
A Young Duplicate Gene Plays Essential Roles in Spermatogenesis by Regulating Several Y-Linked Male Fertility Genes
Gene duplication is supposed to be the major source for genetic innovations. However, how a new duplicate gene acquires functions by integrating into a pathway and results in adaptively important phenotypes has remained largely unknown. Here, we investigated the biological roles and the underlying molecular mechanism of the young kep1 gene family in the Drosophila melanogaster species subgroup to understand the origin and evolution of new genes with new functions. Sequence and expression analysis demonstrates that one of the new duplicates, nsr (novel spermatogenesis regulator), exhibits positive selection signals and novel subcellular localization pattern. Targeted mutagenesis and whole-transcriptome sequencing analysis provide evidence that nsr is required for male reproduction associated with sperm individualization, coiling, and structural integrity of the sperm axoneme via regulation of several Y chromosome fertility genes post-transcriptionally. The absence of nsr-like expression pattern and the presence of the corresponding cis-regulatory elements of the parental gene kep1 in the pre-duplication species Drosophila yakuba indicate that kep1 might not be ancestrally required for male functions and that nsr possibly has experienced the neofunctionalization process, facilitated by changes of trans-regulatory repertories. These findings not only present a comprehensive picture about the evolution of a new duplicate gene but also show that recently originated duplicate genes can acquire multiple biological roles and establish novel functional pathways by regulating essential genes.
Published in the journal:
. PLoS Genet 6(12): e32767. doi:10.1371/journal.pgen.1001255
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001255
Summary
Gene duplication is supposed to be the major source for genetic innovations. However, how a new duplicate gene acquires functions by integrating into a pathway and results in adaptively important phenotypes has remained largely unknown. Here, we investigated the biological roles and the underlying molecular mechanism of the young kep1 gene family in the Drosophila melanogaster species subgroup to understand the origin and evolution of new genes with new functions. Sequence and expression analysis demonstrates that one of the new duplicates, nsr (novel spermatogenesis regulator), exhibits positive selection signals and novel subcellular localization pattern. Targeted mutagenesis and whole-transcriptome sequencing analysis provide evidence that nsr is required for male reproduction associated with sperm individualization, coiling, and structural integrity of the sperm axoneme via regulation of several Y chromosome fertility genes post-transcriptionally. The absence of nsr-like expression pattern and the presence of the corresponding cis-regulatory elements of the parental gene kep1 in the pre-duplication species Drosophila yakuba indicate that kep1 might not be ancestrally required for male functions and that nsr possibly has experienced the neofunctionalization process, facilitated by changes of trans-regulatory repertories. These findings not only present a comprehensive picture about the evolution of a new duplicate gene but also show that recently originated duplicate genes can acquire multiple biological roles and establish novel functional pathways by regulating essential genes.
Introduction
Gene duplication is a fundamental evolutionary process and provides a major source for genetic novelties [1]–[3]. The usual fate of a gene duplicate is pseudogenization, but some duplicates can fortuitously survive through neofunctionalization, in which one copy retains its ancestral function while the other copy acquires a novel function, or subfunctionalization, in which the duplicate and the ancestral copies subdivide the ancestral functions [4], [5]. The two processes, especially neofunctionalization, should have contributed greatly to the biological diversity by providing genetic innovations.
However, how a new duplicate gene acquires functions by integrating into a pathway and results in adaptively important phenotypes has remained largely unknown. Studying the recently originated young genes could be a very informative way to illustrate these processes, as genes at the early stage of evolution should have retained their original features well, which could have changed with time [3]. Currently, a number of young duplicate genes with potential biological functions have been reported [6]–[13]. Among them, three young Drosophila duplicate genes, arisen by retroposition, were reported to have male-related functions: K81 was proposed to be a testes-expressed paternal effect gene [6], mojoless is required for male germline survival [7], and sphinx is an RNA-coding gene responsible for male courtship behavior [8], [14]. Nevertheless, little is known about how these young duplicate genes have been integrated into the molecular pathways and thereby have realized their functions in the host species.
In this study, we systematically characterized a young Drosophila gene of the kep1 gene family, which originated recently in the Drosophila melanogaster (D. melanogaster) species complex (including D. melanogaster, D. simulans, D. mauritiana, and D. sechellia) about 5.4–12.8 million years ago through the duplications of the kep1 gene locus, mediated by the transposon DNAREP1_DM [15]. We performed a comprehensive investigation of its functions within an evolutionary context and successfully revealed its biological roles as well as the underlying molecular mechanism. The results shed novel light on the functional origin of new genes at the pathway level.
Results
Evolutionary Analysis of the kep1 Gene Family
There are 7 members in the kep1 gene family, and their phylogenetic distributions are illustrated in Figure 1A. The parental gene kep1 is present in all Drosophila species. Through the duplications of the kep1 gene locus, the new genes nsr (novel spermatogenesis regulator, CG3875), CG3927, CR9337, and CG4021 originated in the common ancestor of the D. melanogaster species complex, and CR9337-r and CR33318 occurred after the sibling species in the complex diverged [15]. In this study, we focused on the intact new duplicates nsr, CG3927, and CG4021 in D. melanogaster, in which the genetic manipulations are feasible.

The kep1 family copies are located dispersedly on the second chromosome. D. melanogaster kep1 is a pre-mRNA splicing factor, influencing female fertility, eye development, and immune responses to bacterial infection [16]. Consistent with that, the coding sequences of kep1 are conserved throughout the Drosophila phylogeny (Table S1). Multiple alignments of the protein sequences of kep1 family members show that the three intact new genes have a well-retained KH RNA-binding domain but possess highly diverged C-termini (Figure 1B). By sliding window analysis, the ratio of nonsynonymous changes (dN) over synonymous changes (dS) for each kep1-new gene pair was estimated and tested for selection. For all gene pairs, significant purifying selection signals are enriched in the KH domain region (Figure 1C and Figure S1A), revealing functional constraint on the new genes. Most interestingly, the C-termini between the kep1-nsr pair shows significant positive selection signal (dN/dS = 6.11, p-value <0.05) (Figure 1C), which probably arose from accelerated evolution in the nsr as a result of adaptive evolution.
We analyzed the evolutionary patterns along the phylogenetic branches for nsr (Figure 1D), CG3927, and CG4021 (Figure S1B), based on the maximum likelihood estimates of ω values (dN/dS) [17]. If we assume that the duplication events happened when D. melanogaster and D. yakuba diverged 7.4 million years ago [18], even using the most conservative estimate of the synonymous substitution rate for Drosophila [19]–[21], 24.3, 17.9, and 22.6 synonymous substitutions are expected to occur in the ancestral lineage of the D. melanogaster species complex for nsr, CG3927, and CG4021, respectively. These numbers are far beyond our observations, which are 2.6 for nsr, 0 for CG3927, and 9.3 for CG4021 (Figure 1D and Figure S1B). Therefore, the three new duplicate copies must have originated very late in the ancestral lineage, probably close to the split point of the sibling species in the D. melanogaster species complex. In the ancestral lineage, there are many nonsynonymous substitutions in the new genes, and the estimated ω values are 3.192 for nsr (Figure 1D), infinite for CG3927 (there are no synonymous mutations), and 1.149 for CG4021 (Figure S1B), in which the ones for nsr and CG3927 are significantly larger than the neutral expectation (Table S2), indicating that positive selection should have shaped the two new genes, especially nsr. On the branches leading to individual species, the ω values decline, possibly because the new genes might have evolved functions that are under selective constraint.
Tissue-Specific Expression and Subcellular Localization of New Duplicate Genes in the kep1 Family
In D. melanogaster, the kep1 copy is ubiquitously expressed [22], but the new duplicate copies display a male-specific expression pattern, according to our RT-PCR results (Figure S2A). To provide clues for the biological functions of new kep1 family genes, GFP was fused to the coding sequences of each gene to designate their detailed expression patterns in D. melanogaster (Figure S2B). Since the uniform male-specific expression pattern for all of the new duplicate genes is more likely a consequence of a shared regulatory region rather than independently evolved genetic mutations, we used the homologous upstream regulatory sequences of all kep1 family genes as the driving promoter (Figure S2D). As expected, the shared regulatory region is sufficient to drive similar male-specific expression for each of the GFP-tagged kep1 family proteins, which are unexceptionally enriched in the primary spermatocytes of testes (Figure 2A–2D). Previous large-scale profiling of gene expression patterns in D. melanogaster testes demonstrated that all kep1 family genes showed a high level of mitosis and meiosis expression, followed by much-reduced post-meiosis expression [23]. This result is consistent with our observation and also suggests that the kep1 family genes may be expressed in the spermatogonial stage as well.

In the primary spermatocytes, kep1 family proteins are localized in a specked nuclear pattern (Figure 2E–2H), a highly diagnostic feature for spliceosomal components [24], [25]. Considering that D. melanogaster kep1 is a splicing factor responsible for the alternative splicing of the Drosophila caspase molecule dredd [16], the observation above led us to speculate that new kep1 family genes might regulate the pre-mRNA processing of genes required for spermatogenesis and sperm function.
Evolution of novel subcellular localization after duplication is thought to be an important evolutionary mechanism for the origins of genes with novel functions [26]. Though both are distributed in punctuate nuclear structures of primary spermatocytes, the localization of Nsr protein is much broader than the Kep1 protein (Figure 2I). RNase A treatment of testes could lead to the ectopic accumulation and dispersal of GFP-tagged Nsr protein (Figure S2E, S2F, S2G, 2H), indicating that the Nsr protein is localized in an RNA-dependent manner, and its expanded nuclear localization might imply a novel RNA-binding property. CG4021 protein is localized, completely overlapping with the Kep1 protein, in primary spermatocyte nuclei (Figure 2J), and CG3927 protein was found to have a lack of a significant fluorescent signal for the comparison.
Loss-of-Function Analysis for the kep1 Family Genes
To comprehensively understand the biological functions of the kep1 family genes, we have generated null mutants for all four intact gene copies in D. melanogaster by either gene targeting knockout [27] or imprecise P-element excision [28] (Figure 3A and 3B). The wild-type (WT) control flies of the mutants are WT recombinants created by targeted mutagenesis or precisely excised strains of P-element excision, for the sake of an identical genetic background between the mutant and the WT flies. The null males of nsr display significantly reduced fecundity when compared to the WT males (p-value <0.001, Mann-Whitney U test) (Figure 3C). This phenotype can be fully restored by introducing the genomic sequences of nsr back into the genome (Figure 3C). Heterozygous flies of nsr mutants are equally fertile as the WT flies (Figure 3C). We found that the sperm storage tissue (seminal vesicle) of nsr male mutants was empty or contained little sperm, if any (Figure 4A and 4B). During D. melanogaster spermatogenesis, germ cells from gonial precursors differentiate into cysts of 64 syncytial spermatids, which will undergo an actin-based individualization process, in which a bulk of unneeded cytoplasm is eliminated from the spermatids through remodeling of the cyst membrane. Extrusion of the cytoplasm along sperm bundles can form visible cystic bulges, which will migrate to the distal ends and are detached as waste bags. An actin structure, termed the “investment cone (IC),” is formed at the site where each spermatid develops its own membrane [29], [30]. We labeled the sperm bundles together with the cystic bulges and waste bags with GFP under control of the don juan (dj) gene promoter [31], and the ICs are visualized by FITC-conjugated phalloidin. The testis of nsr mutant male contains comparable amounts of spermatids as their WT controls; however, the structures of cystic bulges and waste bags are largely absent (Figure 4C and 4D). In WT flies, ICs in the same cyst move coordinately in clusters (Figure 4E), while they are scattered along the sperm bundles in the nsr mutants (Figure 4F). The phenotypes above are typical features of an impaired individualization process [30]. Electron microscopy examination further confirmed that the spermatids of nsr mutants are unindividualized, with substantial amounts of residual cytoplasm (Figure 4G and 4H). As the final step of spermatogenesis, the spermatids are assembled by coiling at the base of the testis to facilitate their transport into seminal vesicles [29]. Under a phase-contrast microscope, the sperm bundles of nsr mutants are twisted at the distal ends of testis, instead of regular coiling (Figure 4I and 4J). Therefore, nsr is functionally involved in both sperm individualization and coiling.


In contrast, though kep1 is required for female fertility in D. melanogaster [16], no significant difference in male fertility was detected between kep1 mutant males and their WT controls (Figure 3D). Also, we did not observe reduced fertility (Figure 3D) or other obvious defects for the CG3927 and CG4021 mutants. Considering that only nsr exhibits a robust signature of positive selection, this result may not be surprising. Either CG3927 and CG4021 have not acquired new functions or their phenotypic effects are not strong enough to be detected in our phenotyping assay.
Requirement of nsr for the Integrity of Sperm Axoneme Structure by Regulating Several Y-Linked Male Fertility Genes
Microarray comparison of the transcription profiles between nsr WT and mutant testes only identified 14 genes that exhibited at least a 2-fold difference at the expression level, but none of them seemed to be male fertility-related (Table S3). Considering that the background hybridization noise and lack of probes for some genes might limit the power of microarray, we further implemented whole transcriptome shortgun sequencing (RNA-Seq), which is regarded as a more precise way for measurements of transcript levels [32]. Using the Illumina paired-end sequencing platform, we generated 16.3 million reads (75-bp) for WT testes and 9.6 million for nsr mutant testes. Based on these transcriptome data, we identified 10 genes that were significantly differentially expressed (>5-fold) between WT and mutants. Among them, kl-2, kl-3, and kl-5 are known male fertility genes, and the others are either not correlated with male fertility or functionally unknown (Table S4).
The kl-2, kl-3, and kl-5 genes are 12.4-fold, 10.0-fold, and 6.84-fold down-regulated in the mutants, respectively (Table S4), and their sharp reductions in expression were validated by real-time PCR (Figure 5A). Interestingly, the three genes were located adjacently on the Y chromosome, and all encode dynein heavy chain polypeptides of the sperm axoneme [33]–[35]. The phenotypic defect associated with the sterility of kl-2 mutants is not very clear [36], [37], while kl-3 or kl-5 mutations by P-element insertions result in loss of the outer dynein arm of the sperm axoneme and irregular coiling of spermatid tails, and complete deletion of either locus causes defects in sperm individualization [37]–[39]. Electron microscope examination of the spermatid flagellum showed that the outer dynein arms of sperm axonemes were also missing in the nsr mutants (Figure 5B–5F). The deficiencies of nsr mutants, including sperm individualization, coiling, and axonemal structures, fit well with the phenotypes of the kl-3 and kl-5 mutants. This substantial agreement of the loss-of-function phenotypes between the Y-linked genes kl-3, kl-5, and nsr indicates that nsr is involved in male functions by regulating kl-3, kl-5, and, possibly, kl-2 as well. Moreover, it is very likely that nsr regulates the kl-2, kl-3, and kl-5 genes post-transcriptionally, because their primary transcript levels are largely unaltered between the mutants and WT flies, as shown by real-time PCR results (Figure 5A). This is also in accordance with the conserved RNA-binding domain (Figure 1B and 1C) and the splicing factor-like distribution pattern (Figure 2F) of the Nsr protein. More importantly, our co-immunoprecipitation experiment demonstrated that the pre-mRNA cleavage stimulatory factor CstF-64 [40] can be specifically immunoprecipitated by TAP-tagged Nsr protein from testis extracts (Figure S3A and S3B). This result fortifies the idea that nsr might function as an RNA processing factor, although future studies are needed to explore how nsr and CstF-64 collaboratively process the primary transcripts of these male genes.

Functional Status of Ancestral kep1 in the Pre-Duplication Species D. yakuba
We traced the functional status of kep1 in the pre-duplication species D. yakuba by detecting its expression pattern using Kep1 antibody (Figure S3C). Surprisingly, immunocytochemistry with Kep1 antibody showed only background staining of D. yakuba testis (Figure 6D), whereas it is capable of yielding a robust staining pattern in the primary spermatocytes of D. melanogaster (Figure 6B and 6C), exactly as revealed by transgenic GFP localization (Figure 2A). The antibody worked well in detecting Kep1 proteins in ovary extracts from both D. yakuba and D. melanogaster by Western blot (Figure S3D), ruling out the possibility that the antibody sensitivity is not equally sufficient for detecting Kep1 protein of D. yakuba.
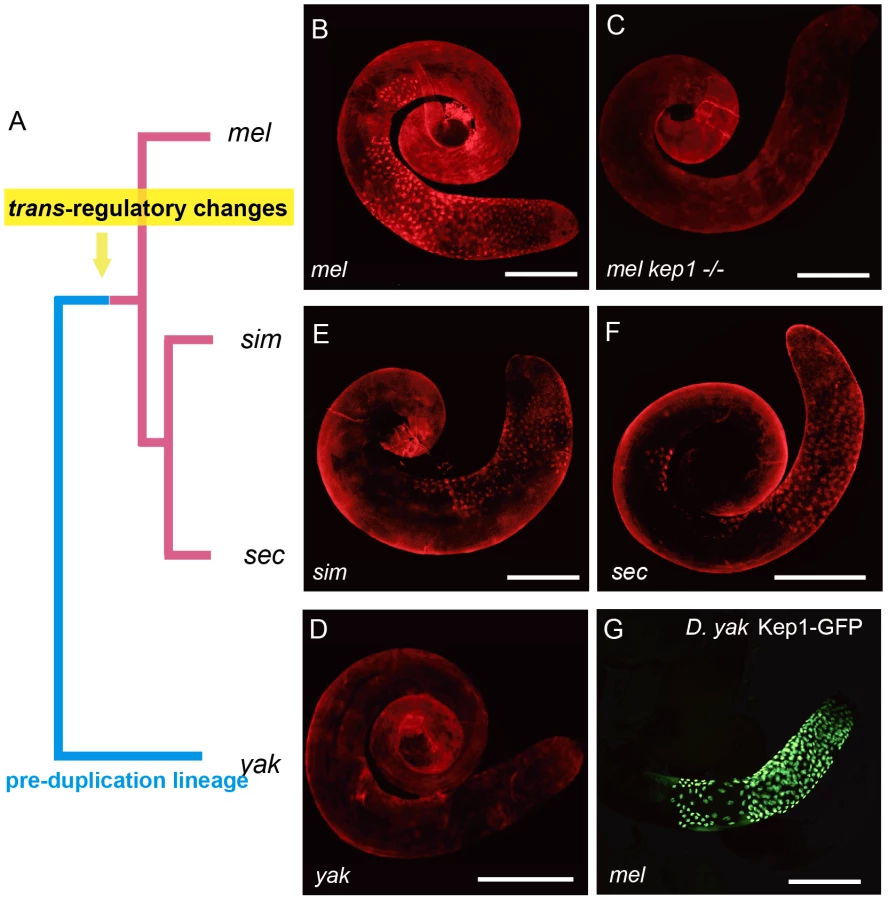
Absence of Kep1 protein in D. yakuba testis suggests that the kep1 gene should not be ancestrally required for male fertility, and it also raises the questions of when and how the novel testicular expression patterns of the kep1 family in D. melanogaster has been evolved. The immunofluorescent signals of Kep1 proteins in the sibling species of D. melanogaster, D. simulans (Figure 6E) and D. sechellia (Figure 6F), suggest that this novel pattern has been established in the common ancestor of the D. melanogaster species complex. This interspecies difference of expression pattern between D. yakuba and D. melanogaster may arise from either cis-acting or trans-acting regulatory changes. The two genetic factors can be distinguished by testing the transcriptional activity of D. yakuba's cis-elements of kep1 in D. melanogaster. Controlled by D. yakuba's cis-elements of kep1, GFP was also found to accumulate in the primary spermatocytes in D. melanogaster (Figure 6G) with the same subcellular localization as with the control of the cis-elements of D. melanogaster kep1 (Figure S3E). This means that the activity of the cis-elements has not been differentiated between D. yakuba and D. melanogaster, and it is the changes in trans-regulatory repertoires that most likely have enabled all kep1 family genes to obtain novel testicular expression patterns.
Discussion
There are two possible scenarios to explain the current functional roles of nsr in D. melanogaster: neofunctionalization and subfunctionalization [4], [5]. Our results tend to support the neofunctionalization scenario, although we cannot completely exclude the possibility of subfunctionalization.
Several pieces of evidence support the neofunctionalization scenario. Firstly, the parental gene kep1 is under strict purifying selection across the Drosophila phylogeny (Table S1). The significant conservation of kep1 and its inessentiality for male fertility in the pre-duplication species D. yakuba is consistent with the reported functions of kep1 in female fertility, eye development, and immune response [16] but not male fertility (Figure 3D) in D. melanogaster. These results suggest that kep1 possibly has retained its ancestral functions without evolving novel male functions after the duplication events, and nsr is free to evolve new functions. Secondly, nsr shows a robust signal of positive selection (Figure 1C and 1D), especially in the C-termini (Figure 1C). As we know, RNA recognition is a complex biological process that may need the collaboration of multiple factors; the RNA-binding domain alone possibly does not contain sufficient information for specific targeting [41], [42]. Thus, the rapidly evolving C-termini of nsr could have contributed to novel RNA-binding ability by mediating co-option with different cofactors, and this idea is further strengthened by the specific immunoprecipitation of the pre-mRNA cleavage stimulatory factor CstF-64 by the Nsr protein (Figure S3B). The subcellular localization pattern of the Nsr protein is also different from the Kep1 protein by displaying a larger localization range in the nuclei of primary spermatocytes (Figure 2I), and cell type-specific expression or subcellular localization is regarded as one of the strategies for RNA-binding proteins to regulate specific splicing events [42]. Although it is still not clear what is the concrete molecular process that the novel distribution pattern of nsr has contributed to its roles in spermatogenesis, it is possible that this novel distribution might allow the spatial-specific assembling between nsr and its cofactors, and the subsequent specific regulation of mRNA substrates. Thirdly, our antibody did not detect obvious expression of Kep1 protein in D. yakuba testis, and thus, the parental gene kep1 should not be ancestrally required for male fertility. After the split of D. yakuba, trans-regulatory changes possibly occurred prior to or accompanied by the duplications of kep1, which enabled the kep1 family genes to obtain novel testicular expression patterns and thereby lend them an opportunity to evolve novel male functions, as nsr has done.
Nevertheless, the alternative subfunctionalization scenario cannot be completely excluded if a recent “gain and loss” turnover of male functions for kep1 did happen or if kep1 has lost its male functions in the D. yakuba lineage for some reason. In the recent “gain and loss” turnover, the parental gene kep1 could have acquired an essential role in spermatogenesis after the split of D. melanogaster and D. yakuba but prior to the duplication events, whereas the new copy nsr has taken over the spermatogenesis role from kep1 after its origination.
The new duplicate gene nsr displays tremendous divergence from kep1 at the levels of biological function and molecular pathway. The kep1 gene participates in female fertility by regulating the apoptosis molecule dredd [16], whereas the new gene nsr is integrated into the spermatogenesis pathway by regulating Y-linked male fertility genes; thus, our findings also provide an unusual case, showing a functional transition in a new gene from a female role to male role. It is interesting that the newly originated genes are often expressed primarily in male reproductive tissues in diverse organisms [43]–[47], and most of the new Drosophila genes with known functions [6]–[8], together with nsr, are associated with male reproduction. This phenomenon pronounces that new genes may tend to be functionally male-biased and suggests a significant role of natural selection and sexual selection in the fixation of beneficial mutations for male reproductive success.
Our study reveals that nsr has been integrated into fundamental developmental processes by regulating pre-existing essential genes. Interestingly, the sperm maturation aspects that nsr participates in are conserved during evolution [48]. For example, the failure of eliminating sperm cytoplasm and loss of the outer axonemal dynein arm can also cause many types of human infertility [49]–[51]. The functional mechanism of nsr indicates that new genes could contribute to the evolutionary turnover of molecular pathways governing essential and conserved developmental processes, which partially explains the phenomenon that the same developmental processes in different organisms are sometimes achieved by a different set of genes. The positive selection signal and biological functions of nsr together strongly suggest that nsr might have contributed to the adaptive evolution of male reproductive pathways in the D. melanogaster species complex.
Materials and Methods
Evolutionary Analysis
Protein sequences of nsr, CG3927, CG4021, and kep1 in D. melanogaster are downloaded from FlyBase (http://flybase.org) and aligned by ClustalW (http://www.ebi.ac.uk/Tools/clustalw). Orthologous coding sequences of kep1 family genes in other Drosophila species (http://flybase.org) were predicted using a combination of BLAT (http://genome.ucsc.edu) and GeneWise (http://www.ebi.ac.uk/Tools/Wise2) and manually checked. Alignments of coding sequences mentioned below are performed by MEGA 3.2 [52], considering the coding structures. To estimate the selective constraint on kep1 through the Drosophila phylogeny, alignments of kep1 coding sequences from different Drosophila species were tested for purifying selection by MEGA 3.2 pairwisely. To detect the selective pressure on the new genes of the kep1 family, alignments of the coding sequences between kep1 and each new gene were performed and calculated for the dN/dS ratio with 120-bp windows and 6-bp slides. For each window, the maximum likelihood method [53] was used to test if the dN/dS ratio was significantly different from one (two-tailed Fisher's exact test).
The ω (dN/dS) values in the phylogeny of new kep1 family genes were estimated using the maximum likelihood approach, implemented by the codeml free-ratio model in the PAML4.2 package (http://abacus.gene.ucl.ac.uk/software/paml.html) [17]. To test if the ω ratio in the ancestral lineage of the D. melanogaster species complex was significantly different from one, the likelihood of the two-ratio model with an estimated ω was compared to an alternative two-ratio model, with ω constrained to be one for this lineage.
Fly Strains
All Drosophila strains were maintained at 25°C using standard cornmeal medium. The transgenic strains were produced by microinjection of w1118 embryos following standard P-element-mediated germline transformation [54]. P-element insertion stocks DG20303 and KG07486 were ordered from Bloomington Stock Center. Strains for P-element excision (Sp/CyO; Δ2-3, Sb/TM6B and Sp/CyO; MKRS/TM6B) are kindly provided by Dr. Yongqing Zhang. Strains for targeted mutagenesis (70FLP70I-SceI, 70FLP and 70I-CreI) were previously described by Xie and Golic (2004).
Transgenic Constructs of kep1 Family Genes
For GFP-tagged vectors, the pH-Stinger plasmid [55] was modified by excision with SpeI/NheI and re-ligation to remove its Hsp70 promoter and nuclear GFP. Gene promoter sequences (plus 5′ UTR) and GFP coding sequences were then cloned into XbaI/EcoRI and EcoRI/KpnI sites of the modified plasmid. Coding sequences of each gene were added into EcoRI sites and selected for correct insertion orientation (Figure S2B). TAP-tagged transgenic vectors were constructed similarly but had GFP replaced with a TAP tag, which consists of two IgG-binding domains of protein A (ProtA) and a calmodulin-binding peptide (CBP) separated by a TEV protease cleavage site [56] (Figure S2C). For all the vectors above, a homologous upstream region of kep1 family genes (including D. yakuba kep1) was adopted as the promoter sequence (Figure S2D). A rescue construct of nsr was prepared by inserting a 2.8-kb DNA fragment, ranging from the end of the upstream gene to the start of the downstream gene, into the NotI site of the pW8 transformation vector (FlyBase). The primer information is available in Table S5.
Generation of Null Flies of Each kep1 Family Gene and Male Fertility Test
P-element excision: The fly strains DG20303 (with a P-element at the 5′ UTR of kep1) and KG07486 (with a P-element annotated to locate at the nsr locus but found to be inserted at the 5′ UTR of CG3927 after PCR validation) were mobilized with Δ2-3 transposase by standard P-element excision, respectively [28]. Excision lines were screened by PCR, and the endpoints were determined by sequencing.
Gene knockout by ends-in targeting: The targeting vectors were designed to create a deletion, spanning from 42-bp downstream of the transcriptional start site to a site within the 3′ UTR of nsr, and a deletion spanning from the start codon to a site within the 3′ UTR of CG4021, respectively. Targeted mutagenesis was performed as previously described [27]. Donor flies bearing the targeting vector were generated and crossed with flies carrying heat shock-activated FLP recombinase and I-SceI endonuclease (70FLP70I-SceI). The 0-3 day old progeny were heat-shocked at 38.5°C for 1 hour, and the enclosed white-eye virgins were crossed with males constitutively expressing FLP recombinase (70FLP). In total, at least 1000 vials were screened for nonmosaic red-eye individuals with successful insertions of the targeted allele at the site of the endogenous allele. Stocks of the recombinant flies were established and crossed with flies carrying heat shock-activated I-CreI endonuclease (70I-CreI). We heat-shocked 0-3 day old progeny at 38.5°C for 1 hour and screened for white-eye adults with recombinant reduction events at the targeted site. The reduction events will lead to either removal of the allele or maintenance of the WT allele. Strains of both genotypes were established to serve as knockout and WT lines, respectively.
For the male fertility test, an individual male of each genotype (<1 day) was placed with three w1118 virgin females, which were collected within 5 hours of enclosure and aged for 2 days. The progeny were counted on the 18th day after the mating and compared between the mutant and WT lines using Mann-Whitney U test.
Antibody Preparation and Immunofluorescence Assay
A polyclonal antibody was raised against the glutathione-S-transferase-Kep1 (amino acids 233–313) recombinant protein in guinea pigs. Testis squashes and immunostaining were performed as previously described [57]. The primary antibodies used are guinea pig anti-Kep1 serum (1∶200 dilution) for Kep1 protein and rabbit peroxidase-antiperoxidase complex (PAP) (1∶1000 dilution, Sigma) for ProtA. The secondary antibodies are Alexa 555-conjugated anti-guinea pig and Alexa 594-conjugated anti-rabbit (Molecular Probes). Testes were co-stained with Hoechst 33342 (1 µg/ml, Molecular Probes) to visualize nuclear DNA if needed. FITC-conjugated phalloidin (1∶100 dilution) was used for IC staining. RNase A treatment was performed as previously described [58] by a 10-min incubation of TBS with 50 µg/ml RNase A (Fermentas), and the controls were incubated in the same buffer, but free of RNase A.
Western Analysis
For sample preparation, adult testes or ovaries from 0–5 day old flies were dissected in PBS, transferred to RIPA buffer, ground, and boiled at 95°C for 10 min for lysis. The primary antibodies used were PAP (1∶2000 dilution, Sigma), mouse anti-β-actin (1∶3000 dilution, Abcam), and guinea pig anti-Kep1 (1∶500 dilution). Peroxidase-conjugated secondary antibodies were used for signal detection (1∶10000 dilution, Santa Cruz).
Immunoprecipitation Assay
Six hundred testes of 0–3 day old flies carrying TAP-tagged Kep1 protein, TAP-tagged Nsr protein, or TAP-tagged CG4021 protein were used for affinity purification, respectively. Testes were ground in 100 µl RIPA buffer plus protease inhibitor cocktail (Roche) with the Sample Grinding Kit (GE Healthcare). The cell suspension was centrifuged at 4°C for 5 min, the supernatant was pre-cleared by 5 µl protein G plus-agarose beads (Santa Cruz), and incubated with 2 µl PAP at 4°C overnight. Then, 10 µl protein G plus-agarose beads were added to the mixture and incubated at 4°C for 1 hour. Complexes of TAP-tagged proteins were liberated from the beads by cleavage of TEV protease as previously described [56], subjected to SDS-PAGE, and visualized by Coomassie blue staining. The protein band of interest was cut out and identified by MALDI-ToF mass spectrometry.
Electronic Microscopy
The dissected testes from WT controls and nsr mutants were fixed in 2.5% glutaraldehyde, washed twice with PBS, post-fixed with OsO4, and dehydrated in an ascending series of ethanol. The resultant specimens were embedded in Araldite, sliced into ultrathin sections (50–100 nm), stained with 1% uranyl acetate, and examined with a JEOL electron microscope.
RT-PCR and Real-Time RT-PCR
Total RNA was isolated from adult testes with Trizol reagent (Invitrogen) and treated with DNase I (Fermentas). Reverse-transcription was performed using the RevertAid First Strand cDNA Synthesis kit (Fermentas) with a no-reverse-transcriptase reaction as the negative control. Real-time PCR was performed in triplicate with SYBR Green PCR Mix (Bio-Rad) and subjected to the ABI 7000 Sequence Detection System. Oligo-dT primer was used to synthesize the cDNA templates for detecting mature transcripts and random hexamer primer for primary transcripts. Information on the PCR primers is available in Table S5. The relative concentration of genes was calculated by analyzing their dissociation curves using the constitutively expressed gene rp49 as the internal control.
Microarray Analysis
With Trizol reagent (Invirtrogen), total RNA was extracted from testes of 0–2 day old nsr mutant and WT flies, respectively. After amplification, mRNA was fluorescently labeled by GeneChip One-Cycle Target Labeling (Affymetrix) and subjected to GeneChip Drosophila Genome 2.0 Arrays (Affymetrix) in duplicate. Image collection was performed in accordance with standard Affymetrix protocols. The significance of gene expression change was estimated using the Significance Analysis of Microarrays (SAM) algorithm, which assigns a score to each gene on the basis of change in gene expression relative to the standard deviation of repeated measurements [59]. The microarray data have been deposited in Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo) under accession number GSE22289.
Paired-End cDNA Library Construction for Illumina Genome Analyzer 2 (GA2) Sequencing
With Trizol reagent (Invirtrogen), 5 µg total RNA was extracted from testes of 0-1 day old nsr mutant and WT flies, respectively. The first-strand cDNA was synthesized with oligo-dT primer by Superscripts II reverse transcriptase (Invitrogen), and second strand cDNA synthesis was followed according to the standard protocol. Then, the double-stranded cDNA was purified with the Qiaquick PCR purification kit (Qiagen) and fragmented with a nebulizer (Invirtrogen), resulting in an average size of 150–250-bp. Overhangs of resultant fragmented cDNAs were blunted with T4 DNA polymerase (NEB) and Klenow polymerase (NEB) and treated with 3′-5′ exonuclease-deficient Klenow polymerase (NEB) to generate 3′ overhangs. After that, cDNA was ligated to an Illumina PE adapter oligo mix by the Quick ligation kit (Qiagen). The adapter-modified cDNA within 200-bp was isolated by agarose gel, extracted with the QIAquick Gel Extraction Kit (NEB), and amplified by PCR reaction. Finally, the library products were sequenced using the Illumina GA2 sequencing machine. Sequence data from this study have been submitted to the NCBI Short Read Archive (http://www.ncbi.nlm.nih.gov/Traces/sra/sra.cgi) under accession number SRA020074.
Measurement of Gene Expression Using Data of Illumina GA2 Sequencing
The generated 75-bp raw reads were mapped to the genomic sequences of D. melanogaster (Ensembl release 55: ftp://ftp.ensembl.org/pub/release-55/fasta/drosophila_melanogaster) using SOAP2 software (http://www.soapmaker.ca) [60]. The count of covering reads for each annotated transcript (Ensembl release 55: ftp://ftp.ensembl.org/pub/release-55/gtf/drosophila_melanogaster) was calculated as the index of their expression level. The alteration of transcript level between nsr mutants and WT flies was estimated and normalized for the variation of the total data size of transcript reads. The significance of expression difference (p-value) for each gene (the longest transcript) was further computed according to the R package “DEGseq” using the MA-plot-based method with a random sampling model and followed by an adjustment with q-values for multiple testing corrections [61].
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. OhnoS
1970 Evolution by gene duplication. New York Springer-Verlag
2. ZhouQ
WangW
2008 On the origin and evolution of new genes–a genomic and experimental perspective. J Genet Genomics 35 639 648
3. LongM
BetranE
ThorntonK
WangW
2003 The origin of new genes: glimpses from the young and old. Nat Rev Genet 4 865 875
4. LynchM
ForceA
2000 The probability of duplicate gene preservation by subfunctionalization. Genetics 154 459 473
5. LynchM
ConeryJS
2000 The evolutionary fate and consequences of duplicate genes. Science 290 1151 1155
6. LoppinB
LepetitD
DorusS
CoubleP
KarrTL
2005 Origin and neofunctionalization of a Drosophila paternal effect gene essential for zygote viability. Curr Biol 15 87 93
7. KalameghamR
SturgillD
SiegfriedE
OliverB
2007 Drosophila mojoless, a retroposed GSK-3, has functionally diverged to acquire an essential role in male fertility. Mol Biol Evol 24 732 742
8. DaiH
ChenY
ChenS
MaoQ
KennedyD
2008 The evolution of courtship behaviors through the origination of a new gene in Drosophila. Proc Natl Acad Sci U S A 105 7478 7483
9. LiD
DongY
JiangY
JiangH
CaiJ
2010 A de novo originated gene depresses budding yeast mating pathway and is repressed by the protein encoded by its antisense strand. Cell Res 20 408 420
10. RogersRL
BedfordT
LyonsAM
HartlDL
2010 Adaptive impact of the chimeric gene Quetzalcoatl in Drosophila melanogaster. Proc Natl Acad Sci U S A 107 10943 10948
11. ZhangJ
DeanAM
BrunetF
LongM
2004 Evolving protein functional diversity in new genes of Drosophila. Proc Natl Acad Sci U S A 101 16246 16250
12. NurminskyDI
NurminskayaMV
De AguiarD
HartlDL
1998 Selective sweep of a newly evolved sperm-specific gene in Drosophila. Nature 396 572 575
13. ZhangJ
ZhangYP
RosenbergHF
2002 Adaptive evolution of a duplicated pancreatic ribonuclease gene in a leaf-eating monkey. Nat Genet 30 411 415
14. WangW
BrunetFG
NevoE
LongM
2002 Origin of sphinx, a young chimeric RNA gene in Drosophila melanogaster. Proc Natl Acad Sci U S A 99 4448 4453
15. YangS
ArguelloJR
LiX
DingY
ZhouQ
2008 Repetitive element-mediated recombination as a mechanism for new gene origination in Drosophila. PLoS Genet 4 e3 doi:10.1371/journal.pgen.0040003
16. Di FruscioM
StyhlerS
WikholmE
BoulangerMC
LaskoP
2003 Kep1 interacts genetically with dredd/caspase-8, and kep1 mutants alter the balance of dredd isoforms. Proc Natl Acad Sci U S A 100 1814 1819
17. YangZ
1998 Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol Biol Evol 15 568 573
18. TamuraK
SubramanianS
KumarS
2004 Temporal patterns of fruit fly (Drosophila) evolution revealed by mutation clocks. Mol Biol Evol 21 36 44
19. LiW
1997 Molecular Evoltuion. Sunderland Sinauer Associates
20. PritchardJK
SchaefferSW
1997 Polymorphism and divergence at a Drosophila pseudogene locus. Genetics 147 199 208
21. HegerA
PontingCP
2007 Evolutionary rate analyses of orthologs and paralogs from 12 Drosophila genomes. Genome Res 17 1837 1849
22. Di FruscioM
ChenT
BonyadiS
LaskoP
RichardS
1998 The identification of two Drosophila K homology domain proteins. Kep1 and SAM are members of the Sam68 family of GSG domain proteins. J Biol Chem 273 30122 30130
23. VibranovskiMD
LopesHF
KarrTL
LongM
2009 Stage-specific expression profiling of Drosophila spermatogenesis suggests that meiotic sex chromosome inactivation drives genomic relocation of testis-expressed genes. PLoS Genet 5 e1000731 doi:10.1371/journal.pgen.1000731
24. LamondAI
SpectorDL
2003 Nuclear speckles: a model for nuclear organelles. Nat Rev Mol Cell Biol 4 605 612
25. SpectorDL
1993 Nuclear organization of pre-mRNA processing. Curr Opin Cell Biol 5 442 447
26. Byun-McKaySA
GeetaR
2007 Protein subcellular relocalization: a new perspective on the origin of novel genes. Trends Ecol Evol 22 338 344
27. XieHB
GolicKG
2004 Gene deletions by ends-in targeting in Drosophila melanogaster. Genetics 168 1477 1489
28. RobertsonHM
PrestonCR
PhillisRW
Johnson-SchlitzDM
BenzWK
1988 A stable genomic source of P element transposase in Drosophila melanogaster. Genetics 118 461 470
29. LindsleyDL
TokuyasuKT
1980 Spermatogensis.
AshburnerM
WrightTR
Genetics and Biology of Drosophila New York Academic Press 225 294
30. FabrizioJJ
HimeG
LemmonSK
BazinetC
1998 Genetic dissection of sperm individualization in Drosophila melanogaster. Development 125 1833 1843
31. SantelA
WinhauerT
BlumerN
Renkawitz-PohlR
1997 The Drosophila don juan (dj) gene encodes a novel sperm specific protein component characterized by an unusual domain of a repetitive amino acid motif. Mech Dev 64 19 30
32. WangZ
GersteinM
SnyderM
2009 RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10 57 63
33. GoldsteinLS
HardyRW
LindsleyDL
1982 Structural genes on the Y chromosome of Drosophila melanogaster. Proc Natl Acad Sci U S A 79 7405 7409
34. GepnerJ
HaysTS
1993 A fertility region on the Y chromosome of Drosophila melanogaster encodes a dynein microtubule motor. Proc Natl Acad Sci U S A 90 11132 11136
35. CarvalhoAB
LazzaroBP
ClarkAG
2000 Y chromosomal fertility factors kl-2 and kl-3 of Drosophila melanogaster encode dynein heavy chain polypeptides. Proc Natl Acad Sci U S A 97 13239 13244
36. HardyRW
LindsleyDL
LivakKJ
LewisB
SiverstenAL
1984 Cytogenetic analysis of a segment of the Y chromosome of Drosophila melanogaster. Genetics 107 591 610
37. HardyRW
TokuyasuKT
LindsleyDL
1981 Analysis of spermatogenesis in Drosophila melanogaster bearing deletions for Y-chromosome fertility genes. Chromosoma 83 593 617
38. ZhangP
StankiewiczRL
1998 Y-Linked male sterile mutations induced by P element in Drosophila melanogaster. Genetics 150 735 744
39. TimakovB
ZhangP
2000 Genetic analysis of a Y-chromosome region that induces triplosterile phenotypes and is essential for spermatid individualization in Drosophila melanogaster. Genetics 155 179 189
40. ZhaoJ
HymanL
MooreC
1999 Formation of mRNA 3′ ends in eukaryotes: mechanism, regulation, and interrelationships with other steps in mRNA synthesis. Microbiol Mol Biol Rev 63 405 445
41. KenanDJ
QueryCC
KeeneJD
1991 RNA recognition: towards identifying determinants of specificity. Trends Biochem Sci 16 214 220
42. SinghR
ValcarcelJ
2005 Building specificity with nonspecific RNA-binding proteins. Nat Struct Mol Biol 12 645 653
43. MarquesAC
DupanloupI
VinckenboschN
ReymondA
KaessmannH
2005 Emergence of young human genes after a burst of retroposition in primates. PLoS Biol 3 e357 doi:10.1371/journal.pbio.0030357
44. VinckenboschN
DupanloupI
KaessmannH
2006 Evolutionary fate of retroposed gene copies in the human genome. Proc Natl Acad Sci U S A 103 3220 3225
45. ZhangY
SturgillD
ParisiM
KumarS
OliverB
2007 Constraint and turnover in sex-biased gene expression in the genus Drosophila. Nature 450 233 237
46. KaessmannH
VinckenboschN
LongM
2009 RNA-based gene duplication: mechanistic and evolutionary insights. Nat Rev Genet 10 19 31
47. VibranovskiMD
ZhangY
LongM
2009 General gene movement off the X chromosome in the Drosophila genus. Genome Res 19 897 903
48. de KresterDM
KerrJB
1994 The cytology of testis. The Physiology of Reproduction New York Raven Press Ltd 1177 1290
49. JouannetP
EscallerD
SerresC
DavidG
1983 Motility of human sperm without outer dynein arms. J Submicrosc Cytol 15 67 71
50. WolfJP
FeneuxD
EscalierD
RodriguesD
FrydmanR
1993 Pregnancy after subzonal insemination with spermatozoa lacking outer dynein arms. J Reprod Fertil 97 487 492
51. KeatingJ
GrundyCE
FiveyPS
ElliottM
RobinsonJ
1997 Investigation of the association between the presence of cytoplasmic residues on the human sperm midpiece and defective sperm function. J Reprod Fertil 110 71 77
52. KumarS
TamuraK
NeiM
2004 MEGA3: Integrated software for Molecular Evolutionary Genetics Analysis and sequence alignment. Brief Bioinform 5 150 163
53. YangZ
NielsenR
2000 Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol Biol Evol 17 32 43
54. RubinGM
SpradlingAC
1982 Genetic transformation of Drosophila with transposable element vectors. Science 218 348 353
55. BaroloS
CarverLA
PosakonyJW
2000 GFP and beta-galactosidase transformation vectors for promoter/enhancer analysis in Drosophila. Biotechniques 29 726, 728, 730, 732
56. PuigO
CasparyF
RigautG
RutzB
BouveretE
2001 The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods 24 218 229
57. WangZ
MannRS
2003 Requirement for two nearly identical TGIF-related homeobox genes in Drosophila spermatogenesis. Development 130 2853 2865
58. HeatwoleVM
HaynesSR
1996 Association of RB97D, an RRM protein required for male fertility, with a Y chromosome lampbrush loop in Drosophila spermatocytes. Chromosoma 105 285 292
59. TusherVG
TibshiraniR
ChuG
2001 Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A 98 5116 5121
60. LiR
YuC
LiY
LamTW
YiuSM
2009 SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25 1966 1967
61. WangL
FengZ
WangX
ZhangX
2010 DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26 136 138
62. MortazaviA
WilliamsBA
McCueK
SchaefferL
WoldB
2008 Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5 621 628
63. KieferBI
1970 Development, organization, and degeneration of the Drosophila sperm flagellum. J Cell Sci 6 177 194
64. MitchisonTJ
MitchisonHM
2010 How cilia beat. Nature 463 308 309
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 12
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Functional Comparison of Innate Immune Signaling Pathways in Primates
- Expression of Linear and Novel Circular Forms of an -Associated Non-Coding RNA Correlates with Atherosclerosis Risk
- Genome-Wide Interrogation of Mammalian Stem Cell Fate Determinants by Nested Chromosome Deletions
- Histone H2A C-Terminus Regulates Chromatin Dynamics, Remodeling, and Histone H1 Binding
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy