
Early Virological and Immunological Events in Asymptomatic Epstein-Barr Virus Infection in African Children
Primary infection with EBV, a common human herpesvirus, is typically asymptomatic in childhood but, if occurring in adolescence or later, often presents as AIM. This febrile illness is characterised by high virus loads in the blood and an exaggerated EBV-specific CD8+ T-cell response that pushes total CD8+ T-cell numbers well above normal levels. By contrast, very little is known about the events of asymptomatic primary infection. We therefore studied young Gambian children at an age at which many acquire EBV, monitoring them over six months for evidence of EBV infection by virus load in the blood, virus-specific IgM and IgG antibody status, and virus-specific CD8+ T-cell responses. Focusing on IgM-positive children with very recent EBV infection but no history of symptoms, we found that they carried a virus load equivalent to that seen in AIM patients and also mounted a classical virus-specific CD8+ T-cell response. However, that response, though it could occupy at least 15% of the circulating CD8+ T-cell pool, occurred without the huge global expansion of CD8 numbers seen in AIM. This work reinforces the idea that the host’s exaggerated CD8+ T-cell response, rather than the virus load per se, leads to the symptoms of AIM.
Published in the journal:
. PLoS Pathog 11(3): e32767. doi:10.1371/journal.ppat.1004746
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004746
Summary
Primary infection with EBV, a common human herpesvirus, is typically asymptomatic in childhood but, if occurring in adolescence or later, often presents as AIM. This febrile illness is characterised by high virus loads in the blood and an exaggerated EBV-specific CD8+ T-cell response that pushes total CD8+ T-cell numbers well above normal levels. By contrast, very little is known about the events of asymptomatic primary infection. We therefore studied young Gambian children at an age at which many acquire EBV, monitoring them over six months for evidence of EBV infection by virus load in the blood, virus-specific IgM and IgG antibody status, and virus-specific CD8+ T-cell responses. Focusing on IgM-positive children with very recent EBV infection but no history of symptoms, we found that they carried a virus load equivalent to that seen in AIM patients and also mounted a classical virus-specific CD8+ T-cell response. However, that response, though it could occupy at least 15% of the circulating CD8+ T-cell pool, occurred without the huge global expansion of CD8 numbers seen in AIM. This work reinforces the idea that the host’s exaggerated CD8+ T-cell response, rather than the virus load per se, leads to the symptoms of AIM.
Introduction
Epstein-Barr Virus (EBV) is a ubiquitous gamma herpesvirus associated with occasional severe primary infections, several malignancies and significant pathology in immunosuppressed hosts. It does not, however, cause significant morbidity in the majority of those infected. In The Gambia most children are infected during childhood, in contrast to most developed countries where the majority of primary infections occur at a later age, often in adolescence [1,2]. It is estimated that between a quarter and up to three quarters of those infected in adolescence will develop a sometimes-severe disease, AIM [3,4]. Paradoxically, those infected during childhood tend to have minor self-limiting illnesses that often go undetected [5]. It is not fully understood why individuals that contract EBV during childhood are usually asymptomatic and do not develop AIM. Of note, most of the published literature regarding the immunopathogenesis of primary EBV infection is derived from studies of AIM, rather than asymptomatic infections.
Many studies in adults have characterised cellular immune responses ex vivo during AIM, both among CD8+ and to a lesser extent CD4+ T-cell subsets [6–13]. The EBV-specific CD8+ T-cell response is hugely amplified, such that total CD8+ T-cell numbers in the blood may reach five to ten-fold higher than usual. Indeed individual lytic antigen reactivities (typically against epitopes within the immediate early (IE) and some early (E) proteins) can account for up to 40% of the highly expanded CD8+ T-cell population, and individual latent antigen reactivities (typically against epitopes from the EBV nuclear antigen 3A, 3B, 3C family) occupying up to 5%. These CD8+ T-cells display a phenotype consistent with recent antigen stimulation, being perforin-positive with direct ex vivo cytotoxic function [14–16] and express the activation marker CD38 and cell cycling marker Ki-67 [8,10,14,17]. What drives these expansions in AIM is unclear, but factors such as an initial lack of natural killer cell control [18], cross-reactive recognition by clonotypes in pre-existing CD8+ T-cell memory [19], and genetic factors [20–22], including polymorphism of the IL-10 promoter [23], have all been proposed.
Whether the cellular response to early EBV infection in asymptomatic children shows features of disruption similar to those described in AIM has been difficult to investigate, mostly because donors without clinical symptoms can’t be readily identified. However, an understanding of how EBV infection is controlled with minimal immunopathology, i.e. without the development of AIM, is important, as AIM is associated with an increased risk of EBV-associated diseases such as EBV-positive Hodgkin lymphoma and multiple sclerosis [24,25]. Of the few published studies on asymptomatic primary EBV infections, Silins et al identified four adult patients undergoing silent seroconversion within a vaccine trial [26]. Interestingly some of these had high EBV loads yet did not have massive T-cell expansions and, where studied, most did not have the distorted T-cell receptor (TcR) repertoires usually seen in AIM; however, their EBV-specific T-cell response was not studied. As to infections during childhood, an early report suggested that this occurs with a serological picture distinct from AIM and without obvious lymphocytosis [27], while another small study of children aged 20–35 months detected EBNA3A, B and C-specific CD8+ T-cell responses in the blood without addressing issues of viral load or hyper-expansion [28].
A detailed study of the EBV-specific immune response and EBV dynamics in asymptomatically infected children utilising modern immunological and virological tools is lacking. The present work follows a cohort of 114 Gambian children longitudinally over six months, using serology and viral load to determine EBV status. It describes the EBV-specific CD8+ T cell responses in those that have had a relatively recent primary EBV infection without any obvious clinical history, and additionally captures six children undergoing silent seroconversion.
Results
Demographics and EBV serostatus of Gambian children
Gambian children of an age, 14–18 months, likely to be undergoing primary EBV infection were recruited for study from an infant vaccination clinic. Blood samples were collected from the children at baseline (visit one), they were vaccinated one week later against Diptheria, Tetanus, Pertussis, Hepatitis B and Haemophilus influenzae B (visit two). A five-millilitre blood sample was collected a week later (visit three) primarily to monitor vaccine responses and a further sample at six months (visit four). Of 120 children screened, six were ineligible due to concurrent illnesses or malnutrition at screening whereas 114 were enrolled, of which 99 remained in the study until completion at six months. The study dropouts did not significantly differ in age, sex, weight, haemoglobin, leucocyte and lymphocyte counts, compared to those who continued to participate (S1 Table).
Initially children were tested for their EBV-VCA antibody status and categorised into one of three groups: non-infected (IgM−IgG−), established infection (IgM−IgG+) or very recently infected (IgM+IgG+/−). At visit one, 71 children had established EBV infection as judged by the presence of VCA-specific antibodies of IgG but not IgM class (Fig. 1). Another four children showed evidence of recent infection, with three having IgM VCA-specific antibodies only and one having both IgM and IgG VCA-specific antibodies. The remaining 39 children appeared to be non-infected, having no detectable VCA-specific antibodies or viral genomes in their peripheral blood mononuclear cells (PBMCs). At visit four (six months later) 17 of these 39 initially EBV non-infected donors had become VCA IgM−IgG+, another two had become VCA IgM+IgG+, 13 remained VCA IgM−IgG−, while seven dropped out of the study. The four initially IgM+ children had now become IgM - and had developed VCA-specific IgG. All children, including those with VCA-specific IgM antibodies, were asymptomatic for classical symptoms of AIM (fever, lymphadenopathy, malaise) prior to recruitment and at subsequent visits, based on maternal history and clinical evaluation. Overall, 62% of children showed serological evidence of being EBV infected at baseline, rising to 86% among those remaining in the study six months later. An analysis of VCA IgG titre in a subset of 25 pairs of samples from children at visit one and four showed no significant difference in titre (p = 0.774, S1A Fig).

Recently-infected asymptomatic Gambian children have EBV genome loads equivalent to donors with acute infectious mononucleosis
Since acquisition of EBV in similar African cohorts begins between six and twelve months after birth [29,30], it is likely that at least some children who were IgG reactive to VCA within the cohort were infected with EBV within the last six months prior to recruitment. To examine for evidence of recent infection among these donors, EBV genome loads in PBMCs were measured by qPCR analysis. Fig. 2 shows viral genome load data from PBMCs collected from 70 IgM−IgG+ donors at baseline, 58 of these donors six months later, and six very recently infected IgM VCA reactive donors (some of whom also had VCA-specific IgG antibodies). Genome load data from Caucasian adolescent patients undergoing primary symptomatic EBV infection, AIM, assessed using the same qPCR assay are also included for comparison.

Almost all of the children who had IgG antibodies to VCA at baseline had high EBV genome levels in their PBMCs, ranging up to two million genomes per million PBMCs. Indeed, for the whole cohort of IgM − IgG+ children sampled at visit 1, the median load of 3000 genomes per million PBMCs (IQR 900 to 8000 genomes per million PBMCs) was not significantly different to that observed in AIM patients. When PBMCs from these same children were assessed for genome loads six months later, a narrower range of values was found and the median load was eight to tenfold lower than at baseline. The decreasing virus loads observed over these two time points suggests that these donors were establishing their virus host balance following recent EBV infection. Comparing virus load to VCA IgG titre in a subset of 25 children at the two time points showed no correlation between load and titre (S1B and C Fig). When the samples from the six IgM+ donors were analysed, these also showed high EBV genome loads with a median value of 8000 genomes per million PBMC, slightly higher than but not significantly different from loads in IgM-IgG+ positive children measured at baseline and again similar to that seen in AIM patients. Such data are consistent with these IgM+ children having been very recently EBV-infected.
Lymphocyte counts are not altered in asymptomatic primary EBV infection in Gambian children
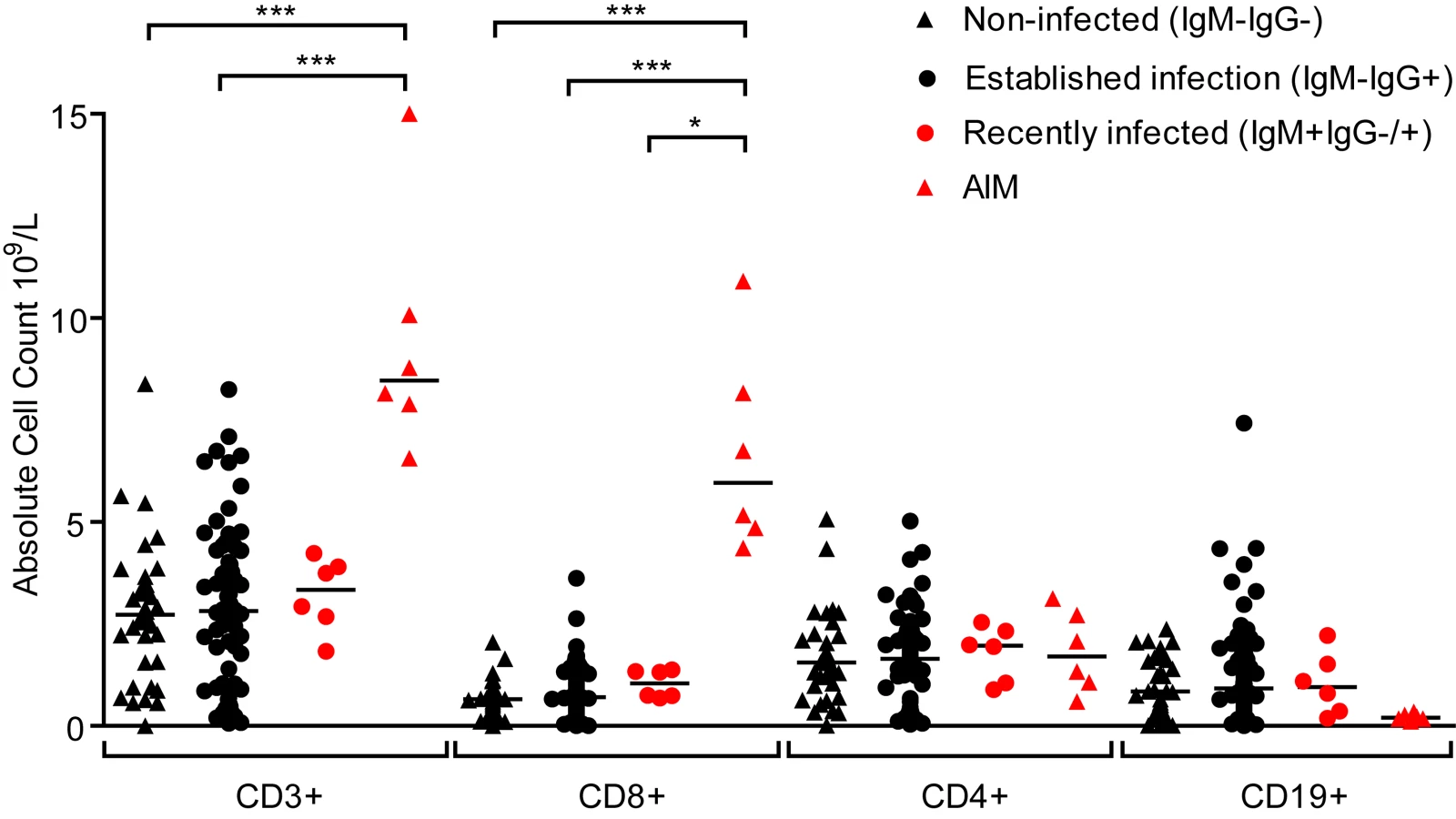
Primary symptomatic infection with EBV is associated with dramatic expansions in the frequency and absolute number of lymphocyte subsets, especially CD8+ lymphocytes [6]. Evidence of disruptions to the lymphocyte compartments of the three groups of children (IgM − IgG−, IgM − IgG+ or IgM+ IgG+/−) were studied by determining absolute numbers of lymphocytes within the CD3, CD4, CD8 and CD19 subsets. Fig. 3 shows results of absolute cell counts and, for comparison, counts of equivalent subsets from six Caucasian AIM patients. Dramatic expansions of the CD3+ and CD8+ (but not CD4+) T-cell numbers and a contraction of B cell numbers were seen in AIM patient samples. However, no obvious or significant expansion of lymphocyte subsets was observed when comparing uninfected children with the two EBV-infected groups. Furthermore, no significant changes in the CD4:CD8 ratios were observed in PBMCs from a subset of 14 children over the six month study period (p = 0.76, S2 Fig). This indicated that there was little disruption to peripheral lymphocyte subsets in children at these different stages of asymptomatic EBV infection.
EBV-specific CD8+ T-cells from children with established EBV infection show evidence of recent activation
To further understand the dynamics of CD8+ T-cell responses and virus loads at early stages of infection, MHC-class I tetramers were used to assess the frequency of EBV-specific T-cells in the PBMCs of children with VCA-specific IgM − IgG+ antibodies at baseline and six months later and compared to their virus loads at these time points. Children with relevant EBV-specific responses were identified by screening PBMCs from visit one for responses by ELISpot to pools of peptides containing peptide-epitopes known to be presented by HLA types frequent within the Gambian population. This identified 14 children with responses which could be assessed with HLA-A*0201, HLA-B*0801 or HLA-B*3501MHC class I tetramers. Virus loads from this subset of donors were representative of the overall population shown in Fig. 2 at the two time points and their loads were significantly decreased at the second time point (Fig. 4A).

Frequencies of EBV-specific responses in each of the children’s paired visit one and visit four PBMCs samples were obtained by staining with an appropriate tetramer which would identify an immunodominant response. These tetramers presented epitopes derived from the immediate early lytic cycle protein BZLF1 protein, either the HLA-B*0801 presented RAKFKQLL peptide or the B*3501 presented EPLPQGQLTAY peptide, or from the early lytic cycle protein BMLF1, the HLA-A*0201 presented GLCTLVAML peptide. Analysis of the frequency of tetramer-specific CD8+ T-cells in PBMCs from this group is shown in Fig. 4B. This shows that at the first time point EBV-specific responses are made, but their frequency of up to 2.5% of CD8+ T cells is not obviously increased as is seen in AIM patients, in whom previous reports have documented up to 40% of the total CD8+ T-cell population as being EBV-specific (Callan et al. 1998). Analysis of the visit four time point, collected six months later, showed that responses were still present, however they were on average significantly lower than those detected at visit one.
This reduction in EBV-specific T-cell numbers with time, coupled with a falling virus load illustrates a pattern consistent with recent EBV infection and establishment of a long term carrier state in these children. In all but one of these 14 children there were sufficient cell numbers to examine the changing phenotype of the T-cell response over time. Both total CD8+ T-cells and tetramer-positive CD8+ T-cells were examined for markers known to be expressed by CD8+ T-cells responding to acute EBV infection; namely activation status as defined by co-expression of CD38 and HLA-DR, cycling status as defined by the expression of Ki-67, and apoptosis sensitivity as indicated by loss of Bcl-2, which is down regulated in activated EBV-specific cells in AIM patients. Of note CD38 and HLA-DR co-staining for activation status was used as lymphocytes from young children may constitutively express CD38 which is progressively lost with age [31]. The graphs on the left hand side of Fig. 5 summarises the results of this analysis while the flow cytometry plots show representative analyses for each stain at the two time points; note that the top flow plots combine data from all CD8+ T cells in black, with the tetramer-positive population in red, while the middle and bottom flow plots show the Ki-67/tetramer and Bcl-2/tetramer profiles. At the first time point there were significantly more HLADR+CD38+ (p = 0.002) and Ki-67+ (p = 0.01) EBV-specific CD8+ T-cells compared to the total CD8+ T-cell population, while tetramer positive CD8+ T-cells expressed lower levels of the anti-apoptotic marker Bcl-2 (p <0.0001). The percentage of HLADR+CD38+ EBV-specific CD8+ T-cells in the IgM−IgG+ children declined significantly over time (p = 0.003), whereas cellular expression of Bcl-2 significantly increased (p = 0.013), with a non-significant decline seen in frequency of cells expressing Ki-67. Again, the phenotypic analysis of these VCA antibody IgG+ donors suggests recent EBV infection and the establishment of a long term carrier state.

Asymptomatic primary EBV-infected Gambian children lack a significant expansion of global T-cell numbers despite substantial activation of EBV-specific CD8+ T-cells
To get the clearest understanding of the early events in EBV-specific CD8+ T-cell responses in children undergoing asymptomatic infection, the EBV tetramer-specific responses and phenotype of these cells were examined in children with IgM VCA-specific antibodies. Of the six IgM+ children found in this study, three were suitable for study with the tetramer panel for frequency and phenotype analysis at the different time points. However the three other donors did not show expanded numbers of CD8+ T cells (Fig. 3) and showed, at most, small changes in the frequencies of CD8+ T cells from 40.9%, 36.8% and 50% at visit one when they were IgM+, to 27.3%, 32.5% and 50.1% at visit four respectively when they were IgG+.
Of the samples from children which could be analysed with tetramers, 082 and 007 were seronegative at the first time point, but had developed IgM+ VCA-specific antibodies at six months. Although there was no significant increase in the absolute numbers of CD8+ T-cells before and after EBV acquisition, both donors 082 and 007 showed increases in the frequencies of CD3+ CD8+ lymphocytes from baseline values of 18.2% and 19.8% to values of 50.3% and 40.8% respectively six months later. As shown in Fig. 6, tetramer analysis of samples at baseline when the donors 082 and 007 were VCA seronegative showed no tetramer-specific staining. However at six months, when the children were VCA IgM+ IgG+, the HLA-B*0801 donor 082 made a substantial response with 6.9% of their CD8+ T-cells being specific to the RAK-epitope while the HLA*0201 donor 007 made a small response of 0.43% CD8+ T-cells to the GLC-epitope. In both cases, tetramer positive cells were highly activated with the majority of EBV-specific cells co-expressing HLA-DR and CD38. In both of these donors a substantial frequency of EBV-specific cells were in cycle; interestingly in the case of child 007 a large proportion of non-tetramer-specific CD8+ T-cells were also in cycle, likely representing other EBV-specificities, possible bystander activation or coincident infection with another pathogen. In the EBV-specific CD8+ T-cells from these children there was little if any expression of Bcl-2 (Fig. 6).

The third child studied, the HLA-B*0801 donor 061, had VCA-specific IgM+ antibodies at the first time point and made a substantial response to the RAK epitope which allowed serial monitoring of this response (Fig. 7). The frequency of PBMC CD8+ T cells did not change over time with levels being 38% at visit one, 37% at three two weeks later, and 37% at visit four with no significant expansion of absolute numbers (see Fig. 1). However, as shown in Fig. 7, tetramer analysis at the first time point detected a large RAK-specific response of 15.9% of CD8+ T-cells, with 32.2% of these being activated, 14.8% in cycle and few expressing Bcl-2. After two weeks the tetramer-specific frequency had decreased to 5.4% of CD8+ T-cells with no associated decrease in activation marker and Ki-67 expression, nor increase in Bcl-2 expression by the tetramer-specific cells. By six months, the frequency of RAK-specific cells had decreased to 1.75% of CD8+ T-cells. Here the RAK-specific cells phenotypically resembled the EBV-specific cells from the VCA-IgG+ donors at visit 4, with only 5% expressing activation markers, 5% of cells in cycle and 15% expressing Bcl-2 (Fig. 7). Overall these findings suggest that in asymptomatic primary EBV infection, the frequency of activated EBV-specific cells in the CD8 population can be substantial but this occurs without significant expansion of the CD8 compartment as a whole.

Discussion
Current understanding of the immunological changes seen during primary EBV infection is almost exclusively derived from studies of AIM in adolescents and adults. These may not apply to the situation in asymptomatic primary EBV in early childhood, when a large proportion of infections occur. In this study, virological and immunological parameters of African children, studied during a time period when they undergo primary asymptomatic EBV infection, were examined to better understand the pathogenesis of primary asymptomatic EBV infection. Children who had EBV infection established for at least some months, as judged by the presence of VCA IgG antibodies but not IgM, have high virus loads, comparable to AIM patients, and their EBV-specific CD8+ T-cells show evidence of recent activation. These loads dropped significantly when tested six months later but were still elevated when compared to loads in other populations such as UK carriers, but were similar to those detected in healthy older children from The Gambia [32]. This suggests that in this situation, the virus set point is established several months after primary infection and that it is higher than in EBV-infected carriers in the UK.
What determines the high virus set point detected in these children is unclear. One factor known to increase EBV loads is exposure to malaria, with children living in malaria endemic regions have higher EBV loads compared to those living in areas of sporadic transmission [32,33]. How EBV loads in children in the present study relate to loads in other African childhood cohorts is difficult to determine due to differences in assays and sample sources used to quantify EBV loads [33,34]. Malaria infection is thought to increase EBV loads either through promoting B cell proliferation [35] or altering T cell responses [36]. However the children in this study showed no evidence of acute malaria and data at the time of the study demonstrated a low prevalence of malaria in The Gambia [37,38]. Others have suggested virus load may be related to the age at infection, with African infants infected shortly after loss of maternal antibodies having higher virus loads than those infected later [34]. In this context, comparing virus loads to western populations should be interpreted with some caution as the timing of infection in these latter populations is relatively delayed and is particularly dependent on the ethnic group studied [39].
A clearer picture of the early immune response dynamics comes from studying children with IgM+ VCA-specific antibodies, who are likely to have had very recent EBV infection. Although these children had high virus loads in their PBMCs, akin to those seen in studies of symptomatic AIM [2,3,40], they showed no obvious physical signs of infection. There was no significant expansion of lymphocytes or the CD3+ or CD8+ T-cell compartment; a finding consistent with those of others who have studied lymphocyte compartments in primary asymptomatic EBV infections [1,26,27]. Nevertheless, there was an ongoing virus-specific CD8+ T cell response in the IgM+ children with, in one case, greater than 15% of CD8+ T cells directed against a single dominant EBV epitope. This increased frequency of EBV-specific T cells found in the periphery in the presence of high virus loads may call in to question their role in control of the infection. However we have previously found that in AIM patients EBV-specific T cells lack expression of lymphoid homing receptors such as CCR7 and CD62L which allow access to tissues such as the tonsil. Virus replication and transformation of B lymphocytes, in AIM patients at least, occurs in this tissue and appears poorly controlled by the inefficient recruitment of EBV-specific T cells to this site [10]. As activated EBV-specific T cells express low levels of CCR7 and CD62L, we propose that the activated EBV-specific T cells in the children are similarly inefficiently recruited to the tonsil and virus replication and transformation poorly controlled at this site, allowing higher loads of virus to be detected in the presence of these strong responses.
The phenotype of the antigen-specific CD8+ T-cells from the IgM+ VCA antibody children was consistent with what has been described in AIM donors, being highly activated (HLADR+CD38+), in cycle (Ki-67+) and pro-apoptotic (low Bcl-2 expression) [8,10,14,17,41,42]. Why these children do not develop the expanded numbers of CD8+ T-cells observed in AIM patients after EBV infection is not clear. Possible reasons for the AIM associated hyper-expansion have included the development of heterologous immunity where an existing response to an epitope coded by a previously encountered pathogen cross reacts with another from EBV, amplifying the pool of T-cells responsive to EBV challenge and potentially inducing an exaggerated response. Children with less antigenic exposure would have a more limited repertoire of T-cells capable of responding in this heterologous manner [19]. Alternatively two recent studies comparing the incidence of AIM in monozygotic compared to dizygotic twins, and first-, second - or third-degree relatives have shown concordance in the development of AIM, suggesting that there may be a genetic component underlying disease development [20,22]. Perhaps more importantly, an emerging concept in the control of early EBV infection comes from studies using immunodeficient mice reconstituted with human haematopoietic cells, which repopulate human NK and T-cell repertoires. Depletion of NK cells in this model followed by challenge with EBV recapitulates an AIM-like response including splenomegaly, increased plasma levels of the pro-inflammatory cytokine IFN-γ and increased CD8+ T-cell numbers and frequencies [18]. Currently there is a lack of clarity in the literature as to the dynamics and role of NK cell responses in AIM and so the immediate relevance of these experimental findings to natural infection remain to be resolved. Some studies have indicated that there are inverse relationships between NK cell numbers and both severity of symptoms and virus load [43], while others have shown a positive correlation between NK cell numbers both with virus load and severity of symptoms in AIM patients [3]. However NK cells are a heterogeneous population and so a key question arising from these studies is whether there is a difference in subsets of NK cells in terms of numbers or function between individuals who go on to develop AIM compared to children or others who don’t develop this disease.
It is important to recognise that the immune system of children is different in comparison to adolescents where AIM is typically seen. Neonates are born with high levels of the immunosuppressive cytokine IL-10, high levels of plasma immunosupressive factors such as adenosine, and have Th2 and Th17 skewed immunity, all of which decline by 1–2 years of age to near adult levels [44]. This “infant-adapted” immune profile evolves in the first few years of life, with a gradual increase in Th1 capacity, maturing of B cell and antibody responses, and development of T and B cell immunological memory [44]. By the time infants were recruited into the study at 14–18 months of age, they would have a considerably matured immune system compared to birth but some differences would persist compared to adolescents. Such immunological differences could have contributed to the asymptomatic primary infection that is seen in the children compared to adolescents.
Throughout this study we have used the VCA serological status as a guide to when infection occurred, consequently the precise timing of the primary EBV infection cannot be determined. However, from previous data on infants at the same study site, only 18% of infants were found to be EBV infected at nine months of age [29] and one can, therefore, assume that the majority of children would have been infected six months to one year prior to recruitment. Furthermore, the IgM+IgG± donors were likely infected within the last 120 days and may be at a different stage of infection. Secondly, ensuring children are truly asymptomatic in this setting can be challenging as reliance on maternal perception may not be reliable and careful clinical studies of infants or children have described AIM like symptoms in some instances [45]. To combat this, the children all underwent a health screen by the study clinician, including baseline clinical observations such as weight, height, temperature and heart rate and where indicated a rapid malaria test. Recent work by Balfour et al. has suggested that 89% of primary EBV infections in a cohort of University students display some symptoms, although they may not fulfil all classic criteria for AIM, which is different to our observations in Gambian children [3].
In summary, this study supports the notion that AIM is an immunopathological disease and that symptoms are caused by the significantly expanded CD8+ T-cell responses to the virus. It provides clear evidence that during primary asymptomatic infection EBV-specific responses are indeed activated and can occupy a significant percentage of the circulating CD8+ T cell pool. However these responses appear able to contain the infection without the massive expansion that characterises AIM. Conversely then, the symptoms of AIM appear to derive from the absolute CD8 expansion rather than from the virus infection per se.
Materials and Methods
Ethics statement
The Gambian Government/ MRC Laboratories Joint Ethics Committee approved this study. Participants were enrolled after individual written informed consent was obtained from the participant's parent/guardian. SCC 1206.
Donors
This study was conducted in a peri-urban Medical Research Council (MRC) UK clinic, Sukuta, situated within the Government Sukuta Health Centre, serving a low-income population living in crowded conditions. A cohort of 120 children aged between 14 and 18 months were screened when they attended the local government health centre for their routine booster vaccination of diphtheria, tetanus, whole cell pertussis (DTwP) combined vaccine. All children were screened by enquiring about a maternal history of recent illness (e.g. fever) and a clinician examination for signs and symptoms of infectious mononucleosis, including weight and baseline observations (temperature, heart rate, weight and length). Any child found to be unwell (observations outside normal clinical range for age or maternal report of recent illness) or had a weight below that specified on the local Infant Welfare Card Growth Chart were not recruited into the study. (n = 6). Of the children not recruited, none of these showed clinical features suggestive of infectious mononucleosis. Five millilitres of blood was collected from each child into vacutainers containing EDTA (BD). A 500μl aliquot was removed and used to obtain a full blood count on each child using a M-series M16/M20 Haematology Analyser (Medonic, Sweden). A further 250μl aliquot was removed and whole blood flow cytometric staining performed. The remaining blood was layered on to 4mls of Lymphoprep (Axis-Shield, UK) in 15ml Leucosep tubes (Greiner Bio-One, UK). Following centrifugation, the plasma layer was removed, and cryopreserved in 2ml aliquots and stored at −70°C for downstream serology. The lymphocyte interphase was harvested and washed. Cells were counted and re-suspended in freezing medium (FCS (Sigma-Aldrich) supplemented with 10% (v/v) dimethyl sulfoxide (DMSO)) at approximately 5 x 106/ml. Children were brought back one week later to receive the Pentavalent vaccination (DTwP, Hep B, Hib) (Easy Five Panacea Biotec). They were subsequently invited to return a week after vaccination and again at six months to undergo further blood sampling.
AIM patients were recruited from a cohort of young adults (18–25 years old) collected at the University of Birmingham, UK. All patients gave written informed consent to donate samples and experiments were approved by the South Birmingham Local Research Ethics Committee (reference number 07/Q2702/24). Patients were defined as AIM by having tonsillitis/sore throat, high lymphocyte counts and being heterophile antibody positive. Mononuclear cells were harvested from blood specimens and stored as described above.
EBV genome loads
EBV genome loads were assayed by quantitative real-time PCR, as described elsewhere [46]. DNA extraction was performed from 1x106 PBMCs using QIAmp DNA Blood Mini kit (Qiagen).
Serology
IgG and IgM reactivity to EBV Viral Capsid Antigen (VCA) were measured using a previously described in-house immunofluorescence assay at the Institute for Cancer Studies, Birmingham [47,48] and the MRC-University of Glasgow Centre for Virus Research, University of Glasgow [49,50] respectively.
Lymphocyte subset analysis by flow cytometric staining of human cells
For children a 100 μl volume of whole blood for each donor was stained with antibodies to the following surface markers: CD3 PE, CD4 PerCP, (BD Biosciences), CD8 efluor450 and CD27 APCalexafluor750 (Ebioscience) for 30 min at 4°C. Red blood cells were then lysed using 1 : 10 FACS Lysing Solution (BD Biosciences) and incubated for 10 min at room temperature. Cells were then washed twice in FACS buffer (PBS, 5% BSA, 5% EDTA) and re-suspended in Cytofix (BD, Biosciences). Samples were acquired on a Cyan ADP flow cytometer using Summit software (Beckman Coulter) at MRC Gambia.
Lymphocyte subsets from AIM patients were identified by staining with antibodies specific to: CD19 FITC, CD4 PE (Biolegend), CD27 APC elfluor 780, CD3 efluor 450 (eBioscience) and CD8 qDot 655 (Invitrogen). Samples were stained for 30 min on ice, washed and analysed immediately on an LSR-II flow cytometer (BD Biosciences). Data was analysed using Flow-Jo software (Treestar Inc).
Cell surface and tetramer flow cytometric staining
Tetramers were used to identify and analyse the surface marker phenotype of epitope-specific CD8+ T-cells. From the aforementioned IFN-γ ELISPOT data we selected the following epitopes, B*0801 RAKFKQLL, B*3501 EPLPQGQLTAY and A*0201 GLCTLVAML for tetramer manufacture, as they were frequent targets of the immune response. Markers of activation (CD38 & HLDR), proliferation (Ki-67) and the anti-apoptotic marker, Bcl-2 were assessed. Tetramers were validated for specificity against HLA-matched and mismatched seropositive and seronegative donors. Tetramer staining was performed on cryopreserved PBMCs as described elsewhere. Cells were thawed and stained with LIVE/DEAD fixable Aqua Dead Cell Stain for 30 min at 4°C, washed and stained with 1μg of tetramer-PE for 15min at 37°C. Following two further washes, surface staining with CD3-Qdot655, CD4-Qdot605, CD8-Qdot705, CD14-V500, CD19-V500, CD38-APC and HLA-DR-Alexafluor700 were performed. Following fixing and permeabilisation as described above, intracellular staining with Ki67-Alexafluor488 and Bcl-2 (B-cell lymphoma 2)-V450 was performed. Fluorescence minus one samples were included to aid gating during subsequent flow cytometric analysis. A comparison of expression of the above phenotypic markers on EBV-specific and the total CD8+ T-cell populations were performed.
Compensation for fluorescence ‘spill-over’ was performed using the BD CompBead Anti-Mouse Ig set (BD Biosciences) and the antibodies described above. Briefly, antibodies were added to separate tubes containing one drop each of Anti-Mouse Ig beads and the negative control beads (which do not bind κ light chain-bearing immunoglobulin). Following a 30 min incubation at 4°C, beads were washed and re-suspended in FACS buffer.
Statistics
All statistical analyses were performed using Graphpad Prism version 5.0 for Macintosh (GraphPad Software, San Diego California, USA, www.graphpad.com). Comparisons between variables were performed using the Mann-Whitney U test (two-tailed) and for non-parametrically distributed data, the Wilcoxon matched pairs test (for comparisons between total CD8+ and virus-specific CD8+ T-cells made within individuals) was used. Correlations between non-normally distributed data were made using the Spearman’s rank correlation coefficient.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Biggar RJ, Henle G, Bocker J, Lennette ET, Fleisher G, et al. (1978) Primary Epstein-Barr virus infections in African infants. II. Clinical and serological observations during seroconversion. Int J Cancer 22 : 244–250. 212370
2. Hislop AD, Taylor GS, Sauce D, Rickinson AB (2007) Cellular responses to viral infection in humans: lessons from Epstein-Barr virus. Annu Rev Immunol 25 : 587–617. 17378764
3. Balfour HH Jr., Odumade OA, Schmeling DO, Mullan BD, Ed JA, et al. (2013) Behavioral, virologic, and immunologic factors associated with acquisition and severity of primary epstein-barr virus infection in university students. J Infect Dis 207 : 80–88. doi: 10.1093/infdis/jis646 23100562
4. Crawford DH, Macsween KF, Higgins CD, Thomas R, McAulay K, et al. (2006) A cohort study among university students: identification of risk factors for Epstein-Barr virus seroconversion and infectious mononucleosis. Clin Infect Dis 43 : 276–282. 16804839
5. Sumaya CV, Henle W, Henle G, Smith MH, LeBlanc D (1975) Seroepidemiologic study of Epstein-Barr virus infections in a rural community. J Infect Dis 131 : 403–408. 163869
6. Callan MF, Steven N, Krausa P, Wilson JD, Moss PA, et al. (1996) Large clonal expansions of CD8+ T cells in acute infectious mononucleosis. Nat Med 2 : 906–911. 8705861
7. Callan MF, Tan L, Annels N, Ogg GS, Wilson JD, et al. (1998) Direct visualization of antigen-specific CD8+ T cells during the primary immune response to Epstein-Barr virus In vivo. J Exp Med 187 : 1395–1402. 9565632
8. Hislop AD, Annels NE, Gudgeon NH, Leese AM, Rickinson AB (2002) Epitope-specific evolution of human CD8(+) T cell responses from primary to persistent phases of Epstein-Barr virus infection. J Exp Med 195 : 893–905. 11927633
9. Hislop AD, Gudgeon NH, Callan MF, Fazou C, Hasegawa H, et al. (2001) EBV-specific CD8+ T cell memory: relationships between epitope specificity, cell phenotype, and immediate effector function. J Immunol 167 : 2019–2029. 11489984
10. Hislop AD, Kuo M, Drake-Lee AB, Akbar AN, Bergler W, et al. (2005) Tonsillar homing of Epstein-Barr virus-specific CD8+ T cells and the virus-host balance. J Clin Invest 115 : 2546–2555. 16110323
11. Woodberry T, Suscovich TJ, Henry LM, Davis JK, Frahm N, et al. (2005) Differential targeting and shifts in the immunodominance of Epstein-Barr virus—specific CD8 and CD4 T cell responses during acute and persistent infection. J Infect Dis 192 : 1513–1524. 16206065
12. Odumade OA, Knight JA, Schmeling DO, Masopust D, Balfour HH Jr., et al. (2012) Primary Epstein-Barr virus infection does not erode preexisting CD8(+) T cell memory in humans. J Exp Med 209 : 471–478. doi: 10.1084/jem.20112401 22393125
13. Long HM, Chagoury OL, Leese AM, Ryan GB, James E, et al. (2013) MHC II tetramers visualize human CD4+ T cell responses to Epstein-Barr virus infection and demonstrate atypical kinetics of the nuclear antigen EBNA1 response. J Exp Med 210 : 933–949. doi: 10.1084/jem.20121437 23569328
14. Callan MF, Fazou C, Yang H, Rostron T, Poon K, et al. (2000) CD8(+) T-cell selection, function, and death in the primary immune response in vivo. J Clin Invest 106 : 1251–1261. 11086026
15. Steven NM, Annels NE, Kumar A, Leese AM, Kurilla MG, et al. (1997) Immediate early and early lytic cycle proteins are frequent targets of the Epstein-Barr virus-induced cytotoxic T cell response. J Exp Med 185 : 1605–1617. 9151898
16. Steven NM, Leese AM, Annels NE, Lee SP, Rickinson AB (1996) Epitope focusing in the primary cytotoxic T cell response to Epstein-Barr virus and its relationship to T cell memory. J Exp Med 184 : 1801–1813. 8920868
17. Catalina MD, Sullivan JL, Bak KR, Luzuriaga K (2001) Differential evolution and stability of epitope-specific CD8(+) T cell responses in EBV infection. J Immunol 167 : 4450–4457. 11591771
18. Chijioke O, Muller A, Feederle R, Barros MH, Krieg C, et al. (2013) Human natural killer cells prevent infectious mononucleosis features by targeting lytic Epstein-Barr virus infection. Cell Rep 5 : 1489–1498. doi: 10.1016/j.celrep.2013.11.041 24360958
19. Clute SC, Watkin LB, Cornberg M, Naumov YN, Sullivan JL, et al. (2005) Cross-reactive influenza virus-specific CD8+ T cells contribute to lymphoproliferation in Epstein-Barr virus-associated infectious mononucleosis. J Clin Invest 115 : 3602–3612. 16308574
20. Hwang AE, Hamilton AS, Cockburn MG, Ambinder R, Zadnick J, et al. (2012) Evidence of genetic susceptibility to infectious mononucleosis: a twin study. Epidemiol Infect 140 : 2089–2095. 22152594
21. McAulay KA, Higgins CD, Macsween KF, Lake A, Jarrett RF, et al. (2007) HLA class I polymorphisms are associated with development of infectious mononucleosis upon primary EBV infection. J Clin Invest 117 : 3042–3048. 17909631
22. Rostgaard K, Wohlfahrt J, Hjalgrim H (2014) A genetic basis for infectious mononucleosis: evidence from a family study of hospitalized cases in denmark. Clin Infect Dis 58 : 1684–1689. doi: 10.1093/cid/ciu204 24696238
23. Helminen M, Lahdenpohja N, Hurme M (1999) Polymorphism of the interleukin-10 gene is associated with susceptibility to Epstein-Barr virus infection. J Infect Dis 180 : 496–499. 10395868
24. Thacker EL, Mirzaei F, Ascherio A (2006) Infectious mononucleosis and risk for multiple sclerosis: a meta-analysis. Ann Neurol 59 : 499–503. 16502434
25. Hjalgrim H, Askling J, Rostgaard K, Hamilton-Dutoit S, Frisch M, et al. (2003) Characteristics of Hodgkin's lymphoma after infectious mononucleosis. N Engl J Med 349 : 1324–1332. 14523140
26. Silins SL, Sherritt MA, Silleri JM, Cross SM, Elliott SL, et al. (2001) Asymptomatic primary Epstein-Barr virus infection occurs in the absence of blood T-cell repertoire perturbations despite high levels of systemic viral load. Blood 98 : 3739–3744. 11739180
27. Fleisher G, Henle W, Henle G, Lennette ET, Biggar RJ (1979) Primary infection with Epstein-Barr virus in infants in the United States: clinical and serologic observations. J Infect Dis 139 : 553–558. 220340
28. Tamaki H, Beaulieu BL, Somasundaran M, Sullivan JL (1995) Major histocompatibility complex class I-restricted cytotoxic T lymphocyte responses to Epstein-Barr virus in children. J Infect Dis 172 : 739–746. 7658067
29. Holder B, Miles DJ, Kaye S, Crozier S, Mohammed NI, et al. (2010) Epstein-Barr virus but not cytomegalovirus is associated with reduced vaccine antibody responses in Gambian infants. PLoS One 5: e14013. doi: 10.1371/journal.pone.0014013 21103338
30. Slyker JA, Casper C, Tapia K, Richardson B, Bunts L, et al. (2013) Clinical and virologic manifestations of primary Epstein-Barr virus (EBV) infection in Kenyan infants born to HIV-infected women. J Infect Dis 207 : 1798–1806. doi: 10.1093/infdis/jit093 23493724
31. de Martino M, Rossi ME, Azzari C, Gelli MG, Galli L, et al. (1998) Different meaning of CD38 molecule expression on CD4+ and CD8+ cells of children perinatally infected with human immunodeficiency virus type 1 infection surviving longer than five years. Pediatr Res 43 : 752–758. 9621984
32. Njie R, Bell AI, Jia H, Croom-Carter D, Chaganti S, et al. (2009) The effects of acute malaria on Epstein-Barr virus (EBV) load and EBV-specific T cell immunity in Gambian children. J Infect Dis 199 : 31–38. doi: 10.1086/594373 19032105
33. Moormann AM, Chelimo K, Sumba OP, Lutzke ML, Ploutz-Snyder R, et al. (2005) Exposure to holoendemic malaria results in elevated Epstein-Barr virus loads in children. J Infect Dis 191 : 1233–1238. 15776368
34. Piriou E, Asito AS, Sumba PO, Fiore N, Middeldorp JM, et al. (2012) Early age at time of primary Epstein-Barr virus infection results in poorly controlled viral infection in infants from Western Kenya: clues to the etiology of endemic Burkitt lymphoma. J Infect Dis 205 : 906–913. doi: 10.1093/infdis/jir872 22301635
35. Donati D, Zhang LP, Chene A, Chen Q, Flick K, et al. (2004) Identification of a polyclonal B-cell activator in Plasmodium falciparum. Infect Immun 72 : 5412–5418. 15322039
36. Chattopadhyay PK, Chelimo K, Embury PB, Mulama DH, Sumba PO, et al. (2013) Holoendemic Malaria Exposure Is Associated with Altered Epstein-Barr Virus-Specific CD8+ T-Cell Differentiation. J Virol 87 : 1779–1788. doi: 10.1128/JVI.02158-12 23175378
37. Ceesay SJ, Casals-Pascual C, Erskine J, Anya SE, Duah NO, et al. (2008) Changes in malaria indices between 1999 and 2007 in The Gambia: a retrospective analysis. Lancet 372 : 1545–1554. doi: 10.1016/S0140-6736(08)61654-2 18984187
38. Ceesay SJ, Casals-Pascual C, Nwakanma DC, Walther M, Gomez-Escobar N, et al. (2010) Continued decline of malaria in The Gambia with implications for elimination. PLoS One 5: e12242. doi: 10.1371/journal.pone.0012242 20805878
39. Condon LM, Cederberg LE, Rabinovitch MD, Liebo RV, Go JC, et al. (2014) Age-Specific Prevalence of Epstein-Barr Virus Infection Among Minnesota Children: Effects of Race/Ethnicity and Family Environment. Clin Infect Dis.
40. Balfour HH Jr., Holman CJ, Hokanson KM, Lelonek MM, Giesbrecht JE, et al. (2005) A prospective clinical study of Epstein-Barr virus and host interactions during acute infectious mononucleosis. J Infect Dis 192 : 1505–1512. 16206064
41. Dunne PJ, Faint JM, Gudgeon NH, Fletcher JM, Plunkett FJ, et al. (2002) Epstein-Barr virus-specific CD8(+) T cells that re-express CD45RA are apoptosis-resistant memory cells that retain replicative potential. Blood 100 : 933–940. 12130505
42. Tamaru Y, Miyawaki T, Iwai K, Tsuji T, Nibu R, et al. (1993) Absence of bcl-2 expression by activated CD45RO+ T lymphocytes in acute infectious mononucleosis supporting their susceptibility to programmed cell death. Blood 82 : 521–527. 8329707
43. Williams H, McAulay K, Macsween KF, Gallacher NJ, Higgins CD, et al. (2005) The immune response to primary EBV infection: a role for natural killer cells. Br J Haematol 129 : 266–274. 15813855
44. Kollmann TR, Levy O, Montgomery RR, Goriely S (2012) Innate immune function by Toll-like receptors: distinct responses in newborns and the elderly. Immunity 37 : 771–783. doi: 10.1016/j.immuni.2012.10.014 23159225
45. Horwitz CA, Henle W, Henle G, Goldfarb M, Kubic P, et al. (1981) Clinical and laboratory evaluation of infants and children with Epstein-Barr virus-induced infectious mononucleosis: report of 32 patients (aged 10–48 months). Blood 57 : 933–938. 6260269
46. Junying J, Herrmann K, Davies G, Lissauer D, Bell A, et al. (2003) Absence of Epstein-Barr virus DNA in the tumor cells of European hepatocellular carcinoma. Virology 306 : 236–243. 12642097
47. Yao QY, Rickinson AB, Epstein MA (1985) A re-examination of the Epstein-Barr virus carrier state in healthy seropositive individuals. Int J Cancer 35 : 35–42. 2981780
48. Yao QY, Rickinson AB, Gaston JS, Epstein MA (1985) In vitro analysis of the Epstein-Barr virus: host balance in long-term renal allograft recipients. Int J Cancer 35 : 43–49. 2981781
49. Macsween KF, Higgins CD, McAulay KA, Williams H, Harrison N, et al. (2010) Infectious mononucleosis in university students in the United kingdom: evaluation of the clinical features and consequences of the disease. Clin Infect Dis 50 : 699–706. doi: 10.1086/650456 20121570
50. Henle G, Henle W (1966) Immunofluorescence in cells derived from Burkitt's lymphoma. J Bacteriol 91 : 1248–1256. 4160230
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 3
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Bacterial Immune Evasion through Manipulation of Host Inhibitory Immune Signaling
- BILBO1 Is a Scaffold Protein of the Flagellar Pocket Collar in the Pathogen
- Antimicrobial-Induced DNA Damage and Genomic Instability in Microbial Pathogens
- Attenuation of Tick-Borne Encephalitis Virus Using Large-Scale Random Codon Re-encoding
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy