
Dependence of Intracellular and Exosomal microRNAs on Viral Oncogene Expression in HPV-positive Tumor Cells
Oncogenic human papillomaviruses (HPVs) are major human carcinogens of broad biomedical importance. The growth of HPV-positive cervical cancer cells is critically dependent on sustained E6/E7 oncogene expression from endogenous viral DNA sequences. We here addressed the question of whether this process is linked to specific, E6/E7-dependent alterations of the cellular micro(mi)RNA network. By comprehensive deep sequencing analyses we show that endogenous E6/E7 expression significantly affects the concentrations of abundant intracellular miRNAs in HPV-positive cervical cancer cells, which are linked to the control of cell proliferation, senescence and apoptosis. These include members of the miR-17~92 cluster, which are expressed at increased levels by sustained E6/E7 expression and repress the anti-proliferative p21 gene in HPV-positive cancer cells. Moreover, we identified an E6/E7-dependent seven-miRNA-signature in exosomes secreted from HPV-positive cancer cells. These small vesicles are involved in intercellular communication and may serve as novel diagnostic markers. Taken together, our results show that continuous E6/E7 expression in HPV-positive cancer cells is linked to significant alterations in the amounts of intracellular and exosomal miRNAs with growth-promoting, anti-senescent and anti-apoptotic potential.
Published in the journal:
. PLoS Pathog 11(3): e32767. doi:10.1371/journal.ppat.1004712
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004712
Summary
Oncogenic human papillomaviruses (HPVs) are major human carcinogens of broad biomedical importance. The growth of HPV-positive cervical cancer cells is critically dependent on sustained E6/E7 oncogene expression from endogenous viral DNA sequences. We here addressed the question of whether this process is linked to specific, E6/E7-dependent alterations of the cellular micro(mi)RNA network. By comprehensive deep sequencing analyses we show that endogenous E6/E7 expression significantly affects the concentrations of abundant intracellular miRNAs in HPV-positive cervical cancer cells, which are linked to the control of cell proliferation, senescence and apoptosis. These include members of the miR-17~92 cluster, which are expressed at increased levels by sustained E6/E7 expression and repress the anti-proliferative p21 gene in HPV-positive cancer cells. Moreover, we identified an E6/E7-dependent seven-miRNA-signature in exosomes secreted from HPV-positive cancer cells. These small vesicles are involved in intercellular communication and may serve as novel diagnostic markers. Taken together, our results show that continuous E6/E7 expression in HPV-positive cancer cells is linked to significant alterations in the amounts of intracellular and exosomal miRNAs with growth-promoting, anti-senescent and anti-apoptotic potential.
Introduction
Oncogenic human papillomaviruses (HPVs), such as HPV16 and HPV18, cause cervical cancer. Infections with oncogenic HPV types are moreover closely linked to the development of additional human malignancies in the oropharynx and anogenital region [1]. The viral E6 and E7 oncoproteins are crucial both for the HPV-associated induction of transformation as well as for the maintenance of the tumorigenic phenotype of HPV-positive cervical cancer cells [2,3]. For example, E6 induces the proteolytic degradation of the p53 tumor suppressor protein [4] and stimulates telomerase activity [5], whereas E7 interferes with the activity of the retinoblastoma tumor suppressor protein, pRb, and other pocket proteins [6]. As a consequence, E6 and E7 deregulate intracellular pathways involved in the control of cellular proliferation, senescence, apoptosis, and genetic stability.
Importantly, at least some of these pathways are not irreversibly impaired by HPVs. Rather, inhibition of viral E6/E7 activities in HPV-positive cancer cells leads to the reactivation of dormant tumor suppressor pathways. For instance, several studies indicate that inhibition of E6 primarily results in apoptosis [7–11], whereas combined inhibition of E6/E7 leads to growth arrest and cellular senescence [12–14]. The reversibility of the malignant phenotype of HPV-positive tumor cells is not only phenomenologically interesting but may also form a rational basis for therapeutic interference. This could, in principle, be achieved by blocking the E6/E7 oncogenes or, alternatively, by correcting downstream cellular pathways that are deregulated by the viral oncogenes. Therefore, it is important to uncover crucial cellular targets that are affected by viral E6/E7 oncogene expression and that support the growth of HPV-positive cancer cells.
Micro(mi)RNAs are short (21–23 nt), non-coding, highly-conserved RNAs that post-transcriptionally regulate gene expression [15]. For several tumor entities, it has been shown that the deregulation of the cellular miRNA network plays a critical role for cancer development and maintenance [16,17]. The oncogenicity of miRNAs has been particularly well demonstrated for members of the miR-17~92 cluster (also called “oncomir-1”; coding for miR-17, miR-20a, miR-18a, miR-19a, miR-19b and miR-92a) and of its paralog cluster miR-106b~25 (coding for miR-106b, miR-93 and miR-25) [18]. Potential cellular target genes for members of the two miRNA clusters include p21, which codes for a cyclin-dependent kinase inhibitor that plays a central role for growth control and induction of the senescence pathway in many cells [19,20].
In contrast to other tumor viruses, such as Epstein-Barr virus (EBV) or Kaposi’s sarcoma-associated herpesvirus (KSHV), oncogenic HPV types presumably do not encode own viral miRNAs [21,22]. However, global miRNA analyses indicate an upregulation of oncogenic miRNAs and a decrease of tumor-suppressive miRNAs in cervical cancer biopsies [23–38] and in cervical cancer cell lines [39–41], in comparison to normal cervical tissue. A contribution of HPVs to the deregulation of miRNA expression in cervical cancer was proposed, mainly based on comparisons between HPV-positive and -negative cell lines [41], and on cell culture models upon introduction of HPV genomes [22,42,43]. More direct evidence that the HPV oncogenes have the potential to influence the cellular miRNA composition was provided by experiments that involved ectopic expression of the viral E6 and/or E7 genes in keratinocytes and subsequent global miRNA analysis [44] or investigation of selected miRNAs [45,46]. However, the miRNA species identified by these different experimental approaches vary substantially (see Discussion). Most importantly, the crucial question whether the E6/E7-dependent maintenance of the growth of HPV-positive cancer cells is linked to specific alterations of the global miRNA network has not been addressed. To resolve this issue, we here performed a comprehensive analysis of the intracellular miRNA composition in HPV-positive cancer cells, upon silencing of endogenous E6/E7 oncogene expression.
An interesting miRNA pool that recently gained interest in cancer research is the miRNA content of exosomes. Exosomes are small extracellular vesicles (50–100 nm in diameter) of endosomal origin that are secreted by a variety of cells, including tumor cells [47]. Exosomes may play an important role for the intercellular communication of tumor cells since they can accelerate cancer growth and invasiveness by horizontally transferring proteins, mRNAs, and non-coding RNAs from tumor cells into recipient cells [48–50]. In the case of miRNAs, several studies showed specific target gene repression in recipient cells upon intercellular transfer of miRNAs via exosomes [51–55]. Also other human tumor viruses, EBV [51,56,57] and possibly KSHV [58], may utilize exosomes to modulate the tumor microenvironment by transporting viral proteins and virus-encoded miRNAs. Due to the facts that exosomes can be isolated from different body fluids (e.g. serum, saliva, urine) and that their content allows conclusions about their cell of origin, exosomes are also intensively investigated as a source of novel biomarkers [59–61].
The above considerations raise two important issues concerning the interplay between HPVs and the miRNA network in cervical cancer cells. First, is the intracellular miRNA pool of HPV-positive tumor cells dependent on the sustained expression of the viral E6/E7 oncogenes? Second, is the miRNA composition of exosomes that are secreted by HPV-positive cancer cells affected by the HPV oncogenes? To answer these questions, we performed a comprehensive deep sequencing study in order to identify the influence of the endogenous E6/E7 oncogene expression on the global miRNA composition of HPV-positive cervical cancer cells, both at the intracellular and at the exosomal level.
Results
Influence of endogenous E6/E7 expression on the intracellular miRNA content of cervical cancer cells
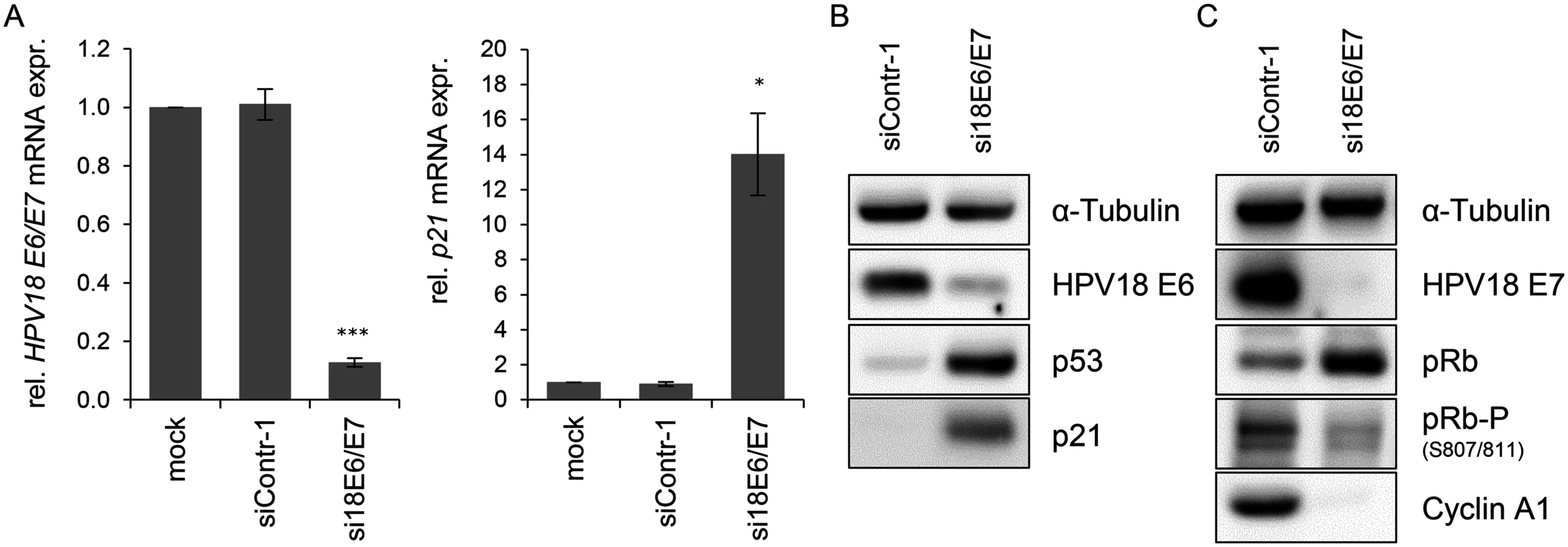
In order to investigate the influence of the HPV oncogenes on the intracellular miRNA composition of HPV-positive cancer cells, endogenous HPV18 E6/E7 expression in HeLa cervical carcinoma cells was blocked by RNA interference (RNAi) for subsequent deep sequencing analyses. Treatment with E6/E7-targeting siRNAs (si18E6/E7) led to efficient downregulation of E6/E7 mRNA levels as shown by qRT-PCR analyses, using primers recognizing all three transcript classes [8] coding for HPV18 E6 and E7 (Fig. 1A, left panel). A substantial reduction of the HPV18 E6 and E7 protein levels 72 h after transfection with si18E6/E7 was observed (Fig. 1B/C). This was linked to increased p53 protein levels (Fig. 1B), as expected from the ability of E6 to induce the degradation of p53 [4], and increased p21 mRNA and protein levels (Fig. 1A/B), representing a downstream transcriptional target gene for p53 [62]. Further, E6/E7 silencing was associated with an increase of total pRb protein levels, consistent with the ability of E7 to induce pRb degradation [6], as well as with decreased amounts of phosphorylated pRb and of Cyclin A1, indicating reactivation of the pRb cascade (Fig. 1C).
cDNA libraries were generated from RNA samples isolated from cells in which the viral oncogene expression was silenced (si18E6/E7) or which underwent control treatment (siContr-1). In order to capture a broad spectrum of E6/E7-modulated miRNAs—unbiased by a pre-selection—analyses of total miRNA contents were accomplished by small RNA deep sequencing using the Illumina platform. Sequencing of the libraries resulted in raw read counts that were pre-processed to remove adapter sequences and filtered to exclude low quality reads (S1 Table).
Mean read counts of cellular sequences mapping to known human miRNAs ranged from 1 to 1,170,975 (S1 Dataset, Fig. 2A for a read count distribution of cellular miRNAs). For cross-library comparison, the read count of a given miRNA was normalized to the total number of uniquely mapped miRNA reads per library and expressed as reads per million (RPM) mapped reads. RPM values of the 15 most frequently sequenced cellular miRNAs are displayed in Fig. 2B.

For stoichiometric reasons, it is assumed that mainly miRNAs with a high intracellular abundance can lead to the repression of their target genes, whereas low-abundant miRNAs, identified in deep sequencing studies with < 100 RPM, are frequently not functional [63]. Therefore, an arbitrary threshold of 1,000 RPM for an individual miRNA in each sample was set and, consequently, 52 cellular miRNAs were further analyzed. Deregulation was defined as a > 1.5-fold change between si18E6/E7 - and siContr-1-treated samples. An overview on the number of deregulated cellular miRNAs is given in Fig. 2C, showing that endogenous E6/E7 silencing affected 23 of the 52 most abundant miRNAs in HeLa cells (15 down - and 8 upregulated). The relative expression upon E6/E7 silencing of each of the 52 miRNAs is displayed in Fig. 2D. The RPMmean values and fold changes of the 23 E6/E7-regulated miRNAs are indicated in S2 Table.The 23 miRNAs comprise several family members of the miR-378 family (miR-378a-3p, miR-378c, miR-378d, miR-378f), as well as members of the miR-17~92 and miR-106b~25 clusters (miR-17–5p, miR-19b-3p, miR-93–5p). Moreover, two seed families, the miR-23 family (miR-23a-3p, miR-23b-3p) and miR-27 family (miR-27a-3p, miR-27b-3p), were upregulated.
The E6/E7-mediated modulation of cellular miRNAs was subsequently validated by qRT-PCR analyses. For normalization, small nuclear RNA (snRNA) RNU6–2 was employed. Ct-values for RNU6–2 were consistent for si18E6/E7 - or siContr-1-treated samples and across experimental conditions. Of the 23 abundant cellular miRNAs that were found to be affected in deep sequencing analyses, deregulation (up or down) upon silencing of endogenous E6/E7 expression was confirmed for 21 miRNAs (91%) by qRT-PCR (dark grey columns in Fig. 2E), 17 of which exhibited a fold change of > 1.5 (Fig. 2E and S2 Table). This shows high agreement between the two methods. Statistical significance of the qRT-PCR data was obtained for ten of these 17 miRNAs: downregulation of miR-17–5p, miR-186–5p, miR-378a-3p, miR-378f, miR-629–5p and miR-7–5p and upregulation of miR-143–3p, miR-23a-3p, miR-23b-3p and miR-27b-3p, upon E6/E7 silencing (Fig. 2E and indicated in bold in S2 Table). In summary, the combination of small RNA deep sequencing and qRT-PCR analyses identified ten abundant cellular miRNAs that are significantly affected in HeLa cells upon silencing of endogenous E6/E7 oncogene expression.
Next, we analyzed the expression of these ten miRNAs upon repressing endogenous E6/E7 expression in SiHa cervical cancer cells (Fig. 3). In contrast to HPV18-positive HeLa cells, which are derived from an adenocarcinoma of the cervix, SiHa cells express HPV16 E6/E7 and originate from a cervical squamous cell carcinoma. As expected, silencing of endogenous HPV16 E6/E7 expression by RNAi in SiHa cells was linked to reduced E6/E7 protein expression and reconstitution of the p53 pathway (Fig. 3A). Notably, both cell lines reveal a substantial overlap in the regulation patterns of the ten selected miRNAs, upon endogenous E6/E7 repression (Fig. 2E and Fig. 3). In specific, with the exception of miR-378f, the levels of nine of the ten selected miRNAs were modulated in the same direction (up or down) in both cell lines upon silencing of E6/E7 expression, with five of these changes also exhibiting statistical significance in SiHa cells (Figs. 3B). Thus, although the two cell lines contain different HPV types and were established from cervical cancers with different histopathological backgrounds, there is a substantial similarity of the regulation of these miRNAs upon endogenous E6/E7 inhibition.

Effect of the p53 status on the regulation of E6/E7-modulated cellular miRNAs
The p53 protein can positively or negatively affect the expression of tumor-associated miRNAs [64]. Since p53 is strongly upregulated following E6/E7 repression (Fig. 1B), we addressed the question whether the miRNA changes observed upon E6/E7 silencing are p53-dependent. For these analyses, we comparatively investigated the miRNA regulation in parental HeLa cells and in “p53-null” HeLa cells. In the latter cells, endogenous p53 expression is silenced by a stably integrated vector expressing a short hairpin (sh)RNA that targets the p53 mRNA [65].
HPV18 E6/E7 expression could be repressed by siRNA in both cell lines with a comparable high efficiency (Fig. 4A, left panel). Basal p53 protein levels were undetectable in “p53-null” cells and remained extremely low even upon endogenous E6/E7 silencing, indicating that the system exhibits only a very minute degree of leakiness (Fig. 4B). Consistently, expression of the p53 target gene p21 is not increased upon E6/E7 silencing in “p53-null” HeLa cells, both at the RNA and protein levels (Fig. 4A, right panel, and Fig. 4B), corroborating that the p53 pathway is efficiently blocked in these cells.

For miRNA analyses, we utilized miR-34a-5p as a positive control since it is well-documented as a p53-inducible miRNA [64]. In line, we found that miR-34a-5p amounts were also increased (6.2-fold) in the deep sequencing analysis upon E6/E7 silencing, however, it did not reach the RPM threshold of > 1,000 (RPMsiContr-1 = 64; RPMsi18E6/E7 = 385). As shown in Fig. 4C, induction of miR-34a-5p upon E6/E7 silencing is clearly detectable by qRT-PCR analyses in parental HeLa cells but virtually abolished in “p53-null” HeLa cells. In contrast to miR-34a-5p, the intracellular levels of eight of the ten miRNAs that were defined as E6/E7-regulated in parental HeLa cells also exhibited statistically significant changes in “p53-null” HeLa cells (Fig. 4C), with congruency in the direction of change (up or down). Only two miRNAs, miR-186–5p and miR-378f, showed a discrepant regulation in that they were no longer repressed in “p53-null” HeLa cells upon E6/E7 silencing, suggesting that p53 may directly or indirectly reduce their expression. Taken together, these experiments indicate that a substantial proportion (eight of ten) of the most abundant and significantly affected cellular miRNAs is modulated by endogenous E6/E7 expression in a p53-independent manner.
HPV E6/E7 increase intracellular levels of members of the oncogenic miR-17~92 cluster that reduce p21 expression in HPV-positive cancer cells
The 52 most abundant cellular miRNAs that were downregulated > 1.5-fold upon E6/E7 silencing in both deep sequencing and qRT-PCR analyses encompassed miR-17–5p and miR-19b-3p, two members of the miR-17~92 cluster, and miR-93–5p, a member of the paralog miR-106b~25 cluster (Fig. 2D/E). Further qRT-PCR analyses revealed that all detectable additional members the miR-17~92 and miR-106b~25 clusters were also downregulated upon silencing of endogenous E6/E7 expression (S3 Table). These findings indicate that continuous E6/E7 expression increases the intracellular concentrations of multiple miRNAs from both oncogenic miRNA clusters in HPV-positive cancer cells.
Four miRNAs encoded by the miR-17~92 and miR-106b~25 clusters (miR-17–5p, miR-20a-5p, miR-106b-5p, miR-93–5p) are grouped into the miR-17 family, according to their identical seed sequence, and target two binding sites in the 3’ UTR of p21 [66,67]. All four miRNAs exhibited a > 1.5-fold downregulation in HeLa cells upon endogenous E6/E7 silencing in deep sequencing and/or qRT-PCR analyses (S3 Table). This raises the question whether the E6/E7-dependent increase of p21-targeting miRNAs may lead to p21 repression in HPV-positive cancer cells and thereby contributes to their proliferative capacity and resistance towards senescence.
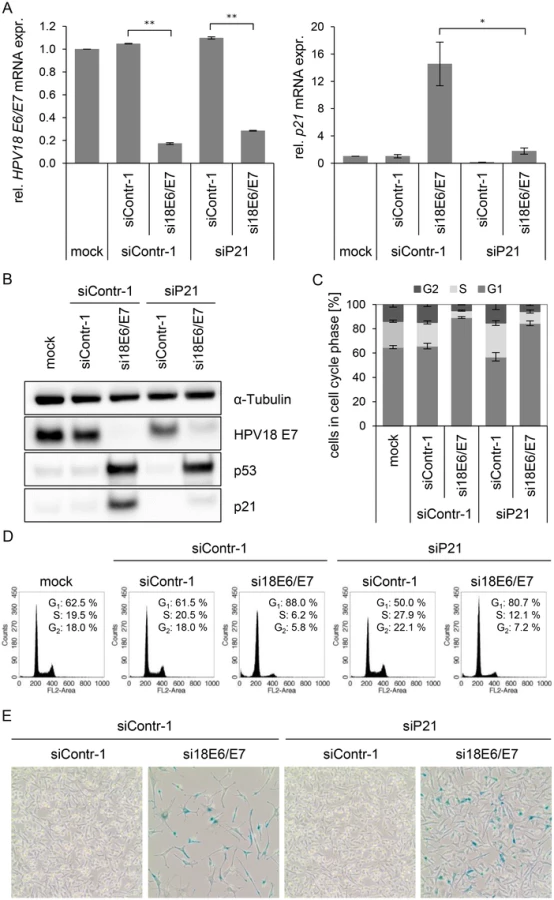
In order to investigate this possibility, we first addressed the controversially discussed issue whether p21 is required for senescence induction upon E6/E7 silencing in HPV-positive cancer cells [13,68]. Therefore, siRNAs (si18E6/E7, siContr-1 and siP21) were utilized to achieve silencing of E6/E7 only, p21 only, or E6/E7 together with p21. Inhibition of p21 alone had no effect on HPV18 E6/E7 expression (Fig. 5A, left panel), whilst it efficiently counteracted the induction of p21 mRNA and protein levels upon E6/E7 inhibition (Fig. 5A, right panel, and Fig. 5B). Cell cycle analyses revealed that E6/E7 silencing alone led to an increase in G1 - and decrease in S-phase populations, indicative for a G1-arrest (Fig. 5C/D). p21 silencing alone, led to an increase in S-phase and reduction in G1-phase populations (Fig. 5C/D). Notably, the G1-arrest observed when inhibiting E6/E7 alone was diminished when silencing p21 in parallel, and S-phase populations doubled (from 6 to 12%, p-value = 0.059), 72 h post transfection. Moreover, 168 h after transfection only a fraction (15%) of the cells stained positive for the senescence marker Senescence-Associated β-Galactosidase (SA-β-Gal) when p21 and E6/E7 were silenced together, whereas the majority of cells remained unstained (Fig. 5E). This was in clear contrast to the results for E6/E7 silencing alone, where almost all cells (85%) exhibited morphological signs of senescence (cellular enlargement and flattening, long cytoplasmic projections and positive staining for SA-β-Gal), 168 h after transfection (Fig. 5E). In conclusion, parallel silencing of E6/E7 and p21 strongly alleviated the induction of senescence that occurs upon mere E6/E7 repression. This indicates that p21 is a contributor to the senescence induction upon E6/E7 silencing in HPV-positive cancer cells.
Next, it was studied whether an experimental increase of mir-17~92 expression, encoding two p21-targeting miRNAs, miR-17–5p and miR-20a-5p [18], can contribute to keep basal levels of p21 expression low in HeLa cells. Transfection of a mir-17~92 expression vector led to an increase of miR-17–5p, miR-20a-5p, miR-19b-3p and miR-92a-3p levels, as expected, but not of miR-34a-5p, which served as a negative control (Fig. 6A). Notably, basal p21 expression in HeLa cells was reduced by overexpression of the miR-17~92 cluster, both at the mRNA and protein level (Fig. 6B/C).

In reciprocal experiments, we investigated whether the downmodulation of miR-17–5p and miR-20a-5p in HeLa cells might result in an upregulation of p21 levels. Therefore, HeLa cells were transfected with a control miRNA inhibitor (“Inhib. control”) that carries no homology to any known mammalian gene or with specific miRNA inhibitors of miR-17–5p, miR-20a-5p, and miR-19b-3p. The latter miRNA inhibitor served as additional control, since its target miRNA does not possess a known binding site in the p21 transcript. The inhibitors of miR-17–5p and miR-20a-5p but neither the inhibitor control nor the miR-19b-3p inhibitor led to a significant upregulation of p21 protein levels (Fig. 6D). Taken together, these results indicate that continuous E6/E7 oncogene expression in HPV-positive cancer cells is required to maintain miRNAs of the oncogenic miR-17~92 cluster at a level that keeps expression of the anti-proliferative p21 gene low.
The E6/E7-dependent miRNA content of exosomes released from cervical cancer cells
To investigate the influence of viral E6/E7 expression on the exosomal miRNA contents, exosomes secreted by HeLa cells were isolated from the cell culture medium by employing a protocol for exosome enrichment involving sequential (ultra-)centrifugation steps [69], with minor modifications (see Material and Methods). A characterization of the exosome preparations used for the deep sequencing studies is presented in Fig. 7. The preparations stained positive for the exosomal markers HSC70, CD63, Annexin-1, β-Actin and CD9, showing the typical exosomal enrichment for the tetraspanins CD63 and CD9 [69] (Fig. 7A). The absence of detectable bands for the endoplasmatic reticulum (ER) marker GRP78 and the early endosome marker EEA1 indicate only minor, or no, contamination with vesicles from other origins. Electron microscopy (EM) revealed the presence of small membrane vesicles with a diameter of 50–100 nm, possessing the typical cup-shaped appearance of exosomes in EM analyses [69] (Fig. 7B).

To investigate possible effects of the HPV oncogenes on the miRNA composition of exosomes, we treated HeLa cells with siRNAs blocking endogenous HPV18 E6/E7 expression or with control siRNA (siContr-1). Forty-eight hours after transfection, cells were allowed to secrete newly formed exosomes for 24 h into the cell culture medium pre-depleted of FBS-derived microvesicles. Inhibition of E6/E7 expression upon transfection of synthetic siRNAs was maintained over the time period required for exosome production and secretion (Fig. 1). Total RNA was extracted from RNase A-treated exosomes and quality and quantity of the isolated RNA samples were assessed using an Agilent Bioanalyzer (Fig. 7C). In parallel, cellular RNA extracted from the respective exosome-producing cells was examined. The total RNA profile (Fig. 7C, upper panel) showed distinct differences between cellular and exosomal RNA, with exosomes lacking discernible 18S and 28S rRNA peaks, in agreement with previous publications [48,70]. Both cellular and exosomal RNA revealed a peak for transfer RNAs (tRNAs) and miRNAs (size range as indicated) in the small RNA profiles (Fig. 7C, lower panel).
Subsequently, the exosomal RNA samples were converted into cDNA libraries, subjected to small RNA deep sequencing and initial analysis was performed as described above for cellular miRNAs. The composition of exosomal and intracellular small RNA fractions differed in that the relative percentage of miRNAs among different classes of small RNAs was approximately 50% lower in exosomes (S1 Fig). E6/E7 silencing only slightly affected the intracellular distribution of small RNA classes, but approximately doubled the relative percentage of miRNAs inside exosomes (S1 Fig). Mean read counts of exosomal sequences mapping to known human miRNAs ranged from 1 to 445,143 in exosomes (S1 Dataset, Fig. 8A for a read count distribution of exosomal miRNAs). RPM values of the 15 most frequently sequenced miRNAs in exosomes are displayed in Fig. 8B. Forty-seven exosomal miRNAs showed > 1,000 RPM in each sample and were subjected to further analysis. Thirty-six of these 47 exosomal miRNAs were also found among the 52 most abundant intracellular miRNAs with > 1,000 RPM (S1 Fig). Relative quantification of si18E6/E7 - versus siContr-1-treated samples revealed that—among the 47 most commonly detected exosomal miRNAs—21 were down - and four upregulated more than 1.5-fold upon intracellular E6/E7 silencing (Fig. 8C). The relative expression of each of these 47 miRNAs upon E6/E7 silencing is displayed in Fig. 8D. The RPMmean values of the 25 E6/E7-modulated exosomal miRNAs are indicated in S4 Table. They encompass several family members with identical seed regions, including the let-7 family (let-7a-5p, let-7d-5p, let-7f-5p, let-7g-5p), miR-378 family (miR-378a-3p, miR-378c), miR-99 family (miR-99a-5p, miR-100–5p), as well as members of the miR-17~92 cluster (miR-20a-5p, miR-92a-3p).

Next, the effects of E6/E7 expression on exosomal miRNAs—as identified by small RNA deep sequencing—were validated by qRT-PCR analyses. So far, there is no common RNA species for normalization of exosomal miRNA levels available. Therefore, two miRNAs, miR-452–5p and miR-183–5p, were chosen as stable endogenous exosomal miRNA controls based on the small RNA deep sequencing data (in analogy to refs. [71,72]). Both miRNAs were frequently sequenced (> 1,000 RPM in each sample) and showed virtually no alterations of their exosomal concentrations upon E6/E7 silencing versus control treatment (miR-452–5p: FCmean = 0.99, miR-183–5p: FCmean = 1.04).
Modulation (up or down) upon inhibition of HPV18 E6/E7 expression was confirmed for roughly three quarters (72%) of the identified exosomal miRNAs by qRT-PCR analyses (Fig. 8E, dark grey columns). A statistically significant and > 1.5-fold decrease upon E6/E7 silencing was detected for exosomal let-7d-5p, miR-20a-5p, miR-378a-3p, miR-423–3p, miR-7–5p, miR-92a-3p, whereas miR-21–5p exhibited a statistically significant and > 1.5-fold increase upon E6/E7 silencing (illustrated in bold in S4 Table). These findings indicate that continuous HPV E6/E7 oncogene expression determines a signature of seven miRNAs in exosomes secreted from HeLa cells in that it leads to significantly increased let-7d-5p, miR-20a-5p, miR-378a-3p, miR-423–3p, miR-7–5p, miR-92a-3p and decreased miR-21–5p levels.
Comparative analyses of the above identified seven miRNAs in exosomes secreted by HPV16-positive SiHa cells revealed that the concentrations of all these are congruently modulated (up or down) upon inhibiting endogenous HPV16 E6/E7 expression (Fig. 9). Six of these seven miRNA alterations were also statistically significant in SiHa cells, with a > 1.5-fold change observed for four of them (Fig. 9). Thus, similar to the results obtained for the regulation of intracellular miRNAs, there is substantial overlap in the E6/E7-dependent regulation of miRNA species in exosomes secreted by HPV16 - and HPV18-positive cancer cells.

Discussion
The growth of HPV-positive cancer cells requires the continuous expression of the viral E6/E7 oncogenes [2,7,13,73–76]. To address the question whether this process is linked to alterations of the miRNA network, we here analyzed the influence of the E6/E7 expression on the intracellular and exosomal miRNA pools of HPV-positive cancer cells. We found that ten of the 52 most abundant intracellular miRNAs identified by deep sequencing analyses of HeLa cells were significantly affected upon silencing of endogenous viral oncogene expression. Notably, they are enriched for miRNAs that are linked to the regulation of cell proliferation, senescence and apoptosis, suggesting that the E6/E7-linked modulation of the miRNA network contributes to the growth of HPV-positive cancer cells. Consistently, we observed that the sustained endogenous E6/E7 expression is linked to an increase of miRNAs with growth promoting potential (e.g. members of the miR-17~92 cluster blocking p21 expression). In addition, we determine the miRNA content of exosomes secreted from HPV-positive cancer cells and delineate specific miRNAs whose exosomal concentrations are dependent on viral oncogene expression.
Several previous studies have identified miRNAs as potential targets for HPVs or have linked specific miRNAs to cervical carcinogenesis. By performing medline searches for the two keywords miRNA/microRNA and HPV/cervical cancer, 258 different publications were found (date: November 26th, 2014). Twenty-one of these reports encompass global miRNA profiling studies, performing analyses of the miRNA expression in tumorous versus normal cervical cancer tissue (13 publications), in different in vitro cell culture models (7 publications), or in both (1 publication). S5 Table provides an overview on these 21 studies and indicates the used platforms for miRNA analysis and the various experimental conditions. Only two of these studies performed comprehensive small RNA deep sequencing analyses [22,43], without a pre-selection of candidate miRNAs (e.g. for microarray design). We withdrew from these 21 publications the miRNAs that were reported to be differentially regulated in cervical cancer tissue or in in vitro models, and updated the original miRNA nomenclature of the publications to the current miRBase entries (release 21, June 2014). As a result, 483 different mature miRNAs have been proposed by these 21 studies to be HPV-dependent and/or deregulated in cervical cancer (S2 Dataset). Out of these 483 miRNAs, 201 were identified in more than one study, but showed substantial discordance with more than half of them (105 miRNAs) being regulated in opposite directions in different reports (S2 Dataset, S5 Table). Importantly, however, none of these experimental approaches have addressed the question whether the actual cellular miRNA composition of HPV-positive cancer cells depends on endogenous E6/E7 expression. This is a critical issue since E6/E7 expression levels are tightly controlled in HPV-positive cancer cells and it is not clear how this relates to the E6/E7 levels obtained, for example, by ectopic E6/E7 expression in keratinocytes. Furthermore, HPV-induced cell transformation requires additional alterations in the host cell, besides viral E6/E7 expression. Thus, it is crucial to investigate the E6/E7-dependence of the cellular miRNA network by analyzing the effects of endogenous E6/E7 expression levels, within the cellular background of HPV-transformed cervical cancer cells, in which the sustained E6/E7 expression leads to the relevant cellular phenotype (maintenance of cell growth).
For these analyses, we chose HPV18-positive HeLa cells as a model for several considerations: (i) as common for HPV-positive cervical cancer cells, HeLa cells express the viral oncogenes from chromosomally integrated HPV copies, using the authentic E6/E7 transcriptional promoter. (ii) HeLa cells mirror known critical mechanisms of HPV-linked carcinogenesis, such as inactivation of the p53 and pRb tumor suppressor proteins by the HPV E6 and E7 proteins, respectively. (iii) The growth of HeLa cells is strictly dependent on viral E6/E7 expression [8,13,73,75,76] as is the case for primary cervical cancer cells freshly isolated from human tumor samples [74]. (iv) E6/E7 silencing in HeLa cells results in the same phenotype as in primary cervical cancer cells, namely growth arrest and induction of cellular senescence [12–14,74,77]. (v) HeLa cells allow functional analyses within the intracellular milieu of an HPV-transformed cancer cell, which has acquired the necessary additional cellular alterations that are required for HPV-induced cervical carcinogenesis. Likely, these are not present in “normal” keratinocytes, which only in very rare instances are transformed to malignancy by the HPV E6/E7 oncogenes alone [78]. (vi) A meta-analysis of mRNA transcriptome studies validated that RNAi-mediated silencing of endogenous E6/E7 expression in HeLa cells [79] is one of the most suitable experimental approaches to predict molecular changes present in cervical cancer tissues [80].
We determined ten abundant intracellular miRNAs as E6/E7-dependent, based on our most stringent selection criteria (RPM values > 1,000; modulated > 1.5-fold upon E6/E7 silencing in both deep sequencing and—statistically significant—in qRT-PCR analyses). Analyses in “p53-null” HeLa cells indicate that eight of the ten miRNAs are modulated upon E6/E7 silencing in a p53-independent manner. These include miR-143–3p, the levels of which have been reported to be increased by p53 via enhanced post-transcriptional miRNA maturation [81]. However, our observation that miR-143–3p levels are very similarly regulated in parental and p53-deficient HeLa cells indicates that this mechanism is not responsible for the miR-143–3p increase upon endogenous E6/E7 silencing.
We included the ten E6/E7-dependent miRNAs in S2 Dataset, resulting in a total number of 485 miRNAs identified in global miRNA expression analyses of cervical cancer biopsies and different in vitro cell culture models. Studies in cervical cancer biopsies reported alterations of six of the ten miRNAs that we identified here as being modulated by E6/E7. Notably, five of these six miRNAs exhibit congruent changes between our functional experiments in vitro and their expression patterns in vivo in at least one study, i.e. reduction upon E6/E7 silencing in HeLa and upregulation in cervical cancer biopsies (miR-7–5p [43], miR-17–5p [27–30,34,37], miR-186–5p [35]) or increase upon E6/E7 silencing in HeLa and downregulation in cervical cancer tissue (miR-23b-3p [28,37] and miR-143–3p [23,29,31,33,37,38,82]) (S2 Dataset). Only for miR-378a-3p, which was downregulated upon E6/E7 silencing, our in vitro data contrasts the reported downregulation of this miRNA in cervical cancer tissue [28,34]. This high concordance with in vivo data provides further strong evidence for the suitability of the functional approach used here to identify E6/E7-dependent miRNA alterations.
Remarkably, multiple of the most abundant miRNAs found to be significantly affected by E6/E7 silencing in HPV-positive cancer cells are known to be involved in the regulation of cell proliferation, senescence and apoptosis. Specifically, continuous E6/E7 expression is necessary to maintain high intracellular levels of miR-7–5p, miR-629–5p, miR-378a-3p, miR378f, miR-17–5p, and miR-186–5p (S2 Table), which all have been linked to pro-tumorigenic activities. For example, miR-7–5p stimulated cell proliferation and tumorigenicity in lung cancer cells [83] and is linked to a more aggressive growth behavior of breast cancers [84]. miR-629–5p also promotes cell growth, has been found to be important for hepatocarcinogenesis via HNF4a repression [85], and targets the NBS1 DNA tumor susceptibility gene [86]. Reduced miR-7–5p or miR-629–5p levels have also been both associated with cellular senescence [87]. Several members of the miR-378 family were among the most frequently sequenced miRNAs that decreased upon E6/E7 silencing (S2 Table). miR-378a-3p can block the tumor-suppressive Fus1 (TUSC2) and SUFU genes, leading to increased cell survival and tumor growth [88]. No functional data is yet available for miR-378f [89], however, it contains the same seed sequence as miR-378a-3p and therefore both miRNAs could regulate overlapping genes. miR-17–5p is discussed in more detail below. Finally, miR-186–5p is an inhibitor of the FOXO1 tumor suppressor gene, which can exert anti-proliferative, pro-apoptotic and pro-senescent activities [90,91].
On the other hand, continuous E6/E7 expression is linked to a decrease of the intracellular concentrations of miR-23a-3p, miR-23b-3p, miR-27b-3p, and miR-143–3p. The E6/E7-dependent reduction of miR-23a-3p is surprising since this miRNA often is elevated in cancers, suggesting that it acts pro-tumorigenic [92]. However, miR-23a-3p possesses pro-senescent potential [93,94] and has also been linked to apoptosis induction [92], indicating that miR-23a-3p can exert context-dependent pro - or anti-tumorigenic activities. miR-23b and miR-27b belong to the miR-23b-27b-24–1 cluster and both exhibit anti-tumorigenic activities [95,96]. Finally, among the 52 most commonly sequenced miRNAs, miR-143–3p represents the most strongly activated miRNA after E6/E7 silencing. It acts anti-proliferative [97], including in cervical cancer cell lines [34], and an increase of miR-143–3p levels has been linked to senescence [97]. It is also remarkable that three of the four abundant miRNAs that are significantly decreased by sustained E6/E7 expression are associated with the metabolic alterations typical for cancer cells. Specifically, lowered levels of miR-23a-3p and miR-23b-3p have been linked to enhanced glutamine catabolism [98] and a decrease of miR-143–3p favors glucose metabolism by aerobic glycolysis (Warburg effect) [99].
The identification of cellular miRNAs in the present work that are dependent on sustained endogenous E6/E7 expression forms a basis for future functional studies. Here, we took a closer look at members of the tumorigenic miR-17~92 cluster, since (i) miR-17–5p was among the top ten hits of abundant miRNAs of which the expression was maintained by the E6/E7 oncogenes, (ii) all other members of this cluster, as well as of the paralog cluster miR-106b~25, were also downregulated by E6/E7 silencing when applying less stringent selection criteria (S3 Table), (iii) several members of the miR-17~92 cluster are well-decumented to be overexpressed in cervical cancer tissues, including the tested miR-17–5p [27–30,34,37] and miR-20a-5p [23,30,31,34,35] (also see S2 Dataset), and (iv) four of these miRNAs (miR-17–5p, miR-20a-5p, miR-93–5p, and miR-106b-5p) possess the same seed sequence and can bind to the 3’ UTR of the p21 mRNA [18]. Our finding that the concomitant inhibition of p21 and E6/E7 expression led to an alleviation of the senescent phenotype, compared to cells in which only E6/E7 was repressed, supports the notion that p21 contributes to the induction of cellular senescence upon E6/E7 inhibition in HPV-positive cancer cells [13]. In view of the strong anti-proliferative and pro-senescent potential of p21, it seems critical for the growth of HPV-positive tumor cells to block p21 function. Our findings that miR-17–5p and miR-20a-5p inhibitors significantly induced endogenous p21 protein levels, indicates that oncogenic HPVs reduce p21 expression in cervical cancer cells by increasing the intracellular concentrations of members of this miRNA seed family. This provides evidence for a third layer of negative regulation of p21 by the HPV oncogenes, in addition to interfering with p53-mediated transcriptional p21 activation via E6-mediated p53 degradation [100] and the inhibitory E7/p21 protein/protein interaction [101,102] (Fig. 10).

Comparative analyses of the ten HPV18 E6/E7-dependent cellular miRNAs (HeLa) in HPV16-positive cells (SiHa) revealed a substantial overlap in their regulation patterns upon inhibition of endogenous E6/E7 expression. This is not necessarily expectable since the two cell lines are derived from a cervical adenocarcinoma and a squamous cell carcinoma, respectively, and the miRNA composition of tumor cells can substantially vary even for the same cancer form, dependent on the histological background or differentiation status [103,104]. Since HPV16 and HPV18 E6/E7 share most of their functions, the overlap in miRNA regulation across tumor cells of different histopathological origin provides further evidence for its E6/E7-dependence.
The present work also represents the first study investigating global changes of the miRNA composition of exosomes released from HPV-positive cancer cells, in dependence on endogenous viral oncogene expression. Consistent with the view that exosomal sorting of miRNAs is a directed process [105], the most commonly sequenced intracellular and exosomal miRNAs exhibited only a partial overlap. We also observed that E6/E7 silencing increases the percentage of exosomal miRNAs relative to other small RNA fractions, raising the possibility that the viral oncogenes affect exosomal sorting of small RNAs. Among the 47 most frequently sequenced exosomal miRNAs, 25 were modulated > 1.5-fold by silencing E6/E7 expression. Seven of those also exhibited statistical significance in the validating qRT-PCR analyses.
Our results show that continuous E6/E7 expression is linked to an upregulation of let-7d-5p, miR-20a-5p, miR-378a-3p, miR-423–3p, miR-7–5p, miR-92a-3p and a downregulation of miR-21–5p, in exosomes secreted from HeLa cells. Interestingly, several of these miRNAs exert cancer-associated activities inside cells. Let-7d-5p belongs to the let-7 miRNA family, which is considered to primarily act tumor-suppressive [106]. However, specific analyses of the let-7d family member also indicate anti-apoptotic activity by targeting the 3’ UTR of caspase 3 [107] and a strong increase of let-7d-5p levels has been observed during progression of breast cancers [108]. The other five of the six abundant exosomal miRNAs that are maintained by continuous E6/E7 expression have been primarily linked to pro-tumorigenic activities. The pro-oncogenic potential of miR-7–5p and miR-378a-3p is discussed above. miR-20a-5p and miR-92a-3p are both members of the miR-17~92 cluster. miR-20a-5p can block oncogene-induced senescence via p21 repression [109], whereas miR-92a-3p possesses anti-apoptotic potential [110]. miR-423–3p has been shown to promote G1/S transition and cell growth. On the other hand, miR-21–5p, which is considered to be pro-tumorigenic [111], was the only miRNA among the 47 most frequently sequenced miRNA species in exosomes that was significantly upregulated > 1.5-fold upon E6/E7 silencing, indicating that continuous E6/E7 expression is associated with reduced exosomal miR-21–5p levels. Thus, taken together, with the exception of miR-21–5p, sustained E6/E7 expression in HPV-positive cancer cells is linked to exosomal miRNA alterations that possess known pro-proliferative or anti-apoptotic potential. These findings complement results indicating that endogenous E6/E7 expression in HPV-positive cancer cells is also linked to exosomal protein alterations with growth promoting and anti-apoptotic potential, e.g. upregulation of Survivin [77]. Comparative analyses of the seven miRNAs in exosomes secreted by HPV16-positive SiHa cells revealed a substantial overlap in their modulation upon endogenous E6/E7 silencing. Thus, as observed for intracellular miRNAs, there is a similar regulation of E6/E7-dependent miRNAs in exosomes secreted by tumor cells that contain different HPV types and that are of different histological origin.
Our observations concerning exosomal miRNA contents could be relevant for intercellular communication in that HPV-positive cells might convey a tumor-promoting message to surrounding cells via exosomes, as has been reported for two of the E6/E7-dependent exosomal miRNAs, miR-92a-3p [55] and miR-378a-3p [52]. Of note, however, studies on the intercellular communication via exosomes have to critically consider technical limitations that are still unresolved. Despite an increasing number of examples showing that an intercellular crosstalk via exosomal miRNAs is possible in cell culture, the physiological significance of these observations is often uncertain [47,112]. Specifically, most studies utilized concentrated exosome preparations and it is not clear how these experimental conditions relate to exosome concentrations in the physiological context [113] which are very low in biological fluids (within 100 fM range). This might be below the threshold for exerting significant physiological effects in vivo [114,115] since it has been estimated that miRNAs require intracellular levels of greater than 1,000 copies per cell to trigger measurable activity on their mRNA targets [115]. These questions could be addressed once experimental systems to test the physiological relevance of exosomes become available, which is a topic of intense ongoing research in the exosome field [47,112].
The identification of an E6/E7-dependent miRNA signature in exosomes may also bear diagnostic potential. Circulating miRNAs are currently intensively investigated as new, minimally invasive biomarkers for early diagnosis, prognosis and prediction of response to specific therapies [116,117]. A significant source of miRNAs in extracellular fluids, like serum or saliva, are exosomes [118]. Major advantages of using exosomal miRNAs as biomarkers include their high stability and the possibility to increase the sensitivity of miRNA amplification from human biologic fluids by exosome isolation and enrichment [71,118]. It thus will be interesting to investigate whether the E6/E7-dependent miRNA changes identified here are mirrored in exosomes isolated from body fluids of patients suffering from HPV-linked diseases, such as in the serum, cervical lavages of cervical cancer patients or saliva of head and neck cancer patients.
Taken together, this study shows that the endogenous E6/E7 expression in HPV-positive cancer cells is linked to increased concentrations of multiple pro-proliferative, anti-senescent and anti-apoptotic miRNAs, while the amounts of anti-proliferative, pro-senescent and pro-apoptotic miRNAs are reduced. This applies to abundant miRNA species both inside the cell and in exosomes. These findings imply that the viral E6/E7 oncogenes affect the growth of HPV-positive cancer cells by manipulating the intracellular and exosomal miRNA compositions. It will be interesting for future studies to further decipher the role of these miRNAs for the proliferation and survival of HPV-positive cancer cells. Moreover, since therapeutic agents acting on the miRNA level are now entering the clinic [117,119] and since therapeutically useful E6/E7 inhibitors are still not available, it will be important to evaluate whether a correction of the E6/E7-dependent miRNA alterations by miRNA mimics or inhibitors possesses therapeutic potential for the treatment of HPV-linked premalignant and malignant lesions.
Materials and Methods
Cell culture, transfections and treatment conditions
HPV18-positive HeLa (obtained from the tumor bank of the German Cancer Research Center, Heidelberg) and HPV16-positive SiHa cervical carcinoma cells (obtained from the American Tissue Culture Collection, ATCC) were cultured in DMEM (Gibco, Life Technologies, Carlsbad, CA, USA) containing 5% fetal bovine serum (Gibco, Life Technologies), 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin (Sigma-Aldrich, Saint Louis, MO, USA). “p53-null” HeLa cells were described in detail in ref. [65].
Synthetic siRNAs (Ambion, Life Technologies, Carlsbad, CA, USA) were transfected with DharmaFECT I (Thermo Fisher Scientific, Waltham, MA, USA), according to the manufacturer’s instructions, to reach a final siRNA concentration of 10 nM. For silencing viral HPV18 or HPV16 E6/E7 oncogene expression, three different siRNAs, which each target all three HPV E6/E7 transcript classes, were generated [8]. To minimize potential off-target effects [120,121] the three siRNAs were pooled at equimolar concentrations (referred to in the text as “si18E6/E7” or “si16E6/E7”, respectively). The siRNA target sequences were as follows: HPV18 E6/E7–1 5’-CCACAACGUCACACAAUGU-3’; HPV18 E6/E7–2 5’-CAGAGAAACACAAGUAUAA-3’; HPV18 E6/E7–3 5’-UCCAGCAGCUGUUUCUGAA-3’, HPV16 E6/E7–1 5’-CCGGACAGAGCCCAUUACA-3’; HPV16 E6/E7–2 5’ - CACCUACAUUGCAUGAAUA-3’; HPV16 E6/E7–3 5’ - CAACUGAUCUCUACUGUUA-3’; p21 (CDKN1A) 5’-CAAGGAGUCAGACAUUUUA-3’. Control siRNA “siContr-1”, 5′-CAGUCGCGUUUGCGACUGG-3′, contains at least four mismatches to all known human genes.
miRNA Inhibitors (Qiagen, Hilden, Germany) and the miScript Inhibitor Negative Control (Qiagen) were transfected with DharmaFECT I (Thermo Fisher Scientific), according to the manufacturer’s instructions, to reach a final concentration of 100 nM for miRNA inhibitors.
Plasmids were transfected by calcium phosphate co-precipitation as described by Chen and Okayama [122]. The plasmid expressing the mir-17~92 cluster (pcDNA3.1/V5-His-TOPO-mir17~92, [123]) was a gift from Joshua Mendell (Addgene plasmid # 21109) and the pcDNA3.1 empty vector (Life Technologies) was used as negative control.
Cell cycle analyses
For cell cycle analysis, cells were trypsinized 72 h after transfection, washed in ice-cold PBS and fixed in 80% cold ethanol overnight at -20°C. Subsequently cells were pelleted, resuspended in phosphate buffered saline (PBS, 137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, 1.4 mM KH2PO4, pH 7.4) containing 1 mg/ml RNase A (Roche Diagnostics) and 25 μg/ml propidium iodide (Sigma-Aldrich) and incubated for 30 min at room temperature (RT). Cell cycle analyses were performed by fluorescence-activated cell sorting (FACS) using a FACSCalibur Flow Cytometer (BD Biosciences, Heidelberg, Germany) with CellQuest Pro software provided by the manufacturer. Quantitation of the percentage of cells in the individual phases was performed using FlowJo software (Tree Star, Ashland, OR, USA), applying the Dean-Jett-Fox model [124].
Senescence assay
HeLa cells were plated on glass cover slips, followed by transfection with siRNAs, as described above. Staining for senescence-associated β-galactosidase (SA-β-Gal) activity was performed 168 h after transfection, as described by Dimri et al. [125].
Generation and purification of exosomes
For exosome production cells were plated on 15 cm dishes to reach 80% confluence 72 h post transfection. Forty-eight h post transfection the cells were washed with DMEM and cultured for 24 h in “vesicle-depleted medium” (complete medium depleted of FBS-derived microvesicles by overnight centrifugation at 100,000 g). The conditioned medium was collected and cleared from intact cells and cellular debris by three rounds of centrifugation at 300 g, 3,000 g, and 10,000 g for 20 min. Subsequently, exosomes were pelleted from the resulting supernatant by ultracentrifugation at 100,000 g for 70 min using a SW28 rotor (Beckman Coulter, Fullerton, CA, USA), resuspended in 36 ml PBS, and re-centrifuged at 100,000 g for 70 min. All centrifugation steps were performed at 4°C. The final pellet was resuspended in 100 μl PBS and an aliquot was analyzed by electron microscopy. For each preparation, the total protein concentration was quantified using the Qubit Protein Assay (Life Technologies). The corresponding exosome-producing cells were harvested and pelleted by centrifugation at 800 g for 3 min, resuspended and washed in 800 μl PBS, and re-pelleted.
Immunoblotting
Cell pellets were lysed in RIPA buffer (10 mM Tris (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1% Nonidet P40, 0.5% Na-Deoxycholat, 0.1% SDS, supplemented with 25 μl/ml Pefabloc (Merck, Whitehouse Station, NJ, USA) and 10 μl/ml of Protease Inhibitor Cocktail (Sigma-Aldrich)) for 30 min on ice and proteins were collected by centrifugation at 12,000 g for 15 min. Protein concentrations were determined using the Bio-Rad Protein Assay (Bio-Rad, Hercules, CA, USA), employing bovine serum albumin as standard. For Western blot analysis, protein extracts and exosome samples were boiled in SDS sample buffer (for reducing conditions: 8% SDS, 250 mM Tris-HCL (pH 6.8), 20% β-mercaptoethanol, 40% glycerol, 0.008% bromphenol blue; for non-reducing conditions without β-mercaptoethanol) for 5 min at 95°C and separated on NuPAGE Novex 4–12% Bis-Tris Mini Gels (Life Technologies). Proteins were electrotransferred onto an Immobilon-P membrane (Millipore, Bedford, MA, USA) using the Trans-Blot Semi-Dry Transfer Cell (Bio-Rad). Membranes were blocked with 5% skim milk powder (Saliter, Obergünzburg, Germany) in PBS-T (PBS supplemented with 0.1% Tween-20) for 1 h at RT. Membranes were probed with primary antibodies over night at 4°C in PBS-T, followed by incubation with the respective HRP-conjugated secondary antibody in PBS-T for 1 h at RT. Proteins were visualized using the ECL Prime Western Blotting Detection Reagent (GE Healthcare, Buckinghamshire, UK). Images were acquired using the Fusion SL Gel Detection System (Vilber Lourmat, Marne-la-Vallée, France), band densities were determined by BioID image analysis software (Vilber Lourmat).
The following primary antibodies were used: mouse anti-α-Tubulin (Merck), CP06, dilution 1 : 5,000; mouse anti-β-Actin (Sigma-Aldrich), A2228, 1 : 10,000; mouse anti-Annexin 1 (BD Transduction, Heidelberg, Germany), #610066, 1 : 10,000; mouse anti-CD63 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), sc-5275, 1 : 400 at non-reducing conditions; mouse anti-CD9 (BD Pharmingen, San Diego, Ca, USA), #555370, 1 : 200 at non-reducing conditions; mouse anti-GRP78 (BD Transduction), #610979, 1 : 1,000; chicken anti-HPV18 E7 (E7C) [126]; mouse anti-HPV18 E6 (Arbor Vita Corporation Sunnyvale, CA, USA) AVC #399; mouse anti-HPV16 E7 (NM2, kind gift of Dr. Martin Müller, German Cancer Research Center, Heidelberg, Germany); mouse anti-HPV16 E6 (Arbor Vita Corporation Sunnyvale, CA, USA) AVC #843; rat anti-Hsc70 (Stressgen, San Diego, CA, USA), ADI-SPA-815, 1 : 1,000; mouse anti-p53 (BD Pharmingen), #554293, 1 : 500; mouse anti-EEA1 (BD Transduction Laboratories), #E41120, 1 : 2,000; mouse anti-pRb (Cell Signaling), #9309, 1 : 1,000; rabbit anti-phospho(Ser807/811)-pRb (Cell Signaling), #9308, 1 : 1,000; rabbit anti-CyclinA1 (Santa Cruz Biotechnology), 1 : 2,000. The following HRP-conjugated secondary antibodies were applied: anti-mouse IgG (Promega, Madison, WI, USA), W4021, 1 : 5,000; anti-chicken IgY (Promega), G1351, 1 : 2,500; anti-rat IgG (Dianova, Hamburg, Germany), #112035003, 1 : 5,000; and anti-goat IgG (Promega), V8051, 1 : 3,000.
Electron Microscopy (EM)
Purified exosomes (6 μl, corresponding to 1 to 4 μg protein, depending on the experiment) were layered onto carbon-coated copper grids (300 mesh, Plano, Wetzlar, Germany) and allowed to dry at RT. Grids were then washed with water for 5 min and stained with 2% uranyl acetate in water (Polysciences, Warrington, PA, USA) for 30 sec to 1 min. Imaging was performed at an acceleration voltage of 80 kV with the EM10 Electron Microscope (Zeiss, Oberkochen, Germany).
RNA extraction, quantification and quality determination
Total cellular RNA, including miRNA, was isolated with the miRNeasy Mini Kit (Qiagen) following the manufacturer’s protocol. All optional washing steps were included and RNA was eluted in a final volume of 30 μl RNase-free water. Total exosomal RNA, including miRNA, was isolated using the protocol described for cells with slight modifications: Exosome samples were pre-treated with 100 ng/μl RNAse A (Roche) for 30 min at 37°C, immediately before extracting RNA. Prior to the addition of chloroform and phase separation, 12 μg glycogen from Mytilus edulis (Sigma) were added to the sample. RNA concentrations were measured with the NanoDrop ND-1000 at 260 nm. RNA quality was assessed with the Agilent 2100 Bioanalyzer (Agilent Technologies, Böblingen, Germany) using the Agilent RNA 6000 Pico Kit (total RNA) and the Agilent Small RNA Kit (small RNA). The 2100 Bioanalyzer Expert Software B.02.08. (Agilent) was applied to generate electropherograms. RNA samples were stored at -80°C until further analysis.
mRNA Quantitative Reverse Transcription-PCR (qRT-PCR)
For mRNA analysis, reverse transcription of 1 μg total RNA was carried out with the ProtoScript First Strand cDNA Synthesis Kit (NEB) according to the manufacturer’s instructions, using oligo-dT primers in an end volume of 20 μl. To assess for genomic DNA contamination of the sample, a no reverse transcriptase control (RT-) was prepared for each experiment by replacing the M-MuLV Enzyme Mix with RNase-free H2O. qRT-PCR reactions were performed with the SYBR Green PCR Master Mix (Applied Biosystems) and a final primer concentration of 500 nM on a 7300 Real-Time PCR System Detector (Applied Biosystems). Two μl of a 1 : 5 dilution of the original cDNA were used for each reaction and samples were run in triplicate for each experiment. A no template control (NTC) to monitor contamination of the reagents was included for each primer pair by adding H2O instead of cDNA template. The forward (fwd) and reverse (rev) primer sequences (Eurofins MWG, Ebersberg, Germany) used for determining mRNA expression levels were as follows: HPV18 E6/E7 fwd 5’-ATGCATGGACCTAAGGCAAC-3’, HPV18 E6/E7 rev 5’-AGGTCGTCTGCTGAGCTTTC-3’, HPV16 E6/E7 fwd 5’-CAATGTTTCAGGACCCACAGG-3’, HPV16 E6/E7 rev 5’-CTCACGTCGCAGTAACTGTTG-3’, p21 (CDKN1A) fwd 5’-GACCATGTGGACCTGTCACT-3’, p21 (CDKN1A) rev 5’-GCGGATTAGGGCTTCCTCTT-3’, ACTB fwd 5’-AGACAGTATACCCCATGCTGCAT-3’, ACTB rev 5’-TCCAATGTGTCTCCATACACAGA-3’. Cycling conditions have been previously described [127]. The sizes of the PCR products were initially analyzed by agarose gel electrophoresis and subsequently checked by melting point analysis after each reaction using the 7300 System SDS Software (Applied Biosystems). Cycle thresholds (Ct) were normalized to the Cts of ACTB using the comparative Ct (2-ΔΔCt) method [128]. Fold enrichments were calculated as compared to the values from the mock control.
miRNA qRT-PCR
miRNA expression was detected using the miScript PCR System (Qiagen) with miRNA specific primers according to the manufacturer’s instructions. Briefly, 1 μg cellular RNA was converted to cDNA using the miScript II Reverse Transcription Kit (Qiagen) in a reaction volume of 20 μl using 5x miScript HiFlex Buffer. Due to the low yield of exosomal RNA and consequently the lack of accurate RNA quantification, identical volumes of exosomal RNA (12 μl) were used for cDNA synthesis. An RT - control was prepared for each experiment. qRT-PCR reactions were performed with the miScript SYBR Green PCR Kit (Qiagen) on a 7300 Real-Time PCR System Detector (Applied Biosystems) using Qiagen’s recommended cycling conditions. Two μl of a 1 : 100 dilution of the original cDNA were used for each reaction and samples were run in triplicate for each experiment. To monitor contamination of the reagents, an NTC was included for each primer pair. Data were analyzed using the comparative Ct (2-ΔΔCt) method [128] with the small nuclear RNA RNU6–2 as endogenous control for cellular samples and respective mock - or control-treated samples (as indicated) as reference. Normalization of exosomal samples was conducted against an average of the expression of miR-452–5p and miR-183–5p. These two miRNAs were chosen as endogenous exosomal miRNAs for normalization since they were among the most frequently sequenced exosomal miRNAs in HeLa cells (> 1,000 RPM in each sample) and showed virtually no regulation in exosomes upon silencing of HPV18 E6/E7 versus control treatment based on the deep sequencing data (miR-452–5p: FCmean = 0.99, miR-183–5p: FCmean = 1.04). miRNAs with Ct values > 35 were below detection limit and excluded from analysis. The following miScript Primer Assays (Qiagen) were applied: Hs_let-7a_2 (hsa-let-7a-5p), Hs_let-7d_1 (hsa-let-7d-5p), Hs_let-7f_1 (hsa-let-7f-5p), Hs_let-7g_2 (hsa-let-7g-5p), Hs_miR-100_2 (hsa-miR-100–5p), Hs_miR-103a_1 (hsa-miR-103a-3p), Hs_miR-106b_1 (hsa-miR-106b-5p), Hs_miR-1246_2 (hsa-miR-1246), Hs_miR-125a_1 (hsa-miR-125a-5p), Hs_miR-128_1 (hsa-miR-128), Hs_miR-1307_1 (hsa-miR-1307–3p), Hs_miR-143_1 (hsa-miR-143–3p), Hs_miR-17_2 (hsa-miR-17–5p), Hs_miR-181b_1 (hsa-miR-181b-5p), Hs_miR-182_2 (hsa-miR-182–5p), Hs_miR-183_2 (hsa-miR-183–5p), Hs_miR-186_1 (hsa-miR-186–5p), Hs_miR-191_1 (hsa-miR-191–5p), Hs_miR-196a_2 (hsa-miR-196a-5p), Hs_miR-19b_2 (hsa-miR-19b-3p), Hs_miR-20a_1 (hsa-miR-20a-5p), Hs_miR-21*_1 (hsa-miR-21–3p), Hs_miR-21_2 (hsa-miR-21–5p), Hs_miR-221_1 (hsa-miR-221–3p), Hs_miR-222_2 (hsa-miR-222–3p), Hs_miR-23a_2 (hsa-miR-23a-3p), Hs_miR-23b_2 (hsa-miR-23b-3p), Hs_miR-25_1 (hsa-miR-25–3p), Hs_miR-25_1 (hsa-miR-25–3p), Hs_miR-26a_2 (hsa-miR-26a-5p), Hs_miR-27a_1 (hsa-miR-27a-3p), Hs_miR-27a*_1 (hsa-miR-27a-5p), Hs_miR-27b_2 (hsa-miR-27b-3p), Hs_miR-30c_2 (hsa-miR-30c-5p), Hs_miR-31_1 (hsa-miR-31–5p), Hs_miR-320a_1 (hsa-miR-320a), Hs_miR-320_2 (hsa-miR-320b), Hs_miR-34a_1 (hsa-miR-34a-5p), Hs_miR-422b_1 (hsa-miR-378a-3p), Hs_miR-378c_1 (hsa-miR-378c), Hs_miR-378d_1 (hsa-miR-378d), Hs_miR-378f_1 (hsa-miR-378f), Hs_miR-423_1 (hsa-miR-423–3p), Hs_miR-452_4 (hsa-miR-452–5p), Hs_miR-629_2 (hsa-miR-629–5p), Hs_miR-7_2 (hsa-mir-7–5p), Hs_miR-92_1 (hsa-miR-92a-3p), Hs_miR-93_1 (hsa-miR-93–5p), Hs_miR-98_1 (hsa-miR-98), Hs_miR-99a_2 (hsa-miR-99a-5p), Hs_RNU6–2_1 (RNU6–2). All analyzed miRNAs are of human (Homo sapiens) origin and therefore the prefix “hsa” was omitted throughout the text.
Small RNA deep sequencing and analysis
For each exosome sample, total RNA (containing the small RNA fraction) was extracted from one entire exosome preparation of 100 μl. Due to the low RNA yield, exosome samples were further concentrated to a volume of 6 μl using the RNeasy MinElute Cleanup Kit (Qiagen). The entire volume of resulting 6 μl concentrated exosomal total RNA (100 to 600 ng) was used as input for the library preparations. For cells, 1 μg total RNA in a volume of 6 μl was applied. Small RNA libraries were prepared using the NEBNext Multiplex Small RNA Library Prep Set for Illumina (NEB, Frankfurt/M., Germany) with custom multiplex adaptors and primers (NEBNext Multiplex Oligos for Illumina, Index Primers Set 1). Essentially, all materials not included in the set were purchased as recommended and the manufacturer’s guidelines were followed with a few modifications. Briefly, the Multiplex 3’ Adaptor was ligated to the RNA at 25°C for 1 h. After hybridization of the Multiplex RT Primer, the Multiplex 5’ Adaptor was ligated to the RNA. Afterwards, reverse transcription was performed using the SuperScript III Reverse Transcriptase (Life Technologies). The cDNA product was amplified by PCR using an optimized cycling protocol: initial denaturation for 3 min at 94°C, thirteen cycles of denaturation for 80 sec at 94°C, annealing for 30 sec at 62°C, extension for 15 sec at 70°C and final extension for 5 min at 70°C. Unique NEBNext Index Primers were applied for each of the six exosome samples (Index 1–6) and the four cellular samples (Index 1–4). Amplicons corresponding to adapter-ligated constructs from 21–30 nt RNA fragments were purified on a 6% TBE polyacrylamide gel (Life Technologies) and eluted at RT for 3 h. The gel slurry was passed through a 5 μm filter tube (IST Engineering, Milipitas, CA, USA) and precipitated overnight at -80°C. The size, DNA concentration and quality of each final small RNA library was determined twice using the High Sensitivity DNA Kit (Agilent) with the BioAnalyzer 2100. The concentration of each sample was adjusted to 10 nM and equal volumes of barcode-labeled samples were pooled for multiplexed sequencing in one lane. Sequencing (50 bp, single read) was performed on an Illumina HiSeq 2000 instrument (San Diego, CA). Raw sequencing reads were pre-processed and mapped using the function mapper.pl of the miRDeep2 package ([129], Max Delbrück Center, Berlin, Germany) as described by Weischenfeldt et al. [130]. Briefly, low quality reads were filtered out, the adaptor sequence was clipped and reads shorter than 18 nt were discarded. Accepted reads were mapped to known human miRNAs based on miRBase v.18.0 (www.mirbase.org/; [131–134]) using the function quantifier.pl in miRDeep2. Since one mismatch was allowed during the mapping procedure, the raw read file of each further analyzed miRNA was manually checked to assure that the reads truly annotated to the respective miRNA. The obtained raw read counts of each sample were normalized by dividing with the total number of reads mapping to known human microRNAs for each sample. Values are expressed as reads per million (RPM). Fold changes (FCs) were obtained by dividing the RPM of the si18E6/E7-treatment by the respective value of the siContr-1-treatment.
Genome mapping was performed on the pre-processed reads with miRDeep2 allowing two mismatches and up to five mapped positions in the genome. Read counts were obtained using python script rpkmforgenes.py (http://sandberg.cmb.ki.se/media/data/rnaseq/rpkmforgenes.py) applied to aligned data along with two major annotation sources: Gencode v.15 (http://www.gencodegenes.org/releases/15.html) for snoRNA; and RepeatMasker track (http://www.repeatmasker.org/) for scRNA, tRNA and snRNA.
Statistical analyses
Statistical significance of the data was evaluated by the paired Student’s t-test using the Sigma Plot software (Systat Software Inc., San Jose, CA). P-values of p ≤ 0.05 (*), p ≤ 0.01 (**), and p ≤ 0.001 (***) were considered statistically significant.
Supporting Information
Zdroje
1. zur Hausen H (2002) Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer 2 : 342–350. 12044010
2. von Knebel Doeberitz M, Oltersdorf T, Schwarz E, Gissmann L (1988) Correlation of modified human papilloma virus early gene expression with altered growth properties in C4–1 cervical carcinoma cells. Cancer Res 48 : 3780–3786. 2837324
3. McLaughlin-Drubin ME, Munger K (2009) Oncogenic activities of human papillomaviruses. Virus Res 143 : 195–208. doi: 10.1016/j.virusres.2009.06.008 19540281
4. Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM (1990) The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 63 : 1129–1136. 2175676
5. Klingelhutz AJ, Foster SA, McDougall JK (1996) Telomerase activation by the E6 gene product of human papillomavirus type 16. Nature 380 : 79–82. 8598912
6. McLaughlin-Drubin ME, Munger K (2009) The human papillomavirus E7 oncoprotein. Virology 384 : 335–344. doi: 10.1016/j.virol.2008.10.006 19007963
7. Butz K, Denk C, Ullmann A, Scheffner M, Hoppe-Seyler F (2000) Induction of apoptosis in human papillomaviruspositive cancer cells by peptide aptamers targeting the viral E6 oncoprotein. Proc Natl Acad Sci U S A 97 : 6693–6697. 10829072
8. Butz K, Ristriani T, Hengstermann A, Denk C, Scheffner M, et al. (2003) siRNA targeting of the viral E6 oncogene efficiently kills human papillomavirus-positive cancer cells. Oncogene 22 : 5938–5945. 12955072
9. Griffin H, Elston R, Jackson D, Ansell K, Coleman M, et al. (2006) Inhibition of papillomavirus protein function in cervical cancer cells by intrabody targeting. J Mol Biol 355 : 360–378. 16324714
10. Yamato K, Yamada T, Kizaki M, Ui-Tei K, Natori Y, et al. (2008) New highly potent and specific E6 and E7 siRNAs for treatment of HPV16 positive cervical cancer. Cancer Gene Ther 15 : 140–153. 18157144
11. Lagrange M, Boulade-Ladame C, Mailly L, Weiss E, Orfanoudakis G, et al. (2007) Intracellular scFvs against the viral E6 oncoprotein provoke apoptosis in human papillomavirus-positive cancer cells. Biochem Biophys Res Commun 361 : 487–492. 17658466
12. Goodwin EC, Yang E, Lee CJ, Lee HW, DiMaio D, et al. (2000) Rapid induction of senescence in human cervical carcinoma cells. Proc Natl Acad Sci U S A 97 : 10978–10983. 11005870
13. Wells SI, Francis DA, Karpova AY, Dowhanick JJ, Benson JD, et al. (2000) Papillomavirus E2 induces senescence in HPV-positive cells via pRB - and p21(CIP)-dependent pathways. EMBO J 19 : 5762–5771. 11060027
14. Hall AH, Alexander KA (2003) RNA interference of human papillomavirus type 18 E6 and E7 induces senescence in HeLa cells. J Virol 77 : 6066–6069. 12719599
15. Ebert MS, Sharp PA (2012) Roles for microRNAs in conferring robustness to biological processes. Cell 149 : 515–524. doi: 10.1016/j.cell.2012.04.005 22541426
16. Cowland JB, Hother C, Gronbaek K (2007) MicroRNAs and cancer. APMIS 115 : 1090–1106. 18042145
17. Iorio MV, Croce CM (2012) MicroRNA dysregulation in cancer: diagnostics, monitoring and therapeutics. A comprehensive review. EMBO Mol Med 4 : 143–159. doi: 10.1002/emmm.201100209 22351564
18. Olive V, Li Q, He L (2013) mir-17–92: a polycistronic oncomir with pleiotropic functions. Immunol Rev 253 : 158–166. doi: 10.1111/imr.12054 23550645
19. Campisi J (2012) Aging, Cellular Senescence, and Cancer. Annu Rev Physiol.
20. Kuilman T, Michaloglou C, Mooi WJ, Peeper DS (2010) The essence of senescence. Genes Dev 24 : 2463–2479. doi: 10.1101/gad.1971610 21078816
21. Kincaid RP, Sullivan CS (2012) Virus-encoded microRNAs: an overview and a look to the future. PLoS Pathog 8: e1003018. doi: 10.1371/journal.ppat.1003018 23308061
22. Wang X, Wang HK, Li Y, Hafner M, Banerjee NS, et al. (2014) microRNAs are biomarkers of oncogenic human papillomavirus infections. Proc Natl Acad Sci U S A 111 : 4262–4267. doi: 10.1073/pnas.1401430111 24591631
23. Villegas-Ruiz V, Juarez-Mendez S, Perez-Gonzalez OA, Arreola H, Paniagua-Garcia L, et al. (2014) Heterogeneity of microRNAs expression in cervical cancer cells: over-expression of miR-196a. Int J Clin Exp Pathol 7 : 1389–1401. 24817935
24. Lin C, Huang F, Zhang YJ, Tuokan T, Kuerban G (2014) Roles of MiR-101 and its target gene Cox-2 in early diagnosis of cervical cancer in Uygur women. Asian Pac J Cancer Prev 15 : 45–48. 24528073
25. Ma D, Zhang YY, Guo YL, Li ZJ, Geng L (2012) Profiling of microRNA-mRNA reveals roles of microRNAs in cervical cancer. Chin Med J (Engl) 125 : 4270–4276. 23217399
26. Cheung TH, Man KN, Yu MY, Yim SF, Siu NS, et al. (2012) Dysregulated microRNAs in the pathogenesis and progression of cervical neoplasm. Cell Cycle 11 : 2876–2884. doi: 10.4161/cc.21278 22801550
27. Lajer CB, Garnaes E, Friis-Hansen L, Norrild B, Therkildsen MH, et al. (2012) The role of miRNAs in human papilloma virus (HPV)-associated cancers: bridging between HPV-related head and neck cancer and cervical cancer. Br J Cancer 106 : 1526–1534. doi: 10.1038/bjc.2012.109 22472886
28. Wilting SM, Snijders PJ, Verlaat W, Jaspers A, van de Wiel MA, et al. (2013) Altered microRNA expression associated with chromosomal changes contributes to cervical carcinogenesis. Oncogene 32 : 106–116. doi: 10.1038/onc.2012.20 22330141
29. Liu L, Yu X, Guo X, Tian Z, Su M, et al. (2012) miR-143 is downregulated in cervical cancer and promotes apoptosis and inhibits tumor formation by targeting Bcl-2. Mol Med Rep 5 : 753–760. doi: 10.3892/mmr.2011.696 22160209
30. Li Y, Wang F, Xu J, Ye F, Shen Y, et al. (2011) Progressive miRNA expression profiles in cervical carcinogenesis and identification of HPV-related target genes for miR-29. J Pathol 224 : 484–495. doi: 10.1002/path.2873 21503900
31. Rao Q, Shen Q, Zhou H, Peng Y, Li J, et al. (2012) Aberrant microRNA expression in human cervical carcinomas. Med Oncol 29 : 1242–1248. doi: 10.1007/s12032-011-9830-2 21264530
32. Li JH, Xiao X, Zhang YN, Wang YM, Feng LM, et al. (2011) MicroRNA miR-886–5p inhibits apoptosis by down-regulating Bax expression in human cervical carcinoma cells. Gynecol Oncol 120 : 145–151. doi: 10.1016/j.ygyno.2010.09.009 20947150
33. Pereira PM, Marques JP, Soares AR, Carreto L, Santos MA (2010) MicroRNA expression variability in human cervical tissues. PLoS One 5: e11780. doi: 10.1371/journal.pone.0011780 20668671
34. Wang X, Tang S, Le SY, Lu R, Rader JS, et al. (2008) Aberrant expression of oncogenic and tumor-suppressive microRNAs in cervical cancer is required for cancer cell growth. PLoS One 3: e2557. doi: 10.1371/journal.pone.0002557 18596939
35. Lee JW, Choi CH, Choi JJ, Park YA, Kim SJ, et al. (2008) Altered MicroRNA expression in cervical carcinomas. Clin Cancer Res 14 : 2535–2542. doi: 10.1158/1078-0432.CCR-07-1231 18451214
36. Witten D, Tibshirani R, Gu SG, Fire A, Lui WO (2010) Ultra-high throughput sequencing-based small RNA discovery and discrete statistical biomarker analysis in a collection of cervical tumours and matched controls. BMC Biol 8 : 58. doi: 10.1186/1741-7007-8-58 20459774
37. Park H, Lee MJ, Jeong JY, Choi MC, Jung SG, et al. (2014) Dysregulated microRNA expression in adenocarcinoma of the uterine cervix: Clinical impact of miR-363–3p. Gynecol Oncol 135 : 565–572. doi: 10.1016/j.ygyno.2014.09.010 25230213
38. Liang S, Tian T, Liu X, Shi H, Tang C, et al. (2014) Microarray analysis revealed markedly differential miRNA expression profiles in cervical intraepithelial neoplasias and invasive squamous cell carcinoma. Future Oncol 10 : 2023–2032. doi: 10.2217/fon.14.38 24559408
39. Tang T, Wong HK, Gu W, Yu MY, To KF, et al. (2013) MicroRNA-182 plays an onco-miRNA role in cervical cancer. Gynecol Oncol 129 : 199–208. doi: 10.1016/j.ygyno.2012.12.043 23313739
40. Lui WO, Pourmand N, Patterson BK, Fire A (2007) Patterns of known and novel small RNAs in human cervical cancer. Cancer Res 67 : 6031–6043. 17616659
41. Martinez I, Gardiner AS, Board KF, Monzon FA, Edwards RP, et al. (2008) Human papillomavirus type 16 reduces the expression of microRNA-218 in cervical carcinoma cells. Oncogene 27 : 2575–2582. 17998940
42. Dreher A, Rossing M, Kaczkowski B, Andersen DK, Larsen TJ, et al. (2011) Differential expression of cellular microRNAs in HPV 11, -16, and-45 transfected cells. Biochem Biophys Res Commun 412 : 20–25. doi: 10.1016/j.bbrc.2011.07.011 21782796
43. Gunasekharan V, Laimins LA (2013) Human papillomaviruses modulate microRNA 145 expression to directly control genome amplification. J Virol 87 : 6037–6043. doi: 10.1128/JVI.00153-13 23468503
44. Yablonska S, Hoskins EE, Wells SI, Khan SA (2013) Identification of miRNAs Dysregulated in Human Foreskin Keratinocytes (HFKs) Expressing the Human Papillomavirus (HPV) Type 16 E6 and E7 Oncoproteins. Microrna 2 : 2–13. 25070710
45. Wang X, Wang HK, McCoy JP, Banerjee NS, Rader JS, et al. (2009) Oncogenic HPV infection interrupts the expression of tumor-suppressive miR-34a through viral oncoprotein E6. RNA 15 : 637–647. doi: 10.1261/rna.1442309 19258450
46. McKenna DJ, McDade SS, Patel D, McCance DJ (2010) MicroRNA 203 expression in keratinocytes is dependent on regulation of p53 levels by E6. J Virol 84 : 10644–10652. doi: 10.1128/JVI.00703-10 20702634
47. Kowal J, Tkach M, Thery C (2014) Biogenesis and secretion of exosomes. Curr Opin Cell Biol 29C: 116–125.
48. Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, et al. (2007) Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 9 : 654–659. 17486113
49. Rak J (2013) Extracellular vesicles—biomarkers and effectors of the cellular interactome in cancer. Front Pharmacol 4 : 21. doi: 10.3389/fphar.2013.00021 23508692
50. Martins VR, Dias MS, Hainaut P (2013) Tumor-cell-derived microvesicles as carriers of molecular information in cancer. Curr Opin Oncol 25 : 66–75. doi: 10.1097/CCO.0b013e32835b7c81 23165142
51. Pegtel DM, Cosmopoulos K, Thorley-Lawson DA, van Eijndhoven MA, Hopmans ES, et al. (2010) Functional delivery of viral miRNAs via exosomes. Proc Natl Acad Sci U S A 107 : 6328–6333. doi: 10.1073/pnas.0914843107 20304794
52. Kogure T, Lin WL, Yan IK, Braconi C, Patel T (2011) Intercellular nanovesicle-mediated microRNA transfer: a mechanism of environmental modulation of hepatocellular cancer cell growth. Hepatology 54 : 1237–1248. doi: 10.1002/hep.24504 21721029
53. Montecalvo A, Larregina AT, Shufesky WJ, Stolz DB, Sullivan ML, et al. (2012) Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes. Blood 119 : 756–766. doi: 10.1182/blood-2011-02-338004 22031862
54. Rana S, Malinowska K, Zoller M (2013) Exosomal tumor microRNA modulates premetastatic organ cells. Neoplasia 15 : 281–295. 23479506
55. Umezu T, Ohyashiki K, Kuroda M, Ohyashiki JH (2013) Leukemia cell to endothelial cell communication via exosomal miRNAs. Oncogene 32 : 2747–2755. doi: 10.1038/onc.2012.295 22797057
56. Meckes DG Jr., Shair KH, Marquitz AR, Kung CP, Edwards RH, et al. (2010) Human tumor virus utilizes exosomes for intercellular communication. Proc Natl Acad Sci U S A 107 : 20370–20375. doi: 10.1073/pnas.1014194107 21059916
57. Pegtel DM, van de Garde MD, Middeldorp JM (2011) Viral miRNAs exploiting the endosomal-exosomal pathway for intercellular cross-talk and immune evasion. Biochim Biophys Acta 1809 : 715–721. doi: 10.1016/j.bbagrm.2011.08.002 21855666
58. Chugh PE, Sin SH, Ozgur S, Henry DH, Menezes P, et al. (2013) Systemically circulating viral and tumor-derived microRNAs in KSHV-associated malignancies. PLoS Pathog 9: e1003484. doi: 10.1371/journal.ppat.1003484 23874201
59. D’Souza-Schorey C, Clancy JW (2012) Tumor-derived microvesicles: shedding light on novel microenvironment modulators and prospective cancer biomarkers. Genes Dev 26 : 1287–1299. doi: 10.1101/gad.192351.112 22713869
60. Properzi F, Logozzi M, Fais S (2013) Exosomes: the future of biomarkers in medicine. Biomark Med 7 : 769–778. doi: 10.2217/bmm.13.63 24044569
61. Taylor DD, Gercel-Taylor C (2013) The origin, function, and diagnostic potential of RNA within extracellular vesicles present in human biological fluids. Front Genet 4 : 142. doi: 10.3389/fgene.2013.00142 23908664
62. el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, et al. (1993) WAF1, a potential mediator of p53 tumor suppression. Cell 75 : 817–825. 8242752
63. Mullokandov G, Baccarini A, Ruzo A, Jayaprakash AD, Tung N, et al. (2012) High-throughput assessment of microRNA activity and function using microRNA sensor and decoy libraries. Nat Methods 9 : 840–846. doi: 10.1038/nmeth.2078 22751203
64. Hermeking H (2012) MicroRNAs in the p53 network: micromanagement of tumour suppression. Nat Rev Cancer 12 : 613–626. doi: 10.1038/nrc3318 22898542
65. Hengstermann A, D’Silva M A, Kuballa P, Butz K, Hoppe-Seyler F, et al. (2005) Growth suppression induced by downregulation of E6-AP expression in human papillomavirus-positive cancer cell lines depends on p53. J Virol 79 : 9296–9300. 15994823
66. Fontana L, Fiori ME, Albini S, Cifaldi L, Giovinazzi S, et al. (2008) Antagomir-17–5p abolishes the growth of therapy-resistant neuroblastoma through p21 and BIM. PLoS One 3: e2236. doi: 10.1371/journal.pone.0002236 18493594
67. Wu S, Huang S, Ding J, Zhao Y, Liang L, et al. (2010) Multiple microRNAs modulate p21Cip1/Waf1 expression by directly targeting its 3′ untranslated region. Oncogene 29 : 2302–2308. doi: 10.1038/onc.2010.34 20190813
68. DeFilippis RA, Goodwin EC, Wu L, DiMaio D (2003) Endogenous human papillomavirus E6 and E7 proteins differentially regulate proliferation, senescence, and apoptosis in HeLa cervical carcinoma cells. J Virol 77 : 1551–1563. 12502868
69. Thery C, Amigorena S, Raposo G, Clayton A (2006) Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol Chapter 3: Unit 3 22.
70. Taylor DD, Gercel-Taylor C (2008) MicroRNA signatures of tumor-derived exosomes as diagnostic biomarkers of ovarian cancer. Gynecol Oncol 110 : 13–21. doi: 10.1016/j.ygyno.2008.04.033 18589210
71. Bellingham SA, Coleman BM, Hill AF (2012) Small RNA deep sequencing reveals a distinct miRNA signature released in exosomes from prion-infected neuronal cells. Nucleic Acids Res.
72. Moldovan L, Batte K, Wang Y, Wisler J, Piper M (2013) Analyzing the circulating microRNAs in exosomes/extracellular vesicles from serum or plasma by qRT-PCR. Methods Mol Biol 1024 : 129–145. doi: 10.1007/978-1-62703-453-1_10 23719947
73. Goodwin EC, DiMaio D (2000) Repression of human papillomavirus oncogenes in HeLa cervical carcinoma cells causes the orderly reactivation of dormant tumor suppressor pathways. Proc Natl Acad Sci U S A 97 : 12513–12518. 11070078
74. Magaldi TG, Almstead LL, Bellone S, Prevatt EG, Santin AD, et al. (2012) Primary human cervical carcinoma cells require human papillomavirus E6 and E7 expression for ongoing proliferation. Virology 422 : 114–124. doi: 10.1016/j.virol.2011.10.012 22056390
75. Hwang ES, Riese DJ 2nd, Settleman J, Nilson LA, Honig J, et al. (1993) Inhibition of cervical carcinoma cell line proliferation by the introduction of a bovine papillomavirus regulatory gene. J Virol 67 : 3720–3729. 8389903
76. Dowhanick JJ, McBride AA, Howley PM (1995) Suppression of cellular proliferation by the papillomavirus E2 protein. J Virol 69 : 7791–7799. 7494290
77. Honegger A, Leitz J, Bulkescher J, Hoppe-Seyler K, Hoppe-Seyler F (2013) Silencing of human papillomavirus (HPV) E6/E7 oncogene expression affects both the contents and the amounts of extracellular microvesicles released from HPV-positive cancer cells. Int J Cancer.
78. Durst M, Gallahan D, Jay G, Rhim JS (1989) Glucocorticoid-enhanced neoplastic transformation of human keratinocytes by human papillomavirus type 16 and an activated ras oncogene. Virology 173 : 767–771. 2556855
79. Kuner R, Vogt M, Sultmann H, Buness A, Dymalla S, et al. (2007) Identification of cellular targets for the human papillomavirus E6 and E7 oncogenes by RNA interference and transcriptome analyses. J Mol Med (Berl) 85 : 1253–1262. 17589817
80. Buitrago-Perez A, Garaulet G, Vazquez-Carballo A, Paramio JM, Garcia-Escudero R (2009) Molecular Signature of HPV-Induced Carcinogenesis: pRb, p53 and Gene Expression Profiling. Curr Genomics 10 : 26–34. doi: 10.2174/138920209787581235 19721808
81. Suzuki HI, Yamagata K, Sugimoto K, Iwamoto T, Kato S, et al. (2009) Modulation of microRNA processing by p53. Nature 460 : 529–533. doi: 10.1038/nature08199 19626115
82. Ding H, Wu YL, Wang YX, Zhu FF (2014) Characterization of the microRNA expression profile of cervical squamous cell carcinoma metastases. Asian Pac J Cancer Prev 15 : 1675–1679. 24641388
83. Chou YT, Lin HH, Lien YC, Wang YH, Hong CF, et al. (2010) EGFR promotes lung tumorigenesis by activating miR-7 through a Ras/ERK/Myc pathway that targets the Ets2 transcriptional repressor ERF. Cancer Res 70 : 8822–8831. doi: 10.1158/0008-5472.CAN-10-0638 20978205
84. Foekens JA, Sieuwerts AM, Smid M, Look MP, de Weerd V, et al. (2008) Four miRNAs associated with aggressiveness of lymph node-negative, estrogen receptor-positive human breast cancer. Proc Natl Acad Sci U S A 105 : 13021–13026. doi: 10.1073/pnas.0803304105 18755890
85. Hatziapostolou M, Polytarchou C, Aggelidou E, Drakaki A, Poultsides GA, et al. (2011) An HNF4alpha-miRNA inflammatory feedback circuit regulates hepatocellular oncogenesis. Cell 147 : 1233–1247. doi: 10.1016/j.cell.2011.10.043 22153071
86. Yang L, Li Y, Cheng M, Huang D, Zheng J, et al. (2012) A functional polymorphism at microRNA-629-binding site in the 3′-untranslated region of NBS1 gene confers an increased risk of lung cancer in Southern and Eastern Chinese population. Carcinogenesis 33 : 338–347. doi: 10.1093/carcin/bgr272 22114071
87. Dhahbi JM, Atamna H, Boffelli D, Magis W, Spindler SR, et al. (2011) Deep sequencing reveals novel microRNAs and regulation of microRNA expression during cell senescence. PLoS One 6: e20509. doi: 10.1371/journal.pone.0020509 21637828
88. Lee DY, Deng Z, Wang CH, Yang BB (2007) MicroRNA-378 promotes cell survival, tumor growth, and angiogenesis by targeting SuFu and Fus-1 expression. Proc Natl Acad Sci U S A 104 : 20350–20355. 18077375
89. Jima DD, Zhang J, Jacobs C, Richards KL, Dunphy CH, et al. (2010) Deep sequencing of the small RNA transcriptome of normal and malignant human B cells identifies hundreds of novel microRNAs. Blood 116: e118–127. doi: 10.1182/blood-2010-05-285403 20733160
90. Zhang Y, Gan B, Liu D, Paik JH (2011) FoxO family members in cancer. Cancer Biol Ther 12 : 253–259. 21613825
91. Kloet DE, Burgering BM (2011) The PKB/FOXO switch in aging and cancer. Biochim Biophys Acta 1813 : 1926–1937. doi: 10.1016/j.bbamcr.2011.04.003 21539865
92. Chhabra R, Dubey R, Saini N (2010) Cooperative and individualistic functions of the microRNAs in the miR-23a~27a~24–2 cluster and its implication in human diseases. Mol Cancer 9 : 232. doi: 10.1186/1476-4598-9-232 20815877
93. Dellago H, Preschitz-Kammerhofer B, Terlecki-Zaniewicz L, Schreiner C, Fortschegger K, et al. (2013) High levels of oncomiR-21 contribute to the senescence-induced growth arrest in normal human cells and its knock-down increases the replicative lifespan. Aging Cell 12 : 446–458. doi: 10.1111/acel.12069 23496142
94. Lee S, Jung JW, Park SB, Roh K, Lee SY, et al. (2011) Histone deacetylase regulates high mobility group A2-targeting microRNAs in human cord blood-derived multipotent stem cell aging. Cell Mol Life Sci 68 : 325–336. doi: 10.1007/s00018-010-0457-9 20652617
95. Goto Y, Kojima S, Nishikawa R, Enokida H, Chiyomaru T, et al. (2014) The microRNA-23b/27b/24–1 cluster is a disease progression marker and tumor suppressor in prostate cancer. Oncotarget.
96. Majid S, Dar AA, Saini S, Arora S, Shahryari V, et al. (2012) miR-23b represses proto-oncogene Src kinase and functions as methylation-silenced tumor suppressor with diagnostic and prognostic significance in prostate cancer. Cancer Res 72 : 6435–6446. doi: 10.1158/0008-5472.CAN-12-2181 23074286
97. Bonifacio LN, Jarstfer MB (2010) MiRNA profile associated with replicative senescence, extended cell culture, and ectopic telomerase expression in human foreskin fibroblasts. PLoS One 5.
98. Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, et al. (2009) c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458 : 762–765. doi: 10.1038/nature07823 19219026
99. Jiang S, Zhang LF, Zhang HW, Hu S, Lu MH, et al. (2012) A novel miR-155/miR-143 cascade controls glycolysis by regulating hexokinase 2 in breast cancer cells. EMBO J 31 : 1985–1998. doi: 10.1038/emboj.2012.45 22354042
100. White EA, Walther J, Javanbakht H, Howley PM (2014) Genus Beta Human Papillomavirus E6 Proteins Vary in Their Effects on the Transactivation of p53 Target Genes. J Virol 88 : 8201–8212. doi: 10.1128/JVI.01197-14 24850740
101. Funk JO, Waga S, Harry JB, Espling E, Stillman B, et al. (1997) Inhibition of CDK activity and PCNA-dependent DNA replication by p21 is blocked by interaction with the HPV-16 E7 oncoprotein. Genes Dev 11 : 2090–2100. 9284048
102. Jones DL, Alani RM, Munger K (1997) The human papillomavirus E7 oncoprotein can uncouple cellular differentiation and proliferation in human keratinocytes by abrogating p21Cip1-mediated inhibition of cdk2. Genes Dev 11 : 2101–2111. 9284049
103. Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, et al. (2005) MicroRNA expression profiles classify human cancers. Nature 435 : 834–838. 15944708
104. Yanaihara N, Caplen N, Bowman E, Seike M, Kumamoto K, et al. (2006) Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell 9 : 189–198. 16530703
105. Villarroya-Beltri C, Gutierrez-Vazquez C, Sanchez-Cabo F, Perez-Hernandez D, Vazquez J, et al. (2013) Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat Commun 4 : 2980. doi: 10.1038/ncomms3980 24356509
106. Boyerinas B, Park SM, Hau A, Murmann AE, Peter ME (2010) The role of let-7 in cell differentiation and cancer. Endocr Relat Cancer 17: F19–36. doi: 10.1677/ERC-09-0184 19779035
107. Nuovo GJ, Garofalo M, Valeri N, Roulstone V, Volinia S, et al. (2012) Reovirus-associated reduction of microRNA-let-7d is related to the increased apoptotic death of cancer cells in clinical samples. Mod Pathol 25 : 1333–1344. doi: 10.1038/modpathol.2012.95 22699519
108. Volinia S, Galasso M, Sana ME, Wise TF, Palatini J, et al. (2012) Breast cancer signatures for invasiveness and prognosis defined by deep sequencing of microRNA. Proc Natl Acad Sci U S A 109 : 3024–3029. doi: 10.1073/pnas.1200010109 22315424
109. Hong L, Lai M, Chen M, Xie C, Liao R, et al. (2010) The miR-17–92 cluster of microRNAs confers tumorigenicity by inhibiting oncogene-induced senescence. Cancer Res 70 : 8547–8557. doi: 10.1158/0008-5472.CAN-10-1938 20851997
110. Li M, Guan X, Sun Y, Shu X, Li C (2014) miR-92a family and their target genes in tumorigenesis and metastasis. Exp Cell Res.
111. Di Leva G, Garofalo M, Croce CM (2014) MicroRNAs in cancer. Annu Rev Pathol 9 : 287–314. doi: 10.1146/annurev-pathol-012513-104715 24079833
112. Raposo G, Stoorvogel W (2013) Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol 200 : 373–383. doi: 10.1083/jcb.201211138 23420871
113. Bobrie A, Colombo M, Raposo G, Thery C (2011) Exosome secretion: molecular mechanisms and roles in immune responses. Traffic 12 : 1659–1668. doi: 10.1111/j.1600-0854.2011.01225.x 21645191
114. Turchinovich A, Weiz L, Langheinz A, Burwinkel B (2011) Characterization of extracellular circulating microRNA. Nucleic Acids Res 39 : 7223–7233. doi: 10.1093/nar/gkr254 21609964
115. Williams Z, Ben-Dov IZ, Elias R, Mihailovic A, Brown M, et al. (2013) Comprehensive profiling of circulating microRNA via small RNA sequencing of cDNA libraries reveals biomarker potential and limitations. Proc Natl Acad Sci U S A 110 : 4255–4260. doi: 10.1073/pnas.1214046110 23440203
116. Nouraee N, Calin GA (2013) MicroRNAs as Cancer Biomarkers. Microrna 2 : 102–117. 25070780
117. Hayes J, Peruzzi PP, Lawler S (2014) MicroRNAs in cancer: biomarkers, functions and therapy. Trends Mol Med 20 : 460–469. doi: 10.1016/j.molmed.2014.06.005 25027972
118. Gallo A, Tandon M, Alevizos I, Illei GG (2012) The majority of microRNAs detectable in serum and saliva is concentrated in exosomes. PLoS One 7: e30679. doi: 10.1371/journal.pone.0030679 22427800
119. Nana-Sinkam SP, Croce CM (2013) Clinical applications for microRNAs in cancer. Clin Pharmacol Ther 93 : 98–104. doi: 10.1038/clpt.2012.192 23212103
120. Parsons BD, Schindler A, Evans DH, Foley E (2009) A direct phenotypic comparison of siRNA pools and multiple individual duplexes in a functional assay. PLoS One 4: e8471. doi: 10.1371/journal.pone.0008471 20041186
121. Jackson AL, Linsley PS (2010) Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat Rev Drug Discov 9 : 57–67. doi: 10.1038/nrd3010 20043028
122. Chen C, Okayama H (1987) High-efficiency transformation of mammalian cells by plasmid DNA. Mol Cell Biol 7 : 2745–2752. 3670292
123. O’Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT (2005) c-Myc-regulated microRNAs modulate E2F1 expression. Nature 435 : 839–843. 15944709
124. Fox MH (1980) A model for the computer analysis of synchronous DNA distributions obtained by flow cytometry. Cytometry 1 : 71–77. 7023881
125. Dimri GP, Lee X, Basile G, Acosta M, Scott G, et al. (1995) A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A 92 : 9363–9367. 7568133
126. Butz K, Geisen C, Ullmann A, Zentgraf H, Hoppe-Seyler F (1998) Uncoupling of p21WAF1/CIP1/SDI1 mRNA and protein expression upon genotoxic stress. Oncogene 17 : 781–787. 9715280
127. Crnkovic-Mertens I, Semzow J, Hoppe-Seyler F, Butz K (2006) Isoform-specific silencing of the Livin gene by RNA interference defines Livin beta as key mediator of apoptosis inhibition in HeLa cells. J Mol Med (Berl) 84 : 232–240. 16437214
128. Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25 : 402–408. 11846609
129. Friedlander MR, Mackowiak SD, Li N, Chen W, Rajewsky N (2012) miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res 40 : 37–52. doi: 10.1093/nar/gkr688 21911355
130. Weischenfeldt J, Simon R, Feuerbach L, Schlangen K, Weichenhan D, et al. (2013) Integrative genomic analyses reveal an androgen-driven somatic alteration landscape in early-onset prostate cancer. Cancer Cell 23 : 159–170. doi: 10.1016/j.ccr.2013.01.002 23410972
131. Griffiths-Jones S (2004) The microRNA Registry. Nucleic Acids Res 32: D109–111. 14681370
132. Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ (2006) miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res 34: D140–144. 16381832
133. Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ (2008) miRBase: tools for microRNA genomics. Nucleic Acids Res 36: D154–158. 17991681
134. Kozomara A, Griffiths-Jones S (2011) miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res 39: D152–157. doi: 10.1093/nar/gkq1027 21037258
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 3
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Bacterial Immune Evasion through Manipulation of Host Inhibitory Immune Signaling
- BILBO1 Is a Scaffold Protein of the Flagellar Pocket Collar in the Pathogen
- Antimicrobial-Induced DNA Damage and Genomic Instability in Microbial Pathogens
- Attenuation of Tick-Borne Encephalitis Virus Using Large-Scale Random Codon Re-encoding
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy