
A Role for ATF2 in Regulating MITF and Melanoma Development
The transcription factor ATF2 has been shown to attenuate melanoma susceptibility to apoptosis and to promote its ability to form tumors in xenograft models. To directly assess ATF2's role in melanoma development, we crossed a mouse melanoma model (NrasQ61K::Ink4a−/−) with mice expressing a transcriptionally inactive form of ATF2 in melanocytes. In contrast to 7/21 of the NrasQ61K::Ink4a−/− mice, only 1/21 mice expressing mutant ATF2 in melanocytes developed melanoma. Gene expression profiling identified higher MITF expression in primary melanocytes expressing transcriptionally inactive ATF2. MITF downregulation by ATF2 was confirmed in the skin of Atf2−/− mice, in primary human melanocytes, and in 50% of human melanoma cell lines. Inhibition of MITF transcription by MITF was shown to be mediated by ATF2-JunB–dependent suppression of SOX10 transcription. Remarkably, oncogenic BRAF (V600E)–dependent focus formation of melanocytes on soft agar was inhibited by ATF2 knockdown and partially rescued upon shMITF co-expression. On melanoma tissue microarrays, a high nuclear ATF2 to MITF ratio in primary specimens was associated with metastatic disease and poor prognosis. Our findings establish the importance of transcriptionally active ATF2 in melanoma development through fine-tuning of MITF expression.
Published in the journal:
. PLoS Genet 6(12): e32767. doi:10.1371/journal.pgen.1001258
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001258
Summary
The transcription factor ATF2 has been shown to attenuate melanoma susceptibility to apoptosis and to promote its ability to form tumors in xenograft models. To directly assess ATF2's role in melanoma development, we crossed a mouse melanoma model (NrasQ61K::Ink4a−/−) with mice expressing a transcriptionally inactive form of ATF2 in melanocytes. In contrast to 7/21 of the NrasQ61K::Ink4a−/− mice, only 1/21 mice expressing mutant ATF2 in melanocytes developed melanoma. Gene expression profiling identified higher MITF expression in primary melanocytes expressing transcriptionally inactive ATF2. MITF downregulation by ATF2 was confirmed in the skin of Atf2−/− mice, in primary human melanocytes, and in 50% of human melanoma cell lines. Inhibition of MITF transcription by MITF was shown to be mediated by ATF2-JunB–dependent suppression of SOX10 transcription. Remarkably, oncogenic BRAF (V600E)–dependent focus formation of melanocytes on soft agar was inhibited by ATF2 knockdown and partially rescued upon shMITF co-expression. On melanoma tissue microarrays, a high nuclear ATF2 to MITF ratio in primary specimens was associated with metastatic disease and poor prognosis. Our findings establish the importance of transcriptionally active ATF2 in melanoma development through fine-tuning of MITF expression.
Introduction
Malignant melanoma is one of the most highly invasive and metastatic tumors [1], and its incidence has been increasing at a higher rate than other cancers in recent years [2]. Significant advances in understanding melanoma biology have been made over the past few years, thanks to identification of genetic changes along the MAPK signaling pathway. Those include mutations in BRAF, NRAS, KIT and GNAQ, all of which result in a constitutively active MAPK pathway [3]–[5]. Consequently, corresponding transcription factor targets such as microphthalmia-associated transcription factor (MITF) [6], AP2 [7], and C-JUN [8] and its heterodimeric partner ATF2 [9] are activated and induce changes in cellular growth, motility and resistance to external stress [10], [11]. In addition, constitutively active MAPK/ERK causes rewiring of other signaling pathways [4]. Among examples of rewired signaling is upregulation of C-JUN expression and activity [8], which potentiates other pathways, including PDK1, AKT and PKC, and plays a critical role in melanoma development [12].
Activating transcription factor 2 (ATF2), a member of the bZIP family, is activated by stress kinases including JNK and p38 and is implicated in transcriptional regulation of immediate early genes regulating stress and DNA damage responses [13]–[15] and expression of cell cycle control proteins [16]. To activate transcription, ATF2 heterodimerizes with bZIP proteins, including C-JUN and CREB [17], [18], both of which are constitutively upregulated in melanomas [8]. ATF2 is also implicated in the DNA damage response through phosphorylation by ATM/ATR [19]. Knock-in mice expressing a form of ATF2 that cannot be phosphorylated by ATM are more susceptible to tumor development [20]. Nuclear localization of ATF2 in melanoma tumor cells is associated with poor prognosis [21], likely due to transcriptional activity of constitutively active ATF2. Indeed, expression of transcriptionally inactive ATF2 or peptides that attenuate endogenous ATF2 activity inhibits melanoma development and progression in xenograft models [22]–[26]. These studies suggest that ATF2 is required for melanoma development and progression.
The transcription factor MITF has been shown to play a central role in melanocyte biology and in melanoma progression [27], [28]. Yet, the role of MITF in early stages of melanoma development remains largely unexplored. Factors controlling MITF transcription have been well documented and include transcriptional activators, such as SOX10, CREB, PAX3, lymphoid enhancer-binding factor 1 (LEF1, also known as TCF), onecut domain 2 (ONECUT-2) and MITF itself [29]–[33], as well as factors that repress MITF transcription, including BRN2 and FOXD3 [34], [35]. In addition, MITF is subject to several post translational modifications which affect its availability and activity, including acetylation, sumoylation and ubiquitination [27], [28].
To directly assess the importance of ATF2 in melanoma development, we employed a mouse melanoma model in which ATF2 is selectively inactivated in melanocytes. We demonstrate that melanoma development is markedly attenuated in mice expressing a transcriptionally inactive form of ATF2 in melanocytes. Surprisingly, ATF2 control of melanoma development was mediated, in part, through its negative regulation of SOX10 and consequently of MITF transcription. Inhibition of ATF2 abolished mutant BRAF-expressing melanocytes' ability to form foci on soft agar, which was partially rescued when expression of MITF was attenuated. The significance of these findings is underscored by our observation of human melanoma tumors, in which high ratio of nuclear ATF2 to MITF expression was associated with poor prognosis. These findings identify a novel mechanism underlying melanocyte transformation and melanoma development.
Results
Generation of melanocyte-specific ATF2 mutant mice
Global Atf2 knockout in mice leads to early post-natal death [36]. Therefore, the Cre-loxP system was utilized to disrupt Atf2 in melanocytes. Deletion of its DNA binding domain and a portion of the leucine zipper motif results in a transcriptionally inactive form of ATF2 (Figure 1a; [36]). To generate loss-of-function mutants, we established mice that would allow CRE-dependent deletion of these domains. Mice homozygous for the loxP-flanked (floxed) Atf2 gene (Atf2f/f) were born at the expected Mendelian ratios and presented no apparent abnormalities. In addition, in several tissues analyzed, ATF2 expression levels were comparable between WT and Atf2f/f mice (data not shown).

To elucidate the role of ATF2 in melanoma, Atf2f/f mice were crossed with mice harboring a 4-hydroxytamoxifen (OHT)-inducible Cre recombinase-estrogen receptor fusion transgene under the control of the melanocyte-specific tyrosinase promoter, designated Tyr::CreER(T2). Upon administration of OHT, we predicted that CRE-mediated recombination would be induced in a spatially and temporally controlled manner in embryonic melanoblasts, melanocytes, and in putative melanocyte stem cells [37]. The resulting Atf2f/f/Tyr-CreER(T2) mice, designated melanocyte-deleted (md) Atf2md), indeed expressed the gene encoding the ATF2 transcriptional mutant in melanocytes. Immunoblot analysis of ATF2 protein confirmed that melanocytes prepared from wild-type TyrCre+::Atf2+/+::Nras+::Ink4a−/− (WT) mice express a 70 kDa band corresponding to full length ATF2, whereas melanocytes of TyrCre+::Atf2md::Nras+::Ink4a−/− mice express only a 55 kDa band, corresponding to the size of ATF2 lacking the DNA binding and leucine zipper domains (Figure 1b).
Disruption of ATF2 in melanocytes inhibits melanoma formation
To address the role of ATF2 in de novo melanoma formation Tyr::CreER::NrasQ61K::Ink4a−/− (KO of exon 2–3 of Cdkn2a locus, which encodes for both p16Ink4a and p19Arf; [38]) mice, which develop spontaneous melanoma (Lynda Chin, unpublished observations), were crossed with Atf2md mice. Similar to findings reported by Ackermann et al. [39], mutant N-Ras/Ink4a−/− mice developed melanoma within 8–12 weeks with metastatic lesions often seen in the lymph nodes. However, the incidence of melanoma was lower in Tyr::CreER::NrasQ61K::Ink4a−/− mice used in the present study (50% penetrance, of which 50% of the tumors were confirmed to be melanoma), probably because expression of mutant NRAS was induced only after birth, as opposed to activation of NRAS during embryogenesis, as reported in [39]). Thus, Atf2md::N-RasQ61K::Ink4a−/− mice were used to assess changes in melanoma incidence in the absence of functional ATF2 over a period of up to 8 months. In all cases, mouse skin was treated with Tamoxifen within 3–5 days after birth to inactivate ATF2 (Figure 1b) and with doxycycline in their drinking water to induce expression of the NRAS mutant transgene (See Materials and Methods for details; Figure 1c). In the control group (Tyr::CreER::Atf2+/+::NrasQ61K::Ink4a−/−), 11/21 mice (52%) developed tumors within 8–16 weeks (Table 1). In ATF2 heterozygotes (Tyr::CreER::Atf2−/+::NrasQ61K::Ink4a−/−), 18/44 mice (41%) developed tumors within 8–16 weeks, and in the Tyr::CreER::Atf2md::NrasQ61K::Ink4a−/− group only 3 of 21 animals (15%) developed tumors within 24–36 weeks (Figure 1d and Table 1). To evaluate tumor type, we examined melanoma markers including DCT and S100 in all tumors (Figure 1e, Figure S1). This analysis identified 55–63% of tumors as melanomas in both the Atf2+/+ (7/11) and Atf2+/− (10/18) groups (Table 2). Only one of the three tumors observed in the Atf2md group was identified as a melanoma. Kaplan Meier curve did not reveal significant differences in survival among the different genotypes, probably since this study was primarily designed to follow tumor incidence. Common to all genotypes, most tumors that were not identified as melanomas were fibrosarcomas and lymphomas, consistent with previous reports [38]. These data suggest that transcriptionally active ATF2 is required for melanoma development in the NrasQ61K::Ink4a−/− mouse melanoma model.


Identification of MITF as an ATF2-regulated gene
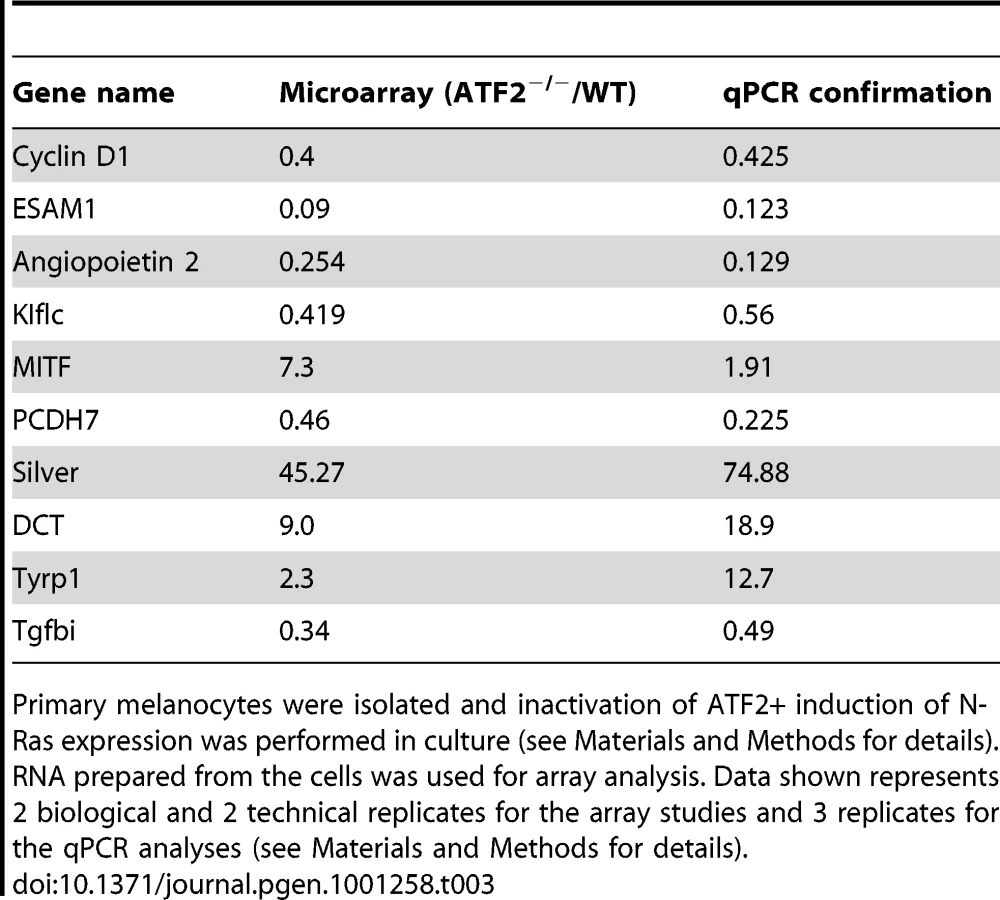
To assess the mechanism underlying ATF2's contribution to melanoma development, we conducted gene profiling array analysis of primary melanocytes prepared from Tyr::Cre+::Atf2+/+::NrasQ61K::Ink4a−/− and Tyr::Cre+::Atf2md::NrasQ61K::Ink4a−/− mice. Analysis was limited to melanocytes, since, as reported above, only one melanoma formed in the ATF2 mutant group. In all cases, ATF2 was inactivated and NRAS was induced in culture within 48h of plating cells, as monitored by western blots (Figure 1b, 1c and data not shown). Melanocytes were enriched, and immunostaining with appropriate markers confirmed that samples were free of keratinocytes and fibroblasts (data not shown; see Materials and Methods for details). RNA was prepared from cultures and two biological and technical replicates were used for data analysis. As shown in Table 3, among transcripts differentially expressed in ATF2 WT and mutant cultures were several factors that play an important role in melanocyte pigmentation, including Mitf, Silver, Tyrp1 and Dct. qPCR analysis, performed on independently prepared RNA samples from melanocytes expressing WT (Tyr::Cre+::Atf2+/+::NrasQ61K::Ink4a−/−) or mutant ATF2 (Tyr::Cre+::Atf2md::NrasQ61K::Ink4a−/−), confirmed altered expression of pigmentation genes (Table 3). These data provide the initial indication that ATF2 negatively regulates Mitf and several other important pigmentation genes. As the pigmentation genes identified in this array are known to be regulated by MITF [27], we focused on regulation of MITF by ATF2.
MITF is negatively regulated by ATF2 in mouse and human melanocytes
To confirm that ATF2 negatively regulates Mitf expression, we assessed MITF transcription in primary mouse melanocytes harboring WT (Tyr::Cre−::Atf2+/+) or mutant (Tyr::Cre+::Atf2md) forms of ATF2. RNA prepared from whole skin of these mice (3 mice per group) was subjected qPCR analysis. Significantly, Mitf expression was inversely correlated to the presence of functional ATF2; samples obtained from ATF2 mutant skin exhibited a greater than 2-fold increase in MITF expression compared with those obtained from WT ATF2 mice (Figure 2a). Likewise, we found that genes transcriptionally regulated by MITF, such as Dct, Silver and Tyrp1, were upregulated in the skin of mutant ATF2 mice (Figure 2a). The degree of altered expression of pigmentation genes was less pronounced in whole skin samples than in cultured melanocytes (Table 3), probably due to confounding effects of in vitro cell culture. To confirm the qPCR data, we performed immunostaining of skin tissue samples obtained from 4 days old WT or ATF2 mutant mice and observed increased MITF expression in melanocytes from Atf2md mice relative to their WT counterparts (Figure 2b). Quantification of MITF staining revealed an approximate 2-fold increase in nuclear MITF expression in Atf2md compared to WT mice (Figure 2c). Of note, the level of S100 staining in the hair matrix was markedly reduced in the skin of Atf2md mice. At a later time point (2 weeks) representing an advanced stage of melanocyte development, S100 staining was similar in both genotypes, while MITF expression remained upregulated in Atf2md mice (not shown). In all, these data confirm our initial observations in primary mouse melanocytes that MITF levels are elevated in ATF2 mutant-expressing cells.

Additional assessment was performed in melan-Ink4a-Arf1 melanocytes, a line derived from black Ink4a-Arf null mice [40], and in primary human melanocytes. In both, ATF2 expression was inhibited by viral infection with the corresponding mouse or human shRNA (shATF2). Infection of either primary human (Figure 3a) or melan-Ink4a-Arf1 melanocytes (Figure 3b) with shATF2 markedly increased MITF transcription and protein expression (Figure 3a, 3b). These findings show that loss of transcriptionally active ATF2 allows higher expression of MITF and strongly suggest that ATF2 negatively regulates MITF expression in melanocytes.

MITF transcription is negatively regulated by ATF2 in about 50% of human melanoma cells
Given that ATF2 negatively regulates MITF in melanocytes of mouse and human tissues and in related melanocyte cell lines, we asked whether ATF2 also regulates MITF in human melanoma cells. Initially, we assessed changes in MITF expression in six human melanoma lines harboring oncogenic mutations in BRAF or NRAS, and in which ATF2 expression was effectively inhibited by corresponding shRNA (shATF2). In all cases, shRNA specificity was confirmed using three independent sequences (data not shown). Surprisingly, the six melanoma lines fell into two classes based on distinct patterns of regulation of MITF by ATF2 (Table 4). The first class comprised four of the six melanoma cultures (1205Lu, WM35, WM793 and WM1361), in which MITF expression was elevated 3–6-fold following inhibition of ATF2 expression (Figure 3c, S2a). Conversely, a second class of cells, including MeWo and 501Mel cells, exhibited decreased MITF expression after ATF2 knockdown (KD), suggesting positive regulation of MITF by ATF2 (Figure 3d, S2b). Notably, this latter group showed high levels of basal MITF expression [41], [42], suggesting that regulation of MITF expression in these cells differs mechanistically from that of the first group. Further, in response to stress (UV or hypoxia) the MeWo and 501Mel lines further reduced MITF expression (Figure 3d and data not shown), providing further evidence for differential regulation of MITF in these cells both prior to and in response to stress stimuli. Additional analyses were performed, employing 12 more melanoma cell lines. Inhibition of ATF2 expression revealed that 4/12 exhibited increase in MITF expression, while 6/12 decreased MITF expression. Two of the 12 lines did not exhibit change in MITF expression following ATF2 KD (Table 4, S5). Collectively, out of 18 melanoma lines we found that 8 (44%) retained similar negative regulation of MITF by ATF2 as observed in the melanocytes. However, another 8 (44%) exhibited positive regulation of MITF by ATF2, pointing to a transcriptional switch that occurred in the course of melanocyte transformation. MITF was not affected by altered ATF2 expression in 2/18 cell lines (Table 4, Figure S5). In all, in about 50% of the melanoma cell lines ATF2 elicits negative regulation of MITF, similar to what was seen in human and mouse melanocytes.

ATF2 regulation of MITF is mediated by discrete promoter elements
MITF transcription is regulated by complex positive and negative cues [27]. For instance, while CREB and SOX10 positively regulate MITF, BRN2 and FOXD3 have been shown to downregulate MITF expression [29], [30], [34], [35]. Hence we used melanocytes and representative melanoma lines to assess mechanisms underlying positive or negative regulation of MITF. Infection of the human melanocyte line Hermes 3A with shATF2 effectively inhibited ATF2 expression, upregulated Mitf transcription and increased transcription of SOX10 and FOXD3 (from 7 - to 10-fold) and to a lesser extent of Pax3 and Brn2 (from 1.5 - to 2-fold) (Figure 4a, S3a). Similarly, inhibition of ATF2 transcription in human melanoma 1361 cells increased SOX10 and FOXD3 transcription, albeit, to a lesser degree (3 - and 1.5-fold, respectively) compared with human melanocytes (Figure S3b). Neither BRN2 nor PAX3 transcription was elevated in melanoma cells in which ATF2 expression was inhibited (Figure S3a). These observations suggest a role for ATF2 in FOXD3 - and SOX10-mediated regulation of MITF transcription in melanocytes and melanoma cells.

To assess the possible role of FOXD3 in regulation of MITF we inhibited FOXD3 expression in melanocytes expressing control shRNA and shATF2. Inhibition of FOXD3 expression increased SOX10 transcription and protein expression, albeit to lower levels compared with inhibition of ATF2 expression (Figure S4). Concomitant increase of MITF RNA and protein levels was also lower, compared with that seen upon inhibition of ATF2 expression. Notably, inhibition of both ATF2 and FOXD3 resulted in additive increase of SOX10 and MITF (Figure S4). These data suggest that FOXD3 may also contribute to negative regulation of MITF in melanocytes, independent of ATF2. Since inhibition of FOXD3 elicited a less pronounced effect compared with ATF2, and since the effect appeared ATF2-independent and furthermore did not appear to mediate similar changes in human melanoma cells (Figure S3b and data not shown), we focused on assessment of direct mechanisms underlying ATF2 effect on MITF transcription.
To this end we first analyzed MITF promoter sequences for ATF2/CRE elements (Cyclic AMP response element), which can be targeted by ATF2, as well as sequences recognized by BRN2 and SOX10 using a luciferase reporter construct (MITF-Luc) [43]. Using either a wild-type (WT) construct or one in which the BRN2 site was mutated, we observed increased luciferase activity following inhibition of ATF2 transcription in WM1361 melanoma (Figure 4b), as well as in LU1205 and WM35 melanoma cells (data not shown). The relative increase in luciferase activity following ATF2 inhibition was equivalent in both constructs, suggesting that an ATF2 effect is not mediated by BRN2 (Figure 4b, left panel). Similarly, MITF transcriptional activities were altered to a similar degree following inactivation of the CRE element (Figure 4b, right panel), suggesting that ATF2 down-regulation of the MITF promoter is indirect. We therefore assessed whether SOX10, which positively regulates MITF and whose transcription markedly increases in melanocytes and melanoma cells in which ATF2 expression is inhibited (Figure 4a, S3b), may mediate ATF2 effect on MITF transcription. Analysis of a MITF-Luc construct harboring a mutant SOX10 binding site revealed that ATF2 inhibition no longer elicited increased MITF transcription in human melanocytes or in melanoma cells (Figure 4c). In agreement, inhibition of SOX10 expression by corresponding siRNA attenuated the increase in MITF transcription seen in shATF2-expressing human melanocytes (Figure 4d) or melanoma cells (Figure 4e). These results suggest that ATF2 regulation of MITF transcription is mediated by SOX10. In agreement, chromatin immunoprecipitation (ChIP) assays confirmed increased binding of SOX10 to the MITF promoter in melanoma cells expressing shATF2 (Figure 5a).

A putative response element for AP1 (which can serve as an ATF2 response element through ATF2 heterodimerization with JUN family members; [9]) has been identified in upstream regions of the Sox10 promoter [44]. We examined potential ATF2 binding to this element by ChIP and found that endogenous ATF2, but not ATFa, binds to that AP1 sequence (−4797–4791) in both human melanocytes and melanoma cells (Figure 5b). We next set to identify ATF2 heterodimeric partner, which could mediate negative regulation of SOX10 transcription. Among members of the JUN family implicated in transcriptional silencing is JunB. Thus, further assessment was performed to determine if JunB functions as an ATF2 heterodimerization partner to regulate SOX10 transcription through the AP1 site. ChIP analysis confirmed that JunB binds to the AP1 site found in SOX10 promoter sequences (Figure 5c). To confirm a possible role for JunB in regulating MITF transcription we asked whether expression of TAM67, a negative regulator of Jun family members, could attenuate the binding and transcriptional activities elicited by JunB. Expression of TAM67 indeed reduced the degree of ATF2 and JunB binding to the AP1 site on SOX10 promoter. Further, KD of ATF2 expression abolished binding of both ATF2 and JunB to the AP1 site on the SOX10 promoter (Figure 5c). These data confirm the presence of ATF2-JunB complex on Sox10 promoter and suggest that ATF2 recruits JunB for binding to the AP1 site on SOX10 promoter. To assess the role of JunB on SOX10 transcription we have monitored changes in Sox10 expression at the protein and RNA levels. Expression of TAM67 caused increased expression of SOX10 in both human melanoma (∼2 folds; Figure 5d) and melanocytes (∼3 folds; Figure 5e), indicating some relief of JunB inhibition. Co-expression of TAM67 with Jun B attenuated this increase, reducing the level of Sox10 expression to basal levels (Figure 5d, 5e). Over-expression of JunB, but not Jun D, effectively inhibited Sox10 expression in both the melanoma and melanocytes cells (Figure 5d, 5e). These data suggest that JunB mediates inhibition of Sox10 expression. To further reveal the role of ATF2 in this inhibition, we assessed the effect of JunB on Sox10 expression in cells expressing control shRNA or shATF2. While ectopic expression of JunB reduced the expression of Sox10 in control shRNA-expressing cells, such decrease was no longer seen in cells expressing shATF2 (Figure 5f). Collectively, these findings suggest that ATF2, in concert with JunB, is responsible for inhibition of Sox10 expression.
We next assessed the effect of ATF2 on SOX10 and MITF expression in 12 additional human melanoma cell lines. In all cases cells were infected with shATF2 and changes in SOX10 and MITF were monitored at the level of RNA. Notably, about 4/12 melanoma lines revealed increase in both SOX10 and MITF expression upon KD of ATF2 (Figure S5, Table 4). In contrast, 6/12 melanoma lines revealed decrease in MITF expression, of which 5 also shown decrease in SOX10 expression, pointing to positive regulation of SOX10 and MITF in these melanoma cells. In two out of the 12 melanoma lines ATF2 affected SOX10 but not MITF transcription (Figure S5). Overall, our cohort of 18 melanoma lines revealed that about 50% of the melanomas retained negative regulation of MITF by ATF2, as seen in the melanocytes (primary and cell lines) (Table 4).
To further assess whether ATF2 regulation of MITF is SOX10-dependent in melanocytes and melanoma cells, we coexpressed SOX10 in shATF2-expressing cells. As seen in earlier analysis, inhibition of ATF2 expression caused increase in MITF transcription in the human melanocytes and 4 melanoma cell lines, (WM1361, WM793, LU1205, WM35; Figure S6). Notably, the melanocytes and 2/4 melanoma cell lines revealed ATF2 effect on MITF expression is SOX10-dependent (WM1361, WM793; Figure S6). Two of the four melanoma cell lines did not reveal increased SOX10 expression, although they retained increased MITF expression, upon inhibition of ATF2 (Lu1205, WM35; Figure S6). These findings confirm that while in melanocytes, expression of SOX10 and MITF is negatively regulated by ATF2, this mechanism is conserved in approximately half of melanomas surveyed.
Along these lines, the two melanoma lines (MeWo and 501 Mel) that exhibit positive regulation of MITF by ATF2 also exhibited positive regulation of SOX10 by ATF2 (Figure S7). Inhibition of ATF2 expression reduced SOX10 and MITF RNA and protein levels (Figure S7a–c). In order to determine whether JunB lost its ability to elicit negative regulation of SOX10 and MITF in melanoma cells where ATF2 no longer inhibited SOX10 or MITF expression, we transfected those cell lines with TAM67 and JunB alone and in combination. In these cells, whereas TAM67 effectively attenuated Sox10 and MITF expression, JunB did not alter expression of these genes, suggesting that positive regulation of MITF and SOX10 by ATF2 depends on other members of the Jun family of transcription factors (Figure S7d). Conversely, TAM67 or JunB had no effect on melanoma cells in which ATF2 inhibits MITF independently of SOX10, suggesting that in these cases, ATF2 likely cooperates with transcription factors other than JunB to elicit negative regulation of SOX10 and MITF (Figure S7d). Consistent with this observation, ChIP assay confirmed ATF2 and CREB, but not JunB, binding to the Sox10 promoter in these cells (Figure S7e). These findings suggest that changes in ATF2 heterodimeric partner (from JunB to CREB) are likely to cause the switch from negative to positive regulation of SOX10, and in turn, MITF (see below). The possibility that altered expression of JunB may account for ATF2 positive or negative regulation of Sox10 and MITF were excluded, as no clear correlation between JunB expression and the ability of ATF2 to elicit negative regulation of Sox10/MITF were seen (Figure S7f).
Among response elements potentially required to upregulate MITF transcription is the CRE element, which is implicated in CREB-mediated upregulation of MITF transcription [45]. Although transcriptional activity from a CRE mutant MITF promoter was lower compared to the WT promoter (30%), it was no longer responsive to inhibition of ATF2 expression in the MeWo cells (Figure S8a). Pull-down assays using biotin-tagged MITF promoter sequences harboring the CRE identified ATF2 and CREB as CRE-bound proteins in MeWo melanoma cells (Figure S8b). In agreement, ChIP analysis confirmed occupancy of the CRE site on MITF promoter by ATF2 (Figure S8c). These findings are consistent with the fact that ATF2 heterodimerizes with CREB [9] and with a report that p38/MAPK14 (which phosphorylates ATF2) plays an important role in MITF transcription dependent on the CRE site [46]. These results establish that ATF2-dependent activation of MITF transcription in these melanoma cells is mediated through the CRE site, likely in cooperation with CREB. Notably, MeWo and 501Mel lines are known to express high MITF levels compared to other melanoma lines [41], [42], suggesting these cells harbor distinct mechanisms that preclude negative regulation of MITF by ATF2.
Inhibition of MITF expression rescues focus formation on soft agar in shATF2-expressing melanocytes
To determine whether the contribution of ATF2 to melanocyte transformation and development is MITF-dependent, we assessed melanocytes' ability to grow and form colonies in soft agar, which is indicative of their transformed potential. Expression of mutant BRAFV600E in immortal melanocytes is reportedly sufficient for growth on soft agar [47]. Thus we infected melan-Ink4a-Arf1 melanocytes with mutant BRAF (Figure 6a) and confirmed their ability to form colonies in soft agar. Mutant BRAF expression effectively caused formation of about 1000 colonies per 5000 cells (Figure 6b, 6c). In contrast, melanocytes infected with BRAF600E and with shATF2 formed on average about 20 colonies, indicative of loss of tumorigenicity (Figure 6b, 6c, 6d) and consistent with our initial observation that the number of melanoma tumors significantly decreases in the absence of transcriptionally functional ATF2 (Tables 1–2). To determine the importance of MITF at this early stage of melanocyte transformation we inhibited MITF expression (using shRNA) in melanocytes expressing mutant BRAF alone or mutant BRAF+shATF2. Significantly, inhibition of MITF expression decreased the number of BRAF-induced foci (from 1000 to about 100 per well). Over-expression of MITF in BRAF-expressing melanocytes also inhibited focus formation, to a degree similar to that seen following inhibition of MITF expression (Figure 6b, 6c, 6d). This observation implies that effective inhibition or overexpression of MITF attenuates melanocyte transformation, consistent with previous reports (52). Remarkably, inhibition of MITF expression in melanocytes expressing both mutant BRAF and shATF2 rescued, at least partially, melanocytes' ability to form foci on soft agar (400 compared with 20 seen in shATF2 cells; Figure 6b, 6c, 6d). These findings suggest that inhibition of MITF expression in melanocytes lacking ATF2 expression can promote transformation. That MITF inhibition in melanocytes expressing ATF2 WT can attenuate their ability to form foci on soft agar is attributable to the relative expression of MITF RNA and protein in each condition (Figure 6d). MITF expression levels in ATF2 KD cells increased 7.5-fold compared with control BRAF-expressing melanocytes. Inhibition of MITF expression in ATF2 KD melanocytes reduced MITF expression 2.5-fold relative to controls, whereas MITF KD alone resulted in lower MITF expression (5-fold; Figure 6d). Thus, complete abrogation of MITF expression attenuates melanocyte transformation, whereas low to moderate levels of MITF expression are sufficient to promote growth on soft agar. Higher MITF expression levels, as seen in ATF2 KD cells, result in a total loss of melanocytes' ability to form foci on soft agar. These findings are in line with the proposed rheostat model in which medium levels of MITF are optimal for growth and melanoma development [48] and in agreement with our observations in a mouse melanoma model.

We next assessed whether inhibition of melanocyte growth on soft agar by altered ATF2 and/or MITF expression can be attributed to decreased proliferation or increased apoptosis. Inhibition of ATF2 expression caused notable accumulation of cells in G2 (60%), with significant cell death induction (22%) compared to controls (4%), (Figure 6e, 6f). Interestingly, such altered cell cycle distribution and cell death rate were associated with a significant increase in MITF protein levels (Figure 6d). In contrast, inhibition of MITF expression did not significantly induce cell death (6.5%) but resulted in fewer cells in G2/M-phase and more cells in G1, compared with inhibition of ATF2 alone. These observations suggest that MITF inhibition is sufficient to reduce the rate of cell cycle progression through G2/M phase and that inhibited growth of BRAF600E-expressing melanocytes on soft agar may be attributed to abrogation of distinct cell cycle-regulatory mechanisms. Combined inhibition of ATF2 and MITF restored cell cycle distribution to that seen in control melanocytes, and reduced cell death from 22.4% to 12.9%. Of interest, MITF overexpression promoted a similar degree of cell death (11.4%) without altering cell cycle distribution, similar to combined inhibition of ATF2 and MITF (Figure 6e, 6f). Together, these observations suggest that simultaneous inhibition of ATF2 and MITF averts cell cycle abrogation induced when expression of either of these factors is perturbed individually, further substantiating regulation of MITF by ATF2.
Low nuclear MITF expression in melanoma tumors that exhibit strong nuclear ATF2 expression is associated with poor prognosis
The availability of a melanoma TMA, consisting of over 500 melanoma samples and in which expression of both ATF2 and MITF in the same tumors had been measured enabled us to assess possible associations between ATF2 and MITF and their correlation with survival and other clinical and pathological factors. Our earlier studies revealed that ATF2 subcellular localization in tumors is significantly correlated with prognosis: nuclear localization, reflecting constitutively active ATF2, was associated with metastasizing tumors and poor outcome [7]. Here we quantitated immunofluorescent staining of TMAs for MITF and ATF2 by employing our automated, quantitative (AQUA) method. To normalize ATF2 and MITF levels, expression of each of the two proteins in individual patients was divided by the median expression level of the respective protein in all patients, and the nuclear ATF2/MITF ratio was calculated and log-transformed. By ANOVA analysis, the ratio was higher in metastatic than in primary specimens (t value = 2.823, P = 0.0051), as shown in Figure 7a. No association was found between nuclear ATF2/MITF ratio and disease-specific survival among patients with metastatic melanoma (not shown). Significantly, a high nuclear ATF2/MITF ratio in primary melanoma specimens was associated with decreased 10-year disease-specific survival (P = 0.0014; Figure 7b). On Cox multivariable analysis, this association with survival was independent of patient age, Breslow thickness or the presence or absence of ulceration (data not shown). Nuclear ATF2 alone in primary specimens was associated with poor survival, but to a lesser degree than the ratio of nuclear ATF2/MITF (P = 0.0118 for ATF2 as a single discriminator versus P = 0.0014 for the ratio of nuclear ATF2/MITF). Nuclear MITF as a single discriminator was not a significant predictor of survival (P = 0.185), as was reported previously using immunohistochemistry [49]. These observations suggest that active (nuclear) ATF2 in melanoma can suppress MITF expression, and that this phenomenon is associated with poor prognosis.

Discussion
Identifying mechanisms underlying early phases of melanocyte transformation and melanoma development is central to understanding the etiology of this devastating tumor, as well as for developing novel treatment approaches. Previous studies indicate the presence of mutant BRAF in melanocytic lesions, as well as its effect on pigment gene expression [6], [50], [51]. The present study enhances our understanding of early events contributing to melanoma development. We demonstrate that loss of a transcriptionally active form of ATF2 in melanocytes inhibits melanoma development in an Nras/Ink4a model. Our quest to understand mechanisms underlying ATF2 activity in this process led us to identify an important role for ATF2 regulation of MITF, an important regulator of melanocyte biogenesis and a factor implicated in melanoma progression [49]. Surprisingly, ATF2 negatively regulated MITF expression in mouse and human melanocytes, suggesting that ATF2 transcriptional activities limit MITF expression. We demonstrate that such negative regulation is elicited through downregulation of SOX10 by ATF2, in cooperation with JunB. A putative AP1 response element has been identified in SOX10 promoter sequences and ChIP analysis of this domain showed ATF2 and JunB binding. Overexpression of JunB efficiently suppressed SOX10 expression in an ATF2-dependent manner and inhibition of Jun transcriptional activities phenocopied the effect of shATF2, suggesting that negative regulation of SOX10 by ATF2 is direct, and is mediated in cooperation with JunB.
Importantly, ATF2-dependent negative regulation of Sox10 and consequently of MITF seen in melanocytes, but only in about 50% of the 18 melanoma cell lines studied here. Correspondingly, JunB, which is required for ATF2-dependent inhibition of Sox10 transcription, is no longer found on the promoter of SOX10 in melanoma cells (i.e. 501Mel) that exhibit positive regulation by ATF2. Rather, CREB and ATF2 are found on SOX10 and MITF promoters, pointing to a switch in ATF2 heterodimeric partners to enable positive regulation of these genes. Notably, melanoma cell lines that exhibit positive regulation of SOX10 and MITF by ATF2, also show high basal levels of MITF expression [41], [42], suggesting that additional genetic or epigenetic changes distinguish these lines from melanocytes and the other melanoma lines in which ATF2 elicits negative regulation of MITF.
Notably, ATF2 control of MITF expression affected the ability of BRAF600E-expressing melanocytes to exhibit transformed phenotype in culture, monitored by their ability to grow on soft agar. Inhibition of ATF2 abolished soft agar growth of BRAF600E-expressing melanocytes, which was partially rescued upon KD of MITF. Interestingly, both the over expression or the KD of MITF resulted in inhibition of melanocytes ability to grow on soft agar, substantiating the notion that a fine balance of MITF expression must be maintained in order to ensure its contribution to cellular proliferation and transformation. We propose that excessively low or high MITF levels block melanocyte transformation, whereas intermediate levels allow transformation to occur. Overall, our observations demonstrate that ATF2 plays an important role in fine-tuning those levels and support the rheostat model proposed for MITF's role in melanoma development and progression [48]. Of importance, ATF2 and MITF affect the ability of BRAF600E-expressing melanocytes to grow on soft agar via distinct mechanisms. Whereas specific inhibition of ATF2 causes both accumulation of cells in G2 and induction of cell death, specific alteration of MITF protein levels—particularly depletion—significantly affects cell proliferation and inhibit growth on soft agar by non-lethally slowing cell cycle progression at G2/M. These observations are consistent with a report from Wellbrock and Marais [52], who showed that altered MITF expression inhibits melanocyte proliferation.
Importantly, inhibiting MITF expression in ATF2 KD melanocytes was sufficient to partially rescue melanocyte growth on soft agar. While supportive of our finding in the Nras::Ink4a mouse melanoma model, where expression of transcriptionally inactive ATF2 inhibits melanoma formation, these observations provide the foundation for a model in which ATF2 inhibition causes increased MITF levels and concomitant inhibition of melanocyte growth, possible induction of cell death and delayed development. The latter is suggested by IHC analysis of mouse skin from ATF2md mice, which shows notably reduced S100 staining indicative of delayed melanocyte development: ATF2 KO melanocytes appear to represent anagen stage IV, whereas WT represent anagen stage VI. This delay was seen at the 4 - but not the 14-day time point, suggesting that an ATF2 effect might be limited to a specific subpopulation or phase of melanocyte development. The early (4 day) time point is within the time frame that allows induction of melanoma development by UV-irradiation of c-Met or H-Ras mutant mice [53]. It is therefore plausible that timely control of MITF expression by ATF2 determines melanocyte susceptibility to transformation.
Our analysis of genes whose expression is altered by ATF2 KD in melanocytes identified a cluster of pigmentation genes, many reportedly regulated by MITF [6], [54]. Therefore, changes in TYRP1, DCT and SILVER expression could be attributed to altered MITF expression. However, initial analysis points to a more complex mechanism since (i) the degree of changes in expression of these genes was often greater than that seen for MITF and (ii) expression of some pigmentation genes was found to be independent of MITF in some melanoma and melanocyte cultures. Hence, further studies are required to address mechanisms underlying ATF2 regulation of these pigmentation genes and the significance of such regulation to melanocyte transformation and melanoma development. While our present studies focused on the ATF2-MITF axis, it is expected that additional ATF2-regulated genes contribute to melanoma development [12]. In agreement, our earlier studies using both human and mouse melanoma lines demonstrate that inhibition of ATF2 effectively inhibits tumorigenesis and blocks metastasis [22]–[26].
Important for ATF2 function is its subcellular localization. While findings presented here position ATF2 as an oncogene functioning in melanocyte transformation and melanoma development, earlier studies from our laboratory and others suggest that in keratinocytes and mammary glands, ATF2 elicits a tumor suppressor function [55], [56]. Of interest, assessing the localization of ATF2 in the melanoma cell lines studied here revealed that all express nuclear ATF2. Interestingly, in most cases the nuclear staining revealed a punctate staining, resembling the localization of ATF2 to DNA repair foci following DNA damage (Figure S9). A possible link between the presence of ATF2 in repair foci in most melanoma cells points to the possible presence of activated DNA damage response which may be associated with genomic instability [19], [20]—aspects that will be explored in future studies. Significantly, the appearance of nuclear ATF2 is correlated with poor prognosis in melanoma, whereas melanomas that exhibit cytosolic ATF2 exhibit a better survival. Notably, cytosolic ATF2 is primarily seen in non-malignant skin tumors [55]. Here we demonstrate that high nuclear ATF2/MITF ratios are associated with poor prognosis in primary melanomas, but not with metastatic melanomas. The latter finding attests for the important role ATF2 plays to control MITF expression in the early phase of melanocyte transformation and melanoma development.
Overall, using the mutant Nras/Ink4a melanoma model we provide genetic evidence for a central role for ATF2 in melanoma development. We demonstrate that in the absence of transcriptionally active ATF2, melanoma formation is largely inhibited. Furthermore, our data point to an unexpected role of ATF2 in fine-tuning of MITF transcription through regulation of its positive regulator SOX10. Mouse melanoma models and in vitro transformation studies indicate that this newly identified regulatory pathway is required for early phases of melanocyte transformation. Given that ATF2 affects activity of the oncogenes N-Ras (mouse model) and BRAF (melanocyte growth on soft agar); we expect that ATF2 play significant roles in melanomas that carry either of these mutations.
Materials and Methods
Ethics statement
Research involving human participants has been approved by the institutional review board at Yale University (where the TMA was prepared and analyzed). All animal work has been conducted according to relevant national and international guidelines in accordance with recommendations of the Weatherall report and approved by the IACUC committee at SBMRI.
Animal treatment and tumor induction protocols
Mice bearing a conditional allele for mutant ATF2 in which the DNA binding domain and part of the leucine zipper domain were deleted, were generated as previously described [36], [55]. To study the function of ATF2 in melanocytes, we utilized the Cre-loxP system for disruption of the ATF2 gene in melanocytes [37]. The Tyr::CreER::Atf2md mice and their littermate controls (WT) were of FVB/129P2/OlaHsd (TyrCreERT mice were FVB, ATF2fl/fl were 129P2/OlaHsd) and N-Ras/Ink4a−/− mice were C57Bl/6/129SvJ. For melanoma studies we have used Tyr::CreER::NrasQ61K::Ink4a−/− mice (developed at HMS by LC) following their cross with the Tyr::CreER::Atf2md mice.
Immunohistochemistry
Skin specimens were fixed in neutral buffered formalin solution and processed for paraffin embedding. Skin sections (5 µm in thickness) were prepared and deparaffinized using xylene. For MITF, DCT and S100 immunostaining, tissue sections were incubated in DAKO antigen retrieval solution, for 20 min in a boiling bath, followed by treatment with 3% hydrogen peroxide for 20 min. Antibodies against MITF (1∶100 from Sigma), DCT (1∶500, kind gift from Dr. Vincent Hearing) and S100 (1∶100, DAKOCytomation; Carpinteria, CA) were allowed to react with tissue sections at 4°C overnight. Biotinylated anti-rabbit IgG was allowed to react for 30 min at room temperature and diaminobenzidineor Nova Red were used for the color reaction. Hematoxylin was used for counterstaining. The control sections were treated with normal mouse serum or normal rabbit serum instead of each antibody.
Cell culture
Immortalized human melanocytes Hermes 3A which has hTERT (puro) and CDK4 (neo) expression [57] were grown in RPMI 1640 medium containing Fetal Bovine Serum (FBS, 10%), 12-O-tetradecanoyl-phorbol-13-acetate (TPA, 200 nM, Sigma, St. Louis, MO), Cholera toxin (200 pM, Sigma), human stem cell factor (10 ng/ml, R&D systems, Minneapolis, MA), and endothelin 1 (10 nM, Bachem Bioscience Inc., Torrance, CA). Primary human melanocytes (NEM-LP; Invitrogen) were grown in medium 254 and HMGS (Cascade Biologics). Mouse melanocytes (melan-Ink4a-Arf1) were grown as for immortalized human melanocytes excluding human stem cell factor and endothelin. Melanoma cell lines were grown in DMEM medium supplemented with 10% FBS and penicillin/streptomycin (P/S; Cellgro). Melanoma cell lines used in this study LU1205, WM793, 501MEL, WM35, WM1361, MeWO (kind gift from Meenhard Herlyn), UACC903 were maintained in DMEM medium supplemented with 10% FBS and Penicillin/Streptomycin. Melanoma cell lines SbCl2, WM9, WM4, WM1650, A2068, WM1366, WM3629, WM1552, SKMEL2, SKMEL5, and SKMEL8 were maintained in RPMI medium supplemented with 10% FBS and Penicillin/Streptomycin. Primary melanocytes cultures were prepared from mice carrying the Atf2 WT or mutant genotypes and N-Ras/Ink4a−/− as follows. Dorsal-lateral skin was removed from one day-old pups, disinfected with 70% ethanol for 1 min and then washed at least twice with sterile PBS. The skin was submerged in 1× Trypsin/EDTA overnight at 4°C and next day, the skin was placed in a Petri dish with mouse melanocyte culture medium (described below). The epidermis and sheared tissue was removed and discarded with forceps. The tissue was transferred to 15 ml centrifuge tubes and vortexed vigorously until solution becomes cloudy (1–2 min). The cell suspension was transferred to tissue culture flasks. After 3 days, melanocyte growth medium containing 0.8 µg/ml geneticin (Sigma-Aldrich) was added to eliminate contaminating fibroblasts (melanocytes are resistant to such treatment). Geneticin-containing medium was removed and replaced with fresh media after 1 day. Media was changed twice a week. Primary mouse melanocytes were grown in F-12 media (Invitrogen) containing 20% L-15 media (Invitrogen), 4% of FBS and Horse serum (Invitrogen), Penicillin (100 units) and streptomycin (50 µg) antibiotics, db-cAMP (40 µM, Sigma-Aldrich), 12-O-tetradecanoyl-phorbol-13-acetate (TPA, 50 ng/ml, Sigma-Aldrich), alpha-Melanocyte stimulating hormone (α-MSH, 80 nM, Sigma-Aldrich), Fungizone (2.5 µg/ml, Sigma-Aldrich) and melanocyte growth supplement (Invitrogen). Primary melanocytes were treated with 4-OHT (10 µM) for 8h followed by addition of doxycycline (2 µg/ml) for 24h to inactivate ATF2 and induce expression of N-Ras.
Constructs
ATF2-specific shRNA clones were obtained from Open Biosystems (catalog no. RHS4533). Five different shRNA were obtained and tested for their efficiency of KD. Clone TRCN0000013714 was more efficient in inhibiting ATF2 in human cell lines while clone TRCN0000013713 was more efficient for knocking down mouse ATF2. For subsequent experiments we used the respective shATF2 clone depending on human or mouse cell lines. We also tested 3 different clones for KD of ATF2 to rule out any off target effect (Data not shown). siRNA control (cat # 4611) and three SOX10-specific siRNA oligonucleotides were obtained from Ambion (cat # 4392420). Four FOXD3 specific siRNA were obtained from Dharmacon (Cat # J-009152-06 -07, -08, -09). These siRNAs were pooled together in equimolar ratio for transient transfection. An MITF specific shRNA, and MITF promoter luciferase constructs (WT and mutant CRE-Luc constructs) were obtained from Dr. David Fisher [58]. pGL3 vectors containing wild-type and BRN2-site-mutated MITF promoters were obtained from Dr. Colin Goding [34]. pGL3 vectors containing wild-type and SOX10-site-mutated MITF promoters were obtained from Dr. Michel Goossens [59]. Retroviral vectors encoding a fusion protein consisting of full length human BRAF and BRAFV600E linked to the T1 form of the human estrogen receptor hormone-binding domain were generously provided by Dr. Martin McMahon [60]. SOX10 expression vector obtained from Dr. Alexey Terskikh, RSV-JunB, RSV-JunD were obtained from Dr. Michael Karin and pBabe-Flag-TAM67 from Dr. Michael Birrer.
Antibodies and immunoblotting
Antibodies against SOX10 and CREB (sc-1734 and sc-186 respectively) were from Santa Cruz Biotechnologies; antibodies against ATF2, pERK and ERK (catalogue # 9226, 4337 and 4695 respectively) were obtained from Cell Signaling; antibodies against MITF (C5) were purchased from Cell Lab vision. Protein extract (40–60 µg) preparation and western blot analysis were done as described previously [8]. Specific bands were detected using fluorescent-labeled secondary antibodies (Invitrogen, Carlsbad, CA) and analyzed using an Odyssey Infrared Scanner (Li-COR Biosciences). β-Actin antibody was used for monitoring loading.
Immunofluorescence
Human melanoma and melanocytes were grown in coverslips, fixed (4% paraformaldehyde and 2% sucrose in 1×PBS), and then permeabilized and blocked (0.4% Triton X-100 and 2% BSA in 1×PBS) at room temperature. The cells were then washed (0.2% Triton X-100 and 0.2% BSA in 1×PBS) and incubated overnight at 4°C with monoclonal anti-rabbit antibody against ATF2 (20F1, 1∶100), followed by five washes and then subsequent incubation at room temperature for 2 h with anti-rabbit IgG (Invitrogen, 1∶300) and Phalloidin (Molecular Probes, 1∶1000). DNA was counterstained with 4,6-diamidino-2-phenylindole (DAPI; Vector Laboratories) containing mounting medium.
Analysis of skin samples
Skin samples were collected from the backs of mice and immediately fixed with Z-fix, processed, and embedded in paraffin. Paraffin sections were routinely stained by H&E. Dewaxed tissue sections (4.0–5.0 µm) were immunostained using rabbit polyclonal antibodies to MITF (Sigma-Aldrich), S100 (S100B; DAKOCytomation; Carpinteria, CA), and DCT (αPEP8, kindly provided by Dr. Vincent Hearing). Application of the primary antibody was followed by incubation with goat anti-rabbit polymer-based EnVision-HRP-enzyme conjugate (DakoCytomation). DAB (DakoCytomation) or SG-Vector (Vector Lab, Inc.; Burlingame, CA) chromogens were applied, yielding brown (DAB) and black (SG) colors, respectively.
Quantitative analysis of immunostaining
Quantitative analysis was performed as described previously [61]. Briefly, all slides were scanned at an absolute magnification of 400× [resolution of 0.25 µm/pixel (100,000 pix/in.)] using the Aperio ScanScope CS system (Aperio Technologies; Vista, CA). The acquired digital images representing whole tissue sections were analyzed applying the Spectrum Analysis algorithm package and ImageScope analysis software (version 9; Aperio Technologies, Inc.) to quantify IHC and histochemical stainings. These algorithms make use of a color deconvolution method [62] to separate stains. Algorithm parameters were set to achieve concordance with manual scoring on a number of high-power fields, including intensity thresholds for positivity and parameters that control cell segmentation using the nuclear algorithm.
Microarray analysis
Primary melanocytes were treated with 4-OHT and Doxycycline before isolation of total RNA. 500 ng of total RNA was used for synthesis of biotin-labeled cRNA using an RNA amplification kit (Ambion). The biotinylated cRNA is labeled by incubation with streptavidin-Cy3 to generate probe for hybridization with the Mouse-6 Expression BeadChip (Illumina MOUSE-6_V1_1_11234304_A) that represents 46.6K mouse gene transcripts. We analyzed the BeadChips using the manufacturers BeadArray Reader and collected primary data using the supplied Scanner software. Data analysis was done as follows. First, expression intensities were calculated for each gene probed on the array for all hybridizations using illumina's BeadStudio 3.0 software. Second, intensity values were quality controlled and normalized: quality control was carried out by using the BeadStudio detection P-value set to <0.01 as a cutoff. This removed genes which were never detected in the arrays. All the arrays were then normalized using the cubic spline routine from the BeadStudio 3.0 software. This procedure accounted for any variation in hybridization intensity between the individual arrays. Finally, these normalized data were analyzed for differentially expressed genes. The groups of 2 biological and 2 technical replicates were described to the BeadStudio 3.0 software and significantly differentially expressed genes were determined on the basis of the difference changes in expression level (Illumina DiffScor>60 or DiffScore<−60) and expression difference p-value<0.01. Microarray data are available under accession number GSE23860.
ShRNA infection and RNA interference
Human embryonic kidney 293T cells were transfected with corresponding retro - or lentiviral shRNA constructs (10 µg), Gag-pol (5 µg) and ENV expression vectors (10 µg) by calcium phosphate transfection into 10 cm plates and supernatant was collected after 48 hours to obtain viral particles. 2 million melanocytes and melanoma cells in 10 cm plates were infected with 5 ml of viral supernatant along with 5 ml of medium in the presence of 8 µg/ml polybrene. The virus was replaced with fresh media after 8 hours of infection. After two days, puromycin (1.5 µg/ml) was used to select cells for 3 days. For human and mouse melanocytes the media was changed to DMEM containing 10% FBS 24 h prior to harvesting cells. 50 nM duplexes of scrambled and SOX10 - or FOXD3 - specific siRNA were transfected into human melanocytes and WM1361 melanoma cells (2 million cells per transfection) by Nucleofection using Amaxa reagents (NHEM-Neo Nucleofector and Solution R respectively) for SOX10 or FOXD3 knock down. Over 90% of the cells transduced were able to resist drug selection, indicating efficient infection of the respective genes. GFP was also used to monitor efficiency of infection, confirming >90% GFP expression by fluorescence microscopy.
Real-time quantitative reverse transcription–PCR (RT–PCR)
Quantitative PCR was performed as described earlier [8]. Total RNA was isolated using an RNeasy mini kit (Sigma, St. Louis, MO) and reverse transcribed using a high cDNA capacity reverse transcription kit (Applied Biosystems, Foster City, CA) following the manufacturer's instructions. Specific primers (Valuegene, San Diego, CA) used for PCR were as follows: Human ATF2, forward: tgtggccagcgttttaccaa, reverse: tgatgtgggctgtgcagttt., human MITF, forward: aaaccccaccaagtaccaca, reverse: acatggcaagctcaggac., human SOX10, forward: caa gtaccagcccaggcggc, reverse: gggtgccggtggtccaagtg., human FOXD3, forward: gcgacgggctggaagag, reverse: gctgtccgtgatggggtgcc., human PAX3, forward: ggaactggagcgtgcttttg, reverse: ggcggttgctaaaccagac., human BRN2, forward: gaaagagcgagcgaggaga, reverse: caggctgtagtggttagacg., mouse MITF, forward: agatttgagatgctcatcccc, reverse: gatgcgtgatgtcatactgga, mouse TYRP1, forward: ccctagcctatatctccctttt, reverse: taccatcgtggggataatggc., mouse DCT, forward: gtcctccactcttttacagacg, reverse: attcggttgtgaccaatgggt, mouse Silver, forward: tgacggtggaccctgcccat, reverse: agctttgcgtggcccgtagc. The reaction mixture was denatured at 95°C for 10 min, followed by 40 cycles of 95°C for 15s, annealing at 60°C for 30s and extension at 72°C for 30s. Reactions were performed using the SYBR Green qPCR reagent (Invitrogen) and run on an MX3000P qPCR machine (Stratagene, La Jolla, CA). The specificity of the products was verified by melting curve analysis and agarose gels. The amount of the target transcript was related to that of a reference gene (Cyclophilin A for both human and mouse) by the Ct method. Each sample was assayed at least in triplicate and was reproduced at least three times.
Chromatin immunoprecipitation
Chromatin immunoprecipitation was performed using the Magna-Chip (Upstate) according to the manufacturer's instructions. Control shRNA and ATF2 knocked down WM1361 cells (one 10 cm plate for each, 80% confluent) were fixed in 37% formaldehyde and sheared chromatin was immunoprecipitated and subjected to PCR for 32 cycles. The following primers corresponding to the MITF promoter, spanning the SOX10 binding site were used, forward: gcagtcggaagtggcag, reverse: caactcactgtcagatcaa. Antibodies against Sox10 and CREB (sc-1734 and sc-186 respectively) were from Santa Cruz Biotechnologies. IgG control, and glyceraldehyde-3-phosphate dehydrogenase oligonucleotides were provided by the kit. Antibody against ATF2 (sc-6233), JunB (sc-8051), JunD (sc-74) were obtained from Santa Cruz. Antibodies against ATFa were provided generated by Nic Jones. For Sox10 promoter, the following primers spanning AP-1 binding site were used; forward: cccagtgctggcctaatagc, reverse: cacccttgatatccccaagtga.
Luciferase assays
MeWo, WM35, WM1361, Lu1205 cells in six-well plates were transiently transfected with 0.5 µg of reporter plasmid containing WT or CRE mutant, BRN2 mutant or SOX10 mutant MITF promoter and 0.1 µg of pSV-β-Galactosidase (Promega, San Luis Obispo, CA) using Lipofectamine 2000 reagent (Invitrogen). Human melanocytes (2 million) were transfected with 2 µg of reporter plasmid containing WT or SOX10 mutant MITF promoter and 0.3 µg of pSV-β-Galactosidase using Amaxa reagent (NHEM-Neo nucleofector kit, Lonza) according to the manufacturer's protocol. Cell lysates were prepared from cells after 48 h. Luciferase activity was measured using the Luciferase assay system (Promega) in a luminometer and normalized to β-galactosidase activity. The data were normalized to β-galactosidase and represent the mean and SD of assays performed in triplicate. All experiments were performed a minimum of 3 times.
Colony formation assay
Melan-Ink4a-Arf1 cells were transduced with a retroviral vector expressing BRAFV600E:ERT1 and selected with puromycin for 3 days. These cells were treated with 200 nM of estrogen receptor antagonist ICI 182780 (ICI, Tocris Bioscience) to induce expression of BRAFV600E. After one day, these cells were transduced with a lentiviral vector expressing either shATF2 or shMITF separately, or in combination. Colony formation was carried out as described by Franken et al. [63]. Briefly, 5,000 cells were plated into each well of a 6-well plate, and cells were grown in mouse melanocyte media containing ICI and puromycin (1.5 µg/ml) for 3 weeks until colonies became visible. The colonies were stained with P-Iodonitrotetrazolium Violet (1 mg/ml Sigma, St. Louis, MO). This experiment was performed in triplicate and reproduced 2 times.
Mouse genotyping
Genomic DNA was isolated from tail tissue was subjected to PCR resulting in amplification of a 549 bp DNA fragment for Atf2 floxed and a 485 bp DNA fragment for wild type mice. PCR conditions included one cycle at 95°C for 3 min; and 30 cycles of 94°C/30 sec, 55°C/30 sec and 72°C/1 min and one cycle at 72°C for 5 min. Primers used for PCR reactions were forward: caatccactgccatggcctt, reverse: tcagataaagccaagtcgaatctgg.
Avidin-biotin DNA–protein binding assay
MeWo cells were left untreated or treated with 20 mJ/cm2 of UV-B for 1 h. The cells were lysed using lysis buffer containing 1% Triton-100 and incubated with 4 µg of biotin-labeled MITF promoter spanning the CRE site oligo (5′-gaaaaaaaagcatgacgtcaagccaggggg-3′) in the presence of poly-(dI-dC) (20 µg/ml) for 2h at 4°c. The oligo-bound proteins were captured using streptavidin-agarose (Invitrogen) for 1 h incubation, followed by extensive washes with washing buffer (20 mm HEPES, 150 mm NaCl, 20% glycerol, 0.5 mm EDTA, and 1% Triton-100) and analyzed using SDS-PAGE and western blots.
BrdU, PI labeling, Annexin V staining
To evaluate the cell cycle index of Melan-Ink4a-Arf1 cells stably overexpressing BRAFV600E:ERT1 alone or in combination with shRNA to the genes indicated in Results, cells were plated in media containing ICI and puromycin (1.5 µg/ml) at 2×106 cells per 10 cm plate O/N. Cells were labeled with 10 µM of 5-bromo-2-deoxyuridine (BrdU; Sigma Chemical Co.), for an hour. Cells were then washed, fixed, and stained with anti-BrdU mAbs and propidium Iodide (BD Biosciences, San Jose, CA) according to the manufacturer's protocol, and analyzed on a BD FACSCanto machine. Cell cycle phase was analyzed using the Mod Fit LT v.2 program (Verity Software, Topsham, ME). In a separate experiment the cells were stained with Annexin V-APC and 7-AAD (BD Pharmingen, San Diego, CA) according to manufacturer's protocol, to enable analysis of early apoptosis and cell death.
Hypoxia treatment
Cells were treated under hypoxia (1% O2) for indicated time points using a hypoxia chamber (In Vivo 400; Ruskin Technologies Ltd, Bridgend, UK).
UV irradiation
Mice were treated with 4-Hydroxytamoxifen (25 mg/ml in DMSO) by swabbing the entire body (excluding the head) on days 1–3 after birth. On day 4 the pups were placed under UVB light source (FL-15E; 320 nm) and exposed to 20 µW/cm2 for 22 seconds. Ninety minutes after UVB treatment mice were sacrificed and entire skin was removed and processed.
TMA and AQUA staining
Tissue microarrays were constructed as previously described [21]. The arrays included a series of 192 sequentially collected primary melanomas and 299 metastatic melanomas. Slides were stained for automated, quantitative analysis (AQUA) for ATF2 and MITF as previously published [49], [64]. The AQUA scores for the two markers were obtained from the AQUAmine database (www.tissuearray.org).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Gray-SchopferV
WellbrockC
MaraisR
2007 Melanoma biology and new targeted therapy. Nature 445 851 857
2. FearsTR
TuckerMA
2005 Re: Sun exposure and mortality from melanoma. J Natl Cancer Inst 97 1789 1790; author reply 1791
3. ChinL
GarrawayLA
FisherDE
2006 Malignant melanoma: genetics and therapeutics in the genomic era. Genes Dev 20 2149 2182
4. Lopez-BergamiP
FitchmanB
RonaiZ
2008 Understanding signaling cascades in melanoma. Photochem Photobiol 84 289 306
5. Van RaamsdonkCD
BezrookoveV
GreenG
BauerJ
GauglerL
2009 Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 457 599 602
6. LevyC
KhaledM
FisherDE
2006 MITF: master regulator of melanocyte development and melanoma oncogene. Trends Mol Med 12 406 414
7. BergerAJ
DavisDW
TellezC
PrietoVG
GershenwaldJE
2005 Automated quantitative analysis of activator protein-2alpha subcellular expression in melanoma tissue microarrays correlates with survival prediction. Cancer Res 65 11185 11192
8. Lopez-BergamiP
HuangC
GoydosJS
YipD
Bar-EliM
2007 Rewired ERK-JNK signaling pathways in melanoma. Cancer Cell 11 447 460
9. Lopez-BergamiP
LauE
RonaiZ
2010 Emerging roles of ATF2 and the dynamic AP1 network in cancer. Nat Rev Cancer 10 65 76
10. MelnikovaVO
Bar-EliM
2008 Transcriptional control of the melanoma malignant phenotype. Cancer Biol Ther 7 997 1003
11. MitraD
FisherDE
2009 Transcriptional regulation in melanoma. Hematol Oncol Clin North Am 23 447 465, viii
12. Lopez-BergamiP
KimH
DewingA
GoydosJ
AaronsonS
2010 c-Jun regulates phosphoinositide-dependent kinase 1 transcription: implication for Akt and protein kinase C activities and melanoma tumorigenesis. J Biol Chem 285 903 913
13. RaingeaudJ
WhitmarshAJ
BarrettT
DerijardB
DavisRJ
1996 MKK3 - and MKK6-regulated gene expression is mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Mol Cell Biol 16 1247 1255
14. GuptaS
CampbellD
DerijardB
DavisRJ
1995 Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science 267 389 393
15. van DamH
WilhelmD
HerrI
SteffenA
HerrlichP
1995 ATF-2 is preferentially activated by stress-activated protein kinases to mediate c-jun induction in response to genotoxic agents. EMBO J 14 1798 1811
16. NakamuraT
OkuyamaS
OkamotoS
NakajimaT
SekiyaS
1995 Down-regulation of the cyclin A promoter in differentiating human embryonal carcinoma cells is mediated by depletion of ATF-1 and ATF-2 in the complex at the ATF/CRE site. Exp Cell Res 216 422 430
17. BenbrookDM
JonesNC
1990 Heterodimer formation between CREB and JUN proteins. Oncogene 5 295 302
18. van DamH
CastellazziM
2001 Distinct roles of Jun : Fos and Jun : ATF dimers in oncogenesis. Oncogene 20 2453 2464
19. BhoumikA
TakahashiS
BreitweiserW
ShilohY
JonesN
2005 ATM-dependent phosphorylation of ATF2 is required for the DNA damage response. Mol Cell 18 577 587
20. LiS
EzhevskyS
DewingA
CatoMH
ScortegagnaM
2010 Radiation Sensitivity and Tumor Susceptibility in ATM phospho-mutant ATF2 mice. Genes & Cancer in press
21. BergerAJ
KlugerHM
LiN
KielhornE
HalabanR
2003 Subcellular localization of activating transcription factor 2 in melanoma specimens predicts patient survival. Cancer Res 63 8103 8107
22. BhoumikA
HuangTG
IvanovV
GangiL
QiaoRF
2002 An ATF2-derived peptide sensitizes melanomas to apoptosis and inhibits their growth and metastasis. J Clin Invest 110 643 650
23. BhoumikA
GangiL
RonaiZ
2004 Inhibition of melanoma growth and metastasis by ATF2-derived peptides. Cancer Res 64 8222 8230
24. BhoumikA
IvanovV
RonaiZ
2001 Activating transcription factor 2-derived peptides alter resistance of human tumor cell lines to ultraviolet irradiation and chemical treatment. Clin Cancer Res 7 331 342
25. BhoumikA
JonesN
RonaiZ
2004 Transcriptional switch by activating transcription factor 2-derived peptide sensitizes melanoma cells to apoptosis and inhibits their tumorigenicity. Proc Natl Acad Sci U S A 101 4222 4227
26. RonaiZ
YangYM
FuchsSY
AdlerV
SardanaM
1998 ATF2 confers radiation resistance to human melanoma cells. Oncogene 16 523 531
27. CheliY
OhannaM
BallottiR
BertolottoC
2010 Fifteen-year quest for microphthalmia-associated transcription factor target genes. Pigment Cell Melanoma Res 23 27 40
28. HoekKS
SchlegelNC
EichhoffOM
WidmerDS
PraetoriusC
2008 Novel MITF targets identified using a two-step DNA microarray strategy. Pigment Cell Melanoma Res 21 665 676
29. MascarenhasJB
LittlejohnEL
WolskyRJ
YoungKP
NelsonM
2010 PAX3 and SOX10 activate MET receptor expression in melanoma. Pigment Cell Melanoma Res
30. YangG
LiY
NishimuraEK
XinH
ZhouA
2008 Inhibition of PAX3 by TGF-beta modulates melanocyte viability. Mol Cell 32 554 563
31. JacqueminP
LannoyVJ
O'SullivanJ
ReadA
LemaigreFP
2001 The transcription factor onecut-2 controls the microphthalmia-associated transcription factor gene. Biochem Biophys Res Commun 285 1200 1205
32. YasumotoK
TakedaK
SaitoH
WatanabeK
TakahashiK
2002 Microphthalmia-associated transcription factor interacts with LEF-1, a mediator of Wnt signaling. EMBO J 21 2703 2714
33. SaitoH
YasumotoK
TakedaK
TakahashiK
FukuzakiA
2002 Melanocyte-specific microphthalmia-associated transcription factor isoform activates its own gene promoter through physical interaction with lymphoid-enhancing factor 1. J Biol Chem 277 28787 28794
34. GoodallJ
CarreiraS
DenatL
KobiD
DavidsonI
2008 Brn-2 represses microphthalmia-associated transcription factor expression and marks a distinct subpopulation of microphthalmia-associated transcription factor-negative melanoma cells. Cancer Res 68 7788 7794
35. ThomasAJ
EricksonCA
2009 FOXD3 regulates the lineage switch between neural crest-derived glial cells and pigment cells by repressing MITF through a non-canonical mechanism. Development 136 1849 1858
36. BreitwieserW
LyonsS
FlennikenAM
AshtonG
BruderG
2007 Feedback regulation of p38 activity via ATF2 is essential for survival of embryonic liver cells. Genes Dev 21 2069 2082
37. BosenbergM
MuthusamyV
CurleyDP
WangZ
HobbsC
2006 Characterization of melanocyte-specific inducible Cre recombinase transgenic mice. Genesis 44 262 267
38. SerranoM
LeeH
ChinL
Cordon-CardoC
BeachD
1996 Role of the INK4a locus in tumor suppression and cell mortality. Cell 85 27 37
39. AckermannJ
FrutschiM
KaloulisK
McKeeT
TrumppA
2005 Metastasizing melanoma formation caused by expression of activated N-RasQ61K on an INK4a-deficient background. Cancer Res 65 4005 4011
40. SviderskayaEV
HillSP
Evans-WhippTJ
ChinL
OrlowSJ
2002 p16(Ink4a) in melanocyte senescence and differentiation. J Natl Cancer Inst 94 446 454
41. KidoK
SumimotoH
AsadaS
OkadaSM
YaguchiT
2009 Simultaneous suppression of MITF and BRAF V600E enhanced inhibition of melanoma cell proliferation. Cancer Sci 100 1863 1869
42. VachtenheimJ
OndrusovaL
BorovanskyJ
2010 SWI/SNF chromatin remodeling complex is critical for the expression of microphthalmia-associated transcription factor in melanoma cells. Biochem Biophys Res Commun 392 454 459
43. GodingC
MeyskensFLJr
2006 Microphthalmic-associated transcription factor integrates melanocyte biology and melanoma progression. Clin Cancer Res 12 1069 1073
44. DealKK
CantrellVA
ChandlerRL
SaundersTL
MortlockDP
2006 Distant regulatory elements in a Sox10-beta GEO BAC transgene are required for expression of Sox10 in the enteric nervous system and other neural crest-derived tissues. Dev Dyn 235 1413 1432
45. PoserI
BosserhoffAK
2004 Transcription factors involved in development and progression of malignant melanoma. Histol Histopathol 19 173 188
46. SahaB
SinghSK
SarkarC
BeraR
RathaJ
2006 Activation of the Mitf promoter by lipid-stimulated activation of p38-stress signalling to CREB. Pigment Cell Res 19 595 605
47. WellbrockC
RanaS
PatersonH
PickersgillH
BrummelkampT
2008 Oncogenic BRAF regulates melanoma proliferation through the lineage specific factor MITF. PLoS ONE 3 e2734 doi:10.1371/journal.pone.0002734
48. VanceKW
GodingCR
2004 The transcription network regulating melanocyte development and melanoma. Pigment Cell Res 17 318 325
49. GarrawayLA
WidlundHR
RubinMA
GetzG
BergerAJ
2005 Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 436 117 122
50. PollockPM
HarperUL
HansenKS
YudtLM
StarkM
2003 High frequency of BRAF mutations in nevi. Nat Genet 33 19 20
51. LinJY
FisherDE
2007 Melanocyte biology and skin pigmentation. Nature 445 843 850
52. WellbrockC
MaraisR
2005 Elevated expression of MITF counteracts B-RAF-stimulated melanocyte and melanoma cell proliferation. J Cell Biol 170 703 708
53. NoonanFP
RecioJA
TakayamaH
DurayP
AnverMR
2001 Neonatal sunburn and melanoma in mice. Nature 413 271 272
54. FernandezLP
MilneRL
PitaG
FloristanU
SendagortaE
2009 Pigmentation-related genes and their implication in malignant melanoma susceptibility. Exp Dermatol 18 634 642
55. BhoumikA
FichtmanB
DerossiC
BreitwieserW
KlugerHM
2008 Suppressor role of activating transcription factor 2 (ATF2) in skin cancer. Proc Natl Acad Sci U S A 105 1674 1679
56. MaekawaT
ShinagawaT
SanoY
SakumaT
NomuraS
2007 Reduced levels of ATF-2 predispose mice to mammary tumors. Mol Cell Biol 27 1730 1744
57. Gray-SchopferVC
CheongSC
ChongH
ChowJ
MossT
2006 Cellular senescence in naevi and immortalisation in melanoma: a role for p16? Br J Cancer 95 496 505
58. HuberWE
PriceER
WidlundHR
DuJ
DavisIJ
2003 A tissue-restricted cAMP transcriptional response: SOX10 modulates alpha-melanocyte-stimulating hormone-triggered expression of microphthalmia-associated transcription factor in melanocytes. J Biol Chem 278 45224 45230
59. BondurandN
PingaultV
GoerichDE
LemortN
SockE
2000 Interaction among SOX10, PAX3 and MITF, three genes altered in Waardenburg syndrome. Hum Mol Genet 9 1907 1917
60. CartlidgeRA
ThomasGR
CagnolS
JongKA
MoltonSA
2008 Oncogenic BRAF(V600E) inhibits BIM expression to promote melanoma cell survival. Pigment Cell Melanoma Res 21 534 544
61. KrajewskaM
SmithLH
RongJ
HuangX
HyerML
2009 Image analysis algorithms for immunohistochemical assessment of cell death events and fibrosis in tissue sections. J Histochem Cytochem 57 649 663
62. RuifrokAC
JohnstonDA
2001 Quantification of histochemical staining by color deconvolution. Anal Quant Cytol Histol 23 291 299
63. FrankenNA
RodermondHM
StapJ
HavemanJ
van BreeC
2006 Clonogenic assay of cells in vitro. Nat Protoc 1 2315 2319
64. Gould RothbergBE
BergerAJ
MolinaroAM
SubtilA
KrauthammerMO
2009 Melanoma prognostic model using tissue microarrays and genetic algorithms. J Clin Oncol 27 5772 5780
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 12
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Functional Comparison of Innate Immune Signaling Pathways in Primates
- Expression of Linear and Novel Circular Forms of an -Associated Non-Coding RNA Correlates with Atherosclerosis Risk
- Genome-Wide Interrogation of Mammalian Stem Cell Fate Determinants by Nested Chromosome Deletions
- Histone H2A C-Terminus Regulates Chromatin Dynamics, Remodeling, and Histone H1 Binding
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy