
Gynekologické léze u hereditárních nádorových syndromů
Gynecological lesions in hereditary cancer predisposition syndromes
Hereditary tumor syndromes with a possible manifestation in the female internal genital tract represent a heterogeneous group of diseases. The two most common entities are the hereditary breast and ovarian cancer syndrome, and the Lynch syndrome. The less common syndromes include the rhabdoid tumor predisposition syndrome, Cowden syndrome, tuberous sclerosis complex, DICER1 syndrome, nevoid basal cell carcinoma syndrome, Peutz-Jeghers syndrome, von Hippel-Lindau disease, and hereditary leiomyomatosis and renal cell cancer syndrome. The goal of this manuscript is to provide a comprehensive overview of those hereditary tumor syndromes which can manifest in the area of the female genital system, with an emphasis on their summary, the characteristics of the tumors which can develop in association with these syndromes, and the approach to the processing of prophylactically removed tissues and organs. The issue of Lynch syndrome screening is also discussed.
Keywords:
mesenchymal uterine tumors – smooth muscle tumors – endometrial stromal tumors – undifferentiated uterine sarcoma – immunohistochemistry – molecular classification – hereditary cancer predisposition syndromes – female genital tract – Lynch syndrome – hereditary breast and ovarian cancer syndrome – Peutz-Jeghers syndrome
Autoři:
Pavel Dundr 1; David Cibula 2; Lenka Foretová 3; Milan Macek jr. 4; Kateřina Kopečková 5; Luboš Petruželka 6; Kristýna Němejcová 1
![]() ; Michaela Bártů 1; Jan Hojný 1; Nikola Hájková 1; Radek Jakša 1; Pavol Janega 7,8; Ivana Stružinská 1
; Michaela Bártů 1; Jan Hojný 1; Nikola Hájková 1; Radek Jakša 1; Pavol Janega 7,8; Ivana Stružinská 1
Působiště autorů:
Ústav patologie 1. LF UK a VFN v Praze
1; Onkogynekologické centrum, Gynekologicko-porodnická klinika 1. LF UK a VFN v Praze
2; Oddělení epidemiologie a genetiky nádorů, Masarykův onkologický ústav, Brno
3; Ústav biologie a lékařské genetiky 2. LF UK a FN Motol
4; Onkologická klinika 2. LF UK a FN Motol
5; Onkologická klinika 1. LF UK a VFN v Praze
6; Ústav patologickejanatómie, Lekárská fakulta, Univerzita Komenského v Bratislave
7; Medirex Group Academy, Trnava, n. o.
8
Vyšlo v časopise:
Čes.-slov. Patol., 57, 2021, No. 2, p. 96-104
Kategorie:
Přehledový článek
Souhrn
Hereditární nádorové syndromy s možnou manifestací v oblasti ženského vnitřního genitálu představují heterogenní skupinu onemocnění. Mezi dva nejčastější syndromy patří syndrom hereditárního karcinomu prsu a ovarií a Lynchův syndrom. Méně časté syndromy zahrnují syndrom predispozice k maligním rabdoidním nádorům, Cowdenův syndrom, komplex tuberózní sklerózy, DICER1 syndrom, syndrom névoidního bazocelulárního karcinomu, Peutz-Jeghersův syndrom, von Hippelova-Lindauova choroba a syndrom hereditární leiomyomatózy a renálního karcinomu. Cílem následujícího sdělení je podat přehled problematiky hereditárních nádorových syndromů s manifestací v oblasti ženského genitálu se zaměřením na jejich přehled, charakteristiky nádorů, které se v souvislosti s jednotlivými syndromy vyskytují, postup při vyšetřování profylakticky odstraněných tkání a orgánů a problematiku screeningu Lynchova syndromu.
Klíčová slova:
hereditární nádorové syndromy – ženský genitál – Lynchův syndrom – syndrom hereditárního karcinomu prsu a ovárií – Peutz-Jeghersův syndrom
Hereditární nádorové syndromy jsou odpovědné za vznik 5-10 % všech maligních nádorů (1-3). V onkogynekologii je nejvýznamnější syndrom hereditárního karcinomu prsu a ovarií (HBOC – „hereditary breast and ovarian cancer“) a Lynchův syndrom (LS) (4-6). Souvislost high grade serózního karcinomu s HBOC a některých typů karcinomu endometria a ovaria s LS je dobře známá a má přímý dopad na stávající diagnostické a léčebné postupy. Většina dalších nádorových syndromů má však v populaci velmi nízkou prevalenci a vlastní nádorová onemocnění související s těmito syndromy jsou tedy celkově vzácná, což může mít přímý dopad na jejich diagnostiku (7). Problémem je také fakt, že nádory obvykle nemají specifickou morfologii. I když některé další klinické rysy (např. věk pacientky v době diagnózy, výskyt více suspektních či diagnostických znaků syndromu, rodinná anamnéza) mohou být sugestivní ze souvislosti s příslušným hereditárním syndromem, všechny nádory se mohou vyskytovat i sporadicky. Pro některé nádory však platí, že pravděpodobnost spojitosti s některým hereditárním syndromem je poměrně vysoká. V následujícím textu je probrán přehled hlavních hereditárních syndromů a s nimi souvisejících nádorů s manifestací v oblasti ženského genitálu (tabulka 1). Diskutována je také problematika případného screeningu a doporučení pro způsob vyšetření profylakticky odstraněných orgánů. Podrobnosti týkající se diferenciální diagnostiky jednotlivých nádorů na úrovni bioptické diagnostiky jsou nad rámec tohoto textu a jsou detailně diskutovány jinde, včetně dostupných monografií.
SYNDROM HEREDITÁRNÍHO KARCINOMU PRSU A OVARIA (HBOC)
Syndrom hereditárního karcinomu prsu a ovaria (HBOC; OMIM PS604370 x OMIM #604370, #612555) je charakterizován přítomností hereditární mutace genu BRCA1 nebo BRCA2. Incidence tohoto syndromu je v naší a západoevropské populaci odhadována asi 1 : 800, nicméně např. u středoevropských židů (Aškenáziů) je výskyt podstatně vyšší, na úrovni 1-2 % (8-10). Celoživotní riziko vzniku karcinomu ovaria je u pacientek s BRCA1 mutací odhadováno na 39-75 % a s BRCA2 mutací na 11-34 % (11-13). Ve srovnání s celoživotním rizikem pro obecnou populaci bez hereditární zátěže, které je asi 1,3-1,6 %, se tedy jedná o zásadní rizikový faktor. Nádory vznikající v souvislosti s BRCA1/BRCA2 mutací jsou zejména high grade serózní karcinomy (HGSC) (14). Pro ostatní histologické typy nejsou data jednoznačná, germinální mutace BRCA1/2 se však vzácně mohou vyskytovat u endometroidních i světlobuněčných karcinomů - ze skupiny všech nádorů ovaria diagnostikovaných u pacientek s germinální mutací genu BRCA1/2 však podle dostupných údajů tyto nádory představují < 6 % (14). Kromě karcinomu prsu a ovarií je tento syndrom spojen i se zvýšeným rizikem vzniku karcinomu prostaty, pankreatu, žlučových cest, kolorektálního karcinomu a melanomu (15,16).
V minulosti byla snaha o nalezení morfologických znaků nádorů, které by upozornili na možnost souvislosti s HBOC syndromem (17-19). Cílem byla selekce pacientek, kterým by na podkladě morfologických rysů nádoru bylo doporučeno genetické poradenství. V současné době však vzhledem k nastaveným algoritmům genetického testování, kdy by každá žena s diagnózou karcinomu ovaria měla být referována k vyšetření klinickým genetikem spojeným s testováním germinálních mutací, snaha o definování morfologických znaků nádorů spojených s HBOC syndromem postrádá s ohledem na selekci pacientek pro genetické testování smysl (20). Přesto však tyto práce měly význam pro zlepšení diagnostiky ovariálních nádorů – ukázalo se totiž, že část nádorů dříve diagnostikovaných jako nediferencované karcinomy, endometroidní karcinomy či tranzicionální karcinomy, jsou ve skutečnosti HGSC (obr. 1). Tyto poznatky definovaly HGSC s tzv. SET rysy (Serózní – pseudoEndometroidní – Tranzicionální) a byly jedním z důvodů zrušení tranzicionálního karcinomu ovaria jako definované jednotky (17). Právě tyto HGSC s tzv. SET rysy jsou častější u pacientek se syndromem HBOC, nejedná se však o morfologii pro uvedený syndrom specifickou. Kombinace více markerů zahrnujících SET rysy, nekrózu (> 25 % objemu nádoru), vysoký mitotický index (> 51 mitóz / 10 HPF) a početné tumor infiltrující lymfocyty (TIL) (> 29 / 1 HPF) se ukázala s ohledem na predikci výskytu germinálních mutací jako přesnější než pouhá SET morfologie, nadále se však nejednalo o spolehlivý přístup a popsané změny se mohou vyskytnout nejen u pacientek se syndromem HBOC, ale i v případě somatických mutací genu BRCA1/2, metylace promotoru BRCA1, aberací jiných genů hrajících roli v procesu homologní rekombinace i u sporadických nádorů (17). Jako spolehlivější znak se ukázal být charakter metastáz – případy s BRCA1/2 abnormalitami vykazují často v metastázách čistě papilární úpravu, či jde o metastázy s expanzivním typem růstu, naopak sporadické případy mají kombinaci různých architektonických vzorů a infiltrativní typ růstu (18).

Zpracování vzorků u profylaktické salpingo-ooforektomie
Pacientky s germinální BRCA1/BRCA2 mutací mohou v rámci profylaktického zákroku podstoupit bilaterální salpingo-ooforektomii (21). Důležité je důkladné vyšetření takovýchto resekátů, které se provádí podle protokolu SEE-FIM („sectioning and extensively examining the fimbriated end“) (22). Cílem vyšetření je detekce serózních tubárních intraepiteliálních karcinomů (STIC) či mikroskopických HGSC a tuby by tedy podle tohoto protokolu měly být zpracovány celé ve 2-3 mm lamelách (obr. 2). S ohledem na zpracování ovaria není jednoznačný konsenzus, doporučováno je ale také jejich zpracování v celém rozsahu.

Vyšetřování somatických mutací BRCA1/2
Kromě germinálních mutací genů BRCA1/2, které jsou podle studie The Cancer Genome Atlas přítomny asi u 17 % HGSC (9 % HGSC mělo v této studii mutaci BRCA1 a 8 % BRCA2) se asi u 6 % HGSC vyskytují somatické mutace genů BRCA1/2 (3 % BRCA1 a 3 % BRCA2) (23). Přítomnost germinální či somatické mutace genu BRCA1/2 je v současné době u pacientek splňujících další indikační kritéria podmínkou pro možnost léčby PARP inhibitory. Vyšetřování somatických mutací genů BRCA1/2je tedy (spolu s testováním germinálních variant prováděných v genetických laboratořích) prediktivním vyšetřením. Testování se v ČR provádí od září 2018 ve vybraných laboratořích ze sítě tzv. „referenčních laboratoří“ na pracovištích patologie na podkladě žádosti ze strany onkologa. Algoritmus testování mutací genů BRCA1/2 byl v obecné úrovni v ČR nastaven tak, že nejprve mělo proběhnout testování germinálních mutací na pracovišti lékařské genetiky (to by v České republice mělo být podle stávajících doporučených postupů provedeno u všech pacientek s diagnostikovaným karcinomem ovaria bez ohledu na jeho histologický typ) a pouze u pacientek s absencí germinální mutace splňujících indikační kritéria léčby PARP inhibitory bylo klinikem požadován testování somatických variant z nádorové tkáně. S ohledem na aktuální situaci je v souvislosti s úhradou PARP inhibitorů v první linii léčby od března roku 2020 schválena možnost souběžného testování germinálních mutací z periferní krve v genetických laboratořích a somatických mutací z nádorové tkáně ve vybraných „referenčních laboratořích“ na pracovištích patologie. Aktuálně se také diskutuje problematika komplexnějšího prediktivního testování pro indikaci léčby PARP inhibitory zahrnujícího buď testování celého panelu genů účastnících se v procesu homologní rekombinace (HRD), nebo vyhodnocení markerů genomické instability sekundárních při HRD, jako je ztráta heterozygosity (LOH), telomerická alelická imbalance (TAI) či tzv. „large scale transitions“ (LST) (24). Tato problematika je však nad rámec stávajícího sdělení.
LYNCHŮV SYNDROM
Lynchův syndrom (LS; OMIM PS120435) je autozomálně dominantní geneticky heterogenní onemocnění s incidencí odhadovanou na 1 : 370 – 1 : 2000 (25). Nejčastěji je LS spojen s mutacemi genů MSH2,MLH1 a MSH6. Ostatní mutace, zahrnující mutace PMS2, MSH6, MLH3 a epigenetickou inaktivaci EPCAM, jsou poměrně vzácné (6,26). Pacienti s LS mají vysoké celoživotní riziko vzniku kolorektálního karcinomu, které se u mužů udává až 75%, u žen asi 30% (průměrný věk vzniku je 41-43 let) – riziko je však odlišné pro mutaci jednotlivých genů (souhrnné počty zastoupení mutací u jednotlivých typů nádorů a celoživotní riziko související s mutací konkrétního genu jsou uvedeny v tabulce 1) (27). Kromě kolorektálního karcinomu je u pacientek s tímto syndromem zvýšené riziko vzniku karcinomu endometria a ovaria (tabulka 1) (6,26,28). Syndrom je spojen i s výskytem dalších maligních nádorů, jako je karcinom prostaty a žaludku (26). Morfologicky nejsou nádory spojené s LS nijak specifické. U karcinomu endometria jde nejčastěji o endometroidní karcinom častěji lokalizovaný v dolním děložním segmentu, častěji s přítomností četnějších tumor infiltrujících lymfocytů, nicméně nejedná se o znaky specifické pro souvislost nádoru s LS (29). Častý je také výskyt dediferencovaného karcinomu či smíšených nádorů. U karcinomu ovaria se většinou jedná o nádory serózní, endometroidní, světlobuněčné, či dle literárních údajů smíšené, nicméně opět bez specifických morfologických rysů (6,29,30). Jako problematické se však při hodnocení zastoupení histologických typů nádorů jeví absence centrálního čtení a proměnlivosti diagnostických kritérií souvisejících i s metodickými možnostmi v jednotlivých obdobích. Např. smíšený typ karcinomu (se složkou mucinózního, endometroidního a světlobuněčného karcinomu) je podle metaanalýzy z roku 2016 u nádorů ovaria souvisejících s LS nejčastější a odpovídá za 33 % případů (6). Vzhledem k vzácnému výskytu smíšených karcinomů je však udávané procento velmi nepravděpodobné.

OC – ovariální karcinom; EC – karcinom endometria; CRC – kolorektální karcinom; NA – data nejsou dostupná
U 50-60 % pacientek karcinom endometria nebo ovaria vzniká před vznikem kolorektálního karcinomu a gynekologická malignita tedy může být tzv. „sentinelovým“ nádorem, který při správné diagnostice a stanovení souvislosti s LS může předejít vzniku závažnější malignity. Problémem nicméně je, že klinická kritéria používaná pro indikaci genetického vyšetření mají u pacientek s gynekologickými malignitami nízkou senzitivitu, a to v případě Amsterdamských, revidovaných Amsterdamským i tzv. Bethesda kritérií (31). V tomto kontextu se tedy diskutuje screening LS u všech pacientek s karcinomy endometria a ovaria, nicméně i přes doporučení odborných společností a mezinárodních guideline (Society of gynecologic Oncologists; American College of Obstetricians and Gynecologist) se zatím algoritmy testování v jednotlivých zemích poměrně výrazně liší (32). Oproti kolorektálnímu karcinomu je u gynekologických malignit mírně odlišná frekvence výskytu jednotlivých mutací s poněkud častějším výskytem mutace zejména MSH6 (6, 26). To se odráží i v metodice screeningu, kdy je u gynekologických malignit preferováno imunohistochemické vyšetření, oproti kterému je fragmentační analýza metodou PCR na abnormality MSH6 méně senzitivní, na úrovni 50-75 % (33). Důvodem je fakt, že mutace genu MSH6 se méně často projevuje mikrosatelitovou instabilitou a při fragmentační analýze tak nádory mohou být mikrosatelitově stabilní (MSS) či vykazují MSI-L výsledek. Imunohistochemické vyšetření je nicméně většinou preferováno i u screeningu kolorektálního karcinomu. Důvody zahrnují cenu, dostupnost ale i snadnou interpretaci a nastavené algoritmy následné indikace genetického vyšetření.
Screening LS
Karcinom endometria
U pacientek s karcinomem endometria je doporučeno automatické screeningové testování, bez ohledu na histologický typ nádoru, jeho morfologické charakteristiky a věk pacientky (34). Podle National Comprehensive Cancer Network (NCCN) guideline a doporučení International Society of Gynecologic Pathologists (ISGyP) by se mělo testování provádět na finálním vzorku z hysterektomie, z kyretáže pouze v případě, že hysterektomie nebude provedena (35,36). Preferenčně je doporučováno testování pomocí imunohistochemického vyšetření, které jak již bylo zmíněno, lépe podchytí i abnormality MSH6 a PMS2 (35). Optimální je současné testování exprese všech 4 proteinů, některé algoritmy však kvůli úsporám v prvé fázi testují pouze 2 vedlejší proteiny (MSH6 a PMS2) s doplněním MSH2 a MLH1 pouze v případě potřeby. Podle některých prací jsou výsledky při využití 2 i 4 proteinů prakticky totožné, jiné studie nicméně poukazují na poněkud nižší senzitivitu danou tím, že jsou popisovány případy se ztrátou exprese hlavního proteinu a zachováním exprese tzv. vedlejšího proteinu (37,38). V rámci IHC vyšetření je důležité správné vyhodnocení exprese proteinů – přesný „návod“ na interpretaci však není k dispozici. Problémem jsou zejména případy, které vykazují pouze fokální a slabou expresi některého proteinu – ta je některými autory interpretována jako zachovalá (tedy normální) exprese, jiní však tyto případy označují jako „indeterminované“ a při následném vyšetření se ukazuje, že 30-50 % z nich je spojeno s germinální mutací příslušného genu (38). Důležitá je přítomnost pozitivní vnitřní kontroly na nenádorové tkáni – při negativitě nádorové tkáně i vnitřní kontroly je nutno nahlížet na výsledek jako nevalidní. Pacientky se ztrátou exprese MLH1 by měly být dále molekulárně testovány k vyloučení metylace promotoru tohoto genu, která je častou příčinou somatické inaktivace MLH1(39). V tomto případě bývá přítomna i mutace BRAF genu, která bývá užívána k vyloučení Lynchova syndromu. Při vyloučení metylace promotoru MLH1 by měly být následně všechny pacientky referovány na vyšetření lékařským genetikem.
Karcinom ovaria
Oproti karcinomu endometria má plošný screening u karcinomu ovaria velmi nízkou výtěžnost a doporučuje se určitá preselekce daná histologickým typem nádoru. HGSC mají velmi nízké zastoupení mikrosatelitově instabilních případů (asi 1 %) a i mikrosatelitově instabilní HGSC obvykle nejsou asociovány s LS. Naopak u endometroidních, světlobuněčných a nediferencovaných karcinomů je mikrosatelitově instabilních > 10 % případů (40). Navrhované algoritmy tedy kombinují histologický typ nádoru a někdy i věk pacientky, který však není přesně navržen – udává „mladý věk“ či např. < 50 let (36).
Vyšetřování profylaktických resekátů
Profylaktická hysterektomie případně i s bilaterální salpingo-ooforektomií je metodou, která má klinické opodstatnění podložené reálnými daty (41). V praxi se však s těmito vzorky setkáváme v současné době poměrně zřídka. Prací, které by se věnovali způsobu vyšetření těchto resekátů s ohledem na doporučený rozsah vyšetření je poměrně málo a data nejsou jednoznačná (29). Obecné doporučení, které však není inkorporováno do mezinárodních guidelines (ta se tím v současné době nijak nezabývají), je následující: děložní tuby a ovaria by se měly zpracovat celé dle protokolu SEE-FIM; dolní děložní segment vyšetřit in toto; děložní hrdlo – reprezentativní řezy z předního, zadního pysku a laterálních oblastí; endometrium – v případě absence makroskopicky patrných abnormalit in toto (29). Práce zaměřující se na histopatologické nálezy v profylaktických resekátech jsou u pacientek s LS velmi vzácné. Ve studii analyzující 25 profylaktických vzorků byl abnormální nález u 8 z nich (32 %) – v 6 případech se jednalo o hyperplazii a ve 2 případech o endometroidní karcinom (29).
COWDENŮV SYNDROM
Cowdenův syndrom (OMIM PS158350) je autozomálně dominantní onemocnění s incidencí asi 1 : 200 000. Ve většině případů (asi 80 %) se jedná o germinální mutaci tumor supresorového genu PTEN (42). Zbylá část případů je geneticky poměrně heterogenní, a kromě mutací některých genů (PIK3CA, AKT1, SEC23B) zahrnuje i vrozenou hypermetylaci genu KLLN (43). Onemocnění je charakterizované výskytem mnohotných hamartomatózních lézí a predispozicí ke vzniku některých nádorů, zahrnujících zejména nádory prsu (celoživotní riziko odhadované na 25-50 %), karcinom endometria (celoživotní riziko 5-28 %), dále karcinomy ledviny, tlustého střeva, štítné žlázy a melanom (42).
Výskyt karcinomu endometria je u pacientek s tímto syndromem poměrně časný a začíná ve věku kolem 25 let, jsou však popsány i vzácné případy u adolescentek (44). Morfologicky se jedná o nádory bez specifický rysů, obvykle dobře diferencované (grade 1) endometroidní karcinomy. Imunohistochemické vyšetření prokáže úplnou ztrátu exprese PTEN, nemá však s ohledem na diagnostiku Cowdenova syndromu žádný význam, jelikož inaktivace a následně ztráta exprese PTEN se vyskytuje asi u 75 % sporadických endometroidních karcinomů a to obvykle v souvislosti se somatickou mutací, delecí či metylací promotoru PTEN (45). Role patologa je u Cowdenova syndromu velmi limitovaná, může na teoretickou možnost tohoto syndromu upozornit zejména v případě výskytu endometroidního karcinomu u mladých žen, při zachovalé expresi MMR proteinů.
SYNDROM PREDISPOZICE K MALIGNÍM RABDOIDNÍM NÁDORŮM (Rhabdoid tumor predisposition syndrome; RTPS)
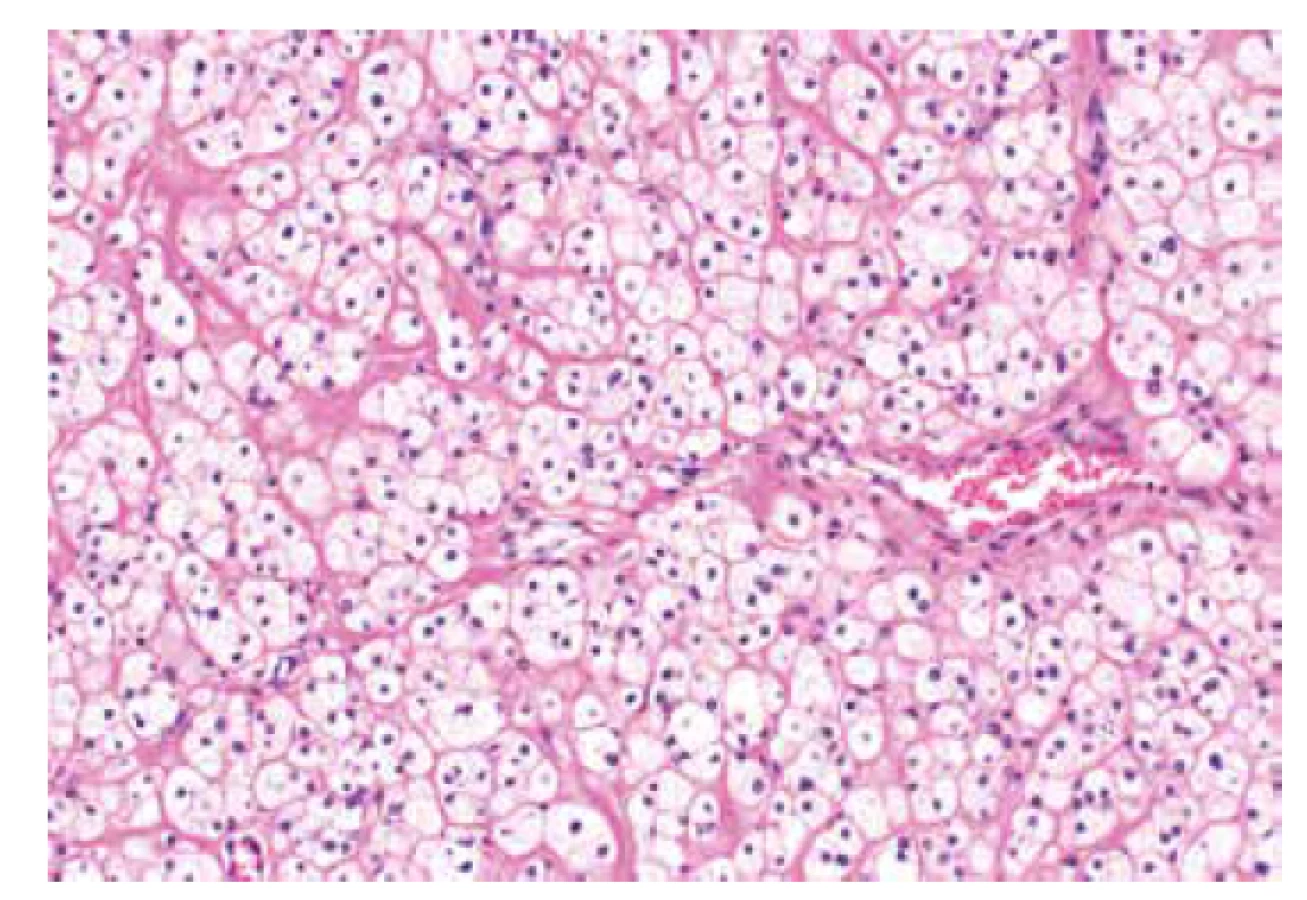
Jedná se o velmi vzácné autozomálně dominantní onemocnění s odhadovanou incidencí 1 : 2000000 které souvisí s germinální mutací genu SMARCB1 (OMIM #609322) nebo SMARCA4 (OMIM #613325). RTPS je spojen se zvýšeným rizikem vzniku rabdoidních nádorů v obvyklých (ledvina, CNS) i neobvyklých lokalitách (oblast hlavy a krku, játra, močový měchýř, mediastinum, retroperitoneum, oblast pánve, srdce a další) (46). V oblasti genitálu je s tímto syndromem spojen výskyt tzv. malobuněčného karcinomu ovaria hyperkalcemického typu (SCCHT), který tedy představuje variantu maligního rabdoidního nádoru s výskytem v ovariu (obr. 3) (47,48). Pro SCCHT je charakteristický výskyt mutace genu SMARCA4 vyskytující se asi u 95 % případů (49). Mutace je doprovázená ztrátou exprese proteinu SMARCA4 (dříve označovaného jako Brg1) na imunohistochemické úrovni (50). Ztráta exprese SMARCA4 je tedy vysoce užitečný senzitivní diagnostický marker, nerozliší však mezi germinální a somatickou inaktivací genu. Jedná se o marker s vysokou specificitou zejména s ohledem na odlišení SCCHT od nádorů ovaria z buněk granulózy, které připadají v diferenciální diagnostice v úvahu nejčastěji a ztráta exprese SMARCA4 u nich doposud nebyla popsána. U jiných nádorů ovaria jako je světlobuněčný karcinom, nediferencovaný či dediferencovaný karcinom či metastáza melanomu se ztráta exprese vzácně může vyskytnout, ve většině případů však tyto nádory nepředstavují diferenciálně diagnostický problém.

Asi 40 % publikovaných případů SCCHT u kterých byla nalezena mutace SMARCA4 mělo germinální mutaci, naprostá většina těchto případů (80 %) však byla u pacientek bez pozitivní rodinné anamnézy a může se tedy jednat o hereditární případy související s gonadálním mozaicismem, inkompletně penetrantní mutace či germinální mutace vzniklé de novo v časné fázi zárodečného vývoje (49). SCCHT spojené s germinální mutací se častěji vyskytují u mladších pacientek, zejména v případě výskytu pod 18 let věku je pravděpodobnost přítomnosti germinální mutace SMARCA4 vysoká. Genetické poradenství by však mělo být v případě této diagnózy indikováno u všech pacientek, bez ohledu na klinicko-patologické charakteristiky.
SYNDROM HEREDITÁRNÍ LEIOMYOMATÓZY A RENÁLNÍHO KARCINOMU (HLRCC)
HLRCC (OMIM#150800) je autozomálně dominantní onemocnění související s germinální mutací genu pro fumarát hydratázu (FH), jehož incidence v populaci není známá (51, 52). Pacienti s tímto onemocněním mají predispozici ke vzniku mnohotných kožních a děložních leiomyomů a renálního karcinomu (obvykle se jedná o agresivně se chovající papilární karcinomy ledviny). Děložní leiomyomy se vyskytují v mladším věku, často jsou mnohotné a velké a mohou (avšak nemusí) vykazovat neobvyklé morfologické rysy buď v podobě tzv. FH-deficientního leiomyomu, nebo leiomyomu s bizarními jádry (LBN) (53,54). FH-deficientní leiomyom je charakterizován prominentními eosinofilními jadérky, hypercelularitou, přítomností jaderných atypií, eosinofilními inkluzními tělísky a parožnatými cévami. Mezi tímto typem leiomyomu a LBN, zejména jeho I. typem, je tedy značný morfologický překryv (55). Mutace genu pro FH je doprovázena ztrátou exprese proteinu na imunohistochemické úrovni. Praktické využití imunohistochemického vyšetření exprese FH je však limitováno faktem, že se jedná o vyšetření s poměrně nízkou specificitou – absence exprese proteinu se vyskytuje nejen v souvislosti s germinálními mutacemi, ale i u somatických mutací, delece FH genu a hypermetylace jeho promotoru. V naší studii na 108 LBN jsme prokázali ztrátu exprese FH u 67 případů a mutaci genu FH u 15 případů (z toho 80 % bylo u LBN I. typu), nerozlišovali jsme však mezi somatickou a germinální mutací (54). Senzitivita imunohistochemického vyšetření s ohledem na detekci přítomnosti mutace byla v naší studii 87 % a specifita 58 %. Praktické využití imunohistochemického vyšetření pro účely jakéhosi „screeningu“ HLRCC je tedy limitované. S ohledem na fakt, že většina FH-deficientních leiomyomů vykazuje somatickou mutaci genu pro FH a morfologické rysy tohoto typu leiomyomu jsou inkonzistentní a překryvné s jinými leiomyocelulárními nádory není u všech pacientek s morfologickou suspekcí na FH-deficientní leiomyom doporučováno genetické poradenství, pokud nesplňují další klinické rizikové faktory zahrnující osobní či rodinnou anamnézu kožních leiomyomů, výskyt karcinomu ledviny v rodině či výskyt symptomatických děložních leiomyomů v časném věku. Role patologa je tedy v případě diagnostiky FH-deficientního leiomyomu v upozornění na to, že tato léze může být spojena se syndromem HLRCC, další postup je však na klinické rozvaze. Ztráta imunohistochemické exprese FH může dále podpořit případnou indikaci genetického testování, zachovalá exprese FH však možnost HLRCC syndromu nevylučuje.
PEUTZ-JEGHERSŮV SYNDROM (PJS)
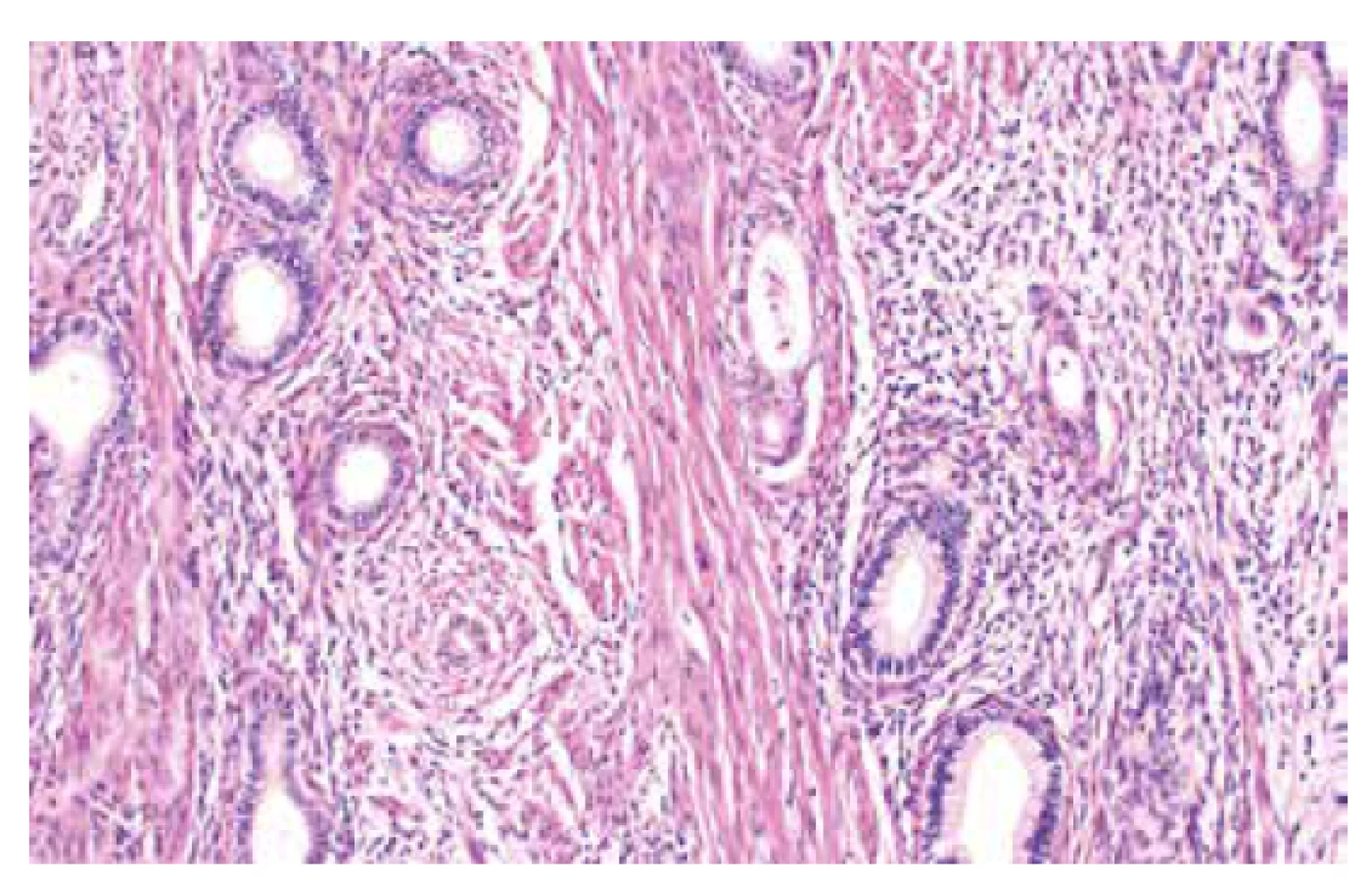
Jedná se o autozomálně dominantní onemocnění spojené s germinální mutací genu STK11 (OMIM #175200), která je detekovatelná asi u 75-95 % pacientů s tímto syndromem (56). Existence dalšího genu, jehož mutace způsobuje PJS je diskutována, doposud však tato možnost nebyla potvrzena (57). Incidence PJS udávána v rozmezí 1 : 8300–1 : 200000. Kromě hamartogenních polypů střeva a typických mukokutánních pigmentací je pro tento syndrom typický výskyt nejrůznějších maligních nádorů, zahrnujících gastrointestinální (žaludek, tlusté i tenké střevo, jícen, pankreas, biliární trakt) i extraintestinální (prso, plíce, varlata) lokality (56, 58). V oblasti ženského genitálu se jedná o celé spektrum lézí (nenádorových i nádorů), které mohou vykazovat určité specifické morfologické rysy, avšak mohou být i morfologicky neodlišitelné od sporadických lézí. Mezi léze s určitými specifickými morfologickými rysy patří gastrický typ mucinózního karcinomu děložního hrdla (zejména dobře diferencovaný, tzv. adenoma malignum) (obr. 4), ovariální sex cord nádor s anulárními tubuly, současný výskyt mucinózní metaplazie a neoplazií ženského genitálu a diskutabilně i lobulární endocervikální glandulární hyperplazie (56,59). Léze bez specifických morfologických rysů zahrnují nádor ze Sertoliho či Sertoliho-Leydigových buněk ovaria (obr. 5 a 6) a ovariální či endometriální karcinomy. Role patologa je tedy zejména rozpoznání lézí vykazujících určité specifické rysy a upozornění na jejich možnou souvislost s Peutz-Jeghersovým syndromem.


DICER1 SYNDROM
Incidence výskytu tohoto syndromu charakterizovaného germinálních mutací genu DICER1 (OMIM #606241) je odhadována asi na 1 : 10600 (60-62). Pro syndrom je typický výskyt multinodulární strumy a predispozice ke vzniku poměrně širokého spektra nádorů zahrnujících karcinom štítné žlázy, pleuropulmonální blastom, cystický nefrom, pinealoblastom a další (60). V oblasti ženského genitálu se jedná o rabdomyosarkom zejména děložního hrdla, nádor ze Sertoliho-Leydigových buněk (středně či málo diferencovaný, retiformní, s heterologními elementy) (obr. 7), sex cord-stromální nádor připomínající juvenilní nádor z buněk granulózy či gynandroblastom a vzácně zřejmě i endometroidní karcinom dělohy a ovariální fibrosarkom (61,63,64). Patolog by měl upozornit na možnost tohoto syndromu a vhodnost genetického poradenství zejména v případě výskytu embryonálního sarkomu děložního hrdla a u všech středně či málo diferencovaných nádorů ze Sertoliho-Leydigových buněk.

KOMPLEX TUBERÓZNÍ SKLERÓZY (TSC)
TSC (OMIM PS191100) je autozomálně dominantní onemocnění s incidencí asi 1 : 6000–1 : 10000 (65). Jedná se o onemocnění spojené s germinální mutací genu TSC1 (kódujícího protein hamartin) nebo TSC2 (kódujícího protein tuberin), až 2/3 mutací (zejména TSC2) však vzniká de novo. Onemocnění má vysokou fenotypickou variabilitu a klinické příznaky jsou velmi heterogenní. Diagnostické znaky zahrnují mnohotné renální angiolipomy, sklerotická ložiska v mozkové kůře, subependymální gliové uzly, retinální hamartomy, angiofibromy obličeje, fibromy v oblasti nehtů a fibrózní plaky hlavy. Mezi suspektní znaky patří mnohotné cysty či nádory ledvin, rabdomyom srdce, projevy v CNS (kalcifikace, hypomyelinizace, hypomelanotické makuly kůže a lymfangioleiomyomatóza (66). Projevy v oblasti ženského genitálu tedy zahrnují extrapulmonální lymfangioleiomyomatózu a výskyt nádorů z perivaskulárních epiteloidních buněk (PEComů) (67). PEComy se vyskytují zejména v těle dělohy (obr. 8), mohou však vznikat i v oblasti adnex, pochvě a děložním hrdle (68,69). Odhaduje se, že asi 10 % pacientů s PEComy má TSC. Na morfologické úrovni jsou projevy v oblasti ženského genitálu spojené s TSC neodlišitelné od lézí sporadických. Platí nicméně, že v případě diagnózy extrapulmonární lymfangioleiomyomatózy či PEComu by měl patolog upozornit na možnou souvislost s TSC a zdůraznit případný benefit genetického poradenství.
SYNDROM NÉVOIDNÍHO BAZOCELULÁRNÍHO KARCINOMU
Syndrom névoidního bazocelulárního karcinomu (Syndrom bazocelulárního névu; Gorlinův syndrom; Gorlin-Goltzův syndrom; OMIM #109400) je syndrom související s germinální mutací genu PTCH1, PTCH2 či SUFU (70). Jedná se o autozomálně dominantní onemocnění s odhadovanou incidencí asi 1 : 50000-150000, přičemž asi 20-30 % mutací vzniká de novo. Fenotypicky se jedná o syndrom velmi heterogenní, který je asi v 60 % případů spojen s faciálním dysmorfismem s makrocefalií a skeletálními abnormalitami. Další možná manifestace zahrnuje časný vznik mnohotných bazaliomů, mnohočetných odontogenních keratocyst, ektopických kalcifikaci falx cerebri, meduloblastomu (asi u 5 %) a srdečních fibromů (71). Asi u 20 % pacientek s tímto syndromem se vyskytují ovariální fibromy (70). Tyto fibromy jsou častěji multifokální, multinodulární, bilaterální a výrazně kalcifikované. Na mikroskopické úrovni však nevykazují žádné specifické rysy. Role patologa spočívá v upozornění na možnost tohoto syndromu při výskytu ovariálního fibromu či vícečetných fibromů s popsanými makroskopickými charakteristikami zejména u mladých pacientek.
VON HIPPELOVA-LINDAUOVA CHOROBA (VHL)
VHL (OMIM #193300) je autozomálně dominantní syndrom s incidencí asi 1 : 36000 související s germinální mutací tumor supresorového genu VHL, která asi ve 20 % případů vzniká de novo (72). Jedná se o fenotypicky velmi heterogenní multisystémové onemocnění charakterizované výskytem cystických lézí a širokého spektra benigních i maligních nádorů. Tyto léze zahrnují hemangioblastom, feochromocytom, karcinom ledviny, cystické léze pankreatu, angiomatózu, tumor endolymphatického vaku („endolymphatic sac tumor“) a bilaterální papilární cystadenom nadvarlete (73). U žen se vyskytuje světlobuněčný papilární cystadenom širokého děložního vazu (74,75). Jedná se o velmi vzácnou lézi, při jejíž diagnostice je ze strany patologa vhodné upozornit na možnou souvislost s VHL chorobou.
ZÁVĚR
Správné rozpoznání a diagnostika hereditárních nádorových syndromů je zásadně důležitá a má přímé dopady na pacienta i celou rodinu. Úloha patologa s ohledem na hereditární nádorové syndromy zahrnuje problematiku bioptické diagnostiky včetně vyšetřování profylakticky odstraněných tkání a případně i screeningu LS. V rámci bioptické diagnostiky patolog provádí vyšetření vzorků u pacientů se známým syndromem, u kterých došlo ke vzniku nádorového onemocnění. Může však mít významnou roli i v tom, že v rámci bioptického vyšetření upozorní na možnou souvislost nádoru s některým z těchto syndromů u pacientek, u nichž syndrom doposud nebyl diagnostikován. Pro patologa je v tomto kontextu důležitá nejen znalost hereditárních syndromů a nádorů, které se v souvislosti s nimi mohou vyskytovat, ale i způsob reportování výsledků a správná formulace diagnostického závěru tak, aby na příslušnou souvislost upozornil. Vlastní rozhodnutí o dalším postupu je pak však vždy na klinickém lékaři, který v rámci komplexního posouzení rozhodne o případném referování pacienta lékařskému genetikovi. Pouze lékařský genetik je oprávněn provést genetické poradenství a případnou indikaci testování germinálního genomu.
S ohledem na bioptickou praxi je důležitá i znalost správného vyšetřování materiálu odebraného během profylaktických zákroků. Vyšetřování materiálu od pacientek s HBOC, u kterých byla provedena profylaktická salpingo-ooforektomie, je standardizované a protokol SEE-FIM je v rutinní diagnostice používám už delší dobu. Oproti tomu u profylaktických zákroků u pacientek s Lynchovým syndromem je situace s ohledem na standardizovaný postup při zpracování materiálu v současné době nedořešena a mezinárodní guidelines doposud kritéria nestanovila, dostupná jsou tedy jen doporučení bez obecně přijatého konsenzu pro standardizované vyšetřování.
Pokud jde o screeningová vyšetření, aktuálně je relevantní pouze vyšetřování mikrosatelitové instability či stavu exprese mismatch repair proteinů (MMR) u pacientek s určitými typy nádorů vyskytujícími se v možné souvislosti s Lynchovým syndromem. Mezinárodní guidelines jsou v podstatě ve shodě v tom, že tento screening z nádorové tkáně by měl být prováděn u všech pacientek s karcinomem endometria a selektovaných pacientek s karcinomem ovaria (34,37,38). Optimálně se provádí imunohistochemicky v kombinaci s vyšetřením hypermetylace promotoru MLH1, lze však využít i jiné metody. V praxi se však v současné době v České republice screeningová vyšetření zaměřená na Lynchův syndrom u pacientek s karcinomem endometria a ovaria provádí nesystematicky, pouze na některých pracovištích. Domníváme se však, že by jeho plošná realizace měla být diskutována na úrovni příslušných odborných společností, tedy patologů, onkologů, genetiků a gynekologů.
PODĚKOVÁNÍ
Práce byla podpořena MZČR (projekt AZV NV19-03-00007 a RVO 64165) a Univerzitou Karlovou (projekt Progress Q28/LF1 a UNCE204065).). Práca bola podporená v rámci OP Výskum a vývoj pre projekt: Centrum pre biomedicínsky výskum – BIOMEDIRES – II. etapa, ITMS 313011W428, spolufinancovaný zo zdrojov Európskeho fondu regionálneho rozvoja.
PROHLÁŠENÍ
Autor práce prohlašuje, že v souvislosti s tématem, vznikem a publikací tohoto článku není ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou. Toto prohlášení se týká i všech spoluautorů.
∗ Adresa pro korespondenci:
Prof. MUDr. Pavel Dundr, Ph.D.
Ústav patologie 1. LF UK a VFN v Praze
Studničkova 2, 128 00 Praha 2
email: pavel.dundr@vfn.cz
Zdroje
- Garber JE, Offit K. Hereditary cancer predisposition syndromes. J Clin Oncol 2005; 23(2): 276-292.
- Foretova L. Hereditary cancer syndromes, their testing and prevention. Cas Lek Cesk 2019; 158(1): 15-21.
- Foretova L, Petrakova K, Palacova M, et al. Genetic testing and prevention of hereditary cancer at the MMCI - over 10 years of experience. Klin Onkol 2010; 23(6): 388-400.
- Pal T, Permuth-Wey J, Betts JA, et al. BRCA1 and BRCA2 mutations account for a large proportion of ovarian carcinoma cases. Cancer 2005; 104(12): 2807-2816.
- Risch HA, Mclaughlin JR, Cole DE, et al. Population BRCA1 and BRCA2 mutation frequencies and cancer penetrances: A kin-cohort study in ontario, canada. J Natl Cancer Inst 2006; 98(23): 1694-1706.
- Helder-Woolderink JM, Blok EA, Vasen HF, et al. Ovarian cancer in lynch syndrome; a systematic review. Eur J Cancer 2016; 55 : 65-73.
- Garg K, Karnezis AN, Rabban JT. Uncommon hereditary gynaecological tumour syndromes: Pathological features in tumours that may predict risk for a germline mutation. Pathology 2018; 50(2): 238-256.
- Struewing JP, Hartge P, Wacholder S, et al. The risk of cancer associated with specific mutations of brca1 and brca2 among ashkenazi jews. N Engl J Med 1997; 336(20): 1401-1408.
- Petrakova K, Palacova M, Schneiderova M, Standara M. [hereditary breast and ovarian cancer syndrome]. Klin Onkol 2016; 29 Suppl 1: S14-21.
- Plevova P, Novotny J, Petrakova K, et al. Hereditary breast and ovarian cancer syndrome. Klin Onkol 2009; 22 Suppl: S8-11.
- Antoniou A, Pharoah PD, Narod S, et al. Average risks of breast and ovarian cancer associated with brca1 or brca2 mutations detected in case series unselected for family history: A combined analysis of 22 studies. Am J Hum Genet 2003; 72(5): 1117-1130.
- King MC, Marks JH, Mandell JB, New York Breast Cancer Study G. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science 2003; 302(5645): 643-646.
- Mavaddat N, Peock S, Frost D, et al. Cancer risks for BRCA1 and BRCA2 mutation carriers: Results from prospective analysis of embrace. J Natl Cancer Inst 2013; 105(11): 812-822.
- Hanley GE, Mcalpine JN, Miller D, et al. A population-based analysis of germline BRCA1 and BRCA2 testing among ovarian cancer patients in an era of histotype-specific approaches to ovarian cancer prevention. BMC Cancer 2018; 18(1): 254.
- Mersch J, Jackson MA, Park M, et al. Cancers associated with BRCA1 and BRCA2 mutations other than breast and ovarian. Cancer 2015; 121(2): 269-275.
- Ibrahim M, Yadav S, Ogunleye F, Zakalik D. Male BRCA mutation carriers: Clinical characteristics and cancer spectrum. BMC Cancer 2018; 18(1): 179.
- Soslow RA, Han G, Park KJ, et al. Morphologic patterns associated with BRCA1 and BRCA2 genotype in ovarian carcinoma. Mod Pathol 2012; 25(4): 625-636.
- Reyes MC, Arnold AG, Kauff ND, Levine DA, Soslow RA. Invasion patterns of metastatic high-grade serous carcinoma of ovary or fallopian tube associated with brca deficiency. Mod Pathol 2014; 27(10): 1405-1411.
- Garg K, Levine DA, Olvera N, et al. BRCA1 immunohistochemistry in a molecularly characterized cohort of ovarian high-grade serous carcinomas. Am J Surg Pathol 2013; 37(1): 138-146.
- Foretova L, Machackova E, Palacova M, et al. Recommended extension of indication criteria for genetic testing of BRCA1 and BRCA2 mutations in hereditary breast and ovarian cancer syndrome. Klin Onkol 2016; 29 Suppl 1: S9-13.
- Zikan M, Kalabova R. Recommendation for prophylactic surgery for decreasing the risk of gynaecological cancer in women with hereditary risk. Klin Onkol 2009; 22 Suppl: S58-59.
- Lee Y, Medeiros F, Kindelberger D, et al. Advances in the recognition of tubal intraepithelial carcinoma: Applications to cancer screening and the pathogenesis of ovarian cancer. Adv Anat Pathol 2006; 13(1): 1-7.
- Cancer Genome Atlas Research N. Integrated genomic analyses of ovarian carcinoma. Nature 2011; 474(7353): 609-615.
- Da Cunha Colombo Bonadio RR, Fogace RN, Miranda VC, Diz M. Homologous recombination deficiency in ovarian cancer: A review of its epidemiology and management. Clinics (Sao Paulo) 2018; 73(suppl 1): e450s.
- Hampel H, De La Chapelle A. How do we approach the goal of identifying everybody with Lynch syndrome? Fam Cancer 2013; 12(2): 313-317.
- Bonadona V, Bonaiti B, Olschwang S, et al. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA 2011; 305(22): 2304-2310.
- Dunlop MG, Farrington SM, Carothers AD, et al. Cancer risk associated with germline DNA mismatch repair gene mutations. Hum Mol Genet 1997; 6(1): 105-110.
- Grindedal EM, Renkonen-Sinisalo L, Vasen H, et al. Survival in women with MMR mutations and ovarian cancer: A multicentre study in Lynch syndrome kindreds. J Med Genet 2010; 47(2): 99-102.
- Downes MR, Allo G, Mccluggage WG, et al. Review of findings in prophylactic gynaecological specimens in Lynch syndrome with literature review and recommendations for grossing. Histopathology 2014; 65(2): 228-239.
- Chui MH, Gilks CB, Cooper K, Clarke BA. Identifying Lynch syndrome in patients with ovarian carcinoma: The significance of tumor subtype. Adv Anat Pathol 2013; 20(6): 378-386.
- Sjursen W, Haukanes BI, Grindedal EM, et al. Current clinical criteria for Lynch syndrome are not sensitive enough to identify msh6 mutation carriers. J Med Genet 2010; 47(9): 579-585.
- Lancaster JM, Powell CB, Chen LM, Richardson DL, Committee SGOCP. Society of gynecologic oncology statement on risk assessment for inherited gynecologic cancer predispositions. Gynecol Oncol 2015; 136(1): 3-7.
- Evaluation of Genomic Applications In P, Prevention Working G. Recommendations from the egapp working group: Genetic testing strategies in newly diagnosed individuals with colorectal cancer aimed at reducing morbidity and mortality from Lynch syndrome in relatives. Genet Med 2009; 11(1): 35-41.
- Dillon JL, Gonzalez JL, Demars L, Bloch KJ, Tafe LJ. Universal screening for Lynch syndrome in endometrial cancers: Frequency of germline mutations and identification of patients with Lynch-like syndrome. Hum Pathol 2017; 70(121-128.
- Cho KR, Cooper K, Croce S, et al. International society of gynecological pathologists (ISGYP) endometrial cancer project: Guidelines from the special techniques and ancillary studies group. Int J Gynecol Pathol 2019; 38 Suppl 1: S114-S122.
- Mills AM, Longacre TA. Lynch syndrome screening in the gynecologic tract: Current state of the art. Am J Surg Pathol 2016; 40(4): e35-44.
- Mcconechy MK, Talhouk A, Li-Chang HH, et al. Detection of DNA mismatch repair (MMR) deficiencies by immunohistochemistry can effectively diagnose the microsatellite instability (MSI) phenotype in endometrial carcinomas. Gynecol Oncol 2015; 137(2): 306-310.
- Sarode VR, Robinson L. Screening for Lynch syndrome by immunohistochemistry of mismatch repair proteins: Significance of indeterminate result and correlation with mutational studies. Arch Pathol Lab Med 2019; 143(10): 1225-1233.
- Goodfellow PJ, Billingsley CC, Lankes HA, et al. Combined microsatellite instability, MLH1 methylation analysis, and immunohistochemistry for lynch syndrome screening in endometrial cancers from GOG210: An NRG oncology and gynecologic oncology group study. J Clin Oncol 2015; 33(36): 4301-4308.
- Lu FI, Gilks CB, Mulligan AM, et al. Prevalence of loss of expression of DNA mismatch repair proteins in primary epithelial ovarian tumors. Int J Gynecol Pathol 2012; 31(6): 524-531.
- Schmeler KM, Lynch HT, Chen LM, et al. Prophylactic surgery to reduce the risk of gynecologic cancers in the Lynch syndrome. N Engl J Med 2006; 354(3): 261-269.
- Gammon A, Jasperson K, Champine M. Genetic basis of Cowden syndrome and its implications for clinical practice and risk management. Appl Clin Genet 2016; 9(83-92.
- Mahdi H, Mester JL, Nizialek EA, et al. Germline PTEN, SDHB-d, and KLLN alterations in endometrial cancer patients with Cowden and Cowden-like syndromes: An international, multicenter, prospective study. Cancer 2015; 121(5): 688-696.
- Baker WD, Soisson AP, Dodson MK. Endometrial cancer in a 14-year-old girl with Cowden syndrome: A case report. J Obstet Gynaecol Res 2013; 39(4): 876-878.
- Djordjevic B, Hennessy BT, Li J, et al. Clinical assessment of PTEN loss in endometrial carcinoma: Immunohistochemistry outperforms gene sequencing. Mod Pathol 2012; 25(5): 699-708.
- Nemes K, Bens S, Bourdeaut F, et al. Rhabdoid tumor predisposition syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K and Amemiya A, eds. Genereviews((r)) ed). Seattle (WA): 1993:
- Kupryjanczyk J, Dansonka-Mieszkowska A, Moes-Sosnowska J, et al. Ovarian small cell carcinoma of hypercalcemic type - evidence of germline origin and SMARCA4 gene inactivation. A pilot study. Pol J Pathol 2013; 64(4): 238-246.
- Hruska L, Sirak I, Laco J, et al. Rare hereditary burden associated with a hypercalcemic small-cell carcinoma of cervix in a young female patient. Klin Onkol 2019; 32(6): 456-462.
- Witkowski L, Goudie C, Foulkes WD, Mccluggage WG. Small-cell carcinoma of the ovary of hypercalcemic type (malignant rhabdoid tumor of the ovary): A review with recent developments on pathogenesis. Surg Pathol Clin 2016; 9(2): 215-226.
- Conlon N, Silva A, Guerra E, et al. Loss of SMARCA4 expression is both sensitive and specific for the diagnosis of small cell carcinoma of ovary, hypercalcemic type. Am J Surg Pathol 2016; 40(3): 395-403.
- Menko FH, Maher ER, Schmidt LS, et al. Hereditary leiomyomatosis and renal cell cancer (HLRCC): Renal cancer risk, surveillance and treatment. Fam Cancer 2014; 13(4): 637-644.
- Plevova P, Hladikova A, Tesarova M. Hereditary leiomyomatosis and renal cell cancer - hlrcc/multiple cutaneous and uterine leimomyomatosis - MCUL. Klin Onkol 2012; 25 Suppl(S55-58.
- Reyes C, Karamurzin Y, Frizzell N, et al. Uterine smooth muscle tumors with features suggesting fumarate hydratase aberration: Detailed morphologic analysis and correlation with s-(2-succino)-cysteine immunohistochemistry. Mod Pathol 2014; 27(7): 1020-1027.
- Gregova M, Hojny J, Nemejcova K, et al. Leiomyoma with bizarre nuclei: A study of 108 cases focusing on clinicopathological features, morphology, and fumarate hydratase alterations. Pathol Oncol Res 2019; 26(3): 1527-1537.
- Ubago JM, Zhang Q, Kim JJ, Kong B, Wei JJ. Two subtypes of atypical leiomyoma: Clinical, histologic, and molecular analysis. Am J Surg Pathol 2016; 40(7): 923-933.
- Van Lier MG, Wagner A, Mathus-Vliegen EM, et al. High cancer risk in Peutz-jeghers syndrome: A systematic review and surveillance recommendations. Am J Gastroenterol 2010; 105(6): 1258-1264; author reply 1265.
- Hearle N, Lucassen A, Wang R, et al. Mapping of a translocation breakpoint in a Peutz-jeghers hamartoma to the putative PJS locus at 19q13.4 and mutation analysis of candidate genes in polyp and stk11-negative pjs cases. Genes Chromosomes Cancer 2004; 41(2): 163-169.
- Chen HY, Jin XW, Li BR, et al. Cancer risk in patients with Peutz-jeghers syndrome: A retrospective cohort study of 336 cases. Tumour Biol 2017; 39(6): 1010428317705131.
- Jones MA, Young RH, Scully RE. Diffuse laminar endocervical glandular hyperplasia. A benign lesion often confused with adenoma malignum (minimal deviation adenocarcinoma). Am J Surg Pathol 1991; 15(12): 1123-1129.
- Stewart DR, Best AF, Williams GM, et al. Neoplasm risk among individuals with a pathogenic germline variant in DICER1. J Clin Oncol 2019; 37(8): 668-676.
- Cowan M, Suntum T, Olivas AD, et al. Second primary rhabdomyosarcoma of the uterine cervix presenting with synchronous ovarian sertoli-leydig cell tumor: An illustrative case of DICER1 syndrome. Gynecol Oncol Rep 2018; 25 : 94-97.
- Kim J, Schultz KaP, Hill DA, Stewart DR. The prevalence of germline DICER1 pathogenic variation in cancer populations. Mol Genet Genomic Med 2019; 7(3): e555.
- De Kock L, Terzic T, Mccluggage WG, et al. DICER1 mutations are consistently present in moderately and poorly differentiated sertoli-leydig cell tumors. Am J Surg Pathol 2017; 41(9): 1178-1187.
- Melendez-Zajgla J, Mercado-Celis GE, Gaytan-Cervantes J, et al. Genomics of a pediatric ovarian fibrosarcoma. Association with the dicer1 syndrome. Sci Rep 2018; 8(1): 3252.
- Hasbani DM, Crino PB. Tuberous sclerosis complex. Handb Clin Neurol 2018; 148(813-822.
- Caban C, Khan N, Hasbani DM, Crino PB. Genetics of tuberous sclerosis complex: Implications for clinical practice. Appl Clin Genet 2017; 10 : 1-8.
- Hayashi T, Kumasaka T, Mitani K, et al. Prevalence of uterine and adnexal involvement in pulmonary lymphangioleiomyomatosis: A clinicopathologic study of 10 patients. Am J Surg Pathol 2011; 35(12): 1776-1785.
- Conlon N, Soslow RA, Murali R. Perivascular epithelioid tumours (PECOMas) of the gynaecological tract. J Clin Pathol 2015; 68(6): 418-426.
- Bennett JA, Braga AC, Pinto A, et al. Uterine pecomas: A morphologic, immunohistochemical, and molecular analysis of 32 tumors. Am J Surg Pathol 2018; 42(10): 1370-1383.
- Evans DG, Farndon PA. Nevoid basal cell carcinoma syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K and Amemiya A, eds. Genereviews((r)) ed). Seattle (WA): 1993.
- Kimonis VE, Goldstein AM, Pastakia B, et al. Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome. Am J Med Genet 1997; 69(3): 299-308.
- Van Leeuwaarde RS, Ahmad S, Links TP, Giles RH. Von hippel-lindau syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K and Amemiya A, eds. Genereviews((r)) ed). Seattle (WA): 1993.
- Van Der Horst-Schrivers ANA, Sluiter WJ, Kruizinga RC, et al. The incidence of consecutive manifestations in von hippel-lindau disease. Fam Cancer 2019; 18(3): 369-376.
- Cox R, Vang R, Epstein JI. Papillary cystadenoma of the epididymis and broad ligament: Morphologic and immunohistochemical overlap with clear cell papillary renal cell carcinoma. Am J Surg Pathol 2014; 38(5): 713-718.
- Brady A, Nayar A, Cross P, et al. A detailed immunohistochemical analysis of 2 cases of papillary cystadenoma of the broad ligament: An extremely rare neoplasm characteristic of patients with von Hippel-Lindau disease. Int J Gynecol Pathol 2012; 31(2): 133-140.
Štítky
Patologie Soudní lékařství ToxikologieČlánek vyšel v časopise
Česko-slovenská patologie

2021 Číslo 2
Nejčtenější v tomto čísle
- Význam imunohistochemických metod v diagnostice karcinomu endometria
- Gynekologické léze u hereditárních nádorových syndromů
- Kazuistika: Postižení ledvin u pacientky s Crohnovou chorobou
- Význam imunohistochemických metod v diagnostice mezenchymálních nádorů dělohy
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy