establishes diverse yet predictable epigenetic states across differentiated cells and tissues. At metastable epialleles (MEs), conversely, epigenotype is established stochastically in the early embryo then maintained in differentiated lineages, resulting in dramatic and systemic interindividual variation in epigenetic regulation. In the mouse, maternal nutrition affects this process, with permanent phenotypic consequences for the offspring. MEs have not previously been identified in humans. Here, using an innovative 2-tissue parallel epigenomic screen, we identified putative MEs in the human genome. In autopsy samples, we showed that DNA methylation at these loci is highly correlated across tissues representing all 3 embryonic germ layer lineages. Monozygotic twin pairs exhibited substantial discordance in DNA methylation at these loci, suggesting that their epigenetic state is established stochastically. We then tested for persistent epigenetic effects of periconceptional nutrition in rural Gambians, who experience dramatic seasonal fluctuations in nutritional status. DNA methylation at MEs was elevated in individuals conceived during the nutritionally challenged rainy season, providing the first evidence of a permanent, systemic effect of periconceptional environment on human epigenotype. At MEs, epigenetic regulation in internal organs and tissues varies among individuals and can be deduced from peripheral blood DNA. MEs should therefore facilitate an improved understanding of the role of interindividual epigenetic variation in human disease.
Published in the journal:
. PLoS Genet 6(12): e32767. doi:10.1371/journal.pgen.1001252
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001252
Summary
Throughout most of the mammalian genome, genetically regulated developmental programming establishes diverse yet predictable epigenetic states across differentiated cells and tissues. At metastable epialleles (MEs), conversely, epigenotype is established stochastically in the early embryo then maintained in differentiated lineages, resulting in dramatic and systemic interindividual variation in epigenetic regulation. In the mouse, maternal nutrition affects this process, with permanent phenotypic consequences for the offspring. MEs have not previously been identified in humans. Here, using an innovative 2-tissue parallel epigenomic screen, we identified putative MEs in the human genome. In autopsy samples, we showed that DNA methylation at these loci is highly correlated across tissues representing all 3 embryonic germ layer lineages. Monozygotic twin pairs exhibited substantial discordance in DNA methylation at these loci, suggesting that their epigenetic state is established stochastically. We then tested for persistent epigenetic effects of periconceptional nutrition in rural Gambians, who experience dramatic seasonal fluctuations in nutritional status. DNA methylation at MEs was elevated in individuals conceived during the nutritionally challenged rainy season, providing the first evidence of a permanent, systemic effect of periconceptional environment on human epigenotype. At MEs, epigenetic regulation in internal organs and tissues varies among individuals and can be deduced from peripheral blood DNA. MEs should therefore facilitate an improved understanding of the role of interindividual epigenetic variation in human disease.
Introduction
Epigenetic mechanisms maintain mitotically heritable differences in gene expression potential without alterations in DNA sequence [1], enabling the diverse cell types of multicellular organisms to stably regulate appropriate patterns of gene expression. The established role of epigenetic mechanisms in cancer and various developmental syndromes has spurred increasing interest in the role of epigenetic dysregulation in a broad range of human diseases including neurological disorders, cardiovascular disease, diabetes, and obesity. A major obstacle to studying epigenetics and human disease, however, is the inherent tissue specificity of epigenetic regulation. In studies of genetic epidemiology, DNA from peripheral blood can be used to assay for a genetic variant present throughout the body. Conversely, epigenetic regulation [2], [3] (and hence dysregulation) may be tissue - and cell-type specific [4], [5]; in many cases, therefore, epigenetic information present in easily obtainable biopsy samples will not provide insights into the epigenetic etiology of disease. Another major obstacle is that interindividual epigenetic variation may often be a consequence of genetic variation [6], making it difficult to disentangle epigenetic and genetic causes of disease.
Hence, genomic loci at which systemic interindividual epigenetic variation occurs independently of genotype would offer major opportunities to advance our understanding of epigenetics and human disease. Such loci have been identified in the mouse; at murine metastable epialleles (MEs) epigenetic regulation is established stochastically in the early embryo then maintained in all germ-layer lineages, resulting in dramatic and systemic interindividual variation in locus-specific epigenetic regulation. Murine MEs cause obvious phenotypic variation among genetically identical mice. For example, the Agouti viable yellow (Avy) ME affects the expression of the Agouti gene which regulates fur pigmentation; isogenic mice heterozygous for Avy range from yellow to mottled to brown [7]. Similarly, the Axin Fused (AxinFu) ME confers epigenetic stochasticity upon Axin, resulting in interindividual variation in tail kinking among isogenic AxinFu heterozygous mice [8]. Rather than affecting fur color or tail development, however, MEs in the human genome could affect individual susceptibility to various diseases. Indeed, because agouti protein binds antagonistically to the melanocortin 4 receptor in the hypothalamus [9], yellow Avy/a mice become hyperphagic and obese, illustrating how epigenetic dysregulation at MEs can result in metabolic disease.
Maternal nutrition and other environmental exposures before and during pregnancy influence the stochastic establishment of epigenetic regulation at murine MEs, with permanent phenotypic consequences [10]–[12]. Hence, if MEs can be identified in humans, they would not only facilitate an advanced understanding of the role of epigenetics in human disease, but also provide excellent candidate loci at which to test epigenetic pathways in the developmental origins hypothesis, which proposes that early environmental influences affect developmental mechanisms, causing permanent metabolic changes that affect risk of adult disease [4], [13]. Of various interacting epigenetic mechanisms including cytosine methylation in DNA, covalent histone modifications, and autoregulatory DNA binding proteins, DNA methylation is recognized as the most stable epigenetic mark [14], making it a prime candidate to mediate the life-long epigenetic changes postulated in the developmental origins paradigm [5], [13].
Here, we have designed an innovative epigenomic screen based upon the epigenetic characteristics of murine MEs, and have screened for MEs in the human genome. We provide evidence that MEs do exist in humans. At the loci we identified, systemic interindividual variation in DNA methylation was confirmed in autopsy samples, and stochastic establishment of epigenotype was supported by epigenetic discordance within monozygotic (MZ) twin pairs. Further, by studying children conceived during different seasons in rural Gambia we show that, as in mice, developmental establishment of DNA methylation at such sites is responsive to maternal environment around the time of conception.
Results
We devised a human genome-scale screening approach based on a definitive characteristic of murine MEs: systemic interindividual variation in DNA methylation [11], [12]. Genomic DNA from peripheral blood leukocytes (PBL) and hair follicles (HF) (mesodermal and ectodermal lineages, respectively) of 8 healthy Caucasian adults was screened for interindividual differences in DNA methylation by methylation-specific amplification microarray [15] (MSAM). We employed a parallel, 2-tissue interindividual cohybridization design: the same four interindividual comparisons (matched for age and sex) were performed in both PBL and HF DNA (Figure 1). Consistent with previous studies [2], [16], most CpG sites assayed did not show measurable interindividual differences in methylation (Table S1). Moreover, interindividual differences were more often observed in a single tissue than in both tissues (Table S1). Nonetheless, our approach identified 107 genomic loci exhibiting concordant interindividual MSAM differences in both tissues (Table S2A).
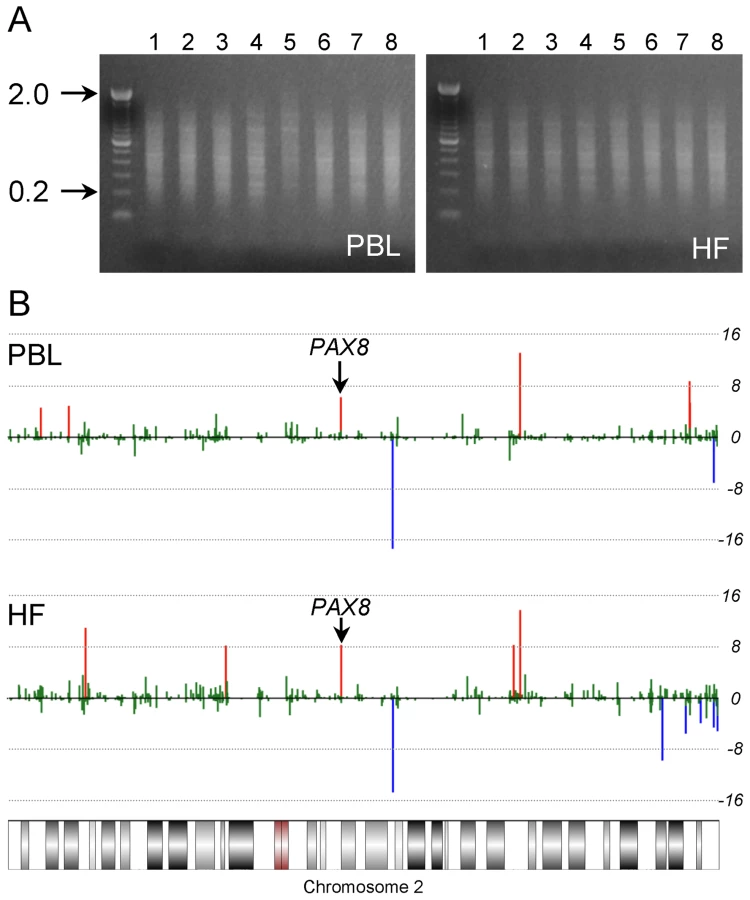
MSAM is based upon serial digestion of genomic DNA with the methylation sensitive/insensitive isoschizomers SmaI and XmaI; our screen could therefore detect genetic variation in addition to systemic epigenetic variation. Indeed, initial attempts to validate several candidates by bisulfite pyrosequencing failed to detect differences in DNA methylation and instead identified single nucleotide polymorphisms (SNPs) within SmaI/XmaI sites. We bioinformatically annotated all potentially informative human SmaI/XmaI intervals with a known SNP within either SmaI/XmaI site (CCCGGG). SNPs that introduce a SmaI/XmaI site within a consensus SmaI/XmaI interval also could affect the MSAM signal, and were likewise annotated. Of 107 SmaI/XmaI intervals originally identified in our screen, 34 were associated with SmaI/XmaI SNPs, a significant over-representation (P = 1.3×10−8). After excluding these (Table S2B), we observed that the remaining 73 candidate MEs tended to localize in subtelomeric regions (Figure S1A). Given the propensity for copy number variation in subtelomeric regions [17], we identified all potentially informative SmaI/XmaI intervals located within known human copy number variants and segmental duplications. Nearly half (35) of the remaining 73 candidate MEs were located within these genetically variable regions, many more than expected by chance (P = 2.1×10−23). After excluding these (Table S2C), the subtelomeric localization was eliminated (Figure S1B).
Excluding all candidate SmaI/XmaI intervals associated with known SNPs, copy number variants, and segmental duplications is extremely conservative, and likely excludes MSAM hits that are in fact caused by interindividual variation in DNA methylation. Indeed, 2 hits in which interindividual DNA methylation differences had already been validated before we performed the bioinformatic filtering were among the affected intervals: the interval at ZNF696 is associated with a SmaI/XmaI SNP, and that at FLJ20433 is within a copy number variant. These 2 loci were retained in the final list of candidate MEs, bringing the number to 40. Of 13 we analyzed by bisulfite pyrosequencing, interindividual variation in PBL and HF DNA methylation was confirmed in 8. (Failure to validate could be caused by uncharacterized SNPs and CNVs, inability to assay both of the informative SmaI/XmaI sites, or low overall methylation levels.)
Our screen was performed using DNA from Caucasians, using 2 tissues that can be sampled relatively non-invasively. To verify concordance across tissues derived from all 3 germ layers, and determine if interindividual epigenetic variation at candidate MEs is conserved across genetically divergent populations, post-mortem liver, kidney and brain tissue was obtained from 8 Vietnamese motor vehicle accident victims (healthy donors). Of the 8 genomic regions with confirmed interindividual variation in DNA methylation in the Caucasian PBL and HF samples, 5 (BOLA3, FLJ20433, PAX8, SLITRK1, and ZFYVE28) showed interindividual variation that was highly correlated among liver, kidney, and brain in the Asian sample (Figure 2A–2E, and Table S3). (SLITRK1 was exceptional in that methylation in brain did not correlate with that in liver and kidney (Figure 2E). This is potentially analogous to the murine AxinFu ME, at which DNA methylation in tail differs from that in all other tissues [11].) For comparison, we similarly analyzed regions within IGF2, GNASAS, and IL10, at which DNA methylation in PBL DNA has been associated with early famine exposure [18], [19]. Although substantial interindividual variation in DNA methylation was confirmed at these loci, not a single statistically significant inter-tissue correlation was found (Figure 2F–2H, and Table S3).

To identify specific genomic characteristics that may confer the special epigenetic behavior of these loci, we bioinformatically compared 6 kb windows encompassing the 40 putative ME SmaI/XmaI intervals and 5000 ‘control’ intervals on the array. We assessed several characteristics of associated CpG islands, as well as the distribution of various classes of tranposable elements (Figures S2, S3, S4, S5, S6, S7, S8, S9). The only significant finding was in the distribution of long-terminal repeat (LTR) retrotransposons; these were depleted at and distributed symmetrically around control intervals, but preferentially localized downstream of putative ME intervals (P = 0.001) (Figure S7). Although clearly insufficient to explain epigenetic metastability, this finding is noteworthy in that nearly all known murine MEs are associated with intracisternal A particle LTR-retrotransposons [20], [21].
Our aim was to identify interindividual epigenetic variation that occurs stochastically; the multiple-tissue screening approach could, however, also detect epigenetic variation associated with genetic variation [6], [22], [23]. Indeed, while performing pyrosequencing validation of one candidate ME, ZNF696, a proximal SNP was identified that explained most of the interindividual variation in methylation (Figure S10). To attempt to rule out such effects, one could map the genomic region flanking each candidate ME to identify haplotype blocks correlated with methylation status. But effects of genetic variation on DNA methylation can occur in cis over tens or even hundreds of kb [23], [24], or in trans [25]. By genetic mapping alone, therefore, it is virtually impossible to exclude that the systemic interindividual epigenetic variation at these select loci is attributable to genetic variation.
Epigenetic discordance within pairs of MZ twins would provide support that interindividual epigenetic variation at our candidate MEs is truly stochastic. We measured DNA methylation at BOLA3, FLJ20433, and PAX8 in buccal DNA from 23 pairs of MZ twins (Figure S11). At PAX8, although there was significant inter-twin correlation, about half of the variance in DNA methylation was not shared by co-twins. At BOLA3 and FLJ20433 there was no inter-twin correlation. These data provide evidence that the interindividual epigenetic variation at our candidate MEs is not genetically mediated.
Another way to determine whether the epigenetic variation at these loci is truly stochastic is to test for an early environmental effect. Unlike interindividual epigenetic variation that is secondary to genetic variation, the stochastic epigenetic variation at bona fide MEs can be influenced by maternal nutrition during early embryonic development [10]–[12]. Demonstrating an effect of periconceptional nutrition on DNA methylation at the identified genomic regions would therefore provide further support that they are MEs. The rural villagers in West Kiang, the Gambia are subsistence farmers whose nutritional status varies dramatically by season. During the rainy season (July–November) depletion of food stores from the previous harvest, combined with an intense agricultural workload, causes negative energy balance and consequent effects on reproductive outcomes [26]. Relative to the dry season, average birth weight during the rainy season is 200–300 g lower and the incidence of small for gestational age infants is doubled [27]. Importantly, seasonal effects on fetal development persist to affect adult mortality in this population [28], but the underlying biologic mechanisms remain unknown.
To test the hypothesis that periconceptional nutrition affects developmental establishment of DNA methylation at candidate MEs, we compared DNA methylation in peripheral blood leukocytes (PBL) of Gambian children conceived during either the dry or the rainy season. Effects of seasonality vary from year to year; we therefore used retrospective birth weight data to identify 1991, 1994, 1995, 1997, and 1998 as years with strong effects of seasonality (Figure S12). Individuals conceived during August–September (rainy season) were compared with those conceived during March–May (dry season), matching for sex and year of conception (n = 30/season). Blood was collected from the children at age 8.9±0.5 years (mean ± sem); age at blood collection did not differ between the season of conception groups. Preliminary analyses of the DNA methylation data showed a highly significant (season of conception) × (year of conception) interaction (P = 0.005), indicating that the effect of seasonality was not consistent in all years. Examining the effects in each year indicated that 1997 was an outlier. Excluding individuals conceived in 1997 eliminated the (season of conception) × (year of conception) interaction (P = 0.17) and left n = 25 individuals per season, representing four years (1991, 1994, 1995, and 1998) in subsequent analyses.
Since maternal supplementation with dietary methyl donors increases DNA methylation at MEs in murine offspring [10]–[12], we anticipated that DNA methylation would be reduced in individuals conceived during the nutritionally challenged rainy season. We found the opposite. At all 5 putative MEs, DNA methylation was significantly higher among individuals conceived during the rainy season (Figure 3A). The overall effect of season of conception on DNA methylation at the 5 MEs combined was highly significant (P = 0.0001). (Detailed statistical analyses provided in Text S1.) Unlike persistent changes in DNA methylation associated with periconceptional famine exposure [18], [19] the effect sizes at the genomic regions we identified were not subtle; rainy season conception was associated with absolute methylation increments of over 10% at both PAX8 and ZFYVE28 (Figure 3A). To determine if the association of season of conception with DNA methylation might be due to chance genetic differences between the groups (such as, for example, differences in one carbon metabolism), we compared DNA methylation at generic LINE1 elements (an indicator of genome-wide methylation [29]) and the same 3 ‘control’ genes studied in the Asian sample (IGF2, GNASAS, and IL10). Contrary to large studies which have associated early famine exposure with subtle persistent changes in DNA methylation at IGF2, GNASAS, and IL10 [18], [19], we found no effect of season of conception in the non-ME control regions, either singly or combined (Figure 3B), indicating that developmental establishment of DNA methylation at MEs is exceptionally sensitive to maternal environment.

The overall effect of season of conception at these putative MEs is especially compelling given that each individual's DNA methylation at one was generally not predictive of methylation at others (Table S4), meaning that stochasticity at these genomic regions is not coordinated. Underscoring the broad relevance of these findings, the genomic loci we identified exhibit similar epigenetic behavior across genetically distinct human populations (Figure S13) indicating that they are ancestral features of the human genome.
Discussion
Murine MEs have attracted extensive study because of their mysterious ability to cause dramatic phenotypic variation among isogenic animals [7], [21], [30], [31]. Viewed by some as an epigenetic oddity, however, their relevance to humans has been questioned [32]. Here, we have for the first time identified elements that are likely to be human MEs, which are characterized by stochastic and systemic interindividual epigenetic variation. These loci exhibit similar interindividual variation in DNA methylation across tissues derived from all 3 germ layers of the early embryo, indicating setting of epigenotype prior to gastrulation. Epigenetic discordance at these genomic loci within MZ twin pairs indicates that establishment of their epigenetic state is determined not genetically, but stochastically. Further, as at murine MEs [10]–[12], developmental establishment of epigenotype at these loci is exquisitely sensitive to maternal periconceptional environment.
Interindividual epigenetic variation that is both systemic and stochastic has not been previously documented in humans. In many cases human interindividual epigenetic variation has been found to be caused by genetic variation [6], [22], [23]. Recent studies of MZ twin pairs have identified epigenetic differences that occur independent of genetic variation [33], [34], but since those differences were studied only in specific tissues it is not clear if they occur systemically. Our results suggest that interindividual epigenetic variation is more often tissue-specific than systemic. Only about half of the SmaI/XmaI intervals showing interindividual variation in PBL, and 15% of those showing interindividual variation in HF, exhibited consistent interindividual variation in both tissues (Table S1).
A key issue is whether establishment of epigenotype at the loci we have identified is truly stochastic. One might argue that the systemic interindividual differences in DNA methylation could be caused by genetic variation, but two pieces of evidence suggest otherwise: the epigenetic discordance within MZ twin pairs, and the effect of season of conception. We must, however, note some caveats. Since we studied DNA methylation in only one tissue from MZ twins, we can not definitively say the observed MZ twin discordance arose in the very early embryo. Future studies should examine ME methylation in MZ twins using DNA from multiple tissues representing the three embryonic germ layers. Likewise, in the Gambian studies, we studied DNA methylation in only one tissue. Hence, although the most parsimonious interpretation of the season of conception effect on PBL DNA methylation is an environmental influence on the early embryo, other interpretations are plausible. For example, if 3 months of age (i.e. 1 year after conception) is a critical window for developmental epigenetics in PBL, there could be a seasonal influence on these processes. Alternatively one could postulate reverse causality, whereby physiological changes induced by seasonal influences on development lead to secondary alterations in DNA methylation. Studying the effect of season of conception on ME DNA methylation in multiple tissues (which is currently underway) will test both of these alternative hypotheses. It is unlikely that postnatal seasonal effects or secondary effects of altered physiology would induce similar epigenetic changes in diverse tissues.
Although DNA methylation at the PAX8 ME was significantly correlated within MZ twin pairs (Figure S11), this does not necessarily indicate a genetic effect on epigenotype. If setting of epigenotype at MEs occurs prior to blastocyst cleavage during MZ twinning, both members of an MZ twin pair could carry concordant epigenetic states at MEs, despite stochastic establishment. Given the different timing of blastocyst cleavage in dichorionic vs. monochorionic MZ twins, examining ME DNA methylation among these different subtypes of MZ twin pairs may prove informative.
The identification of human MEs should advance the study of epigenetics and human disease. Because individually-variable DNA methylation at these loci exhibits little tissue-specificity, epigenetic dysregulation in pathophysiologically relevant tissues such as thyroid and brain, for example, can be inferred from PBL DNA. Indeed, among the putative MEs we identified are genes implicated in hypothyroidism (PAX8) [35], and Tourette's syndrome (SLITRK1) [36]. Such sites therefore represent excellent candidate loci for future studies of epigenetic epidemiology which will utilize existing DNA sample collections to explore associations between epigenetic variation and human disease. Moreover, given their epigenetic lability to early environmental influences, human MEs may enable the elaboration of mechanistic pathways linking early environment to later risk of disease [4], [37]. To the extent that epigenetic variation at MEs is associated with diseases such as cardiovascular disease, type-2 diabetes, and obesity, we may better understand how early nutrition and other environmental exposures predict adult risk of these diseases [5].
By no means should it be inferred that MEs are the sole genomic substrate for early environmental influences on epigenetic regulation. Extensive data from animal [38], [39] and human studies [18], [19] indicate that environmental factors affect epigenetic processes over a broad range of developmental periods, with long-term consequences. Stochastic establishment of epigenotype at MEs, however, does appear to be particularly sensitive to periconceptional environment. For example, by studying 60 famine-exposed humans and their unexposed same sex-siblings, Heijmans et al detected persistent effects of periconceptional famine exposure on PBL DNA methylation at the IGF2 DMR [18], GNASAS, and IL10 [19]. Here, we show that seasonal variation in periconceptional nutrition – likely a milder perturbation – induced significant changes in DNA methylation at all 5 putative MEs studied, but not at the IGF2 DMR, GNASAS, or IL10. Moreover, unlike epigenetic changes that occur only in specific tissues, environmentally-induced epigenetic changes at MEs affect the entire body, and are therefore more likely relevant to human physiology and disease.
Enormous interest in transgenerational epigenetic inheritance has recently been stimulated by provocative data indicating that environmental influences during development might affect the health of subsequent generations [40], [41]. Since transgenerational epigenetic inheritance is known to occur at murine MEs [7], [30], it is logical to consider whether MEs may likewise provide opportunities to understand non-genetic inheritance in humans.
Our findings raise additional questions for future study. First, what causes epigenetic metastability? The stochastic establishment of epigenotype at MEs must fundamentally be a consequence of the genetic sequence in these genomic regions. Indeed, known murine MEs result from transposition of retrotransposons in or nearby genes [21]. But among the genomic loci identified here, no obvious genetic signature of epigenetic metastability was detected. Since our screen was limited to genomic regions containing multiple SmaI/XmaI sites, we detected only a subset of human MEs. Our parallel, 2-tissue screening approach is, however, adaptable to various epigenomic platforms and should enable the identification of many more human MEs. It may then be possible to gain a better understanding of the molecular basis of epigenetic metastability. Second, we still know very little about exactly how maternal nutrition before and during pregnancy affects establishment of epigenotype at MEs. Contrary to our expectations, Gambian individuals conceived in the nutritionally challenged rainy season gained DNA methylation at these putative MEs, emphasizing that our original conjecture that hunger would be associated with a functionally-limiting methyl donor deficiency was overly simplistic. In light of earlier findings that maternal blood folate levels paradoxically increase during the rainy season in the Gambia [42] (potentially due to increased consumption of leafy vegetables), our data suggest that rather than energy intake, availability of one-carbon donors is of key importance. Studies in mouse models and humans are currently underway to improve our understanding of how maternal dietary and other environmental exposures (e.g. insecticides [43] or naturally occurring toxins [44]) affect developmental epigenetics in the preimplantation embryo.
In summary, we have provided strong evidence that stochastic establishment of epigenetic regulation occurs at specific human genomic loci, resulting in interindividual epigenetic variation that affects tissues from all 3 germ layers and persists to adulthood. We have shown that seasonally variable maternal periconceptional exposures affect this stochastic process. Systemic and persistent epigenetic imprints at these loci are likely to be found among diverse human populations that experience seasonal variation in nutritional sufficiency [27], [45], [46] or other environmental exposures during early embryonic development.
Materials and Methods
Sample collection and DNA isolation
Caucasians
Tissue samples from 8 healthy adults (Table S5) were collected in accordance with institutional IRB regulations. Peripheral blood leukocytes were isolated by ficoll gradient centrifugation. Hair follicles (30–50) were obtained by plucking scalp, eyebrow, or shin hair from the same 8 individuals. Tissues were stored at −80°C until isolation of genomic DNA by proteinase-k digestion and phenol-chloroform extraction [47].
Asians
Post-mortem liver, kidney, and brain tissues from 8 Vietnamese motor vehicle accident victims (Table S6) were obtained from a human tissue bank (ILSbio, LLC, Chestertown, MD, USA). The tissues were collected under IRB approved protocols ensuring donor confidentiality. Tissues were flash-frozen upon excision and stored at −80°C until isolation of genomic DNA by proteinase-k digestion and phenol-chloroform extraction [47]. Before DNA isolation, tissue was blotted on absorbent paper to remove excess blood.
MZ twins
Malawian twins were being followed in a special clinic as part of a larger study of the gut microbiota in malnutrition. Permission for sample collection and testing was obtained from the College of Medicine Research and Ethics Committee, University of Malawi. Saliva samples (Table S7) were collected using foam swabs inserted into the buccal cavity until saturated, usually for 3–5 minutes, and then placed in the Oragene preservative (DNA Genotek Inc, Kanata, Ontario). DNA was isolated as recommended by the manufacturer (DNA Genotek).
Gambians
The DNA samples were part of a DNA collection from all the residents of 3 rural villages in West Kiang, namely Keneba, Kantong Kunda and Manduar, the Gambia; the field work and DNA collection have been described [48]. Peripheral blood (5–10 ml) was extracted from consenting subjects (Table S8) according to guidelines established by the Gambia Government/MRC Laboratories Joint Ethics Committee. Children were generally healthy at the time of blood sampling, with no overt clinical infection. DNA extraction was performed in MRC Keneba, by a salting-out procedure [49]. DNA samples were transported to the MRC Human Genetics Laboratory at Fajara for quantification and stored at −20°C.
MSAM screen
MSAM was performed as previously described [15], using a starting quantity of 0.5 µg genomic DNA. MSA products from 2 individuals were differentially dye-labeled and cohybridized to a custom 4×44k array. Array probes were within potentially informative SmaI/XmaI intervals (60–1500 bp) and were selected from Agilent's proximal promoter and CpG island probe libraries (Agilent Technologies, Santa Clara, CA, USA). The 43,222 probes on the array cover 19,187 SmaI/XmaI intervals (average 2.3 probes/interval). Genomic coordinates are based upon hg18 (NCBI Build 36.1). Relevant details of the microarray experiment, including experimental design, microarray probe listing, and hybridization data sets are available in the GEO database (http://www.ncbi.nlm.nih.gov/geo/) (accession # GSE19823).
Four 2-individual MSAM comparisons (Table S5) were performed using PBL DNA, and the same four 2-individual comparisons were performed using HF DNA (incorporating a dye swap) (Table 1).

The analysis was performed at the level of SmaI/XmaI interval; average and median signal intensity, signal ratio, and P value of all probes within each SmaI/XmaI interval were calculated. Candidate MEs were identified as follows. For a given paired comparison (say comparison A) we selected all SmaI/XmaI intervals with both an average A1/A2 signal ratio >1.8 or <0.556 and median P<0.0002 in both PBL and HF. All candidates identified in this manner were further filtered to eliminate those in which the 2 tissues showed discordant interindividual ratios in any of the other pairwise comparisons; only SmaI/XmaI intervals for which the ratio of the PBL:HF signal ratios was >0.445 and <2.25 for all comparisons (A, B, C, D) were retained. (If there is no tissue-specificity in DNA methylation, this ‘ratio of ratios’ equals 1. The maximum departure from this we allowed (2.25) corresponds to a signal ratio of 1.5 in one tissue and 0.667 in the other.) This procedure resulted in the 107 candidate MEs listed in Table S2A.
Bioinformatic analyses
Identification of SmaI/XmaI intervals potentially affected by genetic variation
We identified 90,807 SmaI/XmaI human genomic intervals between 60–1500 bp based on the hg18 genome version. We then used the UCSC Genome Browser SNPs (129) track (http://genome.ucsc.edu/cgi-bin/hgTrackUi?g=snp129) to identify those in which a SNP disrupts a SmaI/XmaI site (N = 10,651), introduces a new XmaI/SmaI site within the consensus interval (N = 867), or both (N = 425). We used the Centre for Applied Genomics Database of Genomic Variants, version variation.hg18.v7.mar.2009.txt (http://projects.tcag.ca/variation/) to identify SmaI/XmaI intervals located within CNVs, and the UCSC Genome Browser Segmental Duplications track (http://genome.ucsc.edu/cgi-bin/hgTrackUi?g=genomicSuperDups) to identify SmaI/XmaI intervals located within segmental duplications. A total of 8631 SmaI/XmaI intervals were found to be located within CNVs or segmental duplications.
Genomic features in vicinity of MEs
The genomic contexts of the ME intervals (n = 40) and control intervals (n = 5026) were investigated by determining the distance from the midpoint of the intervals to genomic annotations within 3000 bp upstream and downstream. Genomic annotations were obtained from the CpG island and RepeatMasker tracks from the UCSC Genome Browser Human hg18 build (http://genome.ucsc.edu/). All CpG islands in addition to SINE, Alu, LINE, LTR, Simple Repeats and Low Complexity repeats were examined.
Quantitative analysis of DNA methylation
Site-specific analysis of CpG methylation was performed by bisulfite pyrosequencing. Genomic DNA (0.5–2 µg) was bisulfite modified [38] and pyrosequencing was performed as previously described [50]. The quantitative performance of each pyrosequencing assay was verified by measuring methylation standards comprised of known proportions of unmethylated (whole genome-amplified) and fully methylated (SssI-treated) genomic DNA [50]. For initial validation of interindividual variation at candidate SmaI/XmaI intervals, DNA methylation was, whenever possible, measured at both SmaI/XmaI sites. Subsequent characterization (measurements in other populations, etc) was performed in the vicinity of the SmaI/XmaI site showing the greatest interindividual variation.
We assessed interindividual variation in the Caucasian samples at 13 of the 40 candidate MEs identified in the MSAM screen: AK098581, AXIN2, BOLA3, FLJ20433, ITPKB, MN1, PAX8, RCC1, SLITRK1, SOX10, ZNF561, ZNF696, and ZFYVE28 (primers listed in Table S9). In 5 of these (AXIN2, ITPKB, MN1, RCC1, and SOX10) the pyrosequencing assays failed to confirm interindividual variation in DNA methylation. ZNF696 was excluded because it exhibited interindividual variation in methylation that was mostly explained by genetic variation at a neighboring SNP (Figure S10). At the remaining 7 loci we examined tissue-specificity of interindividual variation in the Asian liver, kidney, and brain samples. Five (BOLA3, FLJ20433, PAX8, SLITRK1, and ZFYVE28) exhibited significant inter-tissue correlations in DNA methylation consistent with MEs. Two that did not (AK098581 and ZNF561) were excluded from further consideration.
Genotyping
We selected 48 autosomal SNPs with previously demonstrated high reliability for genotyping on the Illumina platform and high minor allele frequency (MAF<0.3) in the Yoruban HapMap population (as the best surrogate we had for the Malawi population). SNPs were selected to be physically distant from each other. These were genotyped on all of the Malawian twin samples. PREST (Pedigree RElationship Statistical Test) was used to estimate the probability of the putative twins sharing 0, 1, or 2 alleles IBD (p0, p1, p2) based on the pairwise analysis of the 48 SNP markers, and the kinship coefficient estimated as phi = 0.25*p1+0.5*p2. In the absence of genotyping error, true MZ twins are expected to have p0 = p1 = 0 and phi = 0.5.
Statistics
Relative enrichment of candidate ME SmaI/XmaI intervals associated with SmaI/XmaI SNPs, CNVs, and segmental duplications was analyzed by chi-square tests. Analysis of CGIs and repetitive elements in the vicinity of MEs and control intervals was performed by analysis of variance (ANOVA) (Proc GLM, SAS Version 9.2). Inter-tissue correlations in interindividual variation in methylation were assessed by Pearson correlation analysis (Proc CORR, SAS).
A REML multifactorial ANOVA (JMP Version 8.0) was used to assess factors affecting average methylation in the Gambian season of conception analysis. Methylation was measured multiple times within each individual for each locus and averaged, for a total of 539 averaged observations. Individual and locus were assessed as random factors, with locus nested within locus type (ME or control). Methylation was arcsine transformed to improve normality. Normality was assessed by Shapiro-Wilk Tests for each sample combination of gene and season of conception (18 combinations consisting of 30 individuals each). All samples were statistically indistinguishable from normal distributions, after sequential Bonferroni correction for carrying out 18 simultaneous tests. One interaction, (season of conception) × (locus type) was investigated as an a priori test.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. JaenischR
BirdA
2003 Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 33 Suppl 245 254
2. IrizarryRA
Ladd-AcostaC
WenB
WuZ
MontanoC
2009 The human colon cancer methylome shows similar hypo - and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet 41 178 186
3. MaunakeaAK
NagarajanRP
BilenkyM
BallingerTJ
D'SouzaC
2010 Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 466 253 257
4. GluckmanPD
HansonMA
BuklijasT
LowFM
BeedleAS
2009 Epigenetic mechanisms that underpin metabolic and cardiovascular diseases. Nat Rev Endocrinol 5 401 408
5. WaterlandRA
MichelsKB
2007 Epigenetic Epidemiology of the Developmental Origins Hypothesis. Annu Rev Nutr 27 363 388
6. RichardsEJ
2006 Inherited epigenetic variation—revisiting soft inheritance. Nat Rev Genet 7 395 401
7. MorganHD
SutherlandHG
MartinDI
WhitelawE
1999 Epigenetic inheritance at the agouti locus in the mouse [see comments]. NatGenet 23 314 318
8. VasicekTJ
ZengL
GuanXJ
ZhangT
CostantiniF
1997 Two dominant mutations in the mouse fused gene are the result of transposon insertions. Genetics 147 777 786
9. WolffGL
RobertsDW
MountjoyKG
1999 Physiological consequences of ectopic agouti gene expression: the yellow obese mouse syndrome. Physiol Genomics 1 151 163
10. DolinoyDC
HuangD
JirtleRL
2007 Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc Natl Acad Sci U S A 104 13056 13061
11. WaterlandRA
DolinoyDC
LinJR
SmithCA
ShiX
2006 Maternal methyl supplements increase offspring DNA methylation at Axin fused. Genesis 44 401 406
12. WaterlandRA
JirtleRL
2003 Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol 23 5293 5300
13. WaterlandRA
GarzaC
1999 Potential mechanisms of metabolic imprinting that lead to chronic disease. AmJClinNutr 69 179 197
14. CedarH
BergmanY
2009 Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet 10 295 304
15. ShenL
KondoY
GuoY
ZhangJ
ZhangL
2007 Genome-wide profiling of DNA methylation reveals a class of normally methylated CpG island promoters. PLoS Genet 3 e181 doi:10.1371/journal.pgen.0030181
16. KatariS
TuranN
BibikovaM
ErinleO
ChalianR
2009 DNA methylation and gene expression differences in children conceived in vitro or in vivo. Hum Mol Genet 18 3769 3778
17. AmbrosiniA
PaulS
HuS
RiethmanH
2007 Human subtelomeric duplicon structure and organization. Genome Biol 8 R151
18. HeijmansBT
TobiEW
SteinAD
PutterH
BlauwGJ
2008 Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci U S A 105 17046 17049
19. TobiEW
LumeyLH
TalensRP
KremerD
PutterH
2009 DNA methylation differences after exposure to prenatal famine are common and timing - and sex-specific. Hum Mol Genet 18 4046 4053
20. DrukerR
BruxnerTJ
LehrbachNJ
WhitelawE
2004 Complex patterns of transcription at the insertion site of a retrotransposon in the mouse. Nucleic Acids Res 32 5800 5808
21. RakyanVK
BlewittME
DrukerR
PreisJI
WhitelawE
2002 Metastable epialleles in mammals. Trends Genet 18 348 351
22. KerkelK
SpadolaA
YuanE
KosekJ
JiangL
2008 Genomic surveys by methylation-sensitive SNP analysis identify sequence-dependent allele-specific DNA methylation. Nat Genet 40 904 908
23. LigtenbergMJ
KuiperRP
ChanTL
GoossensM
HebedaKM
2009 Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3′ exons of TACSTD1. Nat Genet 41 112 117
24. ReikW
WalterJ
2001 Genomic imprinting: parental influence on the genome. Nat Rev Genet 2 21 32
25. LingJQ
LiT
HuJF
VuTH
ChenHL
2006 CTCF mediates interchromosomal colocalization between Igf2/H19 and Wsb1/Nf1. Science 312 269 272
26. PrenticeAM
ColeTJ
FoordFA
LambWH
WhiteheadRG
1987 Increased birthweight after prenatal dietary supplementation of rural African women. Am J Clin Nutr 46 912 925
27. Rayco-SolonP
FulfordAJ
PrenticeAM
2005 Differential effects of seasonality on preterm birth and intrauterine growth restriction in rural Africans. Am J Clin Nutr 81 134 139
28. MooreSE
ColeTJ
PoskittEM
SonkoBJ
WhiteheadRG
1997 Season of birth predicts mortality in rural Gambia. Nature 388 434
29. YangAS
EstecioMR
DoshiK
KondoY
TajaraEH
2004 A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res 32 e38
30. RakyanVK
ChongS
ChampME
CuthbertPC
MorganHD
2003 Transgenerational inheritance of epigenetic states at the murine Axin(Fu) allele occurs after maternal and paternal transmission. Proc Natl Acad Sci U S A 100 2538 2543
31. WolffGL
1965 Body Composition and Coat Color Correlation in Different Phenotypes of “Viable Yellow” Mice. Science 147 1145 1147
32. FeinbergAP
IrizarryRA
2010 Evolution in health and medicine Sackler colloquium: Stochastic epigenetic variation as a driving force of development, evolutionary adaptation, and disease. Proc Natl Acad Sci U S A 107 Suppl 1 1757 1764
33. FragaMF
BallestarE
PazMF
RoperoS
SetienF
2005 Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A 102 10604 10609
34. KaminskyZA
TangT
WangSC
PtakC
OhGH
2009 DNA methylation profiles in monozygotic and dizygotic twins. Nat Genet 41 240 245
35. MacchiaPE
LapiP
KrudeH
PirroMT
MisseroC
1998 PAX8 mutations associated with congenital hypothyroidism caused by thyroid dysgenesis. Nat Genet 19 83 86
36. AbelsonJF
KwanKY
O'RoakBJ
BaekDY
StillmanAA
2005 Sequence variants in SLITRK1 are associated with Tourette's syndrome. Science 310 317 320
37. JirtleRL
SkinnerMK
2007 Environmental epigenomics and disease susceptibility. Nat Rev Genet 8 253 262
38. WaterlandRA
LinJR
SmithCA
JirtleRL
2006 Post-weaning diet affects genomic imprinting at the insulin-like growth factor 2 (Igf2) locus. Hum Mol Genet 15 705 716
39. WeaverIC
CervoniN
ChampagneFA
D'AlessioAC
SharmaS
2004 Epigenetic programming by maternal behavior. Nat Neurosci 7 847 854
40. KaatiG
BygrenLO
EdvinssonS
2002 Cardiovascular and diabetes mortality determined by nutrition during parents' and grandparents' slow growth period. Eur J Hum Genet 10 682 688
41. PembreyME
BygrenLO
KaatiG
EdvinssonS
NorthstoneK
2006 Sex-specific, male-line transgenerational responses in humans. Eur J Hum Genet 14 159 166
42. BatesCJ
FullerNJ
PrenticeAM
1986 Folate status during pregnancy and lactation in a West African rural community. Hum Nutr Clin Nutr 40 3 13
43. RusieckiJA
BaccarelliA
BollatiV
TarantiniL
MooreLE
2008 Global DNA hypomethylation is associated with high serum-persistent organic pollutants in Greenlandic Inuit. Environ Health Perspect 116 1547 1552
44. ZhangYJ
RossnerPJr
ChenY
AgrawalM
WangQ
2006 Aflatoxin B1 and polycyclic aromatic hydrocarbon adducts, p53 mutations and p16 methylation in liver tissue and plasma of hepatocellular carcinoma patients. Int J Cancer 119 985 991
45. LeonardWR
1989 Protection of children from seasonal nutritional stress in an Andean agricultural community. Eur J Clin Nutr 43 597 602
46. RaoS
KanadeAN
YajnikCS
FallCH
2009 Seasonality in maternal intake and activity influence offspring's birth size among rural Indian mothers—Pune Maternal Nutrition Study. Int J Epidemiol 38 1094 1103
47. StraussWM
2001 Preparation of genomic DNA from mammalian tissue.
AusubelFM
Current protocols in molecular biology New York J. Wiley & Sons 2.2.1 2.2.3
48. PetryCJ
Rayco-SolonP
FulfordAJ
SteadJD
WingateDL
2009 Common polymorphic variation in the genetically diverse African insulin gene and its association with size at birth. Hum Genet 126 375 384
49. MillerSA
DykesDD
PoleskyHF
1988 A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16 1215
50. ShenL
GuoY
ChenX
AhmedS
IssaJP
2007 Optimizing annealing temperature overcomes bias in bisulfite PCR methylation analysis. Biotechniques 42 48 58
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 12
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Functional Comparison of Innate Immune Signaling Pathways in Primates
- Expression of Linear and Novel Circular Forms of an -Associated Non-Coding RNA Correlates with Atherosclerosis Risk
- Genome-Wide Interrogation of Mammalian Stem Cell Fate Determinants by Nested Chromosome Deletions
- Histone H2A C-Terminus Regulates Chromatin Dynamics, Remodeling, and Histone H1 Binding
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy