
Sensing Cytosolic RpsL by Macrophages Induces Lysosomal Cell Death and Termination of Bacterial Infection
The death of the host cell during infection can be triggered by one or more microbial molecules; this “live or die” selection provides effective means for the dissection of immune recognition mechanisms as well as for the identification of the microbial molecules responsible for such responses. We found that infection of primary mouse macrophages by Legionella pneumophila strains harboring wild type RpsL, the S12 component of the bacterial ribosome, causes macrophage death by a mechanism independent of the three inflammatory caspases, caspase 1, 7 and 11. Importantly, although both confer resistance to streptomycin at indistinguishable effectiveness, the K88R, but not the K43N mutation in RpsL enables L. pneumophila to replicate in macrophages. Purified RpsL and RpsLK43N physically delivered into macrophages cause cell death by inducing damage to lysosomal membranes and the release of cathepsins. We also found that the lysosomal protease cathepsin B is required for efficient RpsL-induced cell death but its absence is not sufficient for macrophages to support intracellular bacterial replication. Thus, RpsL functions as an immune induction molecule to trigger one or more signaling cascades that leads to lysosomal cell death as well as the termination of bacterial replication.
Published in the journal:
. PLoS Pathog 11(3): e32767. doi:10.1371/journal.ppat.1004704
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004704
Summary
The death of the host cell during infection can be triggered by one or more microbial molecules; this “live or die” selection provides effective means for the dissection of immune recognition mechanisms as well as for the identification of the microbial molecules responsible for such responses. We found that infection of primary mouse macrophages by Legionella pneumophila strains harboring wild type RpsL, the S12 component of the bacterial ribosome, causes macrophage death by a mechanism independent of the three inflammatory caspases, caspase 1, 7 and 11. Importantly, although both confer resistance to streptomycin at indistinguishable effectiveness, the K88R, but not the K43N mutation in RpsL enables L. pneumophila to replicate in macrophages. Purified RpsL and RpsLK43N physically delivered into macrophages cause cell death by inducing damage to lysosomal membranes and the release of cathepsins. We also found that the lysosomal protease cathepsin B is required for efficient RpsL-induced cell death but its absence is not sufficient for macrophages to support intracellular bacterial replication. Thus, RpsL functions as an immune induction molecule to trigger one or more signaling cascades that leads to lysosomal cell death as well as the termination of bacterial replication.
Introduction
Pattern recognition receptors (PRRs) sense pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) generated by infection or endogenous cellular injury or tissue damage to initiate immune responses [1]. The Toll-like receptors (TLRs) were the first identified PRRs that recognize PAMPs and induce the expression of pro-death cytokines and pro-inflammatory molecules through the nuclear factor κB (NF-κB) signaling pathway [1]. These molecules could orchestrate efficient defense against invading pathogens through the induction of cell death, which is an effective means of defense against infections in many microbe-host interaction systems. For example, TNF-α engages the cellular apoptosis or necroptosis pathway to defend against infection [1]. The second group of PRRs contains the NOD-like receptor (NLR), the retinoic-acid inducible gene-I (RIG-I)-like helicase, and the PYHIN (pyrin and HIN200 domain—containing proteins; also known as p200 or HIN200 proteins) protein families [2]. These structurally and functionally heterologous proteins recognize more diverse ligands (including PAMPs) and can be generally divided into two categories based on their downstream signaling events. The first category of receptors promote transcriptional activation of cytokines through pathways controlled by the transcriptional activator NF-κB or IRF3 [2], whereas the second group of receptors initiate the assembly of large cytoplasmic signaling complexes known as the inflammasomes [2]. The inflammasome senses microbial infection and/or danger-associated molecules and activate caspase-1/11-dependent cytokine production and inflammatory cell death (pyroptosis), which is believed to be important in the removal of the replicative niche of intracellular pathogens [3].
Recent studies have identified the ligands for several of these receptors. For example, NAIP5 and NAIP6 are the receptors for bacterial flagellin, and NAIP1 and NAIP2 sense the needle and rod proteins of bacterial type III secretion systems, respectively [4–6]. Cytosolic DNA directly binds to the AIM2 inflammasome [7–9] and intracellular lipopolysaccharides (LPS) directly activate caspase 11, a key component of inflammatory response [10]. However, the ligands for other members of the NAIP family of NLR proteins remain elusive. Similarly, our understanding of the broader mechanisms underlying infection-induced death of immune cells is limited [11].
The pathogen Legionella pneumophila replicates within amoebae hosts in the environment; it also is able to grow in alveolar macrophages in the human lung, which causes Legionnaires’ disease. Intracellular replication of L. pneumophila requires the Dot/Icm type IV secretion system, which delivers hundreds of proteins into host cells to construct a niche supportive of bacterial growth [12–14]. Because its primary evolutionary pressure for virulence derives from life in amoebae hosts, L. pneumophila does not seem to have evolved sophisticated immune-evasive mechanisms. As a result, challenge of immune cells such as macrophages with this bacterium leads to strong immune responses often not seen with more finely-adapted pathogens [15]. Thus, L. pneumophila has emerged as a powerful tool in dissecting novel host immune recognition mechanisms [16]. For example, characterization of mutants capable of replicating in macrophages from incompatible mice led to the identification of flagellin as the bacterial factor responsible for triggering the strong immune response [17,18], which functioned by directly engaging NAIP5 to activate the NLRC4 inflammasome [4,5]. Similarly, mutants lacking effectors with similar biochemical activity have allowed the identification of protein synthesis inhibition as a signal for immune induction [19].
We recently found that an environmental L. pneumophila strain induces extensive cell death in mouse bone marrow-derived macrophages (BMDMs) that are permissive for commonly used laboratory strains [20]. Furthermore, the cell death differed from canonical apoptosis, necrosis or the caspase-1/11-dependent pyroptosis [20]. Here we found that clinical L. pneumophila strains induce similar responses. By identifying mutants capable of replicating in BMDMs, we found that the ribosomal protein RpsL is responsible for the restriction of replication. Although two mutations of RpsL both conferred bacterial resistance to the antibiotic streptomycin, one (K88R) allowed L. pneumophila isolates (both clinical and environmental) to grow in BMDMs, while the other (K43N) did not. Further cell biological studies revealed that RpsL induced cell death by triggering signaling cascades that led to lysosomal membrane permeabilization and subsequent cell death. Our results established RpsL as a ligand capable of triggering a unique immune recognition by inducing lysosomal cell death.
Results
A K88R mutation in rpsL allows strain LPE509 to replicate in mouse primary macrophages
To approach the mechanism underlying cell death induction by strain LPE509 [20], we hypothesized that this strain codes for unique factor(s) capable of triggering a signaling cascade that leads to cell death, and that mutants lacking such factor(s) may enable the bacterium to replicate in macrophages. To identify such factor(s), we attempted to generate an insertion mutant library of LPE509 with a transposon introduced by bi-parental mating. To acquire an antibiotic resistance marker for eliminating the Escherichia coli donor cells used to deliver the transposon, we first isolated spontaneous streptomycin resistant (StrepR) mutants of LPE509, and the transposition mutant pools generated from two such mutants were then tested for intracellular replication in BMDMs from A/J mice, which are permissive for intracellular replication of laboratory L. pneumophila strains. Unexpectedly, robust intracellular growth was observed in all mutant pools derived from one particular StrepR strain. Further analyses indicated that the parental strain used for mutant production had gained the ability to grow in BMDMs (Fig 1A). Sequencing analysis revealed a K88R mutation in the rpsL (lpg0324) gene of the StrepR mutant. To rule out the possibility that additional mutations contributed to this phenotype, we introduced the rpsLK88R allele into strain LPE509 by gene replacement to produce LPE509rpsLK88R; in BMDMs from A/J mice, this strain grew at rates comparable to the laboratory strain Lp02rpsLK88R, which increased almost 1000-fold in 72 hrs (Fig 1B). Consistent with earlier observations [20], wild type bacteria were cleared by macrophages within the experimental duration (Fig 1B). These results indicate that RpsL may regulate intracellular replication of L. pneumophila in BMDMs from A/J mice and that the gain-of-growth phenotype may result from the StrepR property of the bacteria.

Because multiple mutations in the RpsL or the 16S rRNA can result in resistance to streptomycin [21], we assessed the intracellular growth of several independent streptomycin-resistant mutants for intracellular growth in BMDMs to determine whether all mutations that confer StrepR result in the replication phenotype. Among 8 such mutants tested, 4 have overcome the restriction whereas the other 4 still were unable to replicate in macrophages (Figs 1B and S1). Sequencing analysis of the rpsL gene in these mutants revealed that in each of the 4 mutants capable of replicating in BMDMs, an A to G mutation occurred at nucleotide 263, causing a Lys to Arg substitution in the 88th residue (K88R) of RpsL. In each of the 4 StrepR mutants still defective in replicating in BMDMs, an A to T mutation at nucleotide 129 that led to a Lys to Asn substitution (K43N) in residue 43 was detected (Fig 1C). In agreement with the fact that the K88R and the K43N mutations confer indistinguishable resistance to streptomycin in bacteria such as E. coli [22], L. pneumophila mutants harboring these two mutations exhibited similar levels of streptomycin-resistance: both with a minimal inhibitory concentration (MIC) greater than 100 μg/ml (S1 Table). These results suggest that the K88R mutation in RpsL (RpsLK88R) is specific for overcoming the growth restriction of strain LPE509 in BMDMs permissive for commonly used laboratory L. pneumophila strains.
To further determine whether StrepR per se is sufficient to permit intracellular growth in BMDMs, we introduced a plasmid expressing a streptomycin adenyltransferase [23] into strains LPE509. This strain has an MIC of 30 μg/ml (S1 Table), but cannot productively replicate in BMDMs (S2 Fig). Taken together, these results establish that a K88R mutation in the ribosome protein RpsL is sufficient to allow strain LPE509 to replicate in BMDMs and that resistance to Strep alone does not confer on L. pneumophila the ability to overcome the growth restriction imposed by these macrophages.
RpsL is responsible for intracellular growth restriction of L. pneumophila in mouse primary macrophages
Our discovery that the RpsLK88R mutation is required for L. pneumophila to grow in BMDMs is consistent with the fact that commonly used laboratory strains such as Lp02 [24] and JR32 [25] are resistant to streptomycin and that both harbor a K88R mutation in rpsL [26]. These results predicted that the parental streptomycin-sensitive strain Philadelphia 1 (Phil-1) [24] might be unable to replicate in BMDMs. Indeed, unlike its derivatives Lp02 (referred to as Lp02rpsLK88R for clarity), strain Phil-1 was unable to replicate in BMDMs (Fig 2A). Importantly, robust growth occurred when the rpsL gene of strain Phil-1 was replaced by homologous recombination with rpsLK88R but not with rpsLK43N (Fig 2A). Similar growth restriction was observed in strains Paris [27] and Thunder Bay [28], in which a K88R mutation in RpsL allowed both strains to grow robustly in BMDMs (S3 Fig). Furthermore, strain Lp02rpsLWT derived from Lp02rpsLK88R has lost the ability to grow in BMDMs (Fig 2B). This strain, however, retained the ability to replicate in both Hela cells and the human macrophage cell line U937 (S4 Fig). Again, introduction of the streptomycin adenyltransferase gene into strain LPE509 or Lp02rpsLWT conferred antibiotic resistance but not intracellular replication (S1 Table; S2 Fig), further supporting the notion that wild type RpsL is responsible for the restriction of bacterial replication in BMDMs. Taken together, these results indicate that wild type RpsL is the genetic determinant that governs the outcome of L. pneumophila infection in BMDMs; this protein restricts intracellular growth of L. pneumophila in BMDMs and that such restriction occurs in both clinical and environmental isolates. For clarity, we used strains Lp02rpsLK88R and Lp02rpsLWT in all subsequent experiments.

Infection by L. pneumophila expressing wild type RpsL leads to macrophage death
Because host cell death accompanied the restriction of intracellular growth in infections with strain LPE509 [20], we determined whether strain Lp02rpsLWT induces a similar phenotype. Compared to infections with strain Lp02rpsLK88R, significantly higher levels of lactate dehydrogenase (LDH) was detected in culture supernatant of BMDMs challenged with strain Lp02rpsLWT (Fig 3A). Single cell analysis by the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining revealed that at 12 hrs post-infection, close to 50% of the macrophages infected with Lp02rpsLWT stained positively for apoptosis, whereas only about 10% Lp02rpsLK88R infected cells appeared apoptotic (Fig 3B). At this time point, a fraction of the bacterial phagosomes have developed into vacuoles containing more than 10 bacteria. However, unlike Lp02rpsLK88R that can be distinctly stained by the anti-Legionella antibody, the staining signals of Lp02rpsLWT appeared diffuse, which could be the debris from dying bacterial cells detected by the polycolonal antibodies raised against fixed bacterial cells [29] (Fig 3C). Furthermore, although the presence of phagosomes with multiple bacteria is readily detectable in single cell analysis (Fig 3C and S5 Fig), the bacteria cells in these vacuoles appeared not to be viable as the total bacterial counts in samples infected with strains expressing wild type RpsL decreased at the 15 hrs or 24 hrs postinfection time points (Figs 1, 2 and S6).
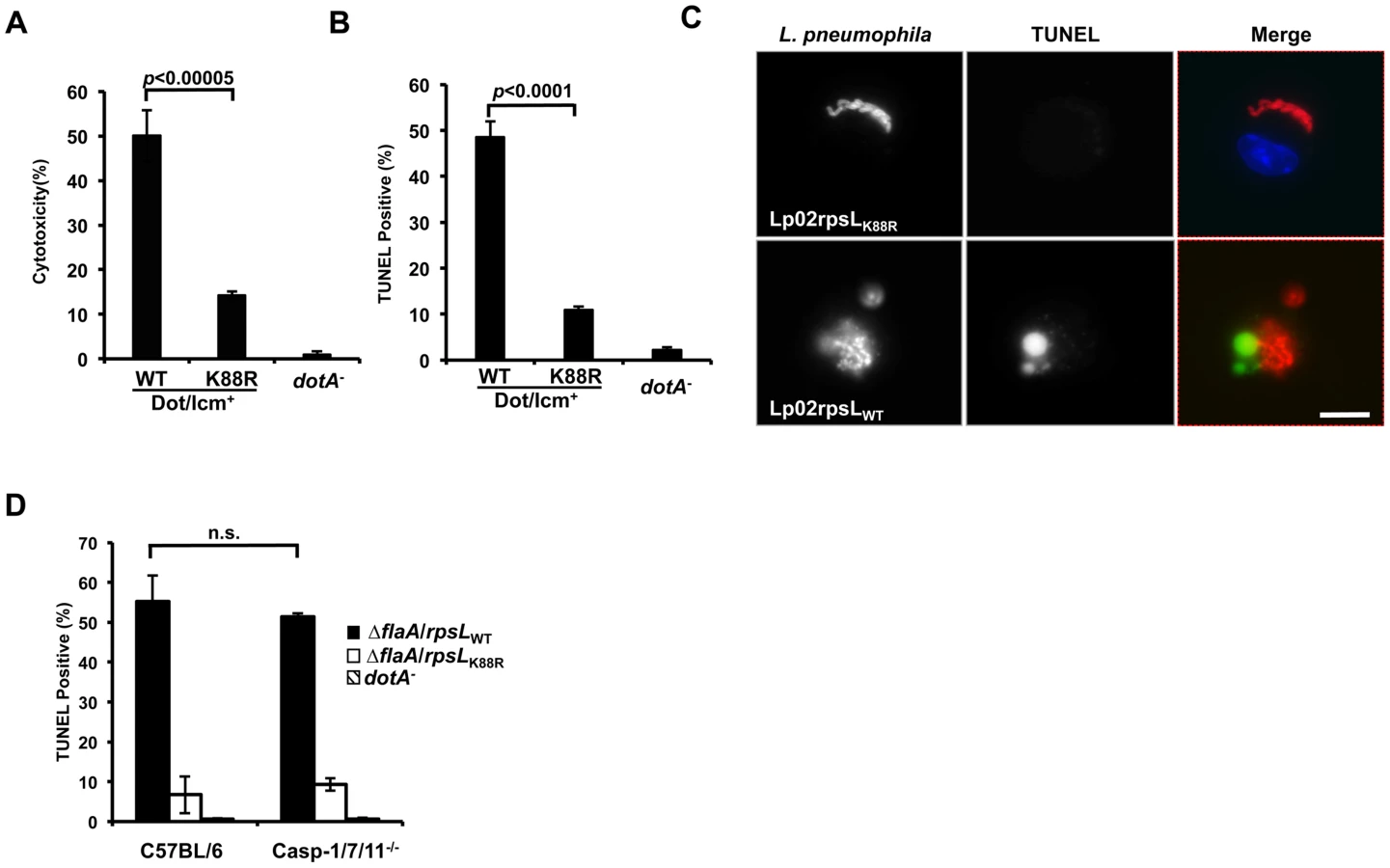
Since inflammatory caspases such as caspase-1, 7 and 11 play important roles in macrophage death induced by bacteria infection [30,31], we thus examined whether these caspases play a role in the cell death induced by L. pneumophila expressing wild type RpsL. We infected macrophages from mice lacking caspase-1, 7 and 11 (caspase-1/7/11-/-) and found that the challenge of macrophages from these mice with strain Lp02rpsLWT∆flaA still led to cell death as measured by TUNEL staining. The reason to delete the flaA gene is that the genetic background of the knockout mice is C57BL/6, which is sensitive to flagellin produced in strain Lp02rpsLWT [17,18]. Again, infection by strain Lp02rpsLK88R∆flaA caused significantly less death in these cells than did infection with strain Lp02rpsLWT∆flaA (Fig 3D). Consistent with the observation of cell death, BMDMs lacking caspase-1/7/11 did not support the growth of strain Lp02rpsLWT∆flaA (S7 Fig). Thus, infections with L. pneumophila expressing wild type RpsL cause macrophage death, which may contribute to the restriction of intracellular growth. Moreover, this cell death occurred in a mechanism independent of caspases-1, 7 and 11.
RpsL directly induces cell death in macrophages
Because the K43N mutation in RpsL imposes higher translation accuracy on the ribosome [32,33], it is unlikely that the cell death is caused by factors such as the potential increase in mistranslated bacterial proteins. The high level of conservation of RpsL among taxonomically diverse bacteria prompted us to hypothesize that this protein is a ligand capable of inducing macrophage death. Thus, we purified recombinant His6-RpsL and His6-RpsLK88R with a procedure employing the extensive isopropanol washing (methods) to exhaustively remove endotoxin (LPS) and delivered the proteins into macrophages by transfection [5]. When directly added to the BMDMs culture, neither protein caused detectable cell death (Fig 4A–B). However, when a transfection reagent was included, samples receiving His6-RpsL released significantly higher amount of LDH than samples receiving His6-RpsLK88R (Fig 4A). Similar results were obtained when cell death was evaluated by TUNEL staining in which close to 30% of cells receiving wild type RpsL were apoptotic, which is significantly higher than the <10% seen in samples transfected with His6-RpsLK88R (Fig 4B–C). Further, adding RNAase III to the protein did not alter its cell death induction activity, indicating that dsRNA [34] did not contribute to the observed phenotype (Fig 4D). On the other hand, treatment of protease K abolished the cell death-inducing ability of the protein (Fig 4D). Consistent with the fact that strains expressing RpsLK43N cannot replicate within BMDMs, His6-RpsLK43N induced macrophage death at rates comparable to those of wild type protein (Fig 4E). In addition, that wild type RpsL but not the K88R mutant from E. coli also induced robust cell death in macrophages (Fig 4F), which is consistent with the high-level conservation (91% identity) of this protein between these two bacteria. Furthermore, the cell death induction is specific for RpsL, as another similarly purified ribosomal protein, His6-RpsM was unable to induce macrophage death (Fig 4F). Finally, consistent with the observation that macrophage death induced by L. pneumophila strains harboring wild type RpsL is independent of caspases 1/7/11 (Fig 3D), His6-RpsLWT induced cell death in macrophages lacking these caspases at rates comparable to those of C57BL/6 (Fig 4G–H). Because caspase-11 directly senses intracellular LPS [10], these results also indicate that the cell death was not caused by LPS potentially present in the RpsL protein preparations. Moreover, the fact that transfection is required for RpsL to exert its effects, this protein must be active only when it is present in the cytosol; this observation also suggested that RpsL was delivered into the cytoplasm of the host cell by L. pneumophila during infection. However, we were unable to detect such translocation using the β-lactamase reporter [12,35] (S8 Fig).

The fact that L. pneumophila strains harboring wild type rpsL were able to replicate in alternative hosts such as Hela and U937 cells (S4 Fig) suggested that RpsL did not induce death in these cells. Indeed, Hela cells transfected with His6-RpsL did not detectably stain positively for apoptosis (S9A Fig). The lack of cell death is not due to deficiency in protein delivery because a similar procedure successfully introduced purified β-galactosidase into these cells (S9B–S9C Fig). Taken together, these results establish that wild type RpsL directly induces cell death and such induction is specific for primary mouse macrophages; the K88R mutant triggers similar responses but in a less potent manner.
RpsL causes lysosome membrane permeabilization in macrophages
Since macrophages from mice lacking the three inflammatory caspases are still sensitive to RpsL, it is very unlikely that the cell death is caused by canonical pyroptosis. We thus explored the involvement of other cell death pathways [36]. The lysosome, an organelle of low pH which contains a variety of hydrolases, has been implicated to play important roles in cell death in response to certain stimuli [37]. Such stimuli could trigger lysosome membrane permeabilization (LMP), leading to the release into the cytosol of hydrolases such as cathepsins (e.g. cathepsin B, D, and L) from the lysosomal lumen, where they induce apoptotic or pyroptotic cell death through caspase activation, or necroptosis when caspase activation was inhibited [38]. If RpsL activates the lysosomal cell death pathway, then inhibiting lysosomal acidification should reduce its cytotoxicity. To test this hypothesis, we inhibited lysosomal acidification by bafilomycin A1 (BFA1), which targets the vacuolar ATPase responsible for regulating organelle pH [39] prior to RpsL transfection. Cell death was almost completely abrogated in samples pre-incubated with BFA1 (Fig 5A).

We next assessed the integrity of lysosomal membranes in macrophages receiving RpsL using acridine orange (AO), a lysotrophic dye that when concentrated in lysosome, emits orange florescence upon blue light excitation, but green florescence when the loss of lysosome membrane integrity redistributes it into the cytosol and nucleus [40]. In samples transfected with RpsL, about 40% of the cells had lost lysosomal membrane integrity, as indicated by the emission of green fluorescence (Fig 5B&D); this dropped to approximately 15% of the cells when RpsLK88R was delivered into the cells (Fig 5B&D). In controls not transfected with the protein, essentially all macrophages emitted orange but not green fluorescence (Fig 5B&D). Similar results were obtained in infections in which strain Lp02rpsLWT caused significantly higher levels of lysosome damage than strain Lp02rpsLK88R (Fig 5C&E). Taken together, we conclude that RpsL induces LMP.
Cathepsin B is required for efficient RpsL-induced macrophage cell death
A direct result of LMP is the release of catabolic hydrolases, particularly cathepsin proteases, which have distinct roles in initiating cell death in different cell types [38]. We first used specific inhibitors to identify which cathepsin(s) are important for RpsL-induced cell death. CA074-Me, an inhibitor for cathepsin B (CtsB) but not pepstatin A, an inhibitor against cathepsin D antagonized RpsL-induced cell death (Fig 6A). Consistently, the cytosolic activity of cathepsins significantly increased upon RpsL delivery in a mechanism that required vacuolar acidification (Fig 6B). Notably, such increase also occurred in response to RpsLK88R but at a significantly lower magnitude (Fig 6B). The strong inhibitory effects of CA074-Me on the cathepsin activity induced by RpsL suggest the selectivity of CtsB release, at least within our experimental duration.

To further examine the role of this protease in RpsL-induced cell death, we prepared macrophages from mice deficient in CtsB (ctsb-/-) or CtsH (ctsH-/-) and transfected them with recombinant RpsL. Six hours after transfection, cell death occurred in macrophages from both wild type and the ctsH-/- mice, although the rates in cells from the latter mouse line were significantly lower (Fig 6C). At the 6-hr time point, consistent with the abolishment of RpsL-induced cell death by inhibition of CtsB, almost no TUNEL positive cells were detected in macrophages from ctsB-/- mice (Fig 6C). These macrophages were not generally defective in apoptosis as staurosporine treatment induced extensive cell death (Fig 6C). Similar results were obtained in infections in which strain Lp02rpsLWT induced significantly less cell death in BMDMs from ctsB-/- mice 14 hrs after uptake (Fig 6D). To determine whether BMDMs that received RpsL maintained their plasma membrane integrity, we measured the level of LDH in similarly treated samples. Intriguingly, although wild type RpsL is consistently more potent than the K88R mutant in inducing LDH release, similar levels of LDH were detected in BMDMs from the three different mouse lines after RpsL transfection (Fig 6E). In light of this observation, we determined the kinetics of RpsL-induced cell death in ctsB-/- BMDMs by TUNEL staining. Again, few TUNEL positive cells were detected in these cells within 6 hrs after transfection (Fig 6F). However, extensive cell death was observed in ctsB-/- BMDMs 12 hrs after transfection; at this time point, the rates of TUNEL positive cells among BMDMs from these three mouse lines became indistinguishable (Fig 6F). Consistent with these results, RpsL is able to induce the release of cathepsins such as aspartic cathepsin into the cytosol in ctsB-/- BMDMs (S10 Fig). These cathepsins may be responsible for the cell death observed when extended induction time was allowed after transfection. Taken together, these results establish that the lack of CtsB does not block but does significantly delays the cell death induced by RpsL.
Cathepsins in the cytoplasm are able to activate cell death by cleaving the BH3-Only protein Bid to produce truncated Bid (tBid), which subsequently inserts into the mitochondrial membranes, leading to cytochrome c (Cyto c) release and caspase activation [41] (Fig 7A). Consistent with this notion, tBid was significantly more abundant in macrophages infected with strain Lp02rpsLWT than in those infected with strain Lp02rpsLK88R or samples infected with the dotA mutant (Fig 7B). Activation of caspase-3 and the subsequent cleavage of poly(ADP-ribose) polymerase (cPARP) also occurred more robustly in macrophages infected with Lp02rpsLWT (Fig 7B). Caspase-3 is activated in permissive macrophages infected with strains harboring RpsLK88R [12,42]; In agreement with the notion that RpsL induces cell death, such activation was significantly more robust in BMDMs infected with strains expressing wild type RpsL (Fig 7B). Furthermore, significantly more activated caspase-3 and cPARP can be detected in macrophages that received recombinant RpsL than those that received RpsLK88R (Fig 7C). Consistent with the involvement of the activation of caspase-3 here, both DEVD-FMK, a cell-permeable, irreversible caspase-3-specific inhibitor [43] and the pan caspase inhibitor z-VAD-FMK [44] significantly dampened the RpsL-induced cell death within the first 6 hrs of protein transfection (Fig 7D–E). Thus, the canonical apoptosis pathway contributes to the cell death induced by RpsL.

We next examined the ability of BMDMs from ctsB-/- mice to support replication of the strain Lp02rpsLWT∆flaA. Consistent with earlier observations [17,18], deletion of flaA allowed strain Lp02rpsLK88R to replicate in BMDMs from the C57BL/6 moue background. Despite the lack of cell death induced by strain Lp02rpsLWT∆flaA at 12 hrs post infection (Fig 6D), ctsB-/- BMDMs were unable to support intracellular growth of this strain (S11 Fig). This suggests that the cell death occurred after bacterial replication has been aborted or that the cell death is one of the branches of the signaling cascade induced by RpsL, which is consistent with the plasma membrane damage observed in ctsB-/- BMDMs that received recombinant RpsL (Fig 6E).
Discussion
Genetically tractable pathogens are effective tools for probing host immune recognition mechanisms. By exploiting the cell death phenotype that restricts the replication of L. pneumophila in primary macrophages, we revealed that such cell death occurs in infections with essentially all wild type (streptomycin-sensitive) L. pneumophila strains, clinical or environmental. More importantly, we found that the ribosomal protein RpsL is directly responsible for such restriction. Our results supports a model in which RpsL activates a pathway that leads to lysosomal membrane permeabilization (LMP) and the subsequent apoptotic cell death, which is accompanied by the termination of intracellular bacteria replication.
It seems clear that RpsL is responsible for the macrophage death caused by L. pneumophila infection. A K88R mutation in RpsL allows the bacteria to replicate proficiently in these cells and the replacement of such mutations with the wild type allele in a widely used replication-competent laboratory strain abolished its ability to grow intracellularly in BMDMs. Although RpsLK88R confers resistance to streptomycin, several lines of evidence indicate that this antibiotic apparently plays no role in intracellular replication. First, although the rpsLK43N mutation confers resistance to streptomycin indistinguishably to that of the K88R mutation, mutants harboring RpsLK43N were unable to grow in BMDMs. Second, expression of a streptomycin inactivation enzyme did not allow the bacteria to grow intracellularly. Third, purified RpsL but not RpsM, another ribosomal protein, induces cell death in macrophages. Fourth, RpsL-induced cell death is independent of caspase 1 or 11, indicating that the potential flagellin [4,5] or LPS [10] contaminants did not contribute to this process. The insensitivity of the RpsL protein preparations to RNase indicates that potential residual RNA [45] is not implicated in the phenotype. Finally, RpsL from other bacteria such as E. coli was able to trigger macrophage cell death, suggesting that this protein is a universal ligand.
Although RpsL is a highly conserved protein, the distinct cell death phenotype has not yet been observed in infections caused by other intracellular pathogens. Such difference most likely results from the nature of the Dot/Icm type IV transporter of L. pneumophila, which recognizes hundreds of substrates with diverse primary sequences in the region essential for translocation [12,46]. Although bacteria lysed by macrophages may generate some cytosolic RpsL, we prefer a model in which the relatively promiscuous substrate recognition nature of the Dot/Icm system “accidentally” delivers a sufficient amount of RpsL into the cytosol of macrophages to trigger the response. The importance of the Dot/Icm transporter in this process is further supported by the fact that deletion of its essential component gene dotA abolishes the ability of the environmental strain LPE509 to cause cell death [20]. This Dot/Icm-dependent phenotype is akin to that of flagellin, which dictates the susceptibility of mouse lines to L. pneumophila infection [17,18]. Flagellin from other bacteria is similarly potent in the activation of the NLRC4 inflammasome but does not exhibit such distinct phenotypes in infections [4,5]. Flagellin has not yet been shown to be translocated into host cells by L. pneumophila with standard reporters. Similarly, we cannot detect Dot/Icm-dependent delivery of RpsL into host cells. Thus, although these proteins can be robustly sensed by their cognate immune recognition system, the amount delivered by the transporter is beyond the sensitivity of current methods for measuring protein translocation.
RpsL is not the first ribosome component shown to be able to trigger immune responses. In plants, EF-Tu, an accessory ribosomal protein is recognized by the receptor kinases EFR, a member of the PRRs, to induce antibacterial immunity [47]. In mice, sensing the 23S rRNA via TRL13 leads to the induction of cytokines such as interleukin-1β [48]. The protozoan profilin-like protein, a non-ribosomal yet integral intracellular component of microorganisms has also been established as the ligand recognized by TLR11 [49].
It is worth noting that methylation of an adenosine in the antibiotic-binding site of 23S rRNA camouflages it from TLR13, and bacteria carrying this modification are resistant to antibiotics such as erythromycin [48]. Similar to this observation, Lysine 88 of RpsL is directly involved in binding to streptomycin, and the mutation of this residue to arginine confers resistance to the antibiotic [21]. Clearly this mutation also renders RpsL less recognizable by macrophages, probably by altering the charging status, conformation or a combination of both. Consistent with this notion, nonpathogenic E. coli strains harboring rpsLK88R survived significantly better than their wild type counterparts in macrophages [50], likely due to less immune activation by RpsLK88R from mutant bacteria. The K88R mutation in RpsL may represent an evolved mechanism that enables the bacteria to gain resistance to both antibiotics and the innate immunity. Alternatively, antibiotics and host innate immune sensors could have converged to recognize the same constrained epitope of the pathogen.
The lysosome is emerging as a signal integration center for various stresses, including those that lead to cell death [38]. Our results indicate that sensing of RpsL leads to the activation of the lysosomal cell death pathway and the induction of LMP, which in turn releases the lysosomal hydrolases, particularly cathepsin B. In agreement with this notion, the cell death cannot be blocked by inhibitors of necrosis or classical apoptosis [20] but can be abrogated by agents targeting vacuolar acidification or cathepsins. LMP can be caused by diverse molecules such as reactive oxygen species (ROS), lipids and certain activated receptors such as TNF-α [38]. It is becoming evident that lysosomes do not indiscriminately release their contents, but rather, certain signals cause selective release of proteins such as cathepsins. For example, selective release of CtsB in response to different signals in different contexts leads to either tumor growth or apoptosis [51–54]. In neutrophils, cytosolic cathepsin D plays a major role in activating caspase-8 to resolve inflammation [55]. One challenge for future studies is to determine the mechanisms that govern the selectivity of cathepsin release in response to specific cues such as RpsL.
The inability of BMDMs from ctsB-/- mice to support intracellular replication of L. pneumophila strains expressing wild type RpsL is consistent with the apparent multiple effects of CtsB in cell death, including its role in the amplification of events of apoptosis [38] and, in some cell types, the timing of apoptosis [56]. It also agrees well with the fact that the plasma membranes of BMDMs from ctsB-/- can still be damaged after RpsL is introduced. CtsB itself is capable of directly causing cell death directly or by serving as a signal amplifier, which has been documented in TNF-α associated LMP [57]. Alternatively, in addition to cell death, sensing RpsL by BMDMs may activate other yet unidentified pathways that lead to the arrest of bacterial replication. This potential multifaceted mechanism of action differs from that of a terminal cell death enzyme such as caspase-1, whose absence leads to limited growth of L. pneumophila strains expressing flagellin [58]. It is also possible that the arrest of bacterial replication occurs prior to macrophage death. One focus of the future studies will be the mechanism underlying both the cell death and the intracellular replication phenotypes.
That the cell death occurs only in primary mouse macrophages suggests that RpsL does not directly damage the lysosomal membranes and that its putative receptor clearly is more abundant in or specifically functions in these cells. Whether similar responses occurs in immortalized mouse macrophage lines remains to be determined. Furthermore, it is possible that the pathway activated by RpsL exists only in mouse cells, perhaps because human cells are defective in one or more components of the pathway such as the putative receptor. Such potential differences may explain the fact that L. pneumophila strains expressing wild type RpsL are able to cause successful infections in humans. It is likely that RpsL engages the putative receptor to activate or to initiate the production of molecules capable of causing LMP. That RpsL-induced macrophage death occurs independent of several inflammatory caspases is consistent with the observation that we did not detect significant induction of inflammatory cytokines in macrophages upon RpsL delivery. The lysosomal cell death process is involved in the activation of the NLRC4 and NLRP3 inflammasomes that are induced by flagellin and cholesterol crystals, respectively [36,59]; whether RpsL-induced cell death is accompanied by any inflammation awaits further investigation. Clearly, identification of the putative receptor for RpsL will greatly facilitate the elucidation of the signaling pathway that leads to LMP. Such results will also aid in the study of the mechanism underlying the differences between mouse and humans in their response to wild type L. pneumophila.
Materials and Methods
Ethics statement
Primary murine macrophages were prepared from indicated mice lines, and the experiments were performed in strict accordance with the regulations of the Public Health Services (PHS) Policy on Humane Care and Use of Laboratory Animals. All animal procedures were performed according to a protocol approved by the Purdue Animal Care and Use Committee (protocol number: 04–081).
Bacterial strains, plasmids and media
Legionella pneumophila strain Philadelphia 1 [24], a generous gift from Dr. Isberg (Tufts Medical School, Boston, MA), its derivatives Lp02 (dot/icm+) (Lp02rpsLK88R) [24], Lp03(dotA-) (rpsLK88R) [24], JR32 [25] were used. Strains Paris [27] and Thunder Bay [28] were from Dr. Alex W. Ensminger (University of Toronto). For strains Lp02 and Lp03 and their derivatives that are thymidine auxotrophic [24], we introduced plasmid pJB908 expressing thyA [60] to make them thymidine autotrophic for all experiments. The environmental strain LPE509 was described earlier [20]. L. pneumophila was cultured on charcoal-yeast extract (CYE) plates or in ACES-buffered yeast extract (AYE) broth [61]. Escherichia coli strains were grown on L-agar plates or L broth, antibiotics were used at the following concentrations: For E. coli, Amp, 100 μg/ml; Km, 30 μg/ml; streptomycin 50 μg/ml. For L. pneumophila, Km was used at 20 μg/ml, Amp was used at 100 μg/ml and streptomycin was used at 100 μg/ml.
Spontaneous streptomycin resistant mutants were isolated by plating cultures of strain LPE509 onto AYE plates containing 100 μg/ml streptomycin. Colonies from independent cultures were purified and used for described experiments. The rpsL locus of each mutant was amplified by PCR and the PCR products were sequenced to determine the mutation. The sequence of the 16S rRNA was also determined via similar methods.
Allele exchange of the rpsL gene was performed using the standard method [62]. Briefly, the open reading frame of rpsL (lpg0324) together with a 300-bp flanking sequence was amplified from genomic DNA of wild type or rpsLK88R mutant strain of LPE509. The DNA digested with appropriate restriction enzymes was ligated to the pir protein-dependent plasmid pSR47s [63], to produce pZLrpsL and pZLrpsLK88R, respectively. The primers used were: 5′-CTGGAGCTCAAAAGAAAACGTGATGGTAG-3′ and 5′-CTGGTCGACGTACTTCCA CACTTGGGCGA-3′. Plasmids were introduced into appropriate L. pneumophila strains by electroporation, and transformants were streaked onto CYE plate supplemented with 5% sucrose and the desired strains were screened by the gain or loss of resistance to streptomycin followed by DNA sequencing of the locus.
Mice, bone marrow-derived macrophage, cell culture and bacterial infection
A/J and C56BL/6 mice were purchased from the Jackson laboratory (Bar Harbor, Maine). Caspase 1/11-/- [64] and the corresponding C56BL/6 control mice were similarly maintained. Mice lacking caspase 1, 7 and 11 were generated by intercrossing Casp1/11-/- mice [65] and Casp7-/-mice [66] (purchased from Jackson Labs, Bar Harbor, ME). The cathepsin B [52] and cathepsin H [67] knock mice (cstB-/-, cstH-/-) and the control mice were maintained in the animal facility of New York University School of Medicine. Bone marrow-derived macrophages were prepared from 6–10-week old female mice with L-supernatant as described previously [68]. Macrophages were seeded in 24-well plate the day before infection. Cell density of 4x105/well was used for intracellular bacterial growth and LDH release assay, whereas 2x105/well was used for immunofluorescence staining and other single cell-based assays. L. pneumophila used for infection was grown in AYE broth to post-exponential phase based on both optical value (OD600 = 3.4–4.0) and bacterial motility.
Intracellular bacterial growth
For intracellular growth curve in mouse macrophages, we infected the cells plated on 24-well at a multiplicity of infection (MOI) of 0.05. Two hours after adding the bacteria, we synchronized the infection by washing the cells with PBS for three times. Infected macrophages were incubated at 37°C with 5% CO2. At the desired time point, the cells were lysed with 0.02% saponin, diluted lysate was plated on CYE plates, and CFU were determined from triplicate infections of each strain.
Antibodies, immunoblotting and immunostaining of infected macrophages
Mouse macrophages seeded on coverslips were infected with L. pneumophila at an MOI of 1, extracellular bacteria were removed by washing the cells with PBS for three times. Infections were terminated at appropriate time points by fixing with 4% paraformeldehyde. After fixation, samples were first stained for extracellular or total bacteria using anti-L. pneumophila antibodies [29] at a dilution of 1 : 10,000 and appropriate secondary antibodies conjugated to distinct fluorescent dyes. Processed coverslips were mounted on glass slides with an anti-fade reagent (Vector laboratories, CA). Samples were examined by visual inspection with an Olympus IX-81 fluorescence microscope.
For western blotting, anti-cleaved PARP was purchased from Abcam (ab2317). Anti-mouse-BID was purchased from R&D System (MAB860). Anti-cleaved caspase-3 was purchased from Cell Signaling Technology (#9664). Anti-tubulin was purchased from Developmental Studies Hybridoma at University of Iowa.
TUNEL staining
2x105 mouse macrophages were infected with relevant L. pneumophila at an MOI of 1, and incubated for 12 hours after infection. Samples were fixed and stained with intracellular and extracellular bacteria as described above, and then stained with TUNEL using the In Situ Cell Death Detection Kit (Roche Diagnostics, Indianapolis). Fifty microliter TUNEL reaction mixture was added to each coverslip and incubated at 37°C for 1 hr. After 3x washes with PBS, coverslips were mounted with anti-fade reagent.
Intracellular protein delivery
Recombinant proteins were purified as previously described [69]. Briefly, recombinant proteins bound to Ni-NTA columns were subjected to extensive organic solvent wash (50x bed volume with wash buffer containing 60% isopropanol) to remove the majority of endotoxin contaminants [29]. β-galactosidase was purchased from Clonetech. If necessary, BMDMs were treated with lysosome acidification inhibitor bafilomycin A1 (20 nM), cathepsin B inhibitor CA-074ME (25 μM), cathepsin D inhibitor pepstatin A (25 μM) or DMSO (vehicle) for 1 hour before protein transfection. The purified proteins were then introduced into macrophages using Lipofectamine 2000 as follows: For transfection of 2x105 macrophages, 12.5-μg recombinant protein suspended in 50-μl RPMI-1640 and 2-μl Lipofectamine 2000 suspended in 50-μl RPMI-1640 were equilibrated at RT for 5 min, then mixed at RT for 30 min. The mixture was then directly applied to the macrophage cultures.
Acridine orange staining
BMDMs plated on glass coverslips at a density of 2x105/well were either transfected with appropriate proteins or infected with different L. pneumophila strains at an MOI of 1. For infection, the samples were washed 3 times with warm PBS to synchronize the infection 2 hrs after adding the bacteria. The transfected/infected samples were incubated for 8 hrs. After centrifugation at 200g for 5 min, the culture medium was removed, and 0.5 ml of the dye solution containing acridine orange (5 μg/ml) was added into each well. The plate was incubated at 37°C for 15 min before imaging using a fluorescence microscope (Olympus IX-81).
LDH release assay
LDH release during infection was determined using CytoTox 96 Assay (Promega, Madison, WI). 4x105 BMDMs were either transfected with appropriate proteins or infected with appropriate L. pneumophila strains at an MOI of 1. Four or six hours post transfection/infection, cell culture was centrifuged at 200g for 5 min, and 50-μl supernatant of each well was transferred to a new 96-well enzymatic assay plate. Fifty microliter reconstituted Substrate Mix was then added to each well of enzymatic assay plate, and incubated in dark at room temperature for 30 min. The enzymatic reaction was stopped by adding 50-μl stop solution to each well. The absorbance at 490 nm was measured using a Biotek microplate reader. Total LDH release was determined by complete lysis of the cells using a lysis solution provided by the manufacturer, and spontaneous LDH release was determined by using the supernatant of cells without infection/transfection. The percentage of LDH release was calculated with the follow formula: LDH release (%) = (Experimental LDH release-Spontaneous LDH release)/(Total LDH release-Spontaneous LDH release)x100.
Cytosolic cathepsin activity
4×105 BMDMs from A/J mice were plated in 24-well plates. RpsL proteins were delivered into the cells as described above. 6 hours post protein transfection, cytosolic cathepsin B activity was determined as previously described [70]. Briefly, macrophages were washed with phenol red and HEPES free DMEM (Invitrogen, OR). 100μg/ml saponin was added to each well and samples are incubated for 10 min on ice. Cell lystae was cleared at 500xg for 10 min at 4°C in flat-bottom 96-well plates. 10 μl of the cell lysate was then added to 90 μl of the cathepsin B substrate buffer (100 μM zRR-AMC, 50 mM sodium acetate [pH 6.0], 4 mM EDTA, 10 mM dithiothreitol, 1 mM Pefablock). The generation of free AMC was determined by recording fluorescence (excitation, 355 nm; emission, 460 nm) at 30° in a TECAN multiplate reader. Cathepsin B activity was determined by measuring the increase in AMC fluorescence. Aspartic cathepsin activity was determined by measuring the aspartic cathepsin activity in total (0.02% Triton) or cytosolic fraction using Mca-KPLGL-Dpa-AR-NH2 Fluorogenic Peptide (R&D, cat# ES010), in a buffer containing 0.1 M NaOAc and 0.2 M NaCl, pH 3.5.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Akira S, Uematsu S, Takeuchi O (2006) Pathogen recognition and innate immunity. Cell 124 : 783–801. 16497588
2. Ting JP, Willingham SB, Bergstralh DT (2008) NLRs at the intersection of cell death and immunity. Nat Rev Immunol 8 : 372–379. doi: 10.1038/nri2296 18362948
3. Schroder K, Tschopp J (2010) The inflammasomes. Cell 140 : 821–832. doi: 10.1016/j.cell.2010.01.040 20303873
4. Kofoed EM, Vance RE (2011) Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature 477 : 592–595. doi: 10.1038/nature10394 21874021
5. Zhao Y, Yang J, Shi J, Gong YN, Lu Q, et al. (2011) The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477 : 596–600. doi: 10.1038/nature10510 21918512
6. Yang J, Zhao Y, Shi J, Shao F (2013) Human NAIP and mouse NAIP1 recognize bacterial type III secretion needle protein for inflammasome activation. Proc Natl Acad Sci U S A 110 : 14408–14413. doi: 10.1073/pnas.1306376110 23940371
7. Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES (2009) AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458 : 509–513. doi: 10.1038/nature07710 19158676
8. Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, et al. (2009) AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458 : 514–518. doi: 10.1038/nature07725 19158675
9. Warren SE, Armstrong A, Hamilton MK, Mao DP, Leaf IA, et al. (2010) Cutting edge: Cytosolic bacterial DNA activates the inflammasome via Aim2. J Immunol 185 : 818–821. doi: 10.4049/jimmunol.1000724 20562263
10. Shi J, Zhao Y, Wang Y, Gao W, Ding J, et al. (2014) Inflammatory caspases are innate immune receptors for intracellular LPS. Nature.
11. Lamkanfi M, Dixit VM (2010) Manipulation of host cell death pathways during microbial infections. Cell Host Microbe 8 : 44–54. doi: 10.1016/j.chom.2010.06.007 20638641
12. Zhu W, Banga S, Tan Y, Zheng C, Stephenson R, et al. (2011) Comprehensive identification of protein substrates of the Dot/Icm type IV transporter of Legionella pneumophila. PLoS One 6: e17638. doi: 10.1371/journal.pone.0017638 21408005
13. Lifshitz Z, Burstein D, Peeri M, Zusman T, Schwartz K, et al. (2013) Computational modeling and experimental validation of the Legionella and Coxiella virulence-related type-IVB secretion signal. Proc Natl Acad Sci U S A 110: E707–715. doi: 10.1073/pnas.1215278110 23382224
14. Huang L, Boyd D, Amyot WM, Hempstead AD, Luo ZQ, et al. The E Block motif is associated with Legionella pneumophila translocated substrates. Cell Microbiol 13 : 227–245. doi: 10.1111/j.1462-5822.2010.01531.x 20880356
15. Fontana MF, Vance RE (2011) Two signal models in innate immunity. Immunol Rev 243 : 26–39. doi: 10.1111/j.1600-065X.2011.01037.x 21884165
16. Vance RE (2010) Immunology taught by bacteria. J Clin Immunol 30 : 507–511. doi: 10.1007/s10875-010-9389-2 20373001
17. Ren T, Zamboni DS, Roy CR, Dietrich WF, Vance RE (2006) Flagellin-deficient Legionella mutants evade caspase-1 - and Naip5-mediated macrophage immunity. PLoS Pathog 2: e18. 16552444
18. Molofsky AB, Byrne BG, Whitfield NN, Madigan CA, Fuse ET, et al. (2006) Cytosolic recognition of flagellin by mouse macrophages restricts Legionella pneumophila infection. J Exp Med 203 : 1093–1104. 16606669
19. Fontana MF, Banga S, Barry KC, Shen X, Tan Y, et al. (2011) Secreted bacterial effectors that inhibit host protein synthesis are critical for induction of the innate immune response to virulent Legionella pneumophila. PLoS Pathog 7: e1001289. doi: 10.1371/journal.ppat.1001289 21390206
20. Tao L, Zhu W, Hu BJ, Qu JM, Luo ZQ (2013) Induction of rapid cell death by an environmental isolate of Legionella pneumophila in mouse macrophages. Infect Immun 81 : 3077–3088. doi: 10.1128/IAI.00252-13 23753633
21. Springer B, Kidan YG, Prammananan T, Ellrott K, Bottger EC, et al. (2001) Mechanisms of streptomycin resistance: selection of mutations in the 16S rRNA gene conferring resistance. Antimicrob Agents Chemother 45 : 2877–2884. 11557484
22. Angst DC, Hall AR (2013) The cost of antibiotic resistance depends on evolutionary history in Escherichia coli. BMC Evol Biol 13 : 163. doi: 10.1186/1471-2148-13-163 23914906
23. Van den Eede G, Deblaere R, Goethals K, Van Montagu M, Holsters M (1992) Broad host range and promoter selection vectors for bacteria that interact with plants. Mol Plant Microbe Interact 5 : 228–234. 1421510
24. Berger KH, Isberg RR (1993) Two distinct defects in intracellular growth complemented by a single genetic locus in Legionella pneumophila. Mol Microbiol 7 : 7–19. 8382332
25. Sadosky AB, Wiater LA, Shuman HA (1993) Identification of Legionella pneumophila genes required for growth within and killing of human macrophages. Infect Immun 61 : 5361–5373. 8225610
26. Rao C, Benhabib H, Ensminger AW (2013) Phylogenetic reconstruction of the Legionella pneumophila Philadelphia-1 laboratory strains through comparative genomics. PLoS One 8: e64129. doi: 10.1371/journal.pone.0064129 23717549
27. Aurell H, Etienne J, Forey F, Reyrolle M, Girardo P, et al. (2003) Legionella pneumophila serogroup 1 strain Paris: endemic distribution throughout France. J Clin Microbiol 41 : 3320–3322. 12843082
28. Khan MA, Knox N, Prashar A, Alexander D, Abdel-Nour M, et al. (2013) Comparative Genomics Reveal That Host-Innate Immune Responses Influence the Clinical Prevalence of Serogroups. PLoS One 8: e67298. 23826259
29. Xu L, Shen X, Bryan A, Banga S, Swanson MS, et al. (2010) Inhibition of host vacuolar H+-ATPase activity by a Legionella pneumophila effector. PLoS Pathog 6: e1000822. doi: 10.1371/journal.ppat.1000822 20333253
30. Erener S, Petrilli V, Kassner I, Minotti R, Castillo R, et al. (2012) Inflammasome-activated caspase 7 cleaves PARP1 to enhance the expression of a subset of NF-kappaB target genes. Mol Cell 46 : 200–211. doi: 10.1016/j.molcel.2012.02.016 22464733
31. Aachoui Y, Leaf IA, Hagar JA, Fontana MF, Campos CG, et al. (2013) Caspase-11 protects against bacteria that escape the vacuole. Science 339 : 975–978. doi: 10.1126/science.1230751 23348507
32. Sharma D, Cukras AR, Rogers EJ, Southworth DR, Green R (2007) Mutational analysis of S12 protein and implications for the accuracy of decoding by the ribosome. J Mol Biol 374 : 1065–1076. 17967466
33. Kurland CG (1992) Translational accuracy and the fitness of bacteria. Annu Rev Genet 26 : 29–50. 1482115
34. Rintahaka J, Lietzen N, Ohman T, Nyman TA, Matikainen S (2011) Recognition of cytoplasmic RNA results in cathepsin-dependent inflammasome activation and apoptosis in human macrophages. J Immunol 186 : 3085–3092. doi: 10.4049/jimmunol.1002051 21257972
35. Charpentier X, Gabay JE, Reyes M, Zhu JW, Weiss A, et al. (2009) Chemical genetics reveals bacterial and host cell functions critical for type IV effector translocation by Legionella pneumophila. PLoS Pathog 5: e1000501. doi: 10.1371/journal.ppat.1000501 19578436
36. Lage SL, Buzzo CL, Amaral EP, Matteucci KC, Massis LM, et al. (2013) Cytosolic flagellin-induced lysosomal pathway regulates inflammasome-dependent and-independent macrophage responses. Proc Natl Acad Sci U S A 110: E3321–3330. doi: 10.1073/pnas.1305316110 23942123
37. Guicciardi ME, Leist M, Gores GJ (2004) Lysosomes in cell death. Oncogene 23 : 2881–2890. 15077151
38. Aits S, Jaattela M (2013) Lysosomal cell death at a glance. J Cell Sci 126 : 1905–1912. doi: 10.1242/jcs.091181 23720375
39. Yoshimori T, Yamamoto A, Moriyama Y, Futai M, Tashiro Y (1991) Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J Biol Chem 266 : 17707–17712. 1832676
40. Boya P, Kroemer G (2008) Lysosomal membrane permeabilization in cell death. Oncogene 27 : 6434–6451. doi: 10.1038/onc.2008.310 18955971
41. Ivanova S, Repnik U, Bojic L, Petelin A, Turk V, et al. (2008) Lysosomes in apoptosis. Methods Enzymol 442 : 183–199. doi: 10.1016/S0076-6879(08)01409-2 18662570
42. Molmeret M, Zink SD, Han L, Abu-Zant A, Asari R, et al. (2004) Activation of caspase-3 by the Dot/Icm virulence system is essential for arrested biogenesis of the Legionella-containing phagosome. Cell Microbiol 6 : 33–48. 14678329
43. Thornberry NA, Lazebnik Y (1998) Caspases: enemies within. Science 281 : 1312–1316. 9721091
44. Slee EA, Zhu H, Chow SC, MacFarlane M, Nicholson DW, et al. (1996) Benzyloxycarbonyl-Val-Ala-Asp (OMe) fluoromethylketone (Z-VAD.FMK) inhibits apoptosis by blocking the processing of CPP32. Biochem J 315 (Pt 1): 21–24. 8670109
45. Kowalinski E, Lunardi T, McCarthy AA, Louber J, Brunel J, et al. (2011) Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell 147 : 423–435. doi: 10.1016/j.cell.2011.09.039 22000019
46. Isberg RR, O’Connor TJ, Heidtman M (2009) The Legionella pneumophila replication vacuole: making a cosy niche inside host cells. Nat Rev Microbiol 7 : 13–24. doi: 10.1038/nrmicro1967 19011659
47. Zipfel C, Kunze G, Chinchilla D, Caniard A, Jones JD, et al. (2006) Perception of the bacterial PAMP EF-Tu by the receptor EFR restricts Agrobacterium-mediated transformation. Cell 125 : 749–760. 16713565
48. Oldenburg M, Kruger A, Ferstl R, Kaufmann A, Nees G, et al. (2012) TLR13 recognizes bacterial 23S rRNA devoid of erythromycin resistance-forming modification. Science 337 : 1111–1115. doi: 10.1126/science.1220363 22821982
49. Yarovinsky F, Zhang D, Andersen JF, Bannenberg GL, Serhan CN, et al. (2005) TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science 308 : 1626–1629. 15860593
50. Miskinyte M, Gordo I (2013) Increased survival of antibiotic-resistant Escherichia coli inside macrophages. Antimicrob Agents Chemother 57 : 189–195. doi: 10.1128/AAC.01632-12 23089747
51. Foghsgaard L, Wissing D, Mauch D, Lademann U, Bastholm L, et al. (2001) Cathepsin B acts as a dominant execution protease in tumor cell apoptosis induced by tumor necrosis factor. J Cell Biol 153 : 999–1010. 11381085
52. Gocheva V, Zeng W, Ke D, Klimstra D, Reinheckel T, et al. (2006) Distinct roles for cysteine cathepsin genes in multistage tumorigenesis. Genes Dev 20 : 543–556. 16481467
53. Vasiljeva O, Papazoglou A, Kruger A, Brodoefel H, Korovin M, et al. (2006) Tumor cell-derived and macrophage-derived cathepsin B promotes progression and lung metastasis of mammary cancer. Cancer Res 66 : 5242–5250. 16707449
54. Bruchard M, Mignot G, Derangere V, Chalmin F, Chevriaux A, et al. (2013) Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat Med 19 : 57–64. doi: 10.1038/nm.2999 23202296
55. Conus S, Perozzo R, Reinheckel T, Peters C, Scapozza L, et al. (2008) Caspase-8 is activated by cathepsin D initiating neutrophil apoptosis during the resolution of inflammation. J Exp Med 205 : 685–698. doi: 10.1084/jem.20072152 18299403
56. Oberle C, Huai J, Reinheckel T, Tacke M, Rassner M, et al. (2010) Lysosomal membrane permeabilization and cathepsin release is a Bax/Bak-dependent, amplifying event of apoptosis in fibroblasts and monocytes. Cell Death Differ 17 : 1167–1178. doi: 10.1038/cdd.2009.214 20094062
57. Werneburg NW, Guicciardi ME, Bronk SF, Gores GJ (2002) Tumor necrosis factor-alpha-associated lysosomal permeabilization is cathepsin B dependent. Am J Physiol Gastrointest Liver Physiol 283: G947–956. 12223355
58. Zamboni DS, Kobayashi KS, Kohlsdorf T, Ogura Y, Long EM, et al. (2006) The Birc1e cytosolic pattern-recognition receptor contributes to the detection and control of Legionella pneumophila infection. Nat Immunol 7 : 318–325. 16444259
59. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, et al. (2010) NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464 : 1357–1361. doi: 10.1038/nature08938 20428172
60. Sexton JA, Pinkner JS, Roth R, Heuser JE, Hultgren SJ, et al. (2004) The Legionella pneumophila PilT homologue DotB exhibits ATPase activity that is critical for intracellular growth. J Bacteriol 186 : 1658–1666. 14996796
61. Fraser DW, Tsai TR, Orenstein W, Parkin WE, Beecham HJ, et al. (1977) Legionnaires’ disease: description of an epidemic of pneumonia. N Engl J Med 297 : 1189–1197. 335244
62. Dumenil G, Isberg RR (2001) The Legionella pneumophila IcmR protein exhibits chaperone activity for IcmQ by preventing its participation in high-molecular-weight complexes. Mol Microbiol 40 : 1113–1127. 11401716
63. Rankin S, Li Z, Isberg RR (2002) Macrophage-induced genes of Legionella pneumophila: protection from reactive intermediates and solute imbalance during intracellular growth. Infect Immun 70 : 3637–3648. 12065505
64. Broz P, Monack DM (2011) Molecular mechanisms of inflammasome activation during microbial infections. Immunol Rev 243 : 174–190. doi: 10.1111/j.1600-065X.2011.01041.x 21884176
65. Li P, Allen H, Banerjee S, Franklin S, Herzog L, et al. (1995) Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock. Cell 80 : 401–411. 7859282
66. Lakhani SA, Masud A, Kuida K, Porter GA Jr, Booth CJ, et al. (2006) Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science 311 : 847–851. 16469926
67. Gocheva V, Chen X, Peters C, Reinheckel T, Joyce JA (2010) Deletion of cathepsin H perturbs angiogenic switching, vascularization and growth of tumors in a mouse model of pancreatic islet cell cancer. Biol Chem 391 : 937–945. doi: 10.1515/BC.2010.080 20731543
68. Swanson MS, Isberg RR (1995) Association of Legionella pneumophila with the macrophage endoplasmic reticulum. Infect Immun 63 : 3609–3620. 7642298
69. Franken KL, Hiemstra HS, van Meijgaarden KE, Subronto Y, den Hartigh J, et al. (2000) Purification of his-tagged proteins by immobilized chelate affinity chromatography: the benefits from the use of organic solvent. Protein Expr Purif 18 : 95–99. 10648174
70. Newman ZL, Leppla SH, Moayeri M (2009) CA-074Me protection against anthrax lethal toxin. Infect Immun 77 : 4327–4336. doi: 10.1128/IAI.00730-09 19635822
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 3
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Bacterial Immune Evasion through Manipulation of Host Inhibitory Immune Signaling
- BILBO1 Is a Scaffold Protein of the Flagellar Pocket Collar in the Pathogen
- Antimicrobial-Induced DNA Damage and Genomic Instability in Microbial Pathogens
- Attenuation of Tick-Borne Encephalitis Virus Using Large-Scale Random Codon Re-encoding
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy