
Kazuistika: Gaucherova choroba v biopsii kostní dřeně 16 leté dívky vyšetřované pro podezření na myelodysplastický syndrom
Case report: Gaucher disease in trepanobiopsy of 16yo woman examined for suspected myelodysplastic syndrome
Gaucher disease is an autosomal recessive disease belonging to the so-called storage diseases. More than 300 mutations of the GBA1 gene encoding the β-glucocerebrosidase enzyme are known. It is a very rare disease in the Czech Republic. Currently 35 patients are treated. In our case report, we present the case of a 16 year old female patient attending the Clinic of Pediatric Medicine at the University Hospital in Ostrava. Since 2007, the patient has suffered prolonged thrombocytopenia, at the time with progression, and splenomegaly, which has not been further investigated. Trepanobiopsy was sent to the Department of Pathology with suspicion of myelodysplastic syndrome in May of 2018. In the biopsy examination, the individual bloodline did not show dysplastic features and the number of blasts was not increased. The marrow interstitium was 70% permeated with gaucher cells with intraplasmatic fibrous material. Cells were in the appearance of „crumpled paper“ and expressed CD68 in immunohistochemical stain and in histochemical examination of PAS and iron (Fe) staining. Based on a morphological finding, Gaucher‘s disease was suspected. Repeated bone marrow aspirates were subsequently captured by gaucher cells, and a next biochemical examination showed a β-glucocerebrosidase enzyme decrease of activity. Gaucher disease is a progressive disease that requires early diagnosis with the onset of therapy.
Keywords:
Gaucher disease – gaucher cells – storage diseases – β-glucocerebrosidase
Autoři:
Vladimír Židlík 1,4; Tomáš Kuhn 2; Pavel Hurník 1,3,4; Mária Wozniaková 1,4; Barbora Mičulková 1,3,4; Dušan Žiak 1,3; Marie Sporková 1; Patricie Delongová 1,4; Jaroslav Horáček 1; Jiří Ehrmann 1
Působiště autorů:
Ústav patologie, Fakultní nemocnice Ostrava, Ostrava
1; Klinika dětského lékařství, Fakultní nemocnice Ostrava, Ostrava
2; CGB laboratoř a. s., Ostrava
3; Lékařská fakulta, Ostravská univerzita, Ostrava
4
Vyšlo v časopise:
Čes.-slov. Patol., 57, 2021, No. 2, p. 105-108
Kategorie:
Původní práce
Souhrn
Gaucherova choroba je autozomálně recesivní onemocnění patřící do skupiny tzv. střádavých nemocí. Je známo více než 300 mutací genu GBA1 kódujícího enzym β-glukocerebrozidázu. V České republice jde o velmi vzácné onemocnění a v současné době je u nás léčených 35 pacientů. V naší kazuistice prezentujeme případ šestnáctileté pacientky sledované na Klinice dětského lékařství ve Fakultní nemocnici v Ostravě. Pacientka trpěla od roku 2007 dlouhotrvající trombocytopénií, toho času s progresí, a splenomegálií, která nebyla blíže došetřena. Na ústav patologie byla v květnu roku 2018 zaslána trepanobiopsie s podezřením na myelodysplastický syndrom. Při bioptickém vyšetření nevykazovaly jednotlivé krevní řady žádné dysplastické rysy a počet blastů nebyl zvýšen. Intersticium dřeně bylo ze 70% prostoupené buňkami gaucherova typu s intraplazmatickým vláknitým materiálem. Buňky nabývaly vzhledu „zmačkaného papíru“, exprimovaly protilátku CD68 v imunohistochemickém vyšetření a v histochemickém vyšetření byly pozitivní v barvení PAS a železo (Fe). Na základě morfologického nálezu bylo vyšetřujícím patologem vysloveno podezření na Gaucherovu chorobu. V opakovaně provedeném aspirátu kostní dřeně byly následně zachyceny gaucherovy buňky a dodatečně provedené biochemické vyšetření prokázalo sníženou aktivitu enzymu β-glukocerebrozidázy. Gaucherova choroba je postupně progredující onemocnění, které vyžaduje včasnou diagnózu se zahájením terapie.
Klíčová slova:
Gaucherova choroba – gaucherovy buňky – střádavé nemoci – β-glukocerebrozidáza
Gaucherova choroba (GD) je vzácné dědičné metabolické onemocnění s multiorgánovým postižením ze skupiny lysozomálních střádavých nemocí (LSD) způsobené vrozeným defektem enzymu β-glukocerebrozidázy, který vede k hromadění jeho běžného substrátu glukocerebrozidu v lysozomech tkáňových makrofágů s postižením kostí a hematologickými a viscerálními komplikacemi.
Historicky jsou popisovány tři typy onemocnění. Typ 1 (non-neuronopatický typ) bez postižení nervového systému a typy 2 a 3 (neuronopatické typy) s postižením nervového systému. Anémie, trombocytopénie, hepatosplenomegálie, kostní abnormity (osteopénie, lytické léze, patologické fraktury, chronická kostní bolest, kostní infarkty, osteonekrózy) jsou typickými manifestacemi nejčastější formy onemocnění typu 1. Stanovení aktivity enzymu β-glukocerebrozidázy v leukocytech nebo fibroblastech je zlatým standardem pro potvrzení diagnózy GD (1).
Koexistence a závažnost různých symptomů je zcela variabilní a kvůli jejich nespecifičnosti zůstává GD poddiagnostikovaná i několik let. Není výjimkou, že se na nemoc přijde i po 12 letech sledování pacienta pro nejednoznačné příznaky. Přitom jde o progredující onemocnění, u kterého je včasná diagnóza se zahájením terapie žádoucí pro zamezení nevratných orgánových změn. V souvislosti s GD je u pacientů popsán častější výskyt Parkinsonovy choroby a dále některé hematologické malignity jako mnohočetný myelom a monoklonální gamapatie nejasného významu (MGUS) (2).
V terapeutickém přístupu jsou metodou volby enzymatická substituční léčba (enzyme replacement therapy, ERT) a substrát redukující léčba (substrate reduction therapy, SRT) (3).
POPIS PŘÍPADU
Šestnáctiletá pacientka byla plánovaně přijata na hematoonkologické oddělení pro dlouhotrvající trombocytopénii s progresí a splenomegálii (od roku 2007), které nebyly diagnosticky došetřeny. Zároveň byl zaznamenán pokles hmotnosti o 9 kg za poslední 3 měsíce. Dívka přestala menstruovat, v poslední době byla více unavená a vybíravá v jídle.
Závěr magnetické rezonance břicha potvrdil výraznou splenomegálii bez obrazu portální hypertenze, bez trombózy v. portae a jaterních žil. Játra byly v normě. Taktéž bez patologických ložiskových změn a bez patologických lymfatických uzlin. Sternální punkce kostní dřeně (9. 5. 2018) potvrdila hypoplastickou kostní dřeň s progredující anémií a trombocytopénií. Hematopoeza byla trilineární s normálním zastoupením v elementech erytropoezy a granulopoezy. Bez morfologických odchylek a bez známek dysplastických změn. Počet blastů nebyl zvýšen. V opakované punkci o dva dny později (11. 5. 2018) popsal vyšetřující hematolog ještě pokročilejší hypocelularitu s nepřítomností megakaryocytární linie a s rysy dysplázie. Na základě těchto zjištění klinik indikoval provedení trepanobiopsie s podezřením na myelodysplastický syndrom.
V biopsii kostní dřeně (25. 5. 2018) bylo intersticium ze dvou třetin (70 %) prostoupené buňkami s bohatší eosinofilní plazmou vyplněnou vláknitým materiálem – Gaucherovy buňky (obr. 1). Buňky nabývaly vzhledu tzv. „zmačkaného papíru“ (obr. 2). Intraplazmatický vláknitý materiál byl dodatečně prokázán také při zobrazení v elektronovém mikroskopu (obr. 3). Patologické histiocytární buňky byly pozitivní v imunohistochemickém barvení CD68 (DAKO, klon KP1) (obr. 4), současně negativní v CD1a (DAKO, klon 010) a S100 (DAKO, kód Z0311). Dále v histochemickém vyšetření vykazovaly pozitivitu v barvení PAS (obr. 5) a železo (obr. 6).





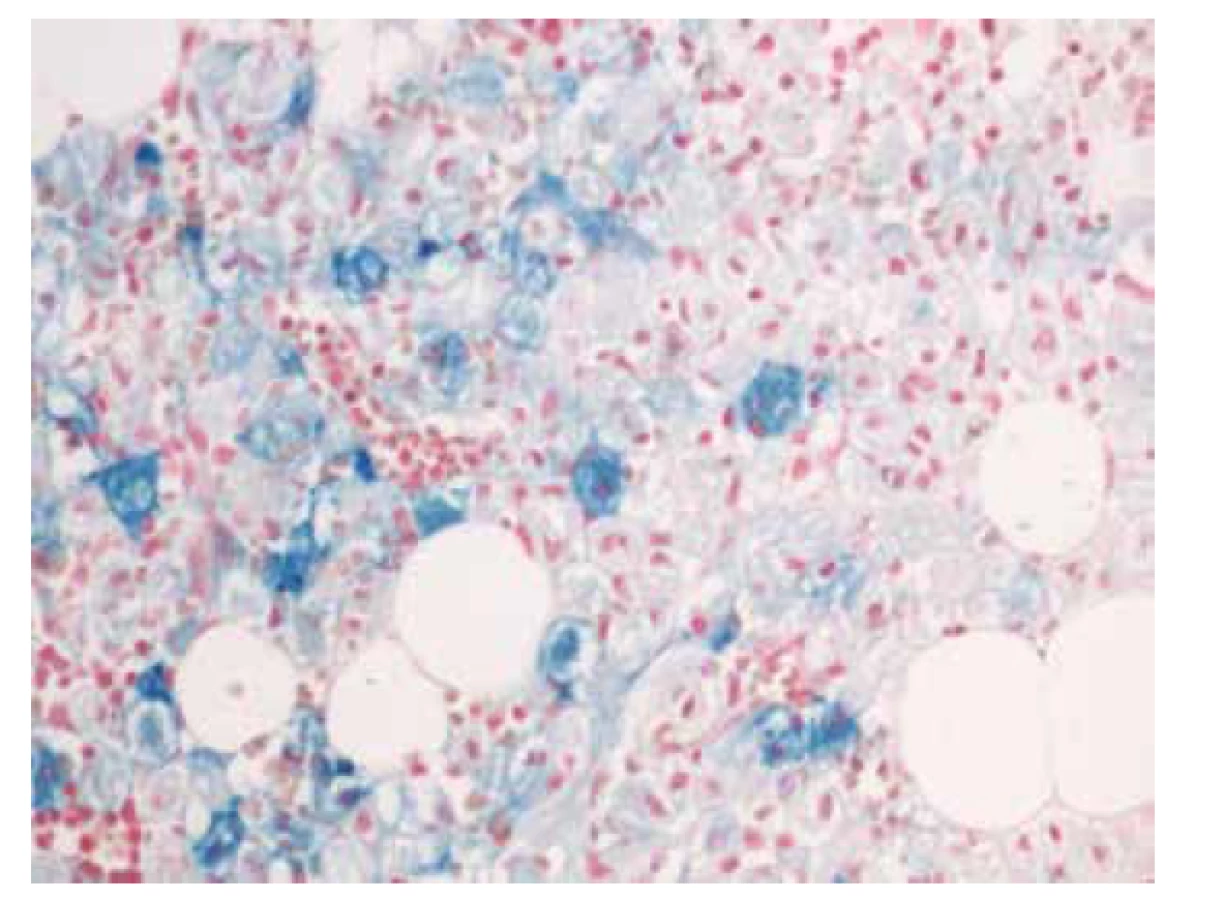
Okolní hematopoeza byla početně redukovaná, morfologicky dislokovaná na periferii shluků histiocytárních buněk. Nebyly zastiženy známky dysplastických změn. Erytropoeza byla tvořená hnízdy normoblastových elementů. Granulopoeza vyzrávala. Megakaryocyty byly početně snížené, morfologicky však v normě.
Na základě popsaného morfologického nálezu bylo vyšetřujícím patologem vysloveno podezření na Gaucherovu chorobu.
V opakovaně provedené punkci kostní dřeně (28. 5. 2018) byly taktéž zastiženy gaucherovy buňky (obr. 7). Enzymologické vyšetření prokázalo sníženou aktivitu enzymu β-glukocerebrozidázy a zvýšené hladiny chitotriosidázy, které diagnózu Gaucherovy choroby potvrdily.

Po potvrzení diagnózy byla pacientka přeložena do lékařské péče na Kliniku dětí a dorostu ve VFN v Praze k MUDr. Malinové, kde je v současné době léčena.
DISKUZE
GD je nejčastější sfingolipidózou, která byla popsána Dr. Phillipem Gaucherem v roce 1882 u pacientky se zvětšenou slezinou bez leukémie. Slezina byla vyplněná neobvyklými většími buňkami, které mylně považoval za maligní. Když byl hlášen podobný případ v roce 1885, jednotka byla nazvána Gaucherovou chorobou. První pacient s GD vykazující neurologické příznaky byl popsán v roce 1920. V roce 1934 bylo ukázáno, že retikuloendotelové orgány jsou infiltrovány buňkami gaucherova typu vyplněnými látkou označenou jako glucerebrozid. Příčina jeho hromadění byla objasněna v roce 1960 objevením chybějícího enzymu glukocerebrozidázy (4,5).
GD se vyskytuje panetnicky s prevalencí udávanou v rozpětí 1 : 57000-1 : 100000 živě narozených dětí. V České republice je prevalence onemocnění 1 : 88000. Vyšší výskyt byl popsán v uzavřených etnických populacích. V židovské populaci Ashkenazi byla zjištěna prevalence 1 : 1000 a u heterozygotů (přenašeči) 1 : 14 (3).
Jedná se o dědičné autozomálně recesivní onemocnění. Gen GBA1 pro enzym glukocerebrozidázu (také označovanou jako glukosylceramidázu nebo kyselou β-glukozidázu) je lokalizován na chromozomu 1 (1q21) a je známo více než 300 mutací tohoto genu. Velmi vzácně je nemoc způsobena defektem aktivátoru glukocerebrozidázy, saponinu C. Nejčastěji mutovanými alelami zodpovědnými v 98% za výskyt GD jsou: c.1226A>G (N370S), c.1448T>C (L444P), c.84dupG (84GG) a c.27+1G>A (IVS2+1). Dvě z těchto mutovaných alel mají určitou prediktivní hodnotu, kdy homozygotní alela N370S je spojována s mírnějším průběhem bez postižení nervového systému, a naopak alela L444P vytváří závažnější průběh s postižením CNS (6,7).
Mutace v genu GBA1 vede ke snížení aktivity glukocerebrozidázy. Její reziduální aktivita pak souvisí se závažností následných symptomů, které jsou důsledkem hromadění meziproduktu metabolismu glykosfingolipidů, glukocerebrozidu. Tento produkt se hromadí v makrofázích a indukuje jejich přeměnu v Gaucherovy buňky, které jsou ve světelném mikroskopu typicky zvětšené, s excentricky uloženým jádrem a cytoplazmou vyplněnou vláknitým materiálem, který jim dodává typický vzhled označovaný jako „zmačkaný papír“. Vláknitá depozita glukocerebrozidu jsou prokazatelná v elektronovém mikroskopu. Buněčná populace monocytů/makrofágů je zřejmě primárně postižena pro jejich roli v eliminaci erytroidních a leukocytárních elementů obsahujících velké množství glykosfingolipidů, které jsou zdrojem glukocerebrozidu (5,8).
V souvislosti s nahromaděním glukocerebrozidu se postupně vyvíjejí klinické příznaky zahrnující splenomegálii, anémii s chronickou únavou, tvorbu modřin a krvácení kvůli trombocytopénii. V pozdějších stádiích onemocnění pak výskyt cytopénie vznikající nahrazením krvetvorné dřeně gaucherovými buňkami, hepatomegálie s poruchou jaterních funkcí, porucha kostního metabolismu, osteonekrózy, zhoršená funkce neutrofilů a sklon k infekcím (2).
Obecně jsou rozlišovány tři typy onemocnění. Typ I (adulní typ, non-neuropatický typ) je charakterizován různou závažností symptomů, včetně asymptomatických pacientů a různou mírou progrese. Nejčastěji je tento typ provázen různě vyvinutou splenomegálií a hepatomegálií. V játrech se objevuje fibróza, k cirhóze, portální hypertenzi a selhání jater však většinou nedochází. Dalšími častými příznaky jsou anémie a trombocytopénie, skeletální postižení, osteolytické léze a patologické fraktury. Typ 2 (akutní neuronopatický typ) je nejzávažnější. Jde o typ s rychle progredující neurologickou symptomatologií. Vyskytuje se u nově narozených dětí, které umírají před druhým rokem věku. Typ 3 (subakutní, chronický neuronopatický typ) je nejméně častý, s kombinací neurologických příznaků projevujících se progredující demencí a viscerální symptomatologií (9,10).
V terapii se uplatňují dva přístupy. Jednak jde o enzymatickou substituční léčbu, která je zásadní zvláště u typu I. Principem je podávání chybějícího enzymu pomocí nitrožilních infuzí. Doporučená dávka je asi 30j/kg/2 týdny. V rozsahu 6-12 měsíců od zahájení terapie je možné pozorovat ústup subjektivních příznaků. Druhým typem terapie je pak substrát redukující léčba založená na snížení syntézy substrátu. Principem je snížení množství glukocerebrozidu v organismu, jehož zbytky jsou pak zpracovávány i při nízkých hladinách glukocerebrozidázy (3).
Navzdory dostupné terapii přibližně čtvrtina pacientů s typem I uniká kvůli pozdnímu stanovení diagnózy. Příčinou mohou být převážně nespecifické příznaky těchto pacientů a také to, že pouze malá část hematoonkologů uvažuje o GD v rámci dif. dg. i při klasických symptomech, kterými jsou splenomegálie, trombocytopénie a anémie. Mnohem častěji jsou tito pacienti vyšetřováni pro leukémii, lymfom a mnohočetný myelom (2).
ZÁVĚR
Gaucherova choroba je progredující onemocnění, které bývá často dlouhá léta poddiagnostikované. Příčinou mohou být nespecifické příznaky pacientů, pro které část z nich diagnóze a tedy i terapii uniká. Navzdory mnohdy „nevýznamné“ symptomatologii postihuje onemocnění celou řadu orgánů s jejich postupným poškozováním. Pro úspěšnou terapii pacientů je tedy nutná včasná diagnostika. Z tohoto důvodu by měla být GD při nejasné splenomegálii a trombocytopénii brána do úvahy v rámci diferenciální diagnózy.
Stav naší pacientky se po šesti měsících od zavedení terapie stabilizoval, subjektivní příznaky vymizely.
PROHLÁŠENÍ
Autor práce prohlašuje, že v souvislosti s tématem, vznikem a publikací tohoto článku není ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou. Toto prohlášení se týká i všech spoluautorů.
∗ Adresa pro korespondenci:
MUDr. Vladimír Židlík, MIAC
Fakultní nemocnice Ostrava
Ústav patologie
17. listopadu 1790/5, Ostrava 708 52
Tel.: +420608820804
email: vldzidlik@gmail.com
Zdroje
1. Linari S, Castaman G. Clinical manifestations and management of Gaucher disease. Clin Cases Miner Bone Metab 2015; 12(2): 157-164.
2. Cappellini MD. A rare condition in haematological practise – Gaucher disease. European Oncology & Haematology 2015; 11(1): 15–20.
3. Malinová V, Mazurová S, Dvořáková L. Gaucherova choroba a lysozomální onemocnění – současné možnosti diagnostiky a léčby. Remedia, ročník 26, číslo 3/2016.
4. Rosenbloom EB, Weinreb JN. Gaucher Disease: a comprehensive review. Crit Rev Oncog 2013; 18(3): 163-175.
5. Stirnemann J, Belmatoug N, Camou F, et al. A review of Gaucher disease pathophysiology, clinical presentation and treatments. Int J Mol Sci 2017; 18(2): 441.
6. Baris NH, Cohen JI, Mistry KP. Gaucher Disease: the metabolic defect, pathophysiology, Phenotypes and natural history. Pediatr Endocrinol Rev 2014; 12(01): 72-81.
7. Huang WJ, Zhang X, Chen WW. Gaucher disease: a lysosomal neurodegenerative disorder. Eur Rev Med Pharmacol Sci 2015; 19(7): 1219-1226.
8. Hruska SK, LaMarca EM, Scott RC, Sindransky E. Gaucher Disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum Mutat 2008; 29(5): 567-583.
9. Bohra V, Nair V. Gaucher’s disease. Indian J Endocrinol Metab 2011; 15(3): 182-186.
10. Jmoudiak M, Futerman AH. Gaucher disease: pathological mechanisms and modern management. Br J Haematol 2005; 129(2): 178-188.
Štítky
Patologie Soudní lékařství ToxikologieČlánek vyšel v časopise
Česko-slovenská patologie

2021 Číslo 2
Nejčtenější v tomto čísle
- Význam imunohistochemických metod v diagnostice karcinomu endometria
- Gynekologické léze u hereditárních nádorových syndromů
- Kazuistika: Postižení ledvin u pacientky s Crohnovou chorobou
- Význam imunohistochemických metod v diagnostice mezenchymálních nádorů dělohy
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy