
Evidence for Ubiquitin-Regulated Nuclear and Subnuclear Trafficking among Matrix Proteins
Elucidating virus-cell interactions is fundamental to understanding viral replication and identifying targets for therapeutic control of viral infection. Paramyxoviruses include human and animal pathogens of medical and agricultural significance. Their matrix (M) structural protein organizes virion assembly at the plasma membrane and mediates viral budding. While nuclear localization of M proteins has been described for some paramyxoviruses, the underlying mechanisms of nuclear trafficking and the biological relevance of this observation have remained largely unexamined. Through comparative analyses of M proteins across five Paramyxovirinae genera, we identify M proteins from at least three genera that exhibit similar nuclear trafficking phenotypes regulated by an NLSbp as well as an NES sequence within M that may mediate the interaction of M with host nuclear transport receptors. Additionally, a conserved lysine within the NLSbp of some M proteins is required for nuclear export by regulating M ubiquitination. Sendai virus engineered to express a ubiquitination-defective M does not produce infectious virus but instead displays extensive cell-cell fusion while M is retained in the nucleolus. Thus, some Paramyxovirinae M proteins undergo regulated and active nuclear and subnuclear transport, a prerequisite for viral morphogenesis, which also suggests yet to be discovered roles for M in the nucleus.
Published in the journal:
. PLoS Pathog 11(3): e32767. doi:10.1371/journal.ppat.1004739
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004739
Summary
Elucidating virus-cell interactions is fundamental to understanding viral replication and identifying targets for therapeutic control of viral infection. Paramyxoviruses include human and animal pathogens of medical and agricultural significance. Their matrix (M) structural protein organizes virion assembly at the plasma membrane and mediates viral budding. While nuclear localization of M proteins has been described for some paramyxoviruses, the underlying mechanisms of nuclear trafficking and the biological relevance of this observation have remained largely unexamined. Through comparative analyses of M proteins across five Paramyxovirinae genera, we identify M proteins from at least three genera that exhibit similar nuclear trafficking phenotypes regulated by an NLSbp as well as an NES sequence within M that may mediate the interaction of M with host nuclear transport receptors. Additionally, a conserved lysine within the NLSbp of some M proteins is required for nuclear export by regulating M ubiquitination. Sendai virus engineered to express a ubiquitination-defective M does not produce infectious virus but instead displays extensive cell-cell fusion while M is retained in the nucleolus. Thus, some Paramyxovirinae M proteins undergo regulated and active nuclear and subnuclear transport, a prerequisite for viral morphogenesis, which also suggests yet to be discovered roles for M in the nucleus.
Introduction
Paramyxoviruses include pathogens of global medical and agricultural concern. These viruses occupy broad ecological niches infecting a wide range of hosts including mammals, reptiles, birds and fish, and they cause diverse outcomes ranging from asymptomatic infection to lethal disease. Measles virus (MeV), mumps virus (MuV), the human parainfluenza viruses (hPIVs), respiratory syncytial virus (RSV), and human metapneumoviruses remain significant causes of human morbidity and mortality [1]. Animal pathogens, such as Newcastle disease virus (NDV) and the recently eradicated Rinderpest virus [2], have caused significant rates of lethal disease in birds and cattle, respectively. The newly emergent zoonotic paramyxoviruses Nipah virus (NiV) and Hendra virus (HeV) are among the most deadly known pathogens, showing case-fatality rates in excess of 70% in humans, and are classified as biosafety level 4 pathogens due to the absence of vaccines or therapeutics approved for human use [3–6].
Paramyxoviruses are released as enveloped virions from the host cell plasma membrane. Virions are ~150–300 nm in diameter and are spherical, pleomorphic or filamentous in shape, depending on the virus and the producer cell-type. The non-segmented, single-strand, negative-sense RNA genomes of paramyxoviruses consist of six principal genes: nucleocapsid (N), phosphoprotein (P), matrix (M), fusion (F) and attachment (HN, H or G) glycoproteins, and polymerase (L) [1,5,7]. The attachment and fusion glycoproteins mediate binding to sialic acid moieties or to specific protein receptors on the cell surface and the fusion of the viral envelope with the host cell plasma membrane [8–10]. Within the virion, the ribonucleoprotein (RNP) consists of the RNA-dependent RNA polymerase complex formed by P and L associated with the N-encapsidated RNA genome. L is required for viral RNA synthesis during viral replication [1,5].
M is the primary viral structural protein [1,5,7]. A number of studies have found that M proteins oligomerize, bind lipids, and form a grid-like array on the inner surface of the viral membrane (_((((xxx))))_)[7,11–25]. M proteins can serve as a molecular scaffold by interacting with the cytoplasmic tails of the transmembrane glycoproteins and the RNP via N [7,17,25–35]. Many paramyxoviral M proteins (NiV-M, MeV-M, NDV-M, SeV-M, and hPIV1-M) can drive viral budding and form virus-like particles (VLPs) in the absence of other viral components [13,31,36–42], albeit with varying efficiencies. However, the budding of some others (PIV5-M and MuV-M) requires coexpression of N and/or the envelope glycoproteins [43,44]. MeV and SeV engineered with budding-defective or deleted M proteins have been found to have severe defects in viral replication [45–47].
Although paramyxoviruses are classic cytoplasmic replicating viruses, some paramyxoviral M proteins have been observed to traffic through the nucleus. For example, SeV-M, NDV-M and RSV-M can be detected in the nucleus at early stages of infection [48–53]. These findings suggest that paramyxoviral M proteins may perform roles beyond viral assembly at the plasma membrane. However, with the exception of RSV, which belongs to the Pneumovirinae subfamily, the cell biology of M protein nuclear trafficking has not been examined in a systematic fashion for most Paramyxovirinae subfamily members. We previously found that NiV-M translocates to the nucleus at early stages of infection. The high homology between NiV-M and HeV-M (~90% amino acid identity) suggests that HeV-M also localizes to the nucleus, and it was recently found that overexpression of ANP32B, a nuclear protein, results in nuclear accumulation of HeV-M and NiV-M [54]. We have shown that nuclear-cytoplasmic trafficking of NiV-M is mediated by a classical bipartite nuclear localization signal (NLSbp), homologous to NDV-M’s NLSbp, and a leucine-rich nuclear export signal (NES) [39,48]. We further demonstrated that nuclear trafficking is regulated by ubiquitination, presumably on a conserved lysine residue (K258) located within the NLSbp of NiV-M (244RR-X10-RRK258). The K258A mutant is defective in nuclear import, while the K258R mutant retains a functional NLS but is defective in nuclear export; both mutants have decreased levels of ubiquitination and have budding defects [39].
The canonical NES and NLSbp that we functionally characterized in NiV-M are highly conserved across most, if not all members of the Paramyxovirinae. Therefore, it is important to resolve whether ubiquitin-dependent nuclear-cytoplasmic trafficking of M is unique to NiV, or to what extent other members of the subfamily also exhibit a nuclear-trafficking phenotype. Uncovering the mechanisms that govern paramyxovirus M protein trafficking has direct bearing on the fundamental biology of paramyxoviral replication, and may reveal host-dependent pathways and factors that can be exploited for antiviral strategies. Here, we specifically analyze ubiquitin-dependent nuclear-cytoplasmic trafficking of M proteins across representative viruses from all five major genera of Paramyxovirinae (Respirovirus, Rubulavirus, Morbillivirus, Henipavirus, and Avulavirus). We use a panoply of methods including quantitative 3D confocal microscopy analysis of M nuclear localization, bimolecular fluorescence complementation (BiFC) assays of M ubiquitination, and introduction of M mutations into live recombinant viruses with the use of reverse genetics. Our findings demonstrate that ubiquitination of M, regulated by a lysine within the second basic patch of the NLSbp, critically modulates the subnuclear and nuclear-cytoplasmic trafficking of M proteins from prototypic viruses of the Henipavirus, Rubulavirus and Respirovirus genera. Proteomic identification of nuclear transport receptors and nuclear pore complex components that copurify with paramyxoviral M proteins further supports a model for active transport of M in and out of the nucleus, and also hints at possible non-structural functions of M proteins.
Results
Nuclear export of some Paramyxovirinae matrix proteins is regulated by the ubiquitin-proteasome system
Since the nuclear-cytoplasmic trafficking of the Nipah virus matrix protein (NiV-M) is regulated by its monoubiquitination [39], we wondered whether the ubiquitin-proteasome system similarly regulates the nuclear sojourn of other Paramyxovirinae M proteins. We cloned 3X-Flag - and GFP-tagged-M from prototypical members of the five Paramyxovirinae genera: NiV-M (genus Henipavirus), Hendra virus M (HeV-M, genus Henipavirus), Sendai virus M (SeV-M, genus Respirovirus), Mumps virus M (MuV-M, genus Rubulavirus), Newcastle disease virus M (NDV-M, genus Avulavirus), and Measles virus M (MeV-M, genus Morbillivirus). To biochemically detect ubiquitination of M proteins, we cotransfected HEK 293T cells with HA-UbK0 and each of 3X-Flag-tagged NiV-M, HeV-M, SeV-M, MuV-M, NDV-M or MeV-M [55]. HA-UbK0 functions as a ubiquitin (Ub) chain terminator or as monoubiquitin because all lysines have been mutated to arginines. We used this construct to visualize discrete ubiquitin bands and to determine if matrix proteins can be monoubiquitinated since this posttranslational modification can regulate the function of proteins without promoting proteasome-dependent protein degradation [56]. Cell lysates were subjected to anti-Flag immunoprecipitation (IP) and immunoblots were simultaneously probed with anti-HA and anti-Flag antibodies. As shown in Fig. 1A, for all the M proteins the majority of M is unmodified (M0) at steady state. However, a detectable minority of M (M1) is size-shifted by the molecular weight of at least one ubiquitin monomer (Ub, ~8.5 kDa) (Fig. 1A, merge). These results indicate that all 3X-Flag-tagged M proteins investigated are ubiquitin substrates.

We have found that proteasome inhibition results in nuclear retention of NiV-M in transfected and in NiV-infected cells (S1 Fig.) [39]. Proteasome inhibition stabilizes polyubiquitinated proteins and depletes the cellular levels of free ubiquitin available for conjugation [57–62]. To determine whether ubiquitination is involved in the nuclear export of the other Paramyxovirinae M proteins, we treated GFP-M-expressing HeLa cells with the proteasome inhibitor MG132 (Fig. 1B-G). We used quantitative 3D confocal microscopy to characterize the subcellular localization of M. The cells were counterstained with DAPI to visualize nuclei, and with fluorescent phalloidin to visualize the entire cell, and the proportion of nuclear M was determined computationally as described in Materials and Methods. As with GFP-NiV-M, ubiquitin depletion via proteasome inhibition resulted in significant nuclear retention of GFP-tagged HeV-M, SeV-M and MuV-M (Fig. 1B-E) [39]. We further confirmed biochemically that MG132 reduces the direct conjugation of ubiquitin to 3X-Flag-tagged NiV-M, HeV-M, SeV-M, MuV-M, MeV-M and NDV-M by co-IP of each and HA-UbK0, as described above, with quantification of immunoblot band integrated intensities as described in Materials and Methods (S2 Fig.). The ubiquitination of the various M proteins were differentially sensitive to proteasome inhibition with NDV-M and MeV-M being the least and most sensitive to proteasome inhibition, respectively (S2E–F Fig.). For NDV-M (Avulavirus) and MeV-M (Morbillivirus), reduction of ubiquitin conjugation did not result in a nuclear retention phenotype under the conditions and cell type examined (Fig. 1F-G), suggesting there is no strict correlation between the degree of matrix ubiquitination per se and M nuclear localization. In contrast, the ubiquitin-proteasome system appears to regulate the nuclear-cytoplasmic trafficking of the Henipavirus (NiV-M, HeV-M), Respirovirus (SeV-M), and Rubulavirus (MuV-M) matrix proteins.
Nuclear export of some Paramyxovirinae matrix proteins is regulated by a putative NES and a lysine within the NLSbp
We have shown that the nuclear-export of NiV-M is regulated by a leucine-rich nuclear export signal (NES) as well as by the K258 lysine residue located within the second basic patch of the bipartite nuclear localization signal (NLSbp; Fig. 2A, blue residues) [39]. A K258A mutation partially disrupts the NLSbp and decreases nuclear localization of NiV-M, while a K258R mutation is unexpectedly retained in the nucleus despite having intact NES sequences and preservation of the positive charge necessary for NLSbp function. However, both mutants are impaired for ubiquitination [39]. Sequence alignment of M proteins indicates that a lysine is present in the homologously aligned position across the Paramyxovirinae genera. Thus, we hypothesized that this residue might be conserved for regulation of M ubiquitin-dependent nuclear export (Fig. 2A, bold and underlined blue residues). To interrogate our hypothesis, we mutated the NLSbp-lysine to an arginine in all M proteins studied and analyzed their subcellular localization by quantitative 3D confocal microcopy as described above (Fig. 2B-G, quantified in Fig. 2H). Since NDV-M contains another lysine adjacent to this position we mutated both (Fig. 2A, bold and underlined blue residues). A lysine to arginine mutation is expected to preserve the nuclear import function of the putative NLSbp, but prevents posttranslational modification at that position. As a comparison for nuclear retention, we also mutated the leucines that correspond to the NES of NiV-M within all M proteins (Fig. 2A, bold and underlined blue residues) [39].

Mutation of the NES of GFP-NiV-M (ML106A L107A) resulted in a significant increase in nuclear localization of the protein, which confirms our previous findings (Fig. 2B, 2H) [39]. Similarly, but to varying degrees, GFP-tagged HeV-ML106A 107A, SeV-ML102A L103A, and MuV-ML106A also exhibited significantly increased nuclear retention compared to their respective wild type (WT) proteins (Fig. 2C-2E, 2H). In contrast, GFP-NDV-ML103A L106A had an apparent nuclear exclusion phenotype (Fig. 2F, 2H) contrary to expectations, while the nuclear localization of GFP-MeV-ML90A 191A was not significantly different than WT (Fig. 2G, 2H), indicating that these motifs are either redundant or non-functional in NDV-M and MeV-M.
Mutation of the NLSbp-lysine resulted in significantly enhanced nuclear localization of GFP-tagged NiV-MK258R, HeV-MK258R, SeV-MK254R, MuV-MK261R, and NDV-MK259R K260R, but not MeV-MK240R (Fig. 2B-2G). These phenotypic differences are quantified in Fig. 2H. Note that the spread in the degree of nuclear localization for any given M mutant also emphasizes the need to score a sufficient number of cells by computationally defined volumetric criteria (see Materials and Methods) in order to obtain robust statistics from inherently variable cell biological data. Thus, each data point in Fig. 2H (as in Fig. 1B-G) represents reconstructed volumetric data from a single cell, acquired from ~20–30 confocal optical Z-stacks (at 0.3–0.5 μm/step) per cell. In contrast to the NLSbp-lysine-to-arginine mutations, our attempt to disrupt the NLSbp consensus sequence through alanine substitutions in the second patch of basic residues (bp2) resulted in diffuse cytoplasmic localization of GFP-tagged NiV-M, HeV-M, SeV-M, and MuV-M (S3 Fig.). However, similar mutations did not appear to disrupt the localization of MeV-M or NDV-M (S3 Fig.) indicating that this motif does not function as the NLSbp in MeV-M or that additional mutations are necessary to fully disrupt the function of the NLSbp as has been previously shown for NDV-M [48].
Ubiquitination of Nipah, Hendra, Sendai and Mumps matrix proteins is dynamically regulated by an NLSbp lysine
Since the proteasome inhibitor MG132 inhibits the nuclear export of GFP-tagged NiV-M, HeV-M, SeV-M, and MuV-M (Fig. 1), we wanted to test whether the NLSbp-lysine that regulates their nuclear export (Fig. 2B-E) also regulates their ubiquitination. In Fig. 3, we first assessed the ability of 3X-Flag-tagged NiV-MK258R, HeV-MK258R, SeV-MK254R, MuV-MK261R, MeV-MK240R and NDV-MK259R K260R to be ubiquitinated biochemically, via co-IP of 3X-Flag-tagged-M with HA-UbK0, as described above (Fig. 3A-D). Although NDV-M and MeV-M did not exhibit a ubiquitin-dependent nuclear trafficking phenotype (Fig. 1F-G), we included NDV-MK259R K260R and MeV-MK240R in this experiment since the ubiquitin conjugation of WT NDV-M and MeV-M was sensitive to MG132 inhibition, albeit to varying degrees (S2E–F Fig.). We controlled for protein abundance by normalizing the integrated intensity of the Ub band by the integrated intensity of the total M (Ub/M0+M1). Using this measure, 3X-Flag-tagged NiV-MK258R (Fig. 3A), HeV-MK258R (Fig. 3B) and MeV-MK240R (Fig. 3E) exhibited the greatest reduction in relative monoubiquitination compared to the WT proteins (>70%), while 3X-Flag-tagged SeV-MK254R (Fig. 3C) and MuV-MK261R (Fig. 3D) showed only a modest to mild impairment in monoubiquitination (36% and ~15% reduction, respectively). 3X-Flag-tagged NDV-MK259R K260R did not display reduced ubiquitination (Fig. 3F). Residual ubiquitination of the matrix mutants indicates that other lysines within the 3X-Flag-tagged M proteins are also targets of ubiquitin conjugation. Table 1 summarizes the results obtained thus far: although ubiquitinated species can be detected for all six matrix proteins examined (Fig. 1A, S2 Fig.), only NiV-M, HeV-M, SeV-M, and, MuV-M displayed a ubiquitin-dependent nuclear-cytoplasmic trafficking phenotype (Fig. 1B-1E, S2B–S2E Fig.) that was also dependent on a lysine in the NLSbp (Fig. 2B-2E).


Ubiquitination is a dynamic process determined in part by the rates of conjugation versus de-conjugation, but our co-IP and immunoblot analysis of HA-UbK0-modified 3X-Flag-tagged M is a steady-state assay. It is possible that this assay for ubiquitinated M might not efficiently detect subtle differences that arise from such dynamic processes since i) ubiquitinated 3X-Flag-tagged M proteins are of low stoichiometry relative to unmodified native protein, ii) 3X-Flag-tagged M proteins might be mono - and/or polyubiquitinated on multiple lysines, further obscuring a contribution of any single lysine to the sum total ubiquitination, iii) ubiquitination is reversible, and iv) HA-UbK0 must compete with endogenous ubiquitin. In an attempt to overcome these issues, and to further assess whether NiV-MK258R, HeV-MK258R, SeV-MK254R and MuV-MK261R are impaired for ubiquitination, we developed a bimolecular fluorescence conjugation (BiFC) ubiquitination assay in which ubiquitin-conjugation of M produces an irreversible fluorescence signal (S4 Fig.) [63,64]. We fused Ub and M to split N - and C-terminal fragments of the fluorescent protein Venus, VN173 and VC155, respectively. Covalent conjugation of Ub to M brings the spilt Venus fragments into close proximity and allows the two otherwise non-fluorescent Venus fragments to reconstitute a functional fluorophore [65,66]. This complemented Venus will remain associated with M (via VC155-M) even if the VN173-Ub moiety is subsequently cleaved from M by a deubiquitinating enzyme (DUB), preserving an atemporal record of ubiquitin conjugation (S4A Fig.). Analysis of total cellular fluorescence showed that the Ub-M BiFC signal was decreased by almost 80% for the Henipavirus-M K258R mutants, confirming the significant role of K258 in ubiquitination of NiV-M (Fig. 3A, Fig. 4A) and HeV-M (Fig. 3B, Fig. 4B). In addition, we determined that the Ub-M BiFC signals for SeV-MK254R (Fig. 4C) and MuV-MK261R (Fig. 4D) were also significantly decreased in BiFC signal by nearly 70% compared to the WT proteins.

Ubiquitination and a lysine within the NLSbp regulate the subnuclear localization of Nipah, Hendra, Sendai and Mumps virus matrix proteins
We observed a punctate localization of GFP-tagged NiV-M, HeV-M, SeV-M, and MuV-M within DNA-void regions of the nucleus when cells were treated with MG132 (Fig. 1B-E). Native untagged NiV-M also exhibited similar subnuclear localization in NiV infected cells treated with bortezomib, an FDA-approved proteasome inhibitor (S1 Fig.). We determined that MG132 redistributes GFP-tagged NiV-M, HeV-M, SeV-M and MuV-M to nucleoli by counterstaining cells with anti-nucleolin antibodies (Fig. 5A-D, second vertical panels). Similarly, treatment of cells with MG132 caused a significant increase in the nucleolar localization of SeV-M during infection with live eGFP-expressing recombinant Sendai virus (rSeV-eGFP). This rSeV-eGFP is derived from a Fushimi strain engineered with mutations that permit replication in mammalian cells without the addition of trypsin as described in Materials and Methods (Fig. 6) [67]. The nucleolar localization of M proteins during ubiquitin depletion predicts that mutations in M that prevent efficient ubiquitination would also cause nucleolar retention. Indeed, GFP-tagged NiV-MK258R, HeV-MK258R, SeV-MK254R and MuV-MK261R phenocopied the MG132-induced nucleolar localization of the WT proteins (Fig. 5A-D, compare the second and third vertical panels). In contrast, the nuclear localized NES mutants, GFP-tagged NiV-ML106A 107A, HeV-ML106A 107A, SeV-ML102A L103A, and MuV-ML106A, were primarily enriched within the nucleoplasm and not the nucleolus. Thus, NES mutants are stalled at a different stage of subnuclear trafficking compared to the NLSbp-lysine mutants (Fig. 5A-D, fourth vertical panels). In sum, for the cognate paramyxovirus matrix proteins that exhibit a consistent nuclear trafficking phenotype that is both ubiquitin - and motif-dependent, our data supports a model where proper matrix ubiquitination is required for efficient nucleolar exit and/or preventing retention in the nucleolus.


Recombinant Sendai virus bearing matrix nuclear export mutants are defective for viral morphogenesis
We previously determined that NiV-ML106A 107A and NiV-MK258R are defective at budding virus like particles (VLPs) [39]. We wanted to compare the effects of the corresponding mutations in 3X-Flag-tagged SeV-M or MuV-M, however these proteins have a poor budding efficiency that is less than 10% of 3X-Flag-tagged Henipavirus-M proteins (Fig. 7A). This may be due to the presence of the 3X-Flag-tag or to the fact that SeV-M and MuV-M do not efficiently bud VLPs without the support of other viral proteins [41–43]. To overcome these technical difficulties and to study these mutations in a biologically relevant context, we engineered SeV-ML102A L103A and SeV-MK254R into a recombinant T7-driven, GFP-expressing Sendai virus genome (rSeV-eGFP) that can be rescued as live virus via the cotransfection of support plasmids expressing N, P, L (comprising the necessary replication complex) and a codon-optimized T7 polymerase. This highly efficient reverse genetics system allows us to quantify the number of rescue events directly in transfected producer cells at early time-points (see Materials and Methods). At two days post-transfection, GFP-positive cells (rescue events) could be observed by epifluorescence and quantified by FACS analysis. As a control for background GFP expression in the absence of virus production, we found that cotransfection of WT rSeV-eGFP and T7 polymerase without the N, P and L support plasmids resulted in no GFP-positive cells. We determined that rSeV-eGFP-ML102A L103A, and rSeV-eGFP-MK254R rescued at similar if not higher efficiencies than rSeV-eGFP-MWT (Fig. 7B). However, only rSeV-eGFP-MWT produced infectious viral titers (~107 I.U./ml) at day 6 post-rescue, while the mutants did not produce detectible infectious virus (<10 I.U./ml) (Fig. 7C).

To determine the nature of the defect in viral replication, we counterstained the viral rescue cells with anti-SeV-M or anti-SeV-F antibodies and analyzed them by 3D confocal microscopy. By day 6 post-rescue of rSeV-eGFP-MWT, infection has spread to all cells without evidence of cell-cell fusion (Fig. 7D). It is known that SeV replication in cell culture does not result in cell-cell fusion [45,47,68], an unusual phenotype as most paramyxovirus infections result in extensive cell-cell fusion (e.g. see S1 Fig. for NiV). Interestingly, although the rSeV-eGFP-ML102A L103A and rSeV-eGFP-MK254R rescue cells did not produce infectious virus, the GFP-positive rescue cells did initiate the formation of large foci of fused cells (Fig. 7D, second and third horizontal panels). Virus-cell and cell-cell fusion require the presence of F and HN [9,10], and we confirmed that SeV-F is expressed on rSeV-eGFP-ML102A L103A and rSeV-eGFP-MK254R foci (Fig. 7E). Recombinant MuV-eGFP genomes engineered with M nuclear export mutants were also unable to efficiently spread beyond the fused cells formed at sites of rescue (S5 Fig.). These data indicate that proper M nuclear-cytoplasmic trafficking is necessary for viral morphogenesis.
To determine the nuclear localization of SeV-M, we counterstained rSeV-eGFP rescue cells with DAPI to visualize nuclei and anti-fibrillarin antibodies to visualize nucleoli. SeV-MWT was primarily extranuclear at the cell periphery (Fig. 7D, 7F). SeV-ML102A L103A did not have an obvious nuclear localization in the viral context, although intracellular inclusions were apparent, suggesting that this mutant nonetheless had an altered localization (Fig. 7F). SeV-MK254R, on the other hand, was strongly nuclear and enriched within the nucleoli (Fig. 7D, 7F). These results are consistent with the previous transient transfection experiments in which SeV-MK254R was also more strongly localized to the nucleus than SeV-ML102A L103A (Fig. 7H). Thus, these live virus results support our model that ubiquitination regulates the nuclear and subnuclear trafficking of SeV-M.
Proteomics analysis supports a model of regulated nuclear transport of Paramyxovirinae matrix proteins that involves a critical nucleolar transit phase
Having characterized determinants of Paramyxovirinae M nuclear-cytoplasmic trafficking encoded within some M proteins, we turned to identifying potential cellular regulators of this process. We generated inducible 3X-Flag-M-expressing stable HEK 293 cell lines to efficiently copurify M-interacting proteins and analyzed their composition using multidimensional protein identification technology (MudPIT) as described in Materials and Methods (S1–S4 Tables). We opted to determine the protein interactomes of NiV-M, HeV-M, SeV-M and NDV-M since these are the Paramyxovirinae M proteins with confirmed nuclear trafficking during live virus infection (Fig. 6, Fig. 7, S10 Fig.) [39,48,51–54,69], and because these proteins cover the widest range of sequence homology to NiV-M: ~90% amino acid identity for HeV-M, ~37% amino acid identity for SeV-M, and ~20% amino acid identity for NDV-M.
S6 Fig. shows our experimental schema and stringent filtering that resulted in our list of putative M protein interactors detailed below and listed in S1–S4 Tables. Nonspecific interactions in MudPIT analyses tend to be independent of the bait of interest. Rather, they are background contaminants related to the cell type and the affinity purification scheme [70]. To remove background contaminants, our putative M interactomes represent only those proteins identified in the sample purifications that are absent in 3 independent negative-control purifications using lysates from the parental/isogenic Flp-In T-REx-293 cells, irrespective of relative abundances (S6 Fig., Worksheet 1 in S1–S4 Tables). As an independent confirmation of stringency, comparison of the putative NiV-M, HeV-M, SeV-M, and NDV-M interactomes to 21 relevant control experiments in the mass spectrometry contaminant repository, CRAPome, revealed relatively few additional proteins that are common sources of contamination (S6 Fig., Worksheet 2 in S1–S4 Tables) [70]. These were primarily actins, tubulins, histones and ribosomal proteins, which are also the most common contaminants across the entire CRAPome (Worksheet 2 in S1–S4 Tables) [70]. During manuscript revisions, another group published the identification of ~130 HeV-M-interacting proteins using affinity-purification, in-gel digestion and mass spectrometric identification, and further characterized AP3B1 as a Henipavirus M interactor that regulates VLP production [71]. A majority of their proteins either went undetected by our global analyses or were excluded as background contaminants because they were present in the control purifications. For the purpose of comparison, the proteins in their paper that are also present in our HeV-M and/or NiV-M interactomes, excluding some ribosomal proteins, are KRI1, RFC1, FAM120A, SMC1A, SART3, UPF1, Nat10, Smarca5, UTP14A, POP1, ZC3HAV1, RAD18, AP3D1, USP7, Tat-SF1, SKIV2L2, PARP-1, and Importin-7 (Worksheet 1 in S1 and S2 Tables). Other than PARP-1, these proteins were unlikely to be present as contaminants in the 21 relevant CRAPome control experiments. We noted that the E3 ubiquitin ligase RAD18 was not present as a contaminant in any of the 21 CRAPome control experiments and was the least likely to be encountered in the entire CRAPome database; it was found in only 4 of 411 experiments with an average of only 1.3 spectra per experiment, whereas we measured 11 unique spectra (22.6% coverage) in the NiV-M affinity purification [70]. We confirmed the interaction of NiV-M with RAD18 by co-IP and immunoblot analysis (S7A Fig.), indicating that a combination of experimental and computational approaches to background contaminant subtraction can facilitate the identification and characterization of bona fide protein-protein interactions (S6 Fig.). Other ubiquitin ligases identified in our proteomics experiments (UBE2O and Cullin ring ligases) were also confirmed for copurification with M proteins by co-IP and immunoblot analysis (S7B–C Fig.).
Confident that our putative M interactomes were largely reflective of true protein-protein interactions, we further analyzed the interactomes bioinformatically and biochemically (Fig. 8, S7–S9 Fig.). Comparisons of our putative NiV-M, HeV-M, SeV-M, and NDV-M interactomes to one another revealed significant overlap; over 60% of the proteins found in any single interactome were also found in the interactomes of one or more of the other three (Fig. 8A). Furthermore, we identified 178 proteins common to all M interactomes, the majority of which are not present in the 21 historical control experiments (Worksheet 3 in S4 Table). This common set of proteins represents 24–48% of all the proteins in any single viral M interactome (Fig. 8A, S1–S4 Tables). Interestingly, proteins associated with the nuclear pore complex were significantly enriched within individual M interactomes as well as the subset of common interacting proteins (Fig. 8B,-log10(p-value)>10). These include nuclear pore complex components (RanBP2, Nup37, Nup93, Nup107, Nup155, Nup205, Sec13, Seh1), nuclear transport receptors (NTRs) required for nuclear import of proteins (α/β-importins), nuclear export of proteins (Exp1/CRM1, Exp2), nuclear export of dsRNA/dsRNA-binding proteins (Exp5), nuclear export of tRNA (Exportin-T), nuclear export of mRNA (Rae1), and a regulator of the RanGTP/GDP cycle that modulates the association/dissociation of cargo with NTRs (RanGAP1) (Fig. 8C, Worksheet 1 in S1–S4 Tables). All of these proteins were unlikely background contaminants (Worksheet 2 in S1–S4 Tables) and we confirmed that NiV-M interacts with α-importins and Exp1/CRM1 (S8 Fig.). Thus Paramyxovirinae M proteins interact with a highly interconnected network of proteins necessary for transport of NLS and NES containing cargo proteins across the nuclear pore.
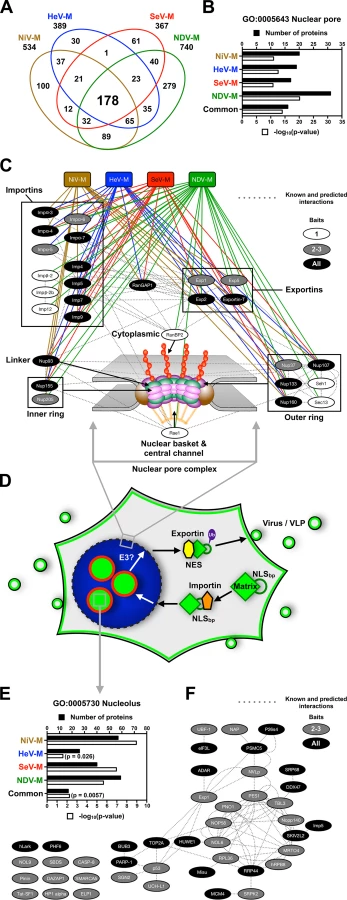
Our cell biological and proteomic findings are synthesized into a working model for ubiquitin-regulated nuclear-cytoplasmic trafficking of M proteins shown in Fig. 8D. Consistent with the nucleolar transit phase exhibited by the Paramyxovirinae M proteins under study, the M interactomes also revealed a significant enrichment of resident or transient nucleolar proteins (Fig. 8E and 8F). One of these, the RNA polymerase I transcription factor UBF-1 (UBTF) had abundant spectral counts in the SeV-M and NiV-M interactomes but was essentially absent in the matched CRAPome control experiments (S1 Table and S3 Table). We determined that UBF-1 sequesters NiV-M in the nucleus and inhibits NiV-M budding when overexpressed (S9 Fig.). Thus, our proteomic and functional data indicate that M proteins interact with an array of nuclear and nucleolar proteins, at least some of which can modulate M nuclear-cytoplasmic trafficking.
Discussion
Whether or not they replicate in the nucleus, many viruses are known to target, modify, and hijack nuclear components and nuclear functions to promote the infectious life cycle. It is generally thought that paramyxoviruses replicate in the cytosol without a nuclear stage. However, it is becoming increasingly clear that nuclear trafficking of M is shared by a number of paramyxoviruses. It was previously observed that SeV-M, NDV-M and RSV-M traffic through the nucleus [49,50,69] and a functional bipartite nuclear localization signal (NLSbp) has been defined within NDV-M [48]. Here, we show that the NLSbp of NDV-M is functionally conserved for nuclear import along with NiV-M, HeV-M, SeV-M and MuV-M (Fig. 8D, S3 Fig.) [39,48]. NLSs specify translocation through the nuclear pore through high-affinity interactions with importins, which in turn interact with cognate nuclear pore components on the cytoplasmic side [72]. In our proteomic analyses we identified numerous importins, exportins and nuclear pore complex components as common candidate interactors of NiV-M, HeV-M, SeV-M and NDV-M (Fig. 8, S8 Fig., S1–S4 Tables). Thus, the size of Paramyxovirinae M proteins (>40 kDa), the presence of a functional NLSbp within M proteins, and the interaction with nuclear transport receptors are strong evidence that the nuclear localization of M proteins is an active and regulated transport process. That the putative M interactomes show such a strong enrichment of proteins involved in nuclear-cytoplasmic transport also suggests that M proteins may antagonize the nuclear-cytoplasmic trafficking of host proteins and RNA to facilitate viral replication [73].
In addition to the NLSbp, we show that a leucine-rich NES sequence is functionally conserved within NiV-M, HeV-M, SeV-M and MuV-M (Fig. 2, Fig. 5, Fig. 8D). We note that mutation of the corresponding region in NDV-M resulted in decreased nuclear localization. However, this sequence is not as well conserved as in the other M proteins (Fig. 2A) and a recent study identified other functional NES motifs within different regions of NDV-M [52]. Thus, Paramyxovirinae M proteins appear to have both shared and unique determinants of nuclear-cytoplasmic trafficking depending on their evolutionary heritage. We also acknowledge that our experimental system utilizing human cells may not fully recapitulate the regulation of NDV-M trafficking since NDV is an avian virus, while NiV, HeV, SeV, MuV and MeV are mammalian viruses.
Nuclear export of NiV-M, HeV-M, SeV-M and MuV-M is also regulated by a lysine within the second basic patch of the NLSbp. Mutating this lysine to an arginine results in decreased ubiquitination and a nuclear retention phenotype that is phenocopied by pharmacological depletion of free ubiquitin with a proteasome inhibitor (Fig. 1B-E, Fig. 2B-E, Fig. 5, Fig. 6). We hypothesize that mutation of this lysine prevents its ubiquitination. Alternately, mutation of this lysine may prevent the ubiquitination of a nearby lysine within the NLSbp by preventing the interaction of M with a ubiquitin ligase. Whether ubiquitination regulates MeV-M or NDV-M function(s) remains indeterminate. The subcellular localization phenotypes of the NES and the various NLSbp mutants for NDV-M and MeV-M are not congruent (compare S3 Fig. and Fig. 2F-G). Furthermore, NDV-M and MeV-M also exhibit divergent sensitivity with regards to ubiquitin conjugation (S2E–F Fig.), and neither were found to be sensitive to MG132 induced nuclear retention (Fig. 1F-G). Altogether, our results suggest that the degree of the nuclear trafficking phenotypes of transfected M proteins mutated at the putative NES, the putative NLSbp, or the homologously aligned lysine within the NLSbp, are strongest for the Henipavirus M proteins, moderate for SeV-M and MuV-M, variable for NDV-M, and inconclusive/absent for MeV-M under the cell type and conditions examined (Table 1). Although we did not find conclusive evidence for ubiquitination of NDV-M on the lysine that corresponds to the Henipavirus M K258, it is probable that NDV-M is a biological target of posttranslational modification on other lysines within the NLSbp. Our proteomic analysis detected a peptide from the NLSbp of NDV-M containing 114.0492 Da mass signatures indicative of the vestigial diglycine of ubiquitin that remains attached to the modified lysine (K[114.04292]) after trypsin cleavage (249GKK[114.04292]VTFDK[114.04292]LEKKIRSLDLSVGLSDVLGPSVLVK281) [74].
How could NLS ubiquitination regulate nuclear trafficking? Protein import into the nucleus is regulated by the affinity of importins for cargo NLSs, which can be modulated by intermolecular or intramolecular masking of the NLS itself [75,76]. For example, ubiquitination of the NLS of p53 by MDM2 has been shown to block p53 nuclear import by preventing the binding of importin-α3 [77]. It was recently shown that the ubiquitin-conjugating enzyme UBE2O multi-monoubiquitinates tumor suppressor BAP1 on its NLSbp to promote cytoplasmic localization. It was further determined that UBE2O specifically binds and ubiquitinates a number of similar bipartite NLSs within nuclear trafficking proteins known to regulate RNA processing, transcription, DNA replication, and chromatin remodeling [78]. We hypothesize that ubiquitination of M on a lysine within the NLSbp itself prevents importin binding as a means to prevent nuclear re-entry once the protein has completed its nuclear sojourn (Fig. 8D, S8A Fig.). Given this model for ubiquitin-dependent nuclear-cytoplasmic trafficking, it is possible that M utilizes a nuclear-resident E3 ubiquitin ligase [79]. Our proteomic analyses identified a number of candidate ubiquitin ligases that interact with M proteins including UBE2O, which was found within the NiV-M and NDV-M interactomes (S7B Fig., S1 Table, S4 Table). Further study of these ubiquitin ligases will help resolve the spatiotemporal dynamics of M ubiquitination vis-à-vis nuclear trafficking (S7 Fig.).
Nuclear-cytoplasmic trafficking is a prerequisite for M budding and viral morphogenesis. We previously showed that NiV-M mutants defective in either nuclear import or nuclear export were also defective at budding VLPs [39]. Alternately, overexpression of a nuclear NiV-M-interacting protein UBF-1 can sequester NiV-M in the nucleus and inhibits efficient budding of VLPs (S9 Fig.). Other viruses have been reported to interact with UBF-1 and utilize or modify UBF-1 function. For example, UBF-1 is inactivated during Poliovirus infection as part of a viral strategy to inhibit host cell transcription globally [80]. The DNA viruses Adenovirus and HSV-1 co-opt UBF-1 into viral DNA replication centers, and it has been hypothesized that UBF-1 is used as a cofactor in viral DNA replication [81–83]. Finally, the SV40 large T antigen and the HCV NS5A protein stimulate RNA Pol I transcriptional activity and enhance rRNA synthesis by hyperphosphorylation of UBF-1 [84,85]. Such rRNA transcriptional activation is thought to contribute to the cell transformation caused by these tumorigenic viruses. For NiV, even though NiV-M clearly interacts with overexpressed UBF-1, and is colocalized with UBF-1 in the nucleus, it is unclear whether NiV utilizes UBF-1 to benefit viral replication under physiological expression levels and conditions (S9 Fig.). Nonetheless, the inhibitory effects of nuclear UBF-1 on VLP budding supports the model that functional trafficking to the plasma membrane requires properly regulated nuclear import and export (Fig. 8D).
Here, we also engineered M nuclear export mutants into recombinant SeV. rSeV-eGFP-ML102A L103A or rSeV-eGFP-MK254R were completely attenuated for production of infectious virus, but formed large foci of fused cells at sites of viral rescue. Moreover, nuclear localization of SeV-MK254R, the ubiquitination mutant, was observed in both virus rescue and transient transfection experiments (Fig. 2D, Fig. 5C, Fig. 7F). It is known that mutations that abrogate the interaction of M with the glycoproteins, including M deletion, can increase cell-cell fusion in SeV and MeV, while mutations that enhance their interaction can decrease cell-cell fusion [33,34,45–47,68]. Since M and F proteins were expressed in the foci of fused cells, our results indicate that rSeV-eGFP-ML102A L103A and rSeV-eGFP-MK254R are defective in proper assembly of viral components at the plasma membrane rather than in expression of viral components necessary for budding per se. The link between viral replication and M ubiquitin-dependent nuclear-cytoplasmic trafficking may explain why proteasome inhibitors that deplete free cellular pools of ubiquitin have been found to inhibit SeV and NiV replication [39,86].
Beyond regulating nuclear import/export itself, we previously found that ubiquitination of NiV-M is necessary for membrane targeting and budding [39]. It is possible that the ubiquitination of M proteins promotes recognition by cellular factors such as ESCRT complexes known to mediate transport and budding of many enveloped viruses [56,87,88], especially in light of known sequence motifs in PIV5-M, SeV-M and MuV-M that can bind ESCRT complex components [27,43,89]. The status of M ubiquitination may also regulate the interactions of M with cellular factors inside the nucleus and within subnuclear compartments such as the nucleolus. A number of cellular proteins become enriched in the nucleolus upon proteasome inhibition, including p53 [90–99]. Similarly, pharmacological or genetic inhibition of NiV-M, HeV-M, SeV-M, and MuV-M ubiquitination sequesters these proteins in the nucleolus (Fig. 5, Fig. 6), and nucleolar localization of SeV-MK254R was also confirmed in the context of rSeV-eGFP rescue (Fig. 7F). Nucleolar localization of M proteins is a natural feature of the nuclear sojourn of some M proteins, with M enriched at nucleoli during the early stage of live NiV and NDV infections (S10 Fig.) [69,100]. Although M proteins do not have an evident nucleolar localization signal (NoLS) that would have predicted this observation [101], our proteomics experiments suggest that Paramyxovirinae M proteins can interact with a number of nucleolar hub proteins (Fig. 8E and F, S1–S4 Tables) [102].
Most, if not all, viral families interact with the nucleolus, often to usurp cellular functions and promote viral replication [103–105], as the nucleolus is a dynamic structure involved in a vast array of biological functions beyond ribosome biogenesis, including tRNA and mRNA processing and export from the nucleus, cell cycle regulation, and response to cellular stress. Additionally, there is growing recognition that NLS containing viral proteins target the nuclear pore complex to alter the export of macromolecules and mRNA, thereby counteracting antiviral responses and promoting viral gene expression at the expense of host gene expression [73]. For example, influenza NS1 is a multifunctional protein known to translocate to the nucleolus and to the nuclear pore where it inhibits host mRNA export factors resulting in impaired immune responses and enhanced viral virulence [106]. Vesicular stomatitis virus M also inhibits mRNA nuclear export through interaction with nuclear pore components [73,107]. Further, RSV-M is shuttled to the host cell nucleus where it inhibits host gene expression and induces cell cycle arrest, indicating that paramyxovirus M proteins also antagonize nuclear functions [108,109]. We hypothesize that ubiquitin-dependent nuclear and subnuclear trafficking of some Paramyxovirinae M proteins is part of a viral strategy to promote viral replication. Therefore, the study of M interactions with the nucleolus and the nuclear pore complex represents an opportunity to gain new insights into the cell biology of the nucleus and to identify novel antiviral targets.
Materials and Methods
Cell culture and transfection
HeLa, Vero, and HEK 293T cells were maintained at 37°C in a 5% CO2 atmosphere in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% 100X penicillin/streptomycin solution (Gibco/Life Technologies, Gaithersburg, MD). For confocal microscopy imaging, cells were seeded on 22 mm #1.5 coverglass coated with Collagen Type I (BD Biosciences, San Jose, California). Cells were transfected using Lipofectamine LTX per the manufacturer’s instructions (Invitrogen/Life Technologies). 3X-Flag-M Flp-In T-REx-293 cell lines, generated as described below, were maintained at 37°C in a 5% CO2 atmosphere in DMEM supplemented with 10% dialyzed FBS and 1% 100X penicillin/streptomycin solution. 3X-Flag-M protein expression was induced and immunoprecipitated as described below.
Plasmids, cell lines and virus reverse genetics constructs
3X-Myc-tagged Cullin constructs (Addgene plasmids 19896, 19892, 19893, 19951, 19922, 19895 and 20695) are described in [110–113]. GFP-UBF-1 (Addgene plasmid 17656) is described in [114]. Flag-tagged Karyopherin constructs were the kind gift of Dr. Christopher F. Basler (Icahn School of Medicine at Mount Sinai, New York, NY). 3XFlag-CRM1 (Addgene plasmid 17647) is described in [115]. The Myc-UBE2O construct was the kind gift of Dr. El Bachir Affar (Maisonneuve-Rosemont Hospital Research Center, Department of Medicine, University of Montreal, Montreal) and is described in [78]. HA-UbK0 (Addgene; plasmid 17603; all lysines mutated to arginines) is described in [55]. Codon optimization and cloning of untagged, 3X-Flag-tagged and 3X-Flag-GFP-tagged Nipah virus matrix (NiV-M) and generation of corresponding NiV-M mutants is described in [39]. We similarly codon optimized and cloned the open reading frames encoding M from Hendra virus (HeV-M, genus Henipavirus), Sendai virus (SeV-M, genus Respirovirus), Mumps virus (MuV-M, genus Rubulavirus), and Measles virus (MeV-M, genus Morbillivirus), and also a non-codon optimized Newcastle disease virus M (NDV-M, genus Avulavirus): briefly, eGFP was fused to the N-terminus of M by overlap extension PCR (OE-PCR). WT or GFP-fused HeV-M, SeV-M, RSV-M, MuV-M, and MeV-M were inserted within the HindIII and XhoI sites of pCMV-3Tag-1, while NDV-M was inserted within HindIII and ApaI sites of pCMV-3Tag-1 (Agilent Technologies, Santa Clara CA) to generate 3X-Flag - and 3X-Flag-GFP-tagged-M constructs. Alignment of M sequences using Clustal Omega identified sequences motifs corresponding to NiV-M’s nuclear export sequence (NES) and bipartite nuclear localization sequence (NLSbp) [39,116]. Mutations were generated using the QuikChange II site-directed mutagenesis kit using PAGE-purified mutagenesis primers designed using the online QuikChange primer design tool (Agilent Technologies). A RAD18 cDNA clone was purchased from Origene (SC323786). 3X-Flag-GFP-RAD18 was generated by replacement of the NiV-M insert in HindIII/XhoI digested 3X-Flag-NiV-M.
The Flp-In T-REx system (Invitrogen) was used to generate doxycycline-inducible 3X-Flag-M cell lines. Codon-optimized 3X-Flag-tagged NiV-M, HeV-M, SeV-M, and NDV-M were inserted within the KpnI and XhoI sites of pcDNA5/FRT/TO. The constructs and pOG44 were cotransfected into Flp-In T-REx-293 cells, and stable cell lines were selected with hygromycin and blasticidin according to the manufacturer’s instructions.
Constructs for bimolecular fluorescence complementation (BiFC) analyses were generated with split Venus residues 1–172 (VN173) and 155–238, A206K (VC155) [65,117]. VN173 and VC155 were PCR amplified from pBiFC-VN173 (Addgene plasmid 22010) and pBiFC-VC155 (Addgene plasmid 22011), and were fused to the N-termini of Ub and M proteins via a flexible linker encoding GGGGSGGGGGR by OE-PCR. VN173-Ub and VC155-M were inserted within the NotI and XhoI sites of pcDNA3.1(+) (Life Technologies). Mutations within VC155-M constructs were generated using the QuikChange II site-directed mutagenesis kit using PAGE-purified mutagenesis primers designed using the online QuikChange primer design tool (Agilent Technologies).
The recombinant Sendai virus (rSeV) anti-genome RGV0, a Fushimi strain construct with F1-R strain mutations in F and M, and helper plasmids encoding SeV N, P and L were the kind gift of Dr. Nancy McQueen and are described in [67]. The encoded virus has the ability to replicate in mammalian cells without the addition of trypsin. We further modified the rSeV anti-genome construct by inserting an eGFP reporter flanked at the 3’ end by a unique NotI site between the N and P genes. A hammerhead ribozyme sequence was inserted between the optimal T7 promoter and the start of the anti-genome. Mutations were introduced into the SeV-M ORF by OE-PCR using primers containing the desired mutations, followed by insertion into NotI and AfeI sites in the parental rSeV-eGFP construct.
The full-length construct encoding recombinant Mumps virus (rMuV) anti-genome of the Jeryl Lynn 5 (JL5) vaccine strain and helper plasmids encoding MuV-JL5 N, P and L proteins were a kind gift from Dr. W. Paul Duprex and are described in [118]. We modified the rMuV anti-genome construct by inserting an eGFP reporter between the NP and P genes. A hammerhead ribozyme sequence was inserted between the optimal T7 promoter and the start of the anti-genome. Mutations were introduced into the MuV-M ORF by OE-PCR using primers containing the desired mutations, followed by insertion into SalI and SbfI sites in the parental rMuV construct.
Multidimensional protein identification technology (MudPIT) analysis of matrix interactomes
3X-Flag-M Flp-In T-REx-293 cell lines were grown to ~80% confluency and induced for protein expression with 100 ng/mL doxycycline for 24h. Cells were washed three times in dPBS and lysed in 100 mM Tris-HCL pH 8, 150 mM NaCL, 5 mM EDTA, 5% glycerol, 0.1% NP40, complete protease cocktail (Roche), PhosSTOP (Roche) and 25 mM N-ethylmaleimide. Cell lysate was clarified by centrifugation at >15,000×g for 15 min at 4°C and incubated with lysis buffer-equilibrated anti-Flag M2 affinity gel (Sigma-Aldrich, St. Louis, MO) for 2 hours at 4°C. The affinity gel was extensively washed with lysis buffer and then with elution buffer consisting of 100 mM Tris-HCL pH 8, 150 mM NaCL, 5 mM EDTA, and 5% glycerol. Bound proteins were eluted from the affinity gel with elution buffer containing 3X-Flag peptide (Sigma-Aldrich), were precipitated with trichloroacetic acid, washed with acetone twice, dried, and stored at -20°C until further processing.
Protein samples were resuspended in 8M urea in 100 mM Tris pH 8.5, reduced, alkylated and digested by the sequential addition of lys-C and trypsin proteases as previously described [119]. The digested peptide solution was fractionated online using strong-cation exchange and reverse phase chromatography and eluted directly into an LTQ-Orbitrap mass spectrometer (Thermofisher) [119,120]. MS/MS spectra were collected and subsequently analyzed using the ProLuCID and DTASelect algorithms [121,122]. Database searches were performed against a human database containing the relevant paramyxovirus M protein sequence. Protein and peptide identifications were further filtered with a false positive rate of less than 5% as estimated by a decoy database strategy [123]. Normalized spectral abundance factor (NSAF) values were calculated as described [124]. Proteins were considered candidate M-interacting proteins if they were identified in the relevant affinity purification but not present in 3 independent control purifications using lysates from the parental Flp-In T-REx-293 cells. Analysis of other potential background contaminants was performed using CRAPome [70]. Venn diagrams were generated using jvenn [125]. Gene-annotation enrichment analysis was performed using DAVID Bioinformatics Resources 6.7 [126,127]. Physical and predicted protein interaction networks were visualized using the GeneMANIA plugin for Cytoscape 3.1 [128,129].
Immunoprecipitations and immunoblot analysis of matrix ubiquitination
Transfected HEK 293T cells were washed once in dPBS and lysed in 100 mM Tris-HCL pH 8, 150 mM NaCL, 5 mM EDTA, 5% glycerol, 0.1% NP40, complete protease cocktail (Roche) and 25 mM N-ethylmaleimide. The cell extract was clarified by centrifugation at >15,000×g for 15 min at 4°C before incubation overnight at 4°C with lysis buffer-equilibrated anti-Flag M2 affinity gel (Sigma-Aldrich, St. Louis, MO). The affinity gel was extensively washed with lysis buffer and then with elution buffer consisting of 100 mM Tris-HCL pH 8, 150 mM NaCL, 5 mM EDTA, and 5% glycerol. Bound proteins were eluted from the affinity gel with elution buffer containing 3X-Flag peptide (Sigma-Aldrich), were subjected to SDS-PAGE and transferred to immobilon-FL PVDF membrane (EMD Millipore, Billerica, MA). To analyze M ubiquitination by immunoblot, HEK 293T cells were cotransfected with 3X-Flag-M and HA-UbK0 for 24h and subjected to immunoprecipitation as described above. Membranes were simultaneously probed with mouse anti-Flag M2 primary antibodies (Sigma-Aldrich) and rabbit anti-HA primary antibodies (Novus Biologicals, Littleton, CO) followed by anti-mouse-680 and anti-rabbit-800 secondary antibodies (LI-COR, Lincoln, Nebraska) and imaged on an Odyssey infrared scanner (LI-COR) according to the manufacturer’s instructions. To quantify relative ubiquitination, the background subtracted integrated fluorescence intensities of the monoubiquitin bands (Ub) normalized to total M (M0+M1) was determined using LI-COR Odyssey software.
Depletion of cellular free ubiquitin
For 3D confocal microscopy analysis, transfected HeLa cells were treated with 50 μM MG132 or 0.5% DMSO at 16 h post-transfection for 8 hours, then fixed and processed for quantitative image analysis as described below. For immunoblot analysis of M ubiquitination during ubiquitin depletion, transfected HEK 293T cells were treated with 10 μM MG132 or 0.1% DMSO at 18 h post-transfection for 6 hours, and 3X-Flag-M was immunoprecipitated as described above.
Quantification of virus-like particle budding
VLP budding assays were performed as described in [39]. Briefly, precleared supernatants from 3X-Flag-M transfected HEK 293T were ultracentrifuged through a 20% (w/v) sucrose at 36,000 rpm for 2 h at 4°C (AH-650 rotor, Thermo Scientific). VLP pellets and cells were resuspended in lysis buffer and subjected to SDS-PAGE and anti-Flag immunoblotting. Relative integrated intensity of VLP/cell lysate bands were quantified and normalized relative to the budding of 3X-Flag-NiV-M.
Nipah virus infection and recombinant virus rescue
HeLa cells were infected with Nipah virus under biosafety level 4 (BSL-4) conditions as described in [39]. For rescue of WT or mutant rSeV-eGFP, 2X106 HEK 293T cells were transfected with recombinant plasmid encoding the anti-genome (4 μg) along with the cognate accessory plasmids encoding SeV NP (1.44 μg), P (0.77 μg), and L (0.07 μg), and a codon optimized T7 RNA polymerase (4 μg) using Lipofectamine LTX (8.9 μL) and Plus Reagent (5.5 μL), according to manufacturer’s instructions. Cells were harvested for FACS analysis at 48 hours post-transfection (the earliest time point when GFP-positive cells can be observed by epifluorescence microscopy, yet when supernatant titer is still not detectable) to quantify rescue efficiency. The number of GFP-positive cells (rescue events) was determined from 500,000 cells analyzed with a FACSCalibur Flow Cytometer (BD Biosciences) and FlowJo software (TreeStar Inc., Ashland, OR). Cells plated on coverslips were fixed at day 6 post-transfection for analysis of rescued virus infection by 3D confocal microscopy. Supernatant was collected from rescue cells at day 6 post-transfection for quantification of viral titers. Briefly, supernatant stored at -80°C was thawed on ice and serial diluted 2-fold in serum-free DMEM. 100 μL of each dilution was used to infect ~60,000 Vero cells in a 24-well plate for 1 hour. After 1 h, 500 μL of DMEM 10% FBS was added to each well and the cells were incubated at 37°C. Cells were harvested for FACS analysis at 24 h post infection and titers were calculated based on percent infection in the linear range of supernatant dilutions. For rescue of WT or mutant rMuV, 4X105 BSR-T7 cells were transfected with recombinant plasmid encoding the anti-genome (5 μg) along with the cognate accessory plasmids encoding MuV NP (0.3 μg), P (0.1 μg), and L (0.2 μg), and a codon optimized T7 RNA polymerase (2 μg) using Lipofectamine LTX (18.75 μL) and Plus Reagent (7.5 μL), according to manufacturer’s instructions.
Microscopy and antibodies
Nipah virus-infected cells were fixed in 10% formalin solution for a minimum of 24 h prior to removal from the BSL-4 laboratory. For all other immunofluorescence microscopy, samples were fixed with 2% paraformaldehyde in 100 mM phosphate buffer (pH 7.4) for 15 min. Fixed cells were permeabilized in blocking buffer containing PBS, 1% saponin, 3% bovine serum albumin, and 0.02% sodium azide. After incubation with antibodies/probes in blocking buffer, samples were extensively washed in blocking buffer and mounted on glass slides with Vectashield mounting medium with DAPI (Vector Laboratories, Burlingame, California, United States). The samples were imaged with a Leica SP5 confocal microscope (Leica Microsystems, Buffalo Grove, IL), acquiring optical Z-stacks of 0.3–0.5 μm steps. Z-stacks were reconstructed and analyzed in three dimensions using Volocity 5.5 software (Perkin Elmer, Waltham, Massachusetts). Widefield microscopy was performed using a Cytation 3 Cell Imaging Multi-Mode Reader (BioTek, Winooski, VT) or a Nikon TE300 microscope. NiV-M was detected with rabbit anti-NiV-M antibodies (1 : 1000) [39]. SeV-M was detected with mouse anti-SeV-M ascites (1 : 200), and SeV-F was detected with mouse anti-SeV-F ascites (1 : 200) kindly provided by Dr. Toru Takimoto [130]. Nucleoli were detected with mouse anti-nucleolin antibodies (1 : 500) (Invitrogen/Life Technologies) or rabbit anti-fibrillarin antibodies (1 : 500) (Abcam, Cambridge, MA). Alexa-fluor conjugated Anti-IgG antibodies of appropriate species reactivity and fluorescence spectra were used for secondary detection (1 : 300–1 : 1000) (Invitrogen/Life Technologies). F-actin was visualized by incubating samples with Alexa-fluor conjugated phalloidins (1 : 300) (Invitrogen/Life Technologies).
Immunoblots were imaged on an Odyssey infrared scanner (LI-COR) using secondary antibodies of appropriate species reactivity and fluorescence spectra (LI-COR). An antibody to the C-terminus of GFP (LS-C51736, LifeSpan BioSciences, Inc., Seattle, WA) was used for immunoblot detection of VC155-fusion proteins. Anti-Flag M2 (F3165, Sigma-Aldrich) was used to for immunoblot detection of Flag-tagged proteins. Anti-HA (NB600–363, Novus Biologicals, Littleton, CO) was used for immunoblot detection HA-tagged proteins. Anti-Myc Tag, clone 4A6 (05–724, Millipore, Temecula, CA) was used for immunoblot detection of Myc-tagged proteins. Anti-UBE2O (NBP1–03336, Novus Biologicals) was used for immunoblot detection of UBE2O. Anti-β-Tubulin (T7816, Sigma-Aldrich) and Anti-COX IV (926–42214, Licor) were used for loading controls.
Quantitative analysis of 3D confocal micrographs
Random 40X fields were imaged using acquisition settings ensuring no under-saturated or over-saturated pixel intensities. Volocity 5.5 software was used for quantitative analysis of 3D confocal images. To determine the quantity of nuclear M, the nuclear compartment was defined with the find objects function within the DAPI-fluorescence channel. Holes in objects (DNA-absent regions such as nucleoli) were filled, and fluorescent objects smaller than nuclei were excluded. The entire cell body was defined by drawing a region of interest (ROI) encompassing all F-actin staining. The sum of voxel intensities in the GFP channel was measured within these defined sets. The average voxel fluorescence of untransfected cells was used for background subtraction. To determine the quantity of nucleolar M, the nucleolus was defined with the find objects function within the Fibrillarin-stained-fluorescence channel. Nucleolar objects were grouped within their respective nuclei, defined as above based on the DAPI-fluorescence channel. To quantify fluorescence from bimolecular fluorescence complementation images, a ROI was drawn around cells fluorescent in the YFP channel. The average voxel fluorescence of untransfected cells was used for background subtraction.
Statistical analysis
For analysis of M nuclear localization, p-values were generated with a Student’s t test when analyzing two sample groups. To analyze three or more sample groups, p-values were generated by ANOVA with Bonferroni correction for multiple comparisons. To analyze BiFC experiments, p-values were generated using a Mann-Whitney test. All graphs and statistical analyses were generated with Prism 6 (GraphPad Software, La Jolla, CA).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. (2011) The Biology of paramyxoviruses. Norfolk, UK: Caister Academic Press.
2. Mariner JC, House JA, Mebus CA, Sollod AE, Chibeu D, et al. (2012) Rinderpest eradication: appropriate technology and social innovations. Science 337 : 1309–1312. doi: 10.1126/science.1223805 22984063
3. Aguilar HC, Lee B (2011) Emerging paramyxoviruses: molecular mechanisms and antiviral strategies. Expert Rev Mol Med 13: e6. doi: 10.1017/S1462399410001754 21345285
4. Eaton BT, Broder CC, Middleton D, Wang LF (2006) Hendra and Nipah viruses: different and dangerous. Nat Rev Microbiol 4 : 23–35. 16357858
5. Lamb RA, Parks GD (2013) Paramyxoviridae: The viruses and their replication. In: Fields BN, Knipe DM, Howley PM, editors. Fields virology. 6th ed. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins. pp. 957–995.
6. Lo MK, Rota PA (2008) The emergence of Nipah virus, a highly pathogenic paramyxovirus. J Clin Virol 43 : 396–400. doi: 10.1016/j.jcv.2008.08.007 18835214
7. Harrison MS, Sakaguchi T, Schmitt AP (2010) Paramyxovirus assembly and budding: building particles that transmit infections. Int J Biochem Cell Biol 42 : 1416–1429. doi: 10.1016/j.biocel.2010.04.005 20398786
8. Jardetzky TS, Lamb RA (2014) Activation of paramyxovirus membrane fusion and virus entry. Curr Opin Virol 5C: 24–33.
9. Lee B, Ataman ZA (2011) Modes of paramyxovirus fusion: a Henipavirus perspective. Trends Microbiol 19 : 389–399. doi: 10.1016/j.tim.2011.03.005 21511478
10. Lamb RA, Paterson RG, Jardetzky TS (2006) Paramyxovirus membrane fusion: lessons from the F and HN atomic structures. Virology 344 : 30–37. 16364733
11. Battisti AJ, Meng G, Winkler DC, McGinnes LW, Plevka P, et al. (2012) Structure and assembly of a paramyxovirus matrix protein. Proc Natl Acad Sci U S A 109 : 13996–14000. doi: 10.1073/pnas.1210275109 22891297
12. Terrier O, Rolland JP, Rosa-Calatrava M, Lina B, Thomas D, et al. (2009) Parainfluenza virus type 5 (PIV-5) morphology revealed by cryo-electron microscopy. Virus Res 142 : 200–203. doi: 10.1016/j.virusres.2008.12.017 19185600
13. Pohl C, Duprex WP, Krohne G, Rima BK, Schneider-Schaulies S (2007) Measles virus M and F proteins associate with detergent-resistant membrane fractions and promote formation of virus-like particles. J Gen Virol 88 : 1243–1250. 17374768
14. Russell PH, Almeida JD (1984) A regular subunit pattern seen on non-infectious Newcastle disease virus particles. J Gen Virol 65 (Pt 6): 1023–1031.
15. Heggeness MH, Smith PR, Choppin PW (1982) In vitro assembly of the nonglycosylated membrane protein (M) of Sendai virus. Proc Natl Acad Sci U S A 79 : 6232–6236. 6292897
16. Hewitt JA, Nermut MV (1977) A morphological study of the M-protein of Sendai virus. J Gen Virol 34 : 127–136. 188976
17. Buechi M, Bachi T (1982) Microscopy of internal structures of Sendai virus associated with the cytoplasmic surface of host membranes. Virology 120 : 349–359. 6285608
18. Bachi T (1980) Intramembrane structural differentiation in Sendai virus maturation. Virology 106 : 41–49. 6251620
19. Manie SN, de Breyne S, Vincent S, Gerlier D (2000) Measles virus structural components are enriched into lipid raft microdomains: a potential cellular location for virus assembly. J Virol 74 : 305–311. 10590118
20. Vincent S, Gerlier D, Manie SN (2000) Measles virus assembly within membrane rafts. J Virol 74 : 9911–9915. 11024118
21. Riedl P, Moll M, Klenk HD, Maisner A (2002) Measles virus matrix protein is not cotransported with the viral glycoproteins but requires virus infection for efficient surface targeting. Virus Res 83 : 1–12. 11864737
22. Subhashri R, Shaila MS (2007) Characterization of membrane association of Rinderpest virus matrix protein. Biochem Biophys Res Commun 355 : 1096–1101. 17336269
23. Stricker R, Mottet G, Roux L (1994) The Sendai virus matrix protein appears to be recruited in the cytoplasm by the viral nucleocapsid to function in viral assembly and budding. J Gen Virol 75 (Pt 5): 1031–1042.
24. Caldwell SE, Lyles DS (1986) Dissociation of newly synthesized Sendai viral proteins from the cytoplasmic surface of isolated plasma membranes of infected cells. J Virol 57 : 678–683. 3003398
25. Henderson G, Murray J, Yeo RP (2002) Sorting of the respiratory syncytial virus matrix protein into detergent-resistant structures is dependent on cell-surface expression of the glycoproteins. Virology 300 : 244–254. 12350355
26. Schmitt AP, He B, Lamb RA (1999) Involvement of the cytoplasmic domain of the hemagglutinin-neuraminidase protein in assembly of the paramyxovirus simian virus 5. J Virol 73 : 8703–8712. 10482624
27. Schmitt AP, Leser GP, Morita E, Sundquist WI, Lamb RA (2005) Evidence for a new viral late-domain core sequence, FPIV, necessary for budding of a paramyxovirus. J Virol 79 : 2988–2997. 15709019
28. Waning DL, Schmitt AP, Leser GP, Lamb RA (2002) Roles for the cytoplasmic tails of the fusion and hemagglutinin-neuraminidase proteins in budding of the paramyxovirus simian virus 5. J Virol 76 : 9284–9297. 12186912
29. Essaidi-Laziosi M, Shevtsova A, Gerlier D, Roux L (2013) Mutation of the TYTLE Motif in the Cytoplasmic Tail of the Sendai Virus Fusion Protein Deeply Affects Viral Assembly and Particle Production. PLoS One 8: e78074. doi: 10.1371/journal.pone.0078074 24339863
30. Ali A, Nayak DP (2000) Assembly of Sendai virus: M protein interacts with F and HN proteins and with the cytoplasmic tail and transmembrane domain of F protein. Virology 276 : 289–303. 11040121
31. Coronel EC, Takimoto T, Murti KG, Varich N, Portner A (2001) Nucleocapsid incorporation into parainfluenza virus is regulated by specific interaction with matrix protein. J Virol 75 : 1117–1123. 11152484
32. Iwasaki M, Takeda M, Shirogane Y, Nakatsu Y, Nakamura T, et al. (2009) The matrix protein of measles virus regulates viral RNA synthesis and assembly by interacting with the nucleocapsid protein. J Virol 83 : 10374–10383. doi: 10.1128/JVI.01056-09 19656884
33. Cathomen T, Naim HY, Cattaneo R (1998) Measles viruses with altered envelope protein cytoplasmic tails gain cell fusion competence. J Virol 72 : 1224–1234. 9445022
34. Tahara M, Takeda M, Yanagi Y (2007) Altered interaction of the matrix protein with the cytoplasmic tail of hemagglutinin modulates measles virus growth by affecting virus assembly and cell-cell fusion. J Virol 81 : 6827–6836. 17442724
35. Ghildyal R, Li D, Peroulis I, Shields B, Bardin PG, et al. (2005) Interaction between the respiratory syncytial virus G glycoprotein cytoplasmic domain and the matrix protein. J Gen Virol 86 : 1879–1884. 15958665
36. Runkler N, Pohl C, Schneider-Schaulies S, Klenk HD, Maisner A (2007) Measles virus nucleocapsid transport to the plasma membrane requires stable expression and surface accumulation of the viral matrix protein. Cell Microbiol 9 : 1203–1214. 17217427
37. Ciancanelli MJ, Basler CF (2006) Mutation of YMYL in the Nipah virus matrix protein abrogates budding and alters subcellular localization. J Virol 80 : 12070–12078. 17005661
38. Patch JR, Crameri G, Wang LF, Eaton BT, Broder CC (2007) Quantitative analysis of Nipah virus proteins released as virus-like particles reveals central role for the matrix protein. Virol J 4 : 1. 17204159
39. Wang YE, Park A, Lake M, Pentecost M, Torres B, et al. (2010) Ubiquitin-regulated nuclear-cytoplasmic trafficking of the Nipah virus matrix protein is important for viral budding. PLoS Pathog 6: e1001186. doi: 10.1371/journal.ppat.1001186 21085610
40. Pantua HD, McGinnes LW, Peeples ME, Morrison TG (2006) Requirements for the assembly and release of Newcastle disease virus-like particles. J Virol 80 : 11062–11073. 16971425
41. Takimoto T, Murti KG, Bousse T, Scroggs RA, Portner A (2001) Role of matrix and fusion proteins in budding of Sendai virus. J Virol 75 : 11384–11391. 11689619
42. Sugahara F, Uchiyama T, Watanabe H, Shimazu Y, Kuwayama M, et al. (2004) Paramyxovirus Sendai virus-like particle formation by expression of multiple viral proteins and acceleration of its release by C protein. Virology 325 : 1–10. 15231380
43. Li M, Schmitt PT, Li Z, McCrory TS, He B, et al. (2009) Mumps virus matrix, fusion, and nucleocapsid proteins cooperate for efficient production of virus-like particles. J Virol 83 : 7261–7272. doi: 10.1128/JVI.00421-09 19439476
44. Schmitt AP, Leser GP, Waning DL, Lamb RA (2002) Requirements for budding of paramyxovirus simian virus 5 virus-like particles. J Virol 76 : 3952–3964. 11907235
45. Inoue M, Tokusumi Y, Ban H, Kanaya T, Shirakura M, et al. (2003) A new Sendai virus vector deficient in the matrix gene does not form virus particles and shows extensive cell-to-cell spreading. J Virol 77 : 6419–6429. 12743299
46. Cathomen T, Mrkic B, Spehner D, Drillien R, Naef R, et al. (1998) A matrix-less measles virus is infectious and elicits extensive cell fusion: consequences for propagation in the brain. EMBO J 17 : 3899–3908. 9670007
47. Irie T, Inoue M, Sakaguchi T (2010) Significance of the YLDL motif in the M protein and Alix/AIP1 for Sendai virus budding in the context of virus infection. Virology 405 : 334–341. doi: 10.1016/j.virol.2010.06.031 20605035
48. Coleman NA, Peeples ME (1993) The matrix protein of Newcastle disease virus localizes to the nucleus via a bipartite nuclear localization signal. Virology 195 : 596–607. 8337834
49. Ghildyal R, Ho A, Dias M, Soegiyono L, Bardin PG, et al. (2009) The respiratory syncytial virus matrix protein possesses a Crm1-mediated nuclear export mechanism. J Virol 83 : 5353–5362. doi: 10.1128/JVI.02374-08 19297465
50. Ghildyal R, Ho A, Wagstaff KM, Dias MM, Barton CL, et al. (2005) Nuclear import of the respiratory syncytial virus matrix protein is mediated by importin beta1 independent of importin alpha. Biochemistry 44 : 12887–12895. 16171404
51. Duan Z, Li Q, He L, Zhao G, Chen J, et al. (2013) Application of green fluorescent protein-labeled assay for the study of subcellular localization of Newcastle disease virus matrix protein. J Virol Methods 194 : 118–122. doi: 10.1016/j.jviromet.2013.08.014 23994149
52. Duan Z, Song Q, Wang Y, He L, Chen J, et al. (2013) Characterization of signal sequences determining the nuclear export of Newcastle disease virus matrix protein. Arch Virol 158 : 2589–2595. doi: 10.1007/s00705-013-1769-5 23807745
53. Yoshida T, Nagai Y'Yoshii S, Maeno K, Matsumoto T (1976) Membrane (M) protein of HVJ (Sendai virus): its role in virus assembly. Virology 71 : 143–161. 179199
54. Bauer A, Neumann S, Karger A, Henning AK, Maisner A, et al. (2014) ANP32B Is a Nuclear Target of Henipavirus M Proteins. PLoS One 9: e97233. doi: 10.1371/journal.pone.0097233 24823948
55. Lim KL, Chew KC, Tan JM, Wang C, Chung KK, et al. (2005) Parkin mediates nonclassical, proteasomal-independent ubiquitination of synphilin-1: implications for Lewy body formation. J Neurosci 25 : 2002–2009. 15728840
56. Komander D, Rape M (2012) The ubiquitin code. Annu Rev Biochem 81 : 203–229. doi: 10.1146/annurev-biochem-060310-170328 22524316
57. Bailey D, O’Hare P (2005) Comparison of the SUMO1 and ubiquitin conjugation pathways during the inhibition of proteasome activity with evidence of SUMO1 recycling. Biochem J 392 : 271–281. 16117725
58. Hjerpe R, Thomas Y, Chen J, Zemla A, Curran S, et al. (2012) Changes in the ratio of free NEDD8 to ubiquitin triggers NEDDylation by ubiquitin enzymes. Biochem J 441 : 927–936. doi: 10.1042/BJ20111671 22004789
59. Mimnaugh EG, Chen HY, Davie JR, Celis JE, Neckers L (1997) Rapid deubiquitination of nucleosomal histones in human tumor cells caused by proteasome inhibitors and stress response inducers: effects on replication, transcription, translation, and the cellular stress response. Biochemistry 36 : 14418–14429. 9398160
60. Schubert U, Ott DE, Chertova EN, Welker R, Tessmer U, et al. (2000) Proteasome inhibition interferes with gag polyprotein processing, release, and maturation of HIV-1 and HIV-2. Proc Natl Acad Sci U S A 97 : 13057–13062. 11087859
61. Patnaik A, Chau V, Wills JW (2000) Ubiquitin is part of the retrovirus budding machinery. Proc Natl Acad Sci U S A 97 : 13069–13074. 11087861
62. Xu Q, Farah M, Webster JM, Wojcikiewicz RJ (2004) Bortezomib rapidly suppresses ubiquitin thiolesterification to ubiquitin-conjugating enzymes and inhibits ubiquitination of histones and type I inositol 1,4,5-trisphosphate receptor. Mol Cancer Ther 3 : 1263–1269. 15486193
63. Fang D, Kerppola TK (2004) Ubiquitin-mediated fluorescence complementation reveals that Jun ubiquitinated by Itch/AIP4 is localized to lysosomes. Proc Natl Acad Sci U S A 101 : 14782–14787. 15469925
64. Lee J, Lee Y, Lee MJ, Park E, Kang SH, et al. (2008) Dual modification of BMAL1 by SUMO2/3 and ubiquitin promotes circadian activation of the CLOCK/BMAL1 complex. Mol Cell Biol 28 : 6056–6065. doi: 10.1128/MCB.00583-08 18644859
65. Kerppola TK (2008) Bimolecular fluorescence complementation (BiFC) analysis as a probe of protein interactions in living cells. Annu Rev Biophys 37 : 465–487. doi: 10.1146/annurev.biophys.37.032807.125842 18573091
66. Kerppola TK (2006) Design and implementation of bimolecular fluorescence complementation (BiFC) assays for the visualization of protein interactions in living cells. Nat Protoc 1 : 1278–1286. 17406412
67. Hou X, Suquilanda E, Zeledon A, Kacsinta A, Moore A, et al. (2005) Mutations in Sendai virus variant F1-R that correlate with plaque formation in the absence of trypsin. Med Microbiol Immunol 194 : 129–136. 15834752
68. Rawling J, Cano O, Garcin D, Kolakofsky D, Melero JA (2011) Recombinant Sendai viruses expressing fusion proteins with two furin cleavage sites mimic the syncytial and receptor-independent infection properties of respiratory syncytial virus. J Virol 85 : 2771–2780. doi: 10.1128/JVI.02065-10 21228237
69. Peeples ME, Wang C, Gupta KC, Coleman N (1992) Nuclear entry and nucleolar localization of the Newcastle disease virus (NDV) matrix protein occur early in infection and do not require other NDV proteins. J Virol 66 : 3263–3269. 1560547
70. Mellacheruvu D, Wright Z, Couzens AL, Lambert JP, St-Denis NA, et al. (2013) The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat Methods 10 : 730–736. doi: 10.1038/nmeth.2557 23921808
71. Sun W, McCrory TS, Khaw WY, Petzing S, Myers T, et al. (2014) Matrix Proteins of Nipah and Hendra Viruses Interact with Beta Subunits of AP-3 Complexes. J Virol 88 : 13099–13110. doi: 10.1128/JVI.02103-14 25210190
72. Kimura M, Imamoto N (2014) Biological Significance of the Importin-beta Family-Dependent Nucleocytoplasmic Transport Pathways. Traffic.
73. Yarbrough ML, Mata MA, Sakthivel R, Fontoura BM (2014) Viral subversion of nucleocytoplasmic trafficking. Traffic 15 : 127–140. doi: 10.1111/tra.12137 24289861
74. Jeram SM, Srikumar T, Pedrioli PG, Raught B (2009) Using mass spectrometry to identify ubiquitin and ubiquitin-like protein conjugation sites. Proteomics 9 : 922–934. doi: 10.1002/pmic.200800666 19180541
75. McLane LM, Corbett AH (2009) Nuclear localization signals and human disease. IUBMB Life 61 : 697–706. doi: 10.1002/iub.194 19514019
76. Terry LJ, Shows EB, Wente SR (2007) Crossing the nuclear envelope: hierarchical regulation of nucleocytoplasmic transport. Science 318 : 1412–1416. 18048681
77. Marchenko ND, Hanel W, Li D, Becker K, Reich N, et al. (2010) Stress-mediated nuclear stabilization of p53 is regulated by ubiquitination and importin-alpha3 binding. Cell Death Differ 17 : 255–267. doi: 10.1038/cdd.2009.173 19927155
78. Mashtalir N, Daou S, Barbour H, Sen NN, Gagnon J, et al. (2014) Autodeubiquitination Protects the Tumor Suppressor BAP1 from Cytoplasmic Sequestration Mediated by the Atypical Ubiquitin Ligase UBE2O. Mol Cell 54 : 392–406. doi: 10.1016/j.molcel.2014.03.002 24703950
79. von Mikecz A (2006) The nuclear ubiquitin-proteasome system. J Cell Sci 119 : 1977–1984. 16687735
80. Banerjee R, Weidman MK, Navarro S, Comai L, Dasgupta A (2005) Modifications of both selectivity factor and upstream binding factor contribute to poliovirus-mediated inhibition of RNA polymerase I transcription. J Gen Virol 86 : 2315–2322. 16033979
81. Lymberopoulos MH, Pearson A (2010) Relocalization of upstream binding factor to viral replication compartments is UL24 independent and follows the onset of herpes simplex virus 1 DNA synthesis. J Virol 84 : 4810–4815. doi: 10.1128/JVI.02437-09 20147409
82. Stow ND, Evans VC, Matthews DA (2009) Upstream-binding factor is sequestered into herpes simplex virus type 1 replication compartments. J Gen Virol 90 : 69–73. doi: 10.1099/vir.0.006353-0 19088274
83. Lawrence FJ, McStay B, Matthews DA (2006) Nucleolar protein upstream binding factor is sequestered into adenovirus DNA replication centres during infection without affecting RNA polymerase I location or ablating rRNA synthesis. J Cell Sci 119 : 2621–2631. 16763197
84. Zhai W, Comai L (1999) A kinase activity associated with simian virus 40 large T antigen phosphorylates upstream binding factor (UBF) and promotes formation of a stable initiation complex between UBF and SL1. Mol Cell Biol 19 : 2791–2802. 10082545
85. Raychaudhuri S, Fontanes V, Barat B, Dasgupta A (2009) Activation of ribosomal RNA transcription by hepatitis C virus involves upstream binding factor phosphorylation via induction of cyclin D1. Cancer Res 69 : 2057–2064. doi: 10.1158/0008-5472.CAN-08-3468 19223538
86. Watanabe H, Tanaka Y, Shimazu Y, Sugahara F, Kuwayama M, et al. (2005) Cell-specific inhibition of paramyxovirus maturation by proteasome inhibitors. Microbiol Immunol 49 : 835–844. 16172538
87. Shields SB, Piper RC (2011) How ubiquitin functions with ESCRTs. Traffic 12 : 1306–1317. doi: 10.1111/j.1600-0854.2011.01242.x 21722280
88. Votteler J, Sundquist WI (2013) Virus budding and the ESCRT pathway. Cell Host Microbe 14 : 232–241. doi: 10.1016/j.chom.2013.08.012 24034610
89. Irie T, Shimazu Y, Yoshida T, Sakaguchi T (2007) The YLDL sequence within Sendai virus M protein is critical for budding of virus-like particles and interacts with Alix/AIP1 independently of C protein. J Virol 81 : 2263–2273. 17166905
90. Moore HM, Bai B, Matilainen O, Colis L, Peltonen K, et al. (2013) Proteasome activity influences UV-mediated subnuclear localization changes of NPM. PLoS One 8: e59096. doi: 10.1371/journal.pone.0059096 23554979
91. Vilotti S, Biagioli M, Foti R, Dal Ferro M, Lavina ZS, et al. (2012) The PML nuclear bodies-associated protein TTRAP regulates ribosome biogenesis in nucleolar cavities upon proteasome inhibition. Cell Death Differ 19 : 488–500. doi: 10.1038/cdd.2011.118 21921940
92. Leljak Levanic D, Horvat T, Martincic J, Bauer N (2012) A novel bipartite nuclear localization signal guides BPM1 protein to nucleolus suggesting its Cullin3 independent function. PLoS One 7: e51184. doi: 10.1371/journal.pone.0051184 23251450
93. Latonen L, Moore HM, Bai B, Jaamaa S, Laiho M (2011) Proteasome inhibitors induce nucleolar aggregation of proteasome target proteins and polyadenylated RNA by altering ubiquitin availability. Oncogene 30 : 790–805. doi: 10.1038/onc.2010.469 20956947
94. Thoms HC, Loveridge CJ, Simpson J, Clipson A, Reinhardt K, et al. (2010) Nucleolar targeting of RelA(p65) is regulated by COMMD1-dependent ubiquitination. Cancer Res 70 : 139–149. doi: 10.1158/0008-5472.CAN-09-1397 20048074
95. Kruger T, Scheer U (2010) p53 localizes to intranucleolar regions distinct from the ribosome production compartments. J Cell Sci 123 : 1203–1208. doi: 10.1242/jcs.062398 20332106
96. Andersen JS, Lam YW, Leung AK, Ong SE, Lyon CE, et al. (2005) Nucleolar proteome dynamics. Nature 433 : 77–83. 15635413
97. Pokrovskaja K, Mattsson K, Kashuba E, Klein G, Szekely L (2001) Proteasome inhibitor induces nucleolar translocation of Epstein-Barr virus-encoded EBNA-5. J Gen Virol 82 : 345–358. 11161273
98. Mattsson K, Pokrovskaja K, Kiss C, Klein G, Szekely L (2001) Proteins associated with the promyelocytic leukemia gene product (PML)-containing nuclear body move to the nucleolus upon inhibition of proteasome-dependent protein degradation. Proc Natl Acad Sci U S A 98 : 1012–1017. 11158586
99. Matafora V, D'Amato A, Mori S, Blasi F, Bachi A (2009) Proteomics analysis of nucleolar SUMO-1 target proteins upon proteasome inhibition. Mol Cell Proteomics 8 : 2243–2255. doi: 10.1074/mcp.M900079-MCP200 19596686
100. Duan Z, Chen J, Xu H, Zhu J, Li Q, et al. (2014) The nucleolar phosphoprotein B23 targets Newcastle disease virus matrix protein to the nucleoli and facilitates viral replication. Virology 452–453 : 212–222.
101. Scott MS, Troshin PV, Barton GJ (2011) NoD: a Nucleolar localization sequence detector for eukaryotic and viral proteins. BMC Bioinformatics 12 : 317. doi: 10.1186/1471-2105-12-317 21812952
102. Emmott E, Hiscox JA (2009) Nucleolar targeting: the hub of the matter. EMBO Rep 10 : 231–238. doi: 10.1038/embor.2009.14 19229283
103. Greco A (2009) Involvement of the nucleolus in replication of human viruses. Rev Med Virol 19 : 201–214. doi: 10.1002/rmv.614 19399920
104. Hiscox JA (2007) RNA viruses: hijacking the dynamic nucleolus. Nat Rev Microbiol 5 : 119–127. 17224921
105. Salvetti A, Greco A (2013) Viruses and the nucleolus: The fatal attraction. Biochim Biophys Acta.
106. Satterly N, Tsai PL, van Deursen J, Nussenzveig DR, Wang Y, et al. (2007) Influenza virus targets the mRNA export machinery and the nuclear pore complex. Proc Natl Acad Sci U S A 104 : 1853–1858. 17267598
107. von Kobbe C, van Deursen JM, Rodrigues JP, Sitterlin D, Bachi A, et al. (2000) Vesicular stomatitis virus matrix protein inhibits host cell gene expression by targeting the nucleoporin Nup98. Mol Cell 6 : 1243–1252. 11106761
108. Ghildyal R, Baulch-Brown C, Mills J, Meanger J (2003) The matrix protein of Human respiratory syncytial virus localises to the nucleus of infected cells and inhibits transcription. Arch Virol 148 : 1419–1429. 12827470
109. Bian T, Gibbs JD, Orvell C, Imani F (2012) Respiratory syncytial virus matrix protein induces lung epithelial cell cycle arrest through a p53 dependent pathway. PLoS One 7: e38052. doi: 10.1371/journal.pone.0038052 22662266
110. Hu J, Zacharek S, He YJ, Lee H, Shumway S, et al. (2008) WD40 protein FBW5 promotes ubiquitination of tumor suppressor TSC2 by DDB1-CUL4-ROC1 ligase. Genes Dev 22 : 866–871. doi: 10.1101/gad.1624008 18381890
111. Andrews P, He YJ, Xiong Y (2006) Cytoplasmic localized ubiquitin ligase cullin 7 binds to p53 and promotes cell growth by antagonizing p53 function. Oncogene 25 : 4534–4548. 16547496
112. Ohta T, Michel JJ, Schottelius AJ, Xiong Y (1999) ROC1, a homolog of APC11, represents a family of cullin partners with an associated ubiquitin ligase activity. Mol Cell 3 : 535–541. 10230407
113. Liu J, Furukawa M, Matsumoto T, Xiong Y (2002) NEDD8 modification of CUL1 dissociates p120(CAND1), an inhibitor of CUL1-SKP1 binding and SCF ligases. Mol Cell 10 : 1511–1518. 12504025
114. Dundr M, Hoffmann-Rohrer U, Hu Q, Grummt I, Rothblum LI, et al. (2002) A kinetic framework for a mammalian RNA polymerase in vivo. Science 298 : 1623–1626. 12446911
115. Wang W, Budhu A, Forgues M, Wang XW (2005) Temporal and spatial control of nucleophosmin by the Ran-Crm1 complex in centrosome duplication. Nat Cell Biol 7 : 823–830. 16041368
116. Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, et al. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7 : 539. doi: 10.1038/msb.2011.75 21988835
117. Shyu YJ, Liu H, Deng X, Hu CD (2006) Identification of new fluorescent protein fragments for bimolecular fluorescence complementation analysis under physiological conditions. Biotechniques 40 : 61–66. 16454041
118. Clarke DK, Sidhu MS, Johnson JE, Udem SA (2000) Rescue of mumps virus from cDNA. J Virol 74 : 4831–4838. 10775622
119. Kaiser P, Wohlschlegel J (2005) Identification of ubiquitination sites and determination of ubiquitin-chain architectures by mass spectrometry. Methods Enzymol 399 : 266–277. 16338362
120. Wohlschlegel JA (2009) Identification of SUMO-conjugated proteins and their SUMO attachment sites using proteomic mass spectrometry. Methods Mol Biol 497 : 33–49. doi: 10.1007/978-1-59745-566-4_3 19107409
121. Tabb DL, McDonald WH, Yates JR 3rd, (2002) DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J Proteome Res 1 : 21–26. 12643522
122. Xu T, Venable J, Park SK, Cociorva D, Lu B, et al. ProLuCID, a fast and sensitive tandem mass spectra-based protein identification program; 2006. AMER SOC BIOCHEMISTRY MOLECULAR BIOLOGY INC 9650 ROCKVILLE PIKE, BETHESDA, MD 20814–3996 USA. pp. S174-S174.
123. Elias JE, Gygi SP (2007) Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods 4 : 207–214. 17327847
124. Florens L, Carozza MJ, Swanson SK, Fournier M, Coleman MK, et al. (2006) Analyzing chromatin remodeling complexes using shotgun proteomics and normalized spectral abundance factors. Methods 40 : 303–311. 17101441
125. Bardou P, Mariette J, Escudie F, Djemiel C, Klopp C (2014) jvenn: an interactive Venn diagram viewer. BMC Bioinformatics 15 : 293. doi: 10.1186/1471-2105-15-293 25176396
126. Huang da W, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4 : 44–57. doi: 10.1038/nprot.2008.211 19131956
127. Huang da W, Sherman BT, Zheng X, Yang J, Imamichi T, et al. (2009) Extracting biological meaning from large gene lists with DAVID. Curr Protoc Bioinformatics Chapter 13: Unit 13 11.
128. Cline MS, Smoot M, Cerami E, Kuchinsky A, Landys N, et al. (2007) Integration of biological networks and gene expression data using Cytoscape. Nat Protoc 2 : 2366–2382. 17947979
129. Montojo J, Zuberi K, Rodriguez H, Kazi F, Wright G, et al. (2010) GeneMANIA Cytoscape plugin: fast gene function predictions on the desktop. Bioinformatics 26 : 2927–2928. doi: 10.1093/bioinformatics/btq562 20926419
130. Stone R, Takimoto T (2013) Critical role of the fusion protein cytoplasmic tail sequence in parainfluenza virus assembly. PLoS One 8: e61281. doi: 10.1371/journal.pone.0061281 23593451
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 3
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Bacterial Immune Evasion through Manipulation of Host Inhibitory Immune Signaling
- BILBO1 Is a Scaffold Protein of the Flagellar Pocket Collar in the Pathogen
- Antimicrobial-Induced DNA Damage and Genomic Instability in Microbial Pathogens
- Attenuation of Tick-Borne Encephalitis Virus Using Large-Scale Random Codon Re-encoding
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy