
Immune Antibodies and Helminth Products Drive CXCR2-Dependent Macrophage-Myofibroblast Crosstalk to Promote Intestinal Repair
To complete their lifecycles, helminth parasites have to migrate through tissues such as the skin, lung, liver and intestine. This migration causes severe tissue damage, resulting in the need for rapid repair to restore the integrity and function of damaged tissues. Protective type 2 immune responses against helminths can repair acute lung damage, but they can also promote liver fibrosis. However, how protective immune mechanisms might contribute to wound healing during enteric nematode infection has remained unclear. Here we show that during a protective antibody response, where helminth larvae are trapped in the intestinal mucosa, macrophages and myofibroblasts secrete chemokines, which promote the repair of helminth-caused lesions. Chemokine secretion by macrophages was triggered by antibodies and helminth products, whilst myofibroblasts produced chemokines directly in response to innate recognition of helminth products. The same chemokines that instructed intestinal repair in mice were also secreted by human macrophages, when co-cultured with immune serum and helminths. Finally, human myofibroblasts closed in vitro scratch wounds more rapidly, when stimulated with the chemokine secretions of helminth-antibody activated human macrophages. Thus, our findings reveal a novel mechanism, by which a protective antibody response can promote the repair of intestinal injury during helminth infection.
Published in the journal:
. PLoS Pathog 11(3): e32767. doi:10.1371/journal.ppat.1004778
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004778
Summary
To complete their lifecycles, helminth parasites have to migrate through tissues such as the skin, lung, liver and intestine. This migration causes severe tissue damage, resulting in the need for rapid repair to restore the integrity and function of damaged tissues. Protective type 2 immune responses against helminths can repair acute lung damage, but they can also promote liver fibrosis. However, how protective immune mechanisms might contribute to wound healing during enteric nematode infection has remained unclear. Here we show that during a protective antibody response, where helminth larvae are trapped in the intestinal mucosa, macrophages and myofibroblasts secrete chemokines, which promote the repair of helminth-caused lesions. Chemokine secretion by macrophages was triggered by antibodies and helminth products, whilst myofibroblasts produced chemokines directly in response to innate recognition of helminth products. The same chemokines that instructed intestinal repair in mice were also secreted by human macrophages, when co-cultured with immune serum and helminths. Finally, human myofibroblasts closed in vitro scratch wounds more rapidly, when stimulated with the chemokine secretions of helminth-antibody activated human macrophages. Thus, our findings reveal a novel mechanism, by which a protective antibody response can promote the repair of intestinal injury during helminth infection.
Introduction
Infections with intestinal helminths affect more than 2 billion people globally [1,2] and drug-resistant helminths pose a considerable threat to agricultural livestock [3,4]. Helminth infections thus present a major global health concern particularly due to the propensity of these parasites to form chronic and repeated infections [5,6]. The potent immune-modulatory capacities of helminth parasites have been shaped by their long co-evolution with their hosts’ immune systems, which has resulted in a fine-tuned balance between inflammation on the one side, and parasite control on the other side [7]. In addition to their anti-inflammatory potential, helminths are increasingly recognized for their capacity to induce rapid tissue repair [8–10].
Considering the tissue migratory potential of the macroscopic larval stages, it is surprising that helminth infections are rarely associated with severe bleeding or bacterial sepsis [11]. This may partially be explained by parasite-mediated immune-modulation including the induction of IL-10 production in settings of bacterial translocation [11]. In addition, tissue dwelling nematodes typically initiate type 2 responses, which have been implicated in promoting tissue repair [12]. Helminths can trigger type 2 responses either directly [13], or through the release of alarmins (IL-25, IL-33, TSLP) or adenosine from epithelial cells following invasion [14,15].
Although the mechanisms by which helminth invasion initiates type 2 responses are increasingly well described, relatively little is known about the effector mechanisms that promote repair of host tissues following larval migration. Models of acute lung damage or liver fibrosis caused by the helminths Nippostrongylus brasiliensis [8,9] or Schistosoma mansoni, respectively have demonstrated important roles for Arginase-1 (Arg1) and insulin-like growth factor 1 (IGF-1) expressing MΦ, and IL-9R-dependent production of innate lymphoid cells (ILCs) in controlling inflammation and tissue damage [9,16,17]. However, intestinal helminth infections are often chronic and proceed without the development of excessive fibrosis, suggesting that distinct mechanisms might contribute to homeostatic wound healing in the intestine. Moreover, little is known about a potential contribution of the immune memory to these processes, yet those living in endemic regions are continuously exposed to helminths and typically suffer repeated infections.
Our previous work identified the helminth-antibody-driven activation of Arg1 expressing MΦ as an important mechanism to limit tissue disruption at early timepoints following repeated infection with the intestinal nematode Hpb [10]. Hpb is a natural parasite of mice with a strictly enteric life cycle that establishes chronic infections upon primary exposure [18], but which can be rapidly targeted by immune antibodies following repeated infections [10,19,20]. Important overlaps between mechanisms of protective immunity and wound healing during helminth infection are beginning to emerge [8], but the role of antibodies in wound healing has not been investigated.
Our previous findings suggested that the combination of immune antibodies and helminth antigens can induce the expression of several MΦ genes with potential roles in tissue repair [10]. This included the CXCR2 ligands Cxcl2 and Cxcl3, which have been implicated in cutaneous wound healing [21,22]. Here we demonstrate that intestinal wound containment was impaired in antibody - and antibody-receptor deficient mice and correlated with reduced expression of CXCR2 ligands. We further show that Fcrg-chain-signaling triggered the release of CXCL2 from MΦ and intestinal MF during helminth infection. Fcrg-dependent chemokine release by MΦ required stimulation by antibodies and helminths, whilst helminth products alone elicited the release of chemokines by MF through the dectin2 pathway, which is known to require Fcrg chain signaling. CXCR2 ligation was required for wound containment during murine helminth infection in vivo, and for efficient wound closure by human MF in vitro. Our findings reveal a dual role for helminths and immune antibodies in the trapping of invasive larvae and the promotion of tissue repair.
Results
Antibody - or Fcrg-deficient mice exhibit dysregulated tissue repair following challenge infection with the intestinal helminth Hpb
We have previously reported an important role for antibody-mediated MΦ activation in limiting parasite-induced tissue disruption and promoting larval trapping following secondary challenge infections with Hpb [10]. We also noted that the same MΦ exhibited a transcriptional profile similar to the one of wound healing MΦ [10,23]. To investigate whether antibodies also impacted on tissue repair we quantified the size of intestinal lesions at various time points post secondary challenge infection (p.i.). A scheme describing the timing of Hpb infections, larval maturation (L3–L5) and antihelminthic treatments is shown in Fig. 1A. At day 10 p.i. we could not observe significant differences in the size of intestinal lesions between wild-type (WT) mice and mice deficient in activation induced cytidine deaminase (Aid) which fail to produce affinity-matured, class-switched antibodies [24] (Fig. 1B, C). We further observed a tendency of Aid-/- mice to develop increased numbers of intestinal lesions (S1A Fig), which might be explained by the higher migratory capacity of larvae in the absence of antibodies. The cellular infiltration in the lesion underwent a dramatic change over time characterized by a shift from monocytic cells (day 4) to granulocytes (day 10) (S1B Fig). However, in agreement with our earlier findings, lesions from Aid-/- and WT mice exhibited similar cellular compositions with the exception of reduced basophil numbers in the absence of Aid [10,25] (S1C-H Fig).
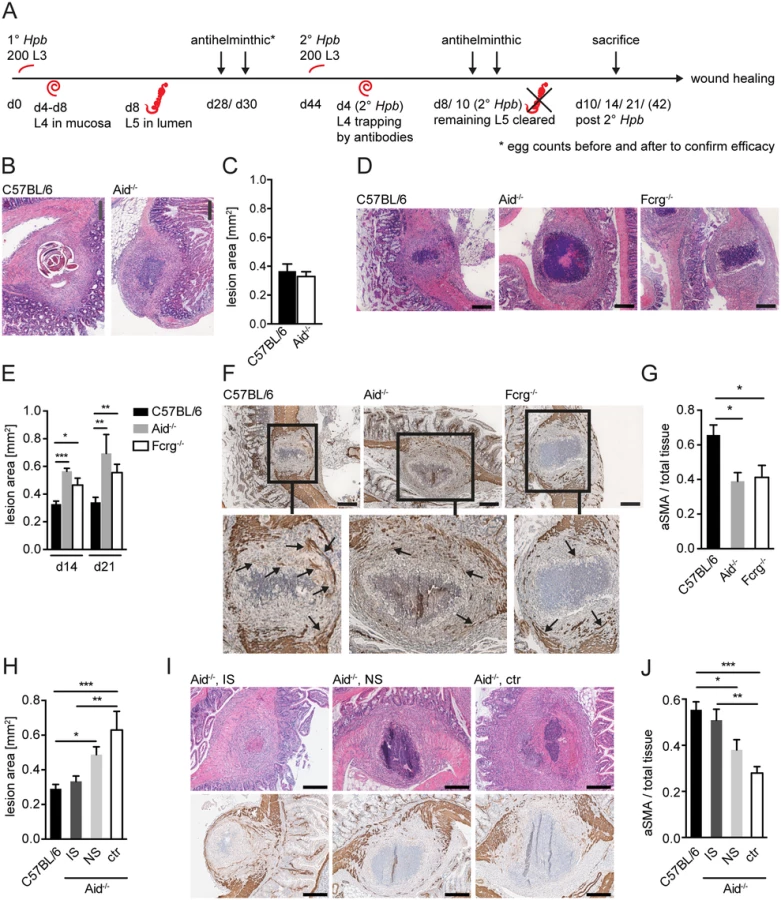
The finding that lesions were of similar size in Aid-/- and WT mice was rather surprising as larvae had already left the intestinal mucosa of Aid-/- mice at this time point, whilst they remained trapped within antibody sufficient WT lesions (Fig. 1B). We therefore additionally quantified lesion sizes in challenge-infected WT and Aid-/- mice, or mice deficient in activating antibody receptors (Fcrg-chain-/-), at later timepoints. Intact larvae were absent from the lesions of most WT mice by day 14 p.i., possibly as a result of degradation by the large numbers of infiltrating granulocytes (Fig. 1D, S1B Fig). Interestingly, lesion size had decreased slightly but not significantly (by around 10%) by day 14–21 p.i. as compared to day 10 p.i. in WT mice whilst Aid-/- and Fcrg-/- mice showed significantly larger lesions (Fig. 1D, E). Impaired lesion contraction in antibody and antibody receptor deficient mice correlated with a reduced accumulation of α smooth muscle actin (aSMA) expressing cells, which include MF, the major cell type involved in wound contraction [26] (Fig. 1F, G). To further investigate the impact of antibodies on the resolution of intestinal lesions we set up experiments to investigate lesion size at even later timepoints (day 42) post secondary challenge infection. However, most of the experiments had to be halted at day 21 post infection as the majority of challenge-infected Aid-/- and Fcrg-/- mice showed symptoms of morbidity (most likely severe peritonitis) around this time point. Indeed only 3 Aid-/- mice survived symptom free until day 42 post challenge infection. When comparing intestinal lesions in WT and these surviving Aid-/- mice, an increased tendency to develop necrosis or fibrosis as well as impaired lesion resolution became apparent in the antibody deficient mice (S2A, B Fig). Increased inducible nitric oxide synthase expression in peritoneal cells in Aid-/- as compared to WT mice already at day 14 post challenge infection hinted at increased peritonitis [27] in the absence of antibodies, possibly explaining the considerable morbidity in these mice late after challenge infection.
Taken together these findings suggest that antibodies act not only to trap helminth larvae but also to promote the accumulation of aSMA+ cells that are important for mucosal lesion contraction. In addition, the efficient (antibody-dependent) resolution of intestinal lesions might by associated with the control of peritoneal inflammation during challenge infection with intestinal helminths.
Hpb-induced type 2 inflammation is intact in challenge-infected antibody deficient or FcRg deficient mice
To investigate if type 2 immunity was intact in antibody and FcRg deficient mice, we analyzed peritoneal eosinophil accumulation and type 2 cytokine (IL-4 and IL-13) production in the duodenum as key parameters of the local memory TH2 response. As shown in S2D Fig, large numbers of eosinophils infiltrated the peritoneal cavity during challenge infection (day 14 and 21) in WT, Aid-/- and Fcrg-/- mice. Moreover, IL-4 and IL-13 were both detectable in duodenal culture supernatants of all challenge-infected mice with minor reductions in Aid-/- or Fcrg-/- mice, respectively (S2E, F Fig). In contrast, during primary infection the levels of both cytokines were close to the levels in naïve mice (limit of detection 4 pg/ ml) (S2E, F Fig). Together these data indicate that the TH2 response in challenge-infected antibody and FcRg deficient mice resembles that in challenge-infected rather than in primary infected WT mice.
We reasoned the increased susceptibility of challenge-infected antibody and FcRg deficient mice to peritonitis may be due to defective lesion repair resulting in increased bacterial translocation across the intestinal epithelium. To test this hypothesis, we quantified soluble CD14 levels in the peritoneum of primary infected WT and challenge-infected WT, Aid-/- or Fcrg-/- mice. As shown in S2G Fig, sCD14 levels did not increase above those found in naïve mice following challenge infection of WT, Aid-/- or Fcrg-/- mice, whilst high levels of peritoneal sCD14 could be detected following primary infection of WT mice (day 14). These data suggest that immune memory may function to prevent bacterial translocation, but through mechanisms distinct from the impact of immune antibodies on wound contraction or peritonitis.
Immune antibodies promote intestinal wound repair following challenge infection with Hpb
As our data suggested a central role for antibodies in lesion contraction during challenge Hpb infection, we sought to confirm a role for “immune memory” in this process. We first compared lesion sizes in primary and secondary Hpb infected WT mice. At day 14 and 21, primary lesions were less dense with fewer infiltrating cells (S2H Fig), but of similar size (S2I Fig) to those seen at the same time-points following challenge infection. However, it should be noted that significantly more lesions were observed in challenge-infected mice compared to primary-infected mice (S2J Fig), probably as a result of the greater type 2 immune response observed following challenge infection (S2D Fig). To determine whether lesion resolution differed between primary or challenge infected mice we next analyzed lesion size at a later time-point, day 42. By this time-point lesions had started to contract in challenge-infected, but not primary infected, mice (S2I Fig), indicating that immune memory can result in more rapid wound contraction.
To further support a role for ‘immune memory’ in lesion contraction we determined lesion size in antibody deficient (Aid-/-) mice receiving serum from naïve or challenge-infected WT mice. As shown in Fig. 1H, intestinal lesions in Aid-/- mice that received immune serum from challenge-infected WT mice exhibited lesions of a similar size to that seen in WT mice. In contrast, transfer of naïve serum into Aid-/- recipients did not result in a significant improvement of intestinal wound repair (Fig. 1H, I top panel). Moreover, the improved lesion contraction in immune serum treated Aid-/- mice correlated with an increased accumulation of aSMA+ cells within the lesions of these mice as compared to lesions in untreated Aid-/- mice (Fig. 1I bottom panel, J). These data indicate that immune memory acts to promote both lesion containment (day 14–21 post-infection) and lesion contraction (day 42 post-infection).
Antibody-trapped helminths promote the expression of CXCL2 and CXCL3 in vivo
Our previous work had identified the chemokines Cxcl2 and Cxcl3 as being amongst the most highly up-regulated MΦ genes following culture of these cells together with helminth larvae and IS [10]. As CXCL2 and CXCL3 have been reported to promote repair processes, including smooth muscle cell migration [21,28], we investigated the expression of these chemokines during helminth infection in vivo. We observed that at day 14 following primary infection or day 10 following challenge infection, CXCL2 & 3 were both expressed at low levels within intestinal lesions (S3A, B Fig). However by day 14 post challenge infection, CXCL2 was abundantly expressed, particularly at the lesion edge where it co-localized with FGFR1 expressing MF and F4/80 expressing MΦ (Fig. 2A). As FGFR1 is also expressed by fibroblasts, we confirmed the co-expression of aSMA in FGFR1 expressing cells in the periphery of intestinal lesions, which identified these cells as myofibroblasts (S3C Fig). We further quantified CXCL2 expression intensity in areas with a comparable MΦ and MF infiltrate (S3D, E Fig) to exclude a potential bias due to lower numbers of MF in lesions of antibody (receptor) deficient mice. As shown in Fig. 2B, Aid-/- and Fcrg-/- mice presented a significantly reduced CXCL2 staining intensity on a per cell basis, which was due to lower intensities in both MΦ and MF (Fig. 2C). Interestingly, CXCL2 expression in lesions of Fcrg-/- mice was significantly reduced as compared to that in Aid-/- lesions, suggesting that antibody independent but FcRg chain dependent signaling events might contribute to CXCL2 production during helminth infection in vivo. In keeping with the observed reduction in aSMA+ cells in lesions of Aid-/- and Fcrg-/- mice (Fig. 1F, G), MF in these lesions expressed lower levels of FGFR1 (S3F Fig), which is required for MF differentiation, including the upregulation of aSMA expression [29]. As our immunofluorescence (IF) analysis could not distinguish between chemokine production or surface binding, we additionally analyzed CXCL2 secretion into supernatants from duodenal organ cultures from challenge-infected mice. Small-intestinal tissue from infected WT mice secreted 3-fold more CXCL2 as compared to naïve mice, whilst Aid-/- or Fcrg-/- tissues secreted levels similar to that of naïve controls (Fig. 2D). Thus, increased levels of CXCL2 might be secreted locally in response to antibody-trapped larvae. To investigate whether antibodies and antibody receptors additionally impacted on the production of the close homologue CXCL3, we performed IF stainings on the corresponding serial sections of small intestinal lesions. As shown in S3G, H Fig, CXCL3 showed a similar pattern of expression as CXCL2 with a significant reduction in Fcrg-/- mice.

Finally, we analyzed CXCL2 in Aid-/- mice that had received WT immune sera during challenge infection. As shown in Fig. 2E,F, transfer of immune but not naïve sera could partially restore chemokine production in antibody deficient mice. These data suggest that the CXCR2 ligands, CXCL2 & 3, are upregulated in intestinal lesions by mechanisms involving Fcrg-chain-mediated activation of MΦ and/or MF by immune antibody-trapped helminth larvae.
Immune serum and helminth larvae trigger Fcrg-dependent CXCL2 release by MΦ in vitro
We had previously demonstrated that the IS-induced trapping of infective helminth larvae (”L3”) by bone marrow derived MΦ (BMM) was associated with a strong transcriptional activation of Cxcl2/3, which occurred independently of Fcrg-chain signaling [10]. To verify a direct effect of larvae and IS on CXCL2 secretion by MΦ, we quantified CXCL2 in supernatants from BMM after co-culture with helminth larvae in the presence or absence of IS. IS alone had no effect on CXCL2 release by WT BMM, whilst larvae induced CXCL2 secretion, which was enhanced by IS (Fig. 3A). Moreover, Fcrg-/- BMM showed a considerably reduced CXCL2 release compared to WT BMM, when co-cultured with larvae in the presence of IS. This suggested that Fcrg-mediated activation of MΦ was responsible for the enhanced release, but not transcription of Cxcl2. Indeed, unstimulated BMM showed considerable amounts of intracellular CXCL2 (Fig. 3B), but no secretion (Fig. 3A), whilst intracellular CXCL2 levels in helminth-IS activated BMM did not reflect the strong mRNA upregulation previously observed (Fig. 3B) [10]. Thus, surface IgG receptors likely play an important role in triggering CXCL2 release by MΦ during helminth infection.

Conditioned media from helminth-immune serum-activated MΦ drive in vitro scratch wound closure by small intestinal myofibroblasts
Crosstalk between MΦ and fibroblasts or MF is integral to the wound healing process [30]. Together, our in vitro and in vivo data suggested, that MΦ, which populate the wound early after infection [31], might be activated by antibody-trapped helminth larvae to enhance MF recruitment to intestinal lesions by secreting CXCL2/3. Hence, we tested a direct effect of MΦ-secreted CXCL2/3 by investigating scratch wound closure by MF in the presence of conditioned medium from helminth-IS activated MΦ. Addition of conditioned media from stimulated BMM significantly improved MF migration into the scratch wound area (Fig. 3C). Both CXCL2 and CXCL3 bind with high affinity to the chemokine receptor CXCR2 [32]. Thus, we studied the involvement of CXCR2 signaling in the positive effect of BMM-secreted factors on scratch wound closure by MF using the CXCR2 receptor antagonist SB265610. As shown in Fig. 3C, addition of the CXCR2 antagonist reduced the improvement of in vitro wound closure in response to conditioned media from helminth-IS activated MΦ (see also S1 Movie). These findings support a paracrine role for MΦ-secreted CXCR2 ligands in MF-mediated wound containment during helminth infection. However, once exposed to larval components, MF may also produce autocrine CXCL2/3.
Helminth larvae induce Fcrg/ dectin2-dependent CXCL2/3 production by intestinal MF in vitro
Previous studies have shown that pro-inflammatory stimuli can elicit CXCL2 expression by stromal cells [33]. To clarify if MF represent a potential additional source of CXCL2/3 during helminth infection, we isolated small intestinal MF and performed co-cultures with helminth larvae in the absence or presence of IS. As shown in Fig. 3D, larvae potently induced Cxcl2 gene expression in MF, which in contrast to MΦ, was not augmented by the addition of IS. Moreover, MF secreted considerable levels of CXCL2 protein when co-cultured with helminth larvae (Fig. 3E), indicating that helminth larvae alone can activate MF to produce CXCL2 independently of antibodies. This was consistent with the finding that only low levels of surface IgG bound to MF cultured in the presence immune serum (Fig. 3F).
We also investigated a potential role of Fcrg-chain signaling in promoting CXCL2 production by MF. Whilst larvae alone, or larvae in combination with immune serum, lead to similar increases in CXCL2 production by WT MF, neither condition could significantly upregulate CXCL2 production by Fcrg-/- MF (Fig. 3E). This suggested that another γ-chain dependent (antibody independent) event was required for the upregulation of MF CXCL2 production by helminth larvae. In addition to surface Fcr-mediated recognition of antibody-coated helminths, helminthic glycan structures can be recognized via pathogen recognition receptors such as dectin1 or 2, the mannose receptor (MR) or DC-SIGN [34,35]. Amongst these receptors, dectin2 is unique in its requirement of the Fcrg-chain for signaling [36]. Thus, we studied a potential involvement of dectin2 in the helminth-driven induction of CXCL2 production by intestinal MF. As shown in Fig. 3G, H dectin2-/- MF showed a significant reduction in helminth-induced Cxcl2 mRNA expression and protein release, suggesting that dectin2 recognition of larval components contributes to the upregulation of chemokine production by MF. Interestingly a more dramatic defect was observed for Fcrg-/- as compared to dectin2-/- MF, which may indicate that additional Fcrg-chain coupling C type lectins are involved [37]. Finally, the pattern of expression of CXCL3 in wildtype and Fcrg-/- and dectin2-/- MF closely paralleled the one of CXCL2 (Fig. 3I-K).
Inhibition of CXCR2 signaling leads to delayed MF accumulation and increased lesion size following helminth infection
Our earlier data suggested increased expression of CXCR2 ligands in helminth-induced lesions in vivo and a role for CXCR2 in MF migration in response to MΦ-secreted chemokines in vitro. To assess a potential role for CXCR2 in intestinal repair during helminth infection in vivo, we treated mice by the orally active CXCR2 antagonist SB265610 [22]. As CXCR2-driven neutrophil recruitment has been reported to contribute to protection against the helminth Strongyloides stercoralis [38], we first enumerated intestinal lesions and luminal parasites at day 14 post Hpb challenge infection. No significant effect of CXCR2 antagonist treatment was noted for either lesion or parasite numbers (Fig. 4A, B). We next compared the size of intestinal lesions between vehicle and SB265610 treated mice at day 14 post challenge infection. As shown in Fig. 4C, D, inhibition of CXCR2 signaling resulted in the development of larger and more irregular intestinal lesions. When analyzing intestinal aSMA expression, we observed a striking defect in the accumulation of aSMA+ cells within intestinal lesions of SB265610 treated mice (Fig. 4E, F), suggesting that CXCR2-ligands might also contribute to MF accumulation and wound containment in vivo. This phenotype observed in SB265610 treated mice closely paralleled the phenotype of Aid-/- and Fcrg-/- mice suggesting that defective CXCL2/3 expression was responsible for impaired wound healing in the absence of immune antibodies or Fcrg chain signaling.

CXCR2-signaling is not required for collagen deposition following helminth infection
In addition to their contractile function, MF promote wound healing by depositing extracellular matrix components, including collagen. Thus, we investigated whether the reduced accumulation of MF associated with CXCR2 blockade also impacted on collagen deposition within intestinal lesions. Mice treated with the CXCR2 antagonist showed normal collagen levels following helminth infection (S4A, B Fig) suggesting that this pathway is not involved in collagen deposition. However antibody (receptor) deficient mice displayed increased lesional collagen levels (S4C, D Fig). This may suggest that collagen production on a per cell basis is increased in Aid-/- and Fcrg-/- mice, which could be explained by the potential of trapped larvae to suppress collagen expression, whilst inducing collagen-remodeling matrix metalloproteinase and anti-fibrotic cyclooxygenase-2 (Cox2) enzymes in intestinal MF (S4 E-I Fig).
CXCR2 signaling is dispensable for granulocyte recruitment to intestinal lesions
Several studies have implicated CXCR2 in granulocyte recruitment during helminth infection [38,39]. We therefore determined the number of neutrophils and eosinophils in lesions of SB265610 treated mice and in Aid-/- or Fcrg-/- mice. We observed considerable levels of neutrophil myeloperoxidase (MPO), a widely used neutrophil marker, in the center of WT lesions at day 14 p.i. (Fig. 4G). However, when comparing MPO expression between WT, SB265610-treated or antibody (receptor) deficient mice, we observed an increase rather than a decrease in neutrophils (Fig. 4G upper panel, H), indicating that CXCR2 ligands are not required for neutrophil recruitment to intestinal lesions during challenge infection.
As neutrophils can contribute to tissue damage during helminth infection [9,40], we attempted to deplete neutrophils in the small intestine of Fcrg-/- mice to test a potential contribution of the increased neutrophil infiltrate to the impaired repair observed in these. Whilst neutrophils were successfully depleted in the peripheral blood of challenge infected Fcrg-/- mice (S5A Fig), high numbers of MPO expressing cells were still observed in intestinal lesions of mice that had been treated with a neutrophil-depleting anti-Ly6G antibody (S5B Fig). However, we could not observe a clear correlation between the quantity of MPO staining within intestinal lesions and lesion size (S5C Fig), suggesting that the increased neutrophil accumulation in antibody deficient mice does not play a major role in their reduced capacity to repair helminth-induced lesions.
Using 12/15-lipoxygenase (12/15LO) as a marker for eosinophils (see S1 Text, S5D Fig), we observed high numbers of eosinophils, many of which appeared de-granulated, within lesions of all mice (Fig. 4G lower panel, I). Similarly to neutrophils, mice treated with a CXCR2 antagonist showed increased eosinophil numbers. Taken together, these findings implicate CXCR2 as an important immune-modulating pathway, which functions to promote wound contraction following helminth infection but does not contribute to granulocyte recruitment.
Conditioned media from helminth-immune serum activated human MΦ promotes in vitro scratch wound closure by human MF in a CXCR2 dependent fashion
To determine whether helminth - and antibody-induced CXCL2/3 production plays a role in wound contraction in response to other helminth species we performed co-cultures of Ascaris suum (A. suum) larvae and IS from challenge-A. suum - infected pigs with human monocyte-derived MΦ (MDM). The pig parasite A. suum and the human parasite A. lumbricoides are closely related and cross-infections are common in endemic areas [41]. When incubated with IS, MDM adhered to A. suum larvae (Fig. 5A upper panel, B), which was associated with partial immobilization of infective larvae (Fig. 5A lower panel). This finding was in keeping with old literature, demonstrating the interaction of MΦ and A. suum after stimulation with IS [42].

Upon stimulation with phorbol esters and calcium ionophores or LPS, human and pig monocytes or MΦ can produce CXCL3, which is homologue to mouse CXCL2 and which binds to CXCR2 with high affinity [43,44]. Thus, we quantified CXCL3 production by human MDM after co-culture with A. suum larvae in the absence or presence of IS by ELISA. As shown in Fig. 5C MDM which had been co-cultured with A. suum larvae and IS secreted significantly larger amounts of CXCL3 as compared to culture with larvae alone. Next, we performed an in vitro wound-healing assay using primary human fibroblasts, which had been differentiated into MF by culture in the presence of TGFβ1 for 48h. Human MF (hMF) cultured in the presence of conditioned media from unstimulated MDM exhibited a reduction in the wound area by around 50% after 48h following scratching (Fig. 5D). In contrast, when conditioned media from A. suum-IS-activated MDM were added, hMF closed the scratch wound significantly faster, reaching complete wound closure after 48h (Fig. 5D, S2 Movie). We also tested if A. suum secreted products directly impacted on scratch wound closure by hMF. However, addition of A. suum products to hMF monolayers after scratch wounding had no significant effect on wound closure (S5E Fig), suggesting that mainly MΦ-secreted factors were responsible for accelerated scratch closure in the presence of conditioned media. Moreover, addition of the CXCR2 antagonist SB265610 abrogated the positive effect of A.suum-IS-activated MDM supernatants on in vitro wound closure by hMF (Fig. 5D, S2 Movie), suggesting that CXCR2 signaling is largely responsible for this effect.
Taken together these findings suggest that helminth-antibody-MΦ interactions can promote CXCR2 dependent wound closure during helminth infection across distinct host and parasite species.
Discussion
The capacity of helminths to induce rapid and efficient tissue repair has long been recognized, but effector mechanisms responsible for wound healing have only recently begun to be determined. Here we demonstrate that antibodies, which contribute to protective immunity against several helminth species [19,45,46], additionally contribute to wound contraction. Helminths damage host tissues not only by breaching physical barriers but also by driving strong inflammatory responses including the recruitment of tissue-destructive granulocytes [9,47]. Inflammatory cell recruitment is particularly large and rapid during memory type 2 responses, when tissue migrating larvae are also trapped and killed [31,48]. Our study reveals a novel mechanism by which the protective antibody response also participates in wound healing. Thus, when mounting a protective memory response, the mammalian host uses parallel mechanisms to ensure the limitation of infection and the repair of tissue damage at the same time.
Of note, delayed type 2 inflammation during primary helminth infection was associated with higher soluble CD14 levels in the peritoneal cavity, thus implicating the rapid and strong inflammatory response during challenge infection in the control of bacterial translocation (in the face of helminth triggered tissue damage). However, the striking morbidity of antibody and FcRg deficient mice at late time points after challenge infection suggested that this strong inflammatory response can (potentially) be harmful if not properly controlled or resolved. Thus, helminth products and antibody-FcRg signaling possibly contribute to wound healing on several levels by promoting lesion containment and contraction whilst also limiting excessive peritoneal inflammation.
Although we only detected low levels of CXCL2/3 following primary helminth infection, our study does not rule out a role for antibodies or CXCL2/3 in promoting tissue repair in the absence of immunological memory. The identification of dectin-2 as an innate signaling event mediating the induction of CXCL2/3 production by helminth products may suggest that these chemokines can indeed be produced even in the absence of antibodies. The finding that FcRg deficient mice showed a stronger reduction in chemokine levels during helminth infection further supports a role for dectin-2/ FcRg chain triggered CXCL2/3. However the similar wound healing phenotype in antibody and FcRg deficient mice suggests that timely, CXCR2-driven lesion contraction during challenge infection may require the concerted activation of MΦ and MF by both antibodies and helminth products. Previous studies have identified several innate immune mechanisms that contribute to the repair of helminth-induced injury following primary infection [9,17,49,50]. Here, we observed that lesions in primary infected mice were of similar size as compared to those in secondary challenge-infected mice at day 14 and 21 post infection, which might be explained by the delayed inflammatory response during primary infection. However, primary lesions failed to contract to the same extent as secondary lesions by day 42 post infection, thus supporting a role for adaptive TH2 immune memory in intestinal wound repair. Moreover, the transfer of immune serum from challenge Hpb infected, but not from naïve, WT mice into challenge-infected Aid-/- recipients could rescue the wound healing defect in these mice. This finding supports the hypothesis that immune antibodies play a major role in the containment and contraction of intestinal lesions following helminth infection. Whether the wound healing-promoting effect of immune serum mainly depends on specificity towards the worm or on different Fc functionality (e.g. through isotype switching or glycosylation) is an open question, which should be addressed in future studies.
Taken together, our work suggests that immune memory activates particularly potent and timely repair responses. It also represents the novel identification of an adaptive immune mechanism, antibody production, that can promote wound healing following helminth infection [8]. Two studies, including our own work, showed that antibody-activated Arg1 expressing MΦ limit the motility of tissue-dwelling helminth larvae, thereby preventing tissue damage during early challenge infection with Hpb or N. brasiliensis, respectively [10,48,51]. Our current study now demonstrates that antibody-trapped helminths modulate gene expression in MΦ and intestinal MF to improve wound contraction also at later stages of helminth infection.
Reduced peripheral MF in Aid-/- and Fcrg-/- mice were associated with increased lesion size and enhanced granulocyte infiltration at late timepoints post-infection, which could be reproduced by CXCR2 blockade. In contrast to the importance of CXCR2 for neutrophil recruitment to other tissues [21,39,52], CXCL2/3 thus appear to play redundant roles in neutrophil recruitment during enteric nematode infection. Instead, other chemoattractants such as complement component C5a, leukotriene B4 or platelet activating factor (PAF) may recruit neutrophils to intestinal lesions [53]. The increased granulocyte infiltrate in CXCR2-inhibited or Aid-/- and Fcrg-/- mice might further suggest that MF, which are recruited to helminth-induced intestinal lesions, can exert an anti-inflammatory function by limiting granulocyte accumulation. Interestingly, intestinal MF cultured with helminth larvae upregulated the expression of cyclooxygenase-2, which serves anti-fibrotic and anti-inflammatory functions in the gut, including limiting neutrophilic inflammation [54,55]. Even if we did not observe a correlation between the extent of neutrophil infiltration and lesion size, our study does not rule out the possibility that the increased granulocyte accumulation in the lesions of antibody and Fcrg deficient or CXCR2-inhibited mice contributes to the impaired repair response in these mice. Future studies could thus address how helminths may modulate MF and granulocyte function to prevent excessive inflammation and instruct homeostatic wound healing.
Our study describes a previously unrecognized function of helminth-induced CXCR2-ligands in intestinal wound containment, thus implicating the CXCL2/3-CXCR2 chemokine axis in repair of mucosal tissues. Whilst little was known about a potential involvement of CXCL2/3 in intestinal repair, the role of CXCR2 signaling in cutaneous wound healing after chemical damage or incision wounding is well-described [21,22]. In such settings of sterile damage, CXCR2 contributed to several of the central processes of skin repair, including keratinocyte migration, re-epithelialization and neovascularization. Moreover, CXCR2 was involved in the IL-33-induced repair of Staphylococcus aureus-infected wounds [56]. Our findings build on this work by additionally demonstrating that CXCR2 ligands act on MF to promote wound healing in the small intestine. Thus, it is likely that CXCR2 signaling is a conserved repair mechanism involved in the restoration of barrier integrity at different body surfaces in the absence or presence of infectious agents. Of note, CXCR2 can bind other ligands (CXCL1, CXCL5, CXCL7 and CXCL8/ IL-8) in addition to CXCL2 and CXCL3. Although these chemokines were not induced in macrophages in response to helminth larvae and immune serum [10], our data does not rule out a contribution of CXCR2 ligands other than CXCL2/3 to lesion contraction during helminth infection in vivo. It should also be noted that we cannot entirely exclude an impact of antibody-FcRγ signaling on the local secretion of type 2 cytokines, e.g. by basophils [57], which might additionally contribute to intestinal wound repair during secondary helminth infection.
Our findings have important clinical implications as CXCR2 has been suggested as a drug target for inflammatory diseases [58]. Together with other studies demonstrating crucial roles for the CXCL2/3-CXCR2 axis in repair and homeostasis [21,22,59], our findings argue for a careful evaluation of the therapeutic inhibition of this pathway. Future studies could address the potential of CXCR2 agonists and/or antibody-helminth-preparations to improve repair in settings of impaired wound healing responses (e.g. diabetic ulcers) or after surgery.
Methods
Ethics statement
All mouse experiments were approved by the office Affaires vétérinaires (1066 Epalinges, Canton Vaud, Switzerland) with the authorization number 2238 according to the guidelines set by the service de la consummation et des affaires vétérinaires federal (Canton Vaud, Switzerland). Mice were euthanized using carbon dioxide. All procedures involving pigs were performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health after review and approval by the Beltsville Area Animal Care and Use Committee under protocol number 10–012.
Mice
Mice (strains see S1 Text) were bred and maintained under specific pathogen free conditions at the École Polytechnique Fédérale (EPFL) de Lausanne, Switzerland.
Infection and parasitology
Mice were infected with 200 H. polygyrus bakeri larvae and two courses of antihelminthic cobantril were administered at days 28 and 30 p.i. Mice were re-infected with 200 larvae at day 44 and two courses of cobantril were administered at days 52 and 54 p.i. in some experiments. The CXCR2 antagonist SB265610 [22,60] (Tocris Bioscience, Bristol, UK) was administered (3 mg/ kg) once daily from day 0 p.i. by oral gavage. Mice were sacrificed at day 4–42 post challenge infection. Ascaris suum larvae were obtained from infective eggs according to published procedures [61]. See also S1 Text.
Pig sera
A. suum-challenge infected pigs (see also S1 Text) were bled at day 14 p.i. for serum collection.
In vitro cultures of MΦ and MF
Murine bone marrow derived MΦ (BMM) or human MDM were prepared as described previously [10,62]. Mouse small intestinal MF were obtained by culturing lamina propria cells isolated from the small intestine of naïve C57BL/6 mice according to previously published methods [63,64]. Human primary gingival fibroblasts were differentiated into MF by culture with TGFβ1 for 48h. See also S1 Text.
Histology
Serial paraffin sections of small intestines (“Swiss rolls”) were stained with hematoxylin and eosin or Sirius red. Lesions were identified by light microscopy and serial sections were used for immune staining and imaging (see also S1 Text).
Flow cytometry
Cells isolated from lesions were stained with fluorescently labeled monoclonal antibodies (see S1 Text) and acquired on a BD LSRII flow cytometer (BD, Franklin Lakes, NJ).
qPCR
RNA extraction and qPCR analysis was performed as described elsewhere [10] (for qPCR primer sequences see Table S1 in S1 Text).
ELISA
Concentrations of mouse CXCL2, IL-4 or IL-13 in cell or intestinal (upper duodenum) culture supernatants were quantified by using an anti-mouse CXCL2 ELISA kit (Sigma Aldrich, Buchs, Switzerland) or eBbioscience Ready-SET-Go! IL-4 or IL-13 kits (eBioscience, San Diego, CA). Human CXCL3 was quantified using an anti-human CXCL3 ELISA Kit (LuBioScience, Luzern, Switzerland).
In vitro wound healing assay
A scratch was introduced into MF monolayers using a 200-μl pipette tip according to published methods [65] and images were recorded using an Olympus Cell’R system with a UPLAN FL 10x phase objective. The scratch area was quantified using ImageJ and normalized to the area in the same position at T = 0.
Statistics
Mann-Whitney test or one-way ANOVA followed by Holm-Sidak’s multiple comparisons test was used to compare the means of two groups or multiple groups, respectively.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Bethony J, Brooker S, Albonico M, Geiger SM, Loukas A, Diemert D, et al. Soil-transmitted helminth infections: ascariasis, trichuriasis, and hookworm. Lancet. 2006;367 : 1521–1532. 16679166
2. Yang G-J, Liu L, Zhu H-R, Griffiths SM, Tanner M, Bergquist R, et al. China’s sustained drive to eliminate neglected tropical diseases. Lancet Infect Dis. 2014;
3. Geerts S, Gryseels B. Drug resistance in human helminths: current situation and lessons from livestock. Clin Microbiol Rev. 2000;13 : 207–222. 10755998
4. Rinaldi M, Geldhof P. Immunologically based control strategies for ostertagiosis in cattle: where do we stand? Parasite Immunol. 2012;34 : 254–264. doi: 10.1111/j.1365-3024.2011.01313.x 21722144
5. Salgame P, Yap GS, Gause WC. Effect of helminth-induced immunity on infections with microbial pathogens. Nat Immunol. 2013;14 : 1118–1126. doi: 10.1038/ni.2736 24145791
6. Urban JF Jr, Steenhard NR, Solano-Aguilar GI, Dawson HD, Iweala OI, Nagler CR, et al. Infection with parasitic nematodes confounds vaccination efficacy. Vet Parasitol. 2007;148 : 14–20. 17587500
7. Allen JE, Maizels RM. Diversity and dialogue in immunity to helminths. Nat Rev Immunol. 2011;11 : 375–388. doi: 10.1038/nri2992 21610741
8. Allen JE, Sutherland TE. Host protective roles of type 2 immunity: Parasite killing and tissue repair, flip sides of the same coin. Semin Immunol. 2014;
9. Chen F, Liu Z, Wu W, Rozo C, Bowdridge S, Millman A, et al. An essential role for T(H)2-type responses in limiting acute tissue damage during experimental helminth infection. Nat Med. 2012;
10. Esser-von Bieren J, Mosconi I, Guiet R, Piersgilli A, Volpe B, Chen F, et al. Antibodies Trap Tissue Migrating Helminth Larvae and Prevent Tissue Damage by Driving IL-4Rα-Independent Alternative Differentiation of Macrophages. PLoS Pathog. 2013;9: e1003771. doi: 10.1371/journal.ppat.1003771 24244174
11. Hübner MP, Layland LE, Hoerauf A. Helminths and their implication in sepsis—a new branch of their immunomodulatory behaviour? Pathog Dis. 2013;69 : 127–141. doi: 10.1111/2049-632X.12080 23929557
12. Gause WC, Wynn TA, Allen JE. Type 2 immunity and wound healing: evolutionary refinement of adaptive immunity by helminths. Nat Rev Immunol. 2013;
13. Massacand JC, Stettler RC, Meier R, Humphreys NE, Grencis RK, Marsland BJ, et al. Helminth products bypass the need for TSLP in Th2 immune responses by directly modulating dendritic cell function. Proc Natl Acad Sci U S A. 2009;106 : 13968–13973. doi: 10.1073/pnas.0906367106 19666528
14. Patel N, Wu W, Mishra PK, Chen F, Millman A, Csóka B, et al. A2B adenosine receptor induces protective antihelminth type 2 immune responses. Cell Host Microbe. 2014;15 : 339–350. doi: 10.1016/j.chom.2014.02.001 24629340
15. Saenz SA, Taylor BC, Artis D. Welcome to the neighborhood: epithelial cell-derived cytokines license innate and adaptive immune responses at mucosal sites. Immunol Rev. 2008;226 : 172–190. doi: 10.1111/j.1600-065X.2008.00713.x 19161424
16. Pesce JT, Ramalingam TR, Mentink-Kane MM, Wilson MS, El Kasmi KC, Smith AM, et al. Arginase-1-expressing macrophages suppress Th2 cytokine-driven inflammation and fibrosis. PLoS Pathog. 2009;5: e1000371. doi: 10.1371/journal.ppat.1000371 19360123
17. Turner J-E, Morrison PJ, Wilhelm C, Wilson M, Ahlfors H, Renauld J-C, et al. IL-9-mediated survival of type 2 innate lymphoid cells promotes damage control in helminth-induced lung inflammation. J Exp Med. 2013;210 : 2951–2965. doi: 10.1084/jem.20130071 24249111
18. Maizels RM, Hewitson JP, Gause WC. Heligmosomoides polygyrus: one species still. Trends Parasitol. 2011;27 : 100–101. doi: 10.1016/j.pt.2010.11.004 21159557
19. McCoy KD, Stoel M, Stettler R, Merky P, Fink K, Senn BM, et al. Polyclonal and specific antibodies mediate protective immunity against enteric helminth infection. Cell Host Microbe. 2008;4 : 362–373. doi: 10.1016/j.chom.2008.08.014 18854240
20. Esser-von Bieren J, Volpe B, Kulagin M, Sutherland DB, Guiet R, Seitz A, et al. Antibody-Mediated Trapping of Helminth Larvae Requires CD11b and Fcγ Receptor I. J Immunol. 2015;194 : 1154–1163. doi: 10.4049/jimmunol.1401645 25548226
21. Devalaraja RM, Nanney LB, Du J, Qian Q, Yu Y, Devalaraja MN, et al. Delayed wound healing in CXCR2 knockout mice. J Invest Dermatol. 2000;115 : 234–244. 10951241
22. Milatovic S, Nanney LB, Yu Y, White JR, Richmond A. Impaired healing of nitrogen mustard wounds in CXCR2 null mice. Wound Repair Regen Off Publ Wound Heal Soc Eur Tissue Repair Soc. 2003;11 : 213–219.
23. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11 : 723–737. doi: 10.1038/nri3073 21997792
24. Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102 : 553–563. 11007474
25. Herbst T, Esser J, Prati M, Kulagin M, Stettler R, Zaiss MM, et al. Antibodies and IL-3 support helminth-induced basophil expansion. Proc Natl Acad Sci U S A. 2012;109 : 14954–14959. doi: 10.1073/pnas.1117584109 22930820
26. Hinz B. The myofibroblast: paradigm for a mechanically active cell. J Biomech. 2010;43 : 146–155. doi: 10.1016/j.jbiomech.2009.09.020 19800625
27. Su F, Huang H, Akieda K, Occhipinti G, Donadello K, Piagnerelli M, et al. Effects of a selective iNOS inhibitor versus norepinephrine in the treatment of septic shock. Shock Augusta Ga. 2010;34 : 243–249. doi: 10.1097/SHK.0b013e3181d75967 20160666
28. Al-Alwan LA, Chang Y, Mogas A, Halayko AJ, Baglole CJ, Martin JG, et al. Differential Roles of CXCL2 and CXCL3 and Their Receptors in Regulating Normal and Asthmatic Airway Smooth Muscle Cell Migration. J Immunol Baltim Md 1950. 2013;191 : 2731–2741. doi: 10.4049/jimmunol.1203421 23904157
29. Perl A-KT, Gale E. FGF signaling is required for myofibroblast differentiation during alveolar regeneration. Am J Physiol Lung Cell Mol Physiol. 2009;297: L299–308. doi: 10.1152/ajplung.00008.2009 19502291
30. Barron L, Wynn TA. Fibrosis is regulated by Th2 and Th17 responses and by dynamic interactions between fibroblasts and macrophages. Am J Physiol Gastrointest Liver Physiol. 2011;300: G723–728. doi: 10.1152/ajpgi.00414.2010 21292997
31. Anthony RM, Urban JF Jr, Alem F, Hamed HA, Rozo CT, Boucher J-L, et al. Memory T(H)2 cells induce alternatively activated macrophages to mediate protection against nematode parasites. Nat Med. 2006;12 : 955–960. 16892038
32. Shibata F, Konishi K, Nakagawa H. Chemokine receptor CXCR2 activates distinct pathways for chemotaxis and calcium mobilization. Biol Pharm Bull. 2002;25 : 1217–1219. 12230122
33. Driscoll KE, Hassenbein DG, Carter J, Poynter J, Asquith TN, Grant RA, et al. Macrophage inflammatory proteins 1 and 2: expression by rat alveolar macrophages, fibroblasts, and epithelial cells and in rat lung after mineral dust exposure. Am J Respir Cell Mol Biol. 1993;8 : 311–318. 8383510
34. Van Liempt E, van Vliet SJ, Engering A, García Vallejo JJ, Bank CMC, Sanchez-Hernandez M, et al. Schistosoma mansoni soluble egg antigens are internalized by human dendritic cells through multiple C-type lectins and suppress TLR-induced dendritic cell activation. Mol Immunol. 2007;44 : 2605–2615. 17241663
35. Ritter M, Gross O, Kays S, Ruland J, Nimmerjahn F, Saijo S, et al. Schistosoma mansoni triggers Dectin-2, which activates the Nlrp3 inflammasome and alters adaptive immune responses. Proc Natl Acad Sci U S A. 2010;107 : 20459–20464. doi: 10.1073/pnas.1010337107 21059925
36. Sato K, Yang X, Yudate T, Chung J-S, Wu J, Luby-Phelps K, et al. Dectin-2 is a pattern recognition receptor for fungi that couples with the Fc receptor gamma chain to induce innate immune responses. J Biol Chem. 2006;281 : 38854–38866. 17050534
37. Graham LM, Brown GD. The Dectin-2 family of C-type lectins in immunity and homeostasis. Cytokine. 2009;48 : 148–155. doi: 10.1016/j.cyto.2009.07.010 19665392
38. Galioto AM, Hess JA, Nolan TJ, Schad GA, Lee JJ, Abraham D. Role of eosinophils and neutrophils in innate and adaptive protective immunity to larval strongyloides stercoralis in mice. Infect Immun. 2006;74 : 5730–5738. 16988250
39. Hall LR, Diaconu E, Patel R, Pearlman E. CXC chemokine receptor 2 but not C-C chemokine receptor 1 expression is essential for neutrophil recruitment to the cornea in helminth-mediated keratitis (river blindness). J Immunol Baltim Md 1950. 2001;166 : 4035–4041. 11238651
40. Sutherland TE, Logan N, Rückerl D, Humbles AA, Allan SM, Papayannopoulos V, et al. Chitinase-like proteins promote IL-17-mediated neutrophilia in a tradeoff between nematode killing and host damage. Nat Immunol. 2014;15 : 1116–1125. doi: 10.1038/ni.3023 25326751
41. Zhou C, Li M, Yuan K, Deng S, Peng W. Pig Ascaris: An important source of human ascariasis in China. Infect Genet Evol J Mol Epidemiol Evol Genet Infect Dis. 2012;12 : 1172–1177
42. Laubach HE. Cytoadherence effects of serum on Ascaris suum infective larvae. Zentralblatt Für Bakteriol Mikrobiol Hyg 1 Abt Orig Med Mikrobiol Infekt Parasitol Int J Microbiol Hyg Med Microbiol Infect Parasitol. 1983;255 : 406–412.
43. Gao Y, Flori L, Lecardonnel J, Esquerré D, Hu Z-L, Teillaud A, et al. Transcriptome analysis of porcine PBMCs after in vitro stimulation by LPS or PMA/ionomycin using an expression array targeting the pig immune response. BMC Genomics. 2010;11 : 292. doi: 10.1186/1471-2164-11-292 20459780
44. Tekamp-Olson P, Gallegos C, Bauer D, McClain J, Sherry B, Fabre M, et al. Cloning and characterization of cDNAs for murine macrophage inflammatory protein 2 and its human homologues. J Exp Med. 1990;172 : 911–919. 2201751
45. Blackwell NM, Else KJ. B cells and antibodies are required for resistance to the parasitic gastrointestinal nematode Trichuris muris. Infect Immun. 2001;69 : 3860–3868. 11349052
46. Gurish MF, Bryce PJ, Tao H, Kisselgof AB, Thornton EM, Miller HR, et al. IgE enhances parasite clearance and regulates mast cell responses in mice infected with Trichinella spiralis. J Immunol Baltim Md 1950. 2004;172 : 1139–1145. 14707089
47. Pearlman E, Hall LR, Higgins AW, Bardenstein DS, Diaconu E, Hazlett FE, et al. The role of eosinophils and neutrophils in helminth-induced keratitis. Invest Ophthalmol Vis Sci. 1998;39 : 1176–1182. 9620077
48. Chen F, Wu W, Millman A, Craft JF, Chen E, Patel N, et al. Neutrophils prime a long-lived effector macrophage phenotype that mediates accelerated helminth expulsion. Nat Immunol. 2014;
49. Hung L-Y, Lewkowich IP, Dawson LA, Downey J, Yang Y, Smith DE, et al. IL-33 drives biphasic IL-13 production for noncanonical Type 2 immunity against hookworms. Proc Natl Acad Sci U S A. 2013;110 : 282–287. doi: 10.1073/pnas.1206587110 23248269
50. Owyang AM, Zaph C, Wilson EH, Guild KJ, McClanahan T, Miller HRP, et al. Interleukin 25 regulates type 2 cytokine-dependent immunity and limits chronic inflammation in the gastrointestinal tract. J Exp Med. 2006;203 : 843–849. 16606667
51. Obata-Ninomiya K, Ishiwata K, Tsutsui H, Nei Y, Yoshikawa S, Kawano Y, et al. The skin is an important bulwark of acquired immunity against intestinal helminths. J Exp Med. 2013;210 : 2583–2595. doi: 10.1084/jem.20130761 24166714
52. Mizutani N, Nabe T, Yoshino S. IL-17A Promotes the Exacerbation of IL-33-Induced Airway Hyperresponsiveness by Enhancing Neutrophilic Inflammation via CXCR2 Signaling in Mice. J Immunol Baltim Md 1950. 2014;192 : 1372–1384. doi: 10.4049/jimmunol.1301538 24446518
53. Harvath L. Neutrophil chemotactic factors. EXS. 1991;59 : 35–52. 1655519
54. Davids JS, Carothers AM, Damas BC, Bertagnolli MM. Chronic cyclooxygenase-2 inhibition promotes myofibroblast-associated intestinal fibrosis. Cancer Prev Res Phila Pa. 2010;3 : 348–358. doi: 10.1158/1940-6207.CAPR-09-0146 20179298
55. Kabashima K, Saji T, Murata T, Nagamachi M, Matsuoka T, Segi E, et al. The prostaglandin receptor EP4 suppresses colitis, mucosal damage and CD4 cell activation in the gut. J Clin Invest. 2002;109 : 883–893. 11927615
56. Yin H, Li X, Hu S, Liu T, Yuan B, Ni Q, et al. IL-33 promotes Staphylococcus aureus-infected wound healing in mice. Int Immunopharmacol. 2013;17 : 432–438. doi: 10.1016/j.intimp.2013.07.008 23892028
57. Schwartz C, Turqueti-Neves A, Hartmann S, Yu P, Nimmerjahn F, Voehringer D. Basophil-mediated protection against gastrointestinal helminths requires IgE-induced cytokine secretion. Proc Natl Acad Sci U S A. 2014;
58. Reutershan J. CXCR2—the receptor to hit? Drug News Perspect. 2006;19 : 615–623. doi: 10.1358/dnp.2006.19.10.1068009 17299604
59. Zenobia C, Luo XL, Hashim A, Abe T, Jin L, Chang Y, et al. Commensal bacteria-dependent select expression of CXCL2 contributes to periodontal tissue homeostasis. Cell Microbiol. 2013;15 : 1419–1426. doi: 10.1111/cmi.12127 23433011
60. Auten RL, Richardson RM, White JR, Mason SN, Vozzelli MA, Whorton MH. Nonpeptide CXCR2 antagonist prevents neutrophil accumulation in hyperoxia-exposed newborn rats. J Pharmacol Exp Ther. 2001;299 : 90–95. 11561067
61. Urban JF Jr, Douvres FW, Tromba FG. A rapid method for hatching Ascaris suum eggs in vitro. Proc Helminthol Soc Wash. 1981;48 : 241–243.
62. Esser J, Gehrmann U, D’Alexandri FL, Hidalgo-Estévez AM, Wheelock CE, Scheynius A, et al. Exosomes from human macrophages and dendritic cells contain enzymes for leukotriene biosynthesis and promote granulocyte migration. J Allergy Clin Immunol. 2010;126 : 1032–1040, 1040.e1–4. doi: 10.1016/j.jaci.2010.06.039 20728205
63. Kamata T, Nogaki F, Fagarasan S, Sakiyama T, Kobayashi I, Miyawaki S, et al. Increased frequency of surface IgA-positive plasma cells in the intestinal lamina propria and decreased IgA excretion in hyper IgA (HIGA) mice, a murine model of IgA nephropathy with hyperserum IgA. J Immunol Baltim Md 1950. 2000;165 : 1387–1394. 10903742
64. Van Tol EA, Holt L, Li FL, Kong FM, Rippe R, Yamauchi M, et al. Bacterial cell wall polymers promote intestinal fibrosis by direct stimulation of myofibroblasts. Am J Physiol. 1999;277: G245–255. 10409173
65. Iwanaga K, Okada M, Murata T, Hori M, Ozaki H. Prostaglandin E2 promotes wound-induced migration of intestinal subepithelial myofibroblasts via EP2, EP3, and EP4 prostanoid receptor activation. J Pharmacol Exp Ther. 2012;340 : 604–611. doi: 10.1124/jpet.111.189845 22138372
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 3
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Bacterial Immune Evasion through Manipulation of Host Inhibitory Immune Signaling
- BILBO1 Is a Scaffold Protein of the Flagellar Pocket Collar in the Pathogen
- Antimicrobial-Induced DNA Damage and Genomic Instability in Microbial Pathogens
- Attenuation of Tick-Borne Encephalitis Virus Using Large-Scale Random Codon Re-encoding
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy