
The Inflammasome Pyrin Contributes to Pertussis Toxin-Induced IL-1β Synthesis, Neutrophil Intravascular Crawling and Autoimmune Encephalomyelitis
Microbial agents can aggravate inflammatory diseases, such as multiple sclerosis (MS) and its animal model, experimental autoimmune encephalomyelitis (EAE). An example is pertussis toxin (PTX), which is used to promote EAE by an obscure mechanism. We have reported that PTX triggers an IL-6-mediated signaling cascade that increases the number of leukocytes that patrol the vasculature by crawling on its luminal surface. We show here that PTX, through its ADP-ribosyltransferase activity, induces: 1) TLR4 signaling in myeloid cells, leading to pro-IL-1β synthesis; and 2) a pyrin-dependent inflammasome that cleaves pro-IL-1β into its active form. Then, IL-1β stimulates nearby stromal cells to secrete IL-6. Without pyrin, PTX does not induce neutrophil adhesion to cerebral capillaries and is less effective at inducing EAE in mice with encephalitogenic T lymphocytes. This study identifies the first microbial molecule that activates pyrin, a mechanism by which infections may influence MS and a potential therapeutic target for immune disorders.
Published in the journal:
. PLoS Pathog 10(5): e32767. doi:10.1371/journal.ppat.1004150
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004150
Summary
Microbial agents can aggravate inflammatory diseases, such as multiple sclerosis (MS) and its animal model, experimental autoimmune encephalomyelitis (EAE). An example is pertussis toxin (PTX), which is used to promote EAE by an obscure mechanism. We have reported that PTX triggers an IL-6-mediated signaling cascade that increases the number of leukocytes that patrol the vasculature by crawling on its luminal surface. We show here that PTX, through its ADP-ribosyltransferase activity, induces: 1) TLR4 signaling in myeloid cells, leading to pro-IL-1β synthesis; and 2) a pyrin-dependent inflammasome that cleaves pro-IL-1β into its active form. Then, IL-1β stimulates nearby stromal cells to secrete IL-6. Without pyrin, PTX does not induce neutrophil adhesion to cerebral capillaries and is less effective at inducing EAE in mice with encephalitogenic T lymphocytes. This study identifies the first microbial molecule that activates pyrin, a mechanism by which infections may influence MS and a potential therapeutic target for immune disorders.
Introduction
MS is a T lymphocyte-mediated autoimmune demyelinating disease of the central nervous system (CNS), whose development is dictated by complex interactions between genetic and environmental factors [1], [2]. Among the latter are microbes and their toxins. Although the search for a causative microbial agent has proved fruitless so far, many epidemiological studies have shown associations between infections and MS. For example, the risk of developing MS is at least doubled in individuals with a clinical history of infectious mononucleosis [3]–[11], while the risk of MS exacerbation is higher after common infections of the upper respiratory tract [3], [12]–[17].
Several non-exclusive mechanisms have been proposed to explain the influence of microbial agents on MS [18], [19]. One hypothesis, called bystander activation, states that such agents trigger adjuvant effects that can non-specifically promote the activation of autoreactive lymphocytes and their trafficking across the blood-brain barrier. This idea is supported by the observation that transgenic mice with a large number of myelin-reactive T cells do not develop spontaneous encephalomyelitis when housed in pathogen-free conditions, but do so after injection of bacterial toxins or when raised in a non-sterile environment [20]. Furthermore, recent studies show that the intestinal microbial flora contributes to the development of EAE [21]–[23], the most widely used animal model of MS.
To induce EAE, mice immunized with myelin antigens or transplanted with myelin-reactive T cells are commonly injected with PTX, a hexameric protein produced by the bacteria causing whooping cough and used as a surrogate for infection to increase EAE incidence and severity [24], [25]. The mechanism by which PTX promotes EAE is still not fully understood and seems paradoxical considering the ability of PTX to block leukocyte migration by interfering with chemokine receptor signaling [26]. This effect results from its largest subunit, called S1 or A protomer, which is endocytosed and transported to the endoplasmic reticulum [27], where it catalyzes the ADP-ribosylation of G-protein αi subunits [28], [29], thereby preventing their interaction with G protein-coupled receptors. Another mechanism has been proposed, in which the other subunits that form the B oligomer (i.e., the subunit S5 resting on two dimers, S4–S2 and S4–S3) would interact with transmembrane receptors, leading to the induction of inflammatory signaling pathways [30]. Although this hypothesis remains to be proved, it is indirectly supported by several observations. For example, Kubes and colleagues have reported that PTX stimulates P-selectin expression and leukocyte adhesion on the cerebral endothelium by acting through Toll-like receptor 4 (TLR4) [31]. Furthermore, we have shown that PTX increases the plasma level of IL-6, which in turn increases the expression of endothelial adhesion molecules and chemokines, leading to the recruitment of leukocytes that patrol the cerebral vasculature by crawling on the luminal endothelial surface [32], [33].
The mechanism responsible for detecting PTX and triggering the IL-6-mediated response described above is not elucidated. The theory holds that innate immune cells sense pathogen - and damage-associated molecules through a variety of membrane-bound and cytoplasmic receptors, such as those of the Toll-like and Nod-like families [34], [35]. Many of them engage signaling pathways that drive transcription of proinflammatory cytokines (e.g., IL-1β, TNF and IL-6). IL-1β differs from most of the others in that it is synthetized as an inactive form that must be cleaved to be activated and secreted. This cleavage is typically catalyzed by caspase-1 (Casp1), which is assembled into a mutiprotein complex called the inflammasome [36], [37]. The latter also includes a protein that serves as both a sensor for danger signals and a Casp1 activator. About ten inflammasome sensors have been characterized so far and most belong to the Nod-like receptor (NLR) family [38]. Whether such a sensor is involved in the response to PTX is not obvious a priori, since no increase in IL-1β is detectable in the plasma of PTX-treated mice [32].
In the present study, we sought to clarify how PTX is detected by the immune system and influences EAE development. We found that PTX induces IL-6 secretion from stromal cells indirectly via IL-1β, which is synthetized in myeloid cells under the control of the TLR4 pathway and the putative non-NLR inflammasome sensor pyrin (encoded by the Mefv gene). We also found that PTX requires pyrin to induce neutrophil adhesion to the cerebral endothelium of otherwise normal mice, and to effectively promote EAE in transgenic mice containing myelin-reactive T cells. Notably, these observations help us to appreciate the proinflammatory role of pyrin, which is currently a matter of debate, because divergent studies suggest that it either interacts with Casp1 through the adaptor protein ASC to form an active inflammasome [39]–[44] or acts as an inhibitor of Casp1 [45]–[48].
Results
PTX induces an inflammatory cascade involving myeloid cell-derived IL-1β and stromal cell-derived IL-6
We have reported that PTX promotes leukocyte adhesion to the brain vasculature by increasing the plasma level of IL-6 [32]. However, the molecular mechanism responsible for this upregulation and the cellular source of IL-6 remain unknown. To answer these questions, we first compared the IL-6 mRNA levels obtained by qRT-PCR in various tissues from mice killed 3 or 6 h after intraperitoneal injection of PTX (20 µg/kg). For comparison, we also measured the expression of IL-1β, a known regulator of IL-6 that does not increase in the blood of PTX-treated mice [32]. As shown in Figure 1a, PTX induced the transcription of both cytokines in peritoneal leukocytes and tissues surrounding the peritoneal cavity, including the mesentery and abdominal wall. To confirm these results at the protein level, we measured by ELISA the amounts of IL-6 and IL-1β in peritoneal fluid and blood samples collected from the same animals. As expected from our qRT-PCR data, and in agreement with our previous study [32], we found that both cytokines were secreted in the peritoneal fluid of PTX-treated mice, but that only IL-6 reached the circulation in significant amounts (Fig. 1b).

We next wanted to determine whether IL-1β mediates the effect of PTX on IL-6 expression and to identify the cell type(s) expressing these cytokines. We therefore generated chimeras by transplanting bone marrow cells into lethally irradiated mice using different combinations of donors and recipients expressing or lacking IL-1β or IL-6. In these mice, non-hematopoietic cells are radioresistant, while leukocytes are radiosensitive and replaced by donor-derived hematopoietic cells. ELISA on plasma and peritoneal fluid samples revealed that PTX could induce IL-6 secretion in wild-type mice reconstituted with IL-6-deficient bone marrow cells, but not in IL-6-deficient mice reconstituted with wild-type cells (Fig. 2a, left graphs). In contrast, PTX induced neither IL-6 nor IL-1β secretion in wild-type mice reconstituted with IL-1β-deficient bone marrow cells, but induced both cytokines in IL-1β-deficient mice reconstituted with wild-type cells (Fig. 2a, right graphs). These results indicate that IL-1β derives from radiosensitive leukocytes and regulates IL-6 expression in radioresistant cells.

We next performed a series of experiments to further pinpoint the cellular source of IL-1β and IL-6. First, we used CD11b-TKmt-30 transgenic mice expressing a mutated form of the human herpesvirus 1 thymidine kinase gene under the control of the myeloid-specific CD11b promoter [49]. In these mice, it is possible to deplete CD11b+ cells by the administration of ganciclovir (GCV), a prodrug that is converted by viral TK to a toxic nucleotide that kills proliferating cells. We found that CD11b-TKmt-30 mice responded normally to PTX by producing IL-1β, but not after myeloid cell depletion achieved through a 6-day treatment with GCV (Fig. 2b and Fig. S1). Second, we measured IL-1β mRNA in peritoneal leukocytes freshly harvested and purified by flow cytometry using the myeloid cell marker CD11b, the macrophage marker F4/80 and the neutrophil marker Ly6G. PTX upregulated IL-1β mRNA in CD11b+F4/80+ and CD11b+Ly6G+ leukocytes, but not in those negative for all these markers (Fig. 2c). Third, we injected PTX to transgenic mice expressing the fluorescent protein DsRed under the control of the IL-1β promoter [50], and analyzed their peritoneal leukocytes by flow cytometry. We observed that PTX increased DsRed expression almost exclusively in CD11b+F4/80+ and CD11b+Ly6G+ leukocytes (Fig. 2d and Fig. S2). Fourth, knowing that IL-6 derives from radioresistant cells, abdominal wall sections from PTX-treated mice were analyzed for the presence of IL-6 mRNA by radioisotopic in situ hybridization and immunostained or not for the mesenchymal/fibroblastic marker S100A4 (also called fibroblast-specific protein-1). Strong hybridization signals were detected in the connective tissue surrounding the muscle bundles (Fig. 2e, main image), where they colocalized with S100A4 immunostaining (Fig. 2e, insert). No signal was detected in sections from PBS-treated mice (data not shown). Finally, we used mouse abdominal wall biopsies to prepare primary cultures of S100A4+ stromal cells. The aim was to determine whether these cells had the ability to produce IL-6 when exposed for 3 h to PTX or IL-1β. The results showed a strong upregulation of IL-6 at the mRNA and protein levels in the presence of IL-1β, but not of PTX (Fig. 2f). Overall, these results demonstrate that PTX stimulates peritoneal macrophages and neutrophils to produce IL-1β, which in turn stimulates IL-6 synthesis in abdominal stromal cells.
PTX induces the IL-1β−IL-6 cascade through the TLR4 pathway and pyrin inflammasome in a manner dependent on its ADP-ribosyltransferase activity
To clarify the immune recognition mechanism through which PTX induces the IL-1β−IL-6 cascade, we examined mice lacking either TLR4 or inflammasome components for a defect in IL-6 expression after intraperitoneal injection of PTX. ELISA results revealed a normal increase of plasma IL-6 after PTX injection in mice lacking the ASC-dependent inflammasome sensors NLRP3 and NLRP6, but no increase in mice lacking TLR4, Casp1, ASC or pyrin (Fig. 3a and Fig. S3). We next further examined the latter mice for a defect in IL-1β synthesis. Consistently, none of them had an increased amount of IL-1β in their peritoneal fluid (Fig. 3b), despite the fact that those lacking Casp1, ASC or pyrin exhibited a normal increase of IL-1β mRNA in their peritoneal leukocytes (Fig. 3c). In contrast to PTX, the TLR4 agonist lipopolysaccharide (LPS) could induce IL-1β and IL-6 secretion in pyrin-deficient mice (Fig. S4), indicating that the absence of pyrin causes a stimulus-specific defect in IL-1β production. Altogether, these results support the concept that PTX-driven inflammation operates under multiple levels of control; it induces IL-1β transcription through the TLR4 signaling pathway and then requires function of the pyrin inflammasome to induce IL-1β post-translational processing.

To test whether the ability of PTX to induce the IL-1β−IL-6 cascade depends on the integrity of its multimeric structure and ADP-ribosyltransferase activity, we compared the expression levels of these cytokines in mice exposed to different forms of PTX, including the A protomer (PTX-A) and B oligomer (PTX-B) separately, and an enzymatically inactivated mutant (PTXmut) with 2 amino acid substitutions (R9K and E129A). The latter has an antigenic structure comparable to that of the wild-type form, but an ADP-ribosyltransferase activity below 0.01% [51]. We found that all these forms, except the wild-type one, were unable to activate IL-1β transcription in peritoneal leukocytes and secretion of IL-1β and IL-6 in the peritoneal fluid (Fig. 4a, b). Western blots confirmed and extended these results by showing increased levels of cleaved Casp1, mature IL-1β, TLR4 and MyD88 only in mice challenged with wild-type PTX (Fig. 4c, d). These observations suggest that PTX depends on both its multimeric structure and its ADP-ribosyltransferase activity to induce IL-1β production.

Because PTX strongly induces neutrophil recruitment into the peritoneum (Fig. S1 and S2), we asked whether the reduced expression of cytokines observed in pyrin-knockout mice could be attributable to a change in the number of infiltrating leukocytes. We therefore analyzed peritoneal leukocytes from pyrin-knockout and wild-type mice by flow cytometry using various leukocytic markers. The most noticeable effect of PTX was a marked increase in the number of CD11b+Ly6G+ neutrophils, but no intergenotype difference was recorded (Fig. S5). These results indicate not only that the differences in cytokine expression were not the consequence of a difference in the cellular composition, but also that neutrophil infiltration into the peritoneum in response to PTX is neither dependent on pyrin nor the IL-1β−IL-6 cascade.
Finally, we asked whether the in vivo effect of PTX on IL-1β production could be reproduced ex vivo by using freshly isolated peritoneal leukocytes, which were stimulated for 6 h at 37°C. ELISA results showed that the amount of IL-1β was strongly increased in cell extracts after treatment with PTX (20 µg/ml), but weakly with heat-inactivated PTX and not at all with PTXmut (Fig. S6a). However, only a very small increase in IL-1β was detected in the supernatant of cells treated with PTX. Similar results were obtained using pyrin-deficient leukocytes (Fig. S6b). The deficient secretion of IL-1β from wild-type leukocytes was not due to an absence of pyrin expression, which was normally increased in response to PTX and correlated with IL-1β expression (Fig. S6c), as previously observed in vivo (Fig. 5b). These observations suggest that PTX can actually trigger IL-1β and pyrin synthesis ex vivo, but not pyrin activation, which is required for IL-1β processing and release and which depends on more complex conditions found in vivo.

PTX induces pyrin expression in myeloid cells via TLR4
To determine whether and how PTX regulates pyrin expression, we performed qRT-PCR on mRNA from peritoneal leukocytes of TLR4-knockout and wild-type mice harvested 6 h after exposure or not to PTX. The results showed that the levels of pyrin mRNA were low in non-stimulated leukocytes, but 18 times higher in PTX-exposed leukocytes (Fig. 5a), in which they positively correlated with the levels of IL-1β mRNA (Fig. 5b). However, this upregulation was not observed in TLR4-deficient leukocytes (Fig. 5a). Consistent with the results obtained for IL-1β (Fig. 4b), a repetition of the experiment using PTXmut revealed no change in pyrin mRNA (303±151 copies versus 365±145 for the PBS group and 1722±221 for the PTX group). To determine the cellular source of pyrin, we measured its mRNA by qRT-PCR in sorted peritoneal cells. We found that PTX increased pyrin mRNA expression in CD11b+F4/80+ and CD11b+Ly6G+ leukocytes, but not in the CD11b− population (Fig. 5c). Finally, to confirm the inducible nature of pyrin at the protein level, we analyzed peritoneal cell lysates by Western blotting using a polyclonal antibody directed against the N-terminal 410 amino acids of mouse pyrin and recognizing two major splicing variants [45]. A band doublet at the expected molecular weight of ∼100 kDa was detected in peritoneal cell lysates from mice treated with PTX, but not from control mice (Fig. 5d). We conclude from these results that the ADP-ribosyltransferase activity of PTX leads to de novo expression of pyrin in macrophages and neutrophils via a TLR4-dependent mechanism.
Pyrin contributes to PTX-induced neutrophil intravascular patrolling and EAE
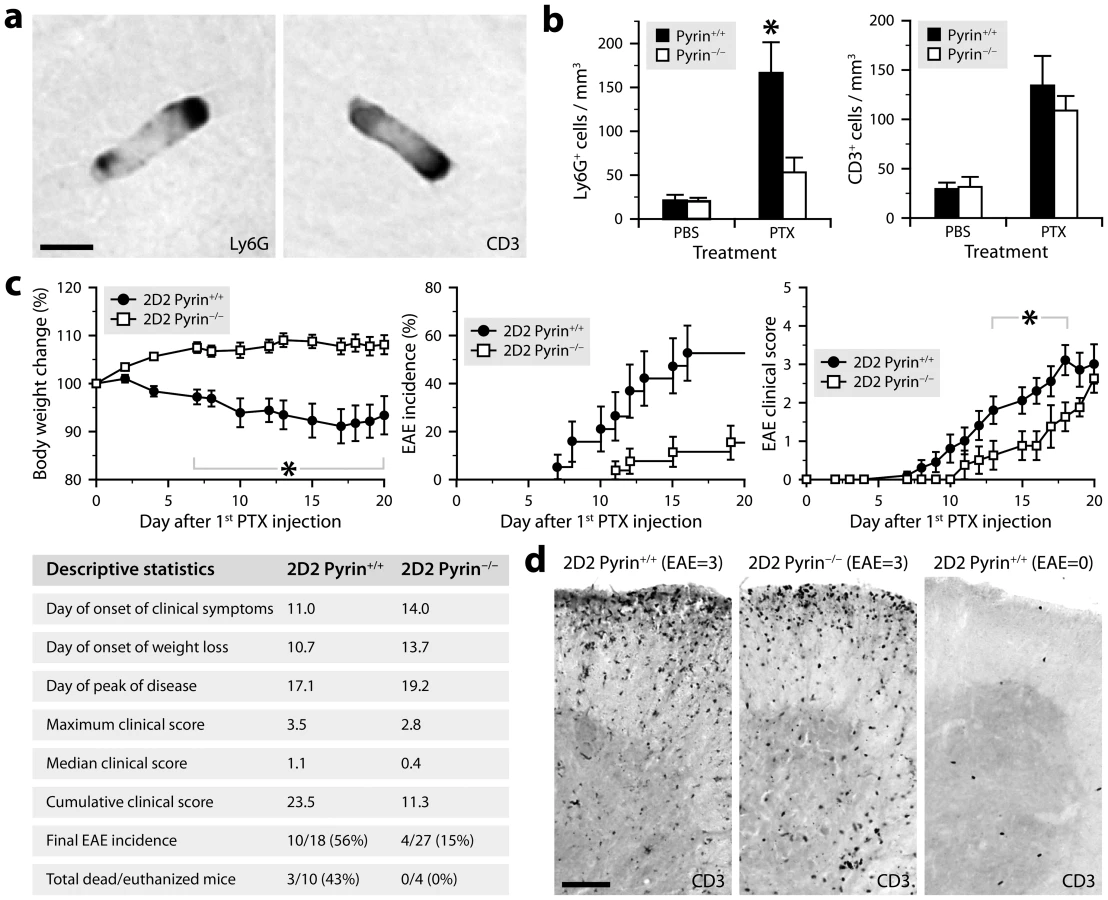
The CNS microvasculature is constantly patrolled by rod-shaped leukocytes that crawl on the luminal endothelial surface [52], [53]. This cell population is mainly composed of monocytes and neutrophils under normal conditions and within a few hours after exposure to bacterial toxins, but also includes a significant proportion of lymphocytes during EAE [32]. We have shown that PTX increases the number of crawling neutrophils in brain capillaries through an IL-6-dependent mechanism, but we have failed to demonstrate a similar effect on lymphocytes, at least during the first 6 h after PTX injection [32]. To examine the possibility that such an increase occurs after a longer period, we administered PTX to mice according to a protocol commonly used for EAE induction (i.e., 2 intraperitoneal injections of 20 µg/kg given 2 days apart). Twenty-four hours after the last injection, the brains were perfused to remove non-adherent leukocytes and collected to assess the presence of rod-shaped CD3+ T cells in the capillary network of the cerebral cortex. Stereological analysis revealed a 3.5-fold increase of these cells in PTX-treated mice, reaching a density of approximately 141 cells per mm3 compared to 40 in control mice (n = 3 per group). As expected, the effect was not limited to lymphocytes, because a concomitant 4.2-fold increase of rod-shaped Ly6G+ neutrophils was observed (139 cells per mm3 versus 33 in controls).
Having found that the regimen of PTX used to promote EAE increases intravascular patrolling by both lymphocytes and neutrophils, we wondered whether pyrin: 1) is involved in the underlying mechanism; and 2) contributes to the effect of PTX on EAE, that is, a CD4+ T cell-mediated disease in which neutrophils play an important role [33], [54]–[56]. To answer these questions, we used pyrin-deficient mice in a C57BL/6 or 2D2 background. 2D2 mice express a transgenic myelin oligodendrocyte glycoprotein (MOG)-specific T cell receptor in most of their CD4+ lymphocytes and can develop EAE at a reported incidence of 39% if exposed to PTX under specific pathogen-free housing conditions [57]. First, we found that pyrin-deficient C57BL/6 mice exhibited no increase in the recruitment of rod-shaped Ly6G+ neutrophils on the cerebral vasculature after treatment with PTX, contrary to wild-type controls and despite a normal increase in the number of adherent CD3+ lymphocytes (Fig. 6a,b). Second, the cohort of pyrin-deficient 2D2 mice developed EAE in response to PTX at an incidence ∼3 times lower compared to pyrin-expressing 2D2 mice (Fig. 6c). The pyrin-deficient 2D2 mice that developed EAE did it with a 4-day delay and with less severity during the progressive phase of the disease (Fig. 6c, right graph). However, comparable infiltration of CD3+ cells was observed in the spinal cord of mice with similar clinical scores killed after 21 days, regardless of their genotype at the Mefv locus (Fig. 6d). This suggested that pyrin might not be essential for the effector phase of EAE, when myelin-reactive lymphocytes have been activated and recruited to the CNS parenchyma. This hypothesis was confirmed by the observation that pyrin-deficient and wild-type mice developed similar EAE when adoptively transferred with encephalitogenic T cells isolated from MOG-immunized wild-type mice and reactivated in vitro (Fig. 7). Altogether, these results demonstrate that pyrin contributes to two different, but potentially related phenomena: the induction of neutrophil adhesion to CNS capillaries and the activation of the initial phase of EAE by PTX.

Discussion
This study provides answers to the long-lasting question of how PTX produces an adjuvant effect that promotes EAE. First, contrary to the prevailing concept [30], our results suggest that PTX does not act directly via membrane-bound receptors such as TLR4, but rather by catalyzing the ADP-ribosylation of G-protein αi subunits, a phenomenon that requires the internalization and disassembling of PTX to release its A protomer in the cytoplasm. Indeed, mutant PTX, containing only 2 amino acid substitutions in one of its 6 subunits, was unable to activate IL-1β transcription (Fig. 4), which was dependent on TLR4 (Fig. 3). The mutations affect the enzymatic activity of the A-protomer, but not the antigenic structure of the whole toxin [51]. The protein binding properties of mutant PTX are thus likely to be comparable to that of wild-type PTX. Therefore, we propose that PTX causes perturbations in G protein-coupled receptor signaling (e.g., cAMP-dependent pathways), leading to the release of an endogenous danger signal called alarmin that activates TLR4 on myeloid cells and drives IL-1β transcription. Several TLR4-interacting alarmins have been identified so far, including HMGB1, S100A8/A9 and heat-shock proteins [58]. Additional work will be needed to further clarify the mechanism by which TLR4 is activated in response to PTX. Meanwhile, we speculate that genetic defects or other factors that may disrupt G protein-coupled receptor signaling in the manner of PTX might contribute to MS and MS-like diseases.
A second answer is that PTX promotes the formation of a pyrin-dependent inflammasome that cleaves pro-IL-1β into its active form (and perhaps other Casp1-dependent cytokines such as IL-18). To our knowledge, PTX represents the first microbial molecule known to activate pyrin. Our observations are in agreement with the view that pyrin exerts a proinflammatory function, as supported by studies showing that pyrin interacts with ASC and activates Casp1 [39]–[44]. Mutations in the gene encoding pyrin (MFEV) are known to cause familial Mediterranean fever [59]–[61], a hereditary inflammatory disorder characterized by recurrent fevers and painful inflammation. Interestingly, some of these mutations have been linked to a higher susceptibility of developing a more progressive or severe form of MS [62]–[65]. The question of whether defects in inflammasome components such as pyrin could reduce the activation threshold of autoreactive T cells should fuel future investigations.
A third answer may reside in the systemic innate immune response triggered by IL-1β. Indeed, after being secreted by myeloid cells exposed to PTX, IL-1β stimulates nearby stromal cells to produce IL-6, which passes into the bloodstream. As previously shown [32], [33], IL-6 induces the expression of ICAM1 and CXCR2 ligands at the surface of cerebral endothelial cells, leading to the adhesion and crawling of neutrophils through interaction between CD11b and ICAM1. The question arises as to whether neutrophils mediate the effect of PTX on EAE. Mounting evidence actually suggests a major involvement of neutrophils in this model. It has been reported that neutrophils massively infiltrate the CNS of EAE mice [32], [54], [66]–[73], and that EAE is attenuated in mice lacking the neutrophil receptor CXCR2 or treated with antibodies against CXCR2, its ligand CXCL1 or the neutrophil-specific protein Ly6G [33], [54], [56], [74], [75]. Although their functions in EAE are unknown, it is hypothesized that neutrophils may initiate the disease by altering the permeability of the blood-brain barrier, thereby allowing access to other immune cells [55]. It is also proposed that neutrophils secrete proinflammatory molecules that promote the maturation of monocytic cells into professional antigen-presenting cells [56], which are essential for local restimulation of myelin-reactive T cells. In human, neutrophils play a role in neuromyelitis optica [76], an autoimmune disorder that resembles MS in some ways, but the possibility that they contribute to MS is questionable. While neutrophils have not been traditionally observed in increased number in fluids and postmortem tissues from MS patients, more recent studies show that neutrophils with an activated phenotype are more numerous in the blood of MS patients and that the CXCR2 ligand IL-8 is elevated [77], [78].
Of course, it is also possible that the IL-1β−IL-6 cascade exerts neutrophil-independent effects, such as the bystander activation of autoreactive T lymphocytes, defined as a process that does not require specific TCR stimulation (e.g., it can occur via soluble mediators or cross-linking of membrane-bound receptors such as CD2 and CD28 [79]). IL-6 is a multifunctional proinflammatory cytokine that is essential in the development of autoimmune diseases, including EAE [80]–[85]. It is believed to act by stimulating the generation of autoreactive Th17 lymphocytes in the presence of TGFβ [86]–[89]. IL-6 and IL-17 have been shown to be engaged in a positive feedback loop that sustains inflammation and contributes to autoimmune responses [85], [90], [91]. However, IL-6 is not sufficient in itself and most likely requires another factor, because CD3+ cells increase normally in number in the CNS vasculature of pyrin-KO mice challenged with PTX, despite a lack of IL-6 expression. This observation suggests the existence of another PTX-induced pathway that regulates lymphocyte trafficking independently of pyrin, IL-1β and IL-6. This pathway could also be involved in the PTX-induced recruitment of neutrophils into the peritoneum, which is independent on pyrin, contrary to the recruitment of crawling neutrophils at the surface of the CNS vasculature.
The effects of PTX are paradoxical: how can a chemotaxis-blocking agent induce leukocyte recruitment? A possible explanation is that these effects are mediated by distinct mechanisms that are separated in space and time. Indeed, early after injection, PTX is likely to be internalized by different cells, including peritoneal leukocytes, whose migratory ability would be blocked, as previously documented [26]. A few hours later, affected cells would release an alarm signal that would trigger pyrin-dependent and independent mechanisms, leading to the recruitment of neutrophils into the peritoneum and of crawling leukocytes onto the luminal surface of the systemic vasculature. The recruitment of these cells would not be blocked by PTX, because it would occur at a distant site or when PTX is likely to have been cleared from the extracellular peritoneal milieu (e.g., by cellular internalization and passive diffusion).
An important question to address in future work is how PTX activates pyrin. This protein comprises: 1) a N-terminal pyrin domain that binds to ASC [92], an adaptor that links pyrin to Casp1; 2) a B-box type zinc finger domain that interacts with other proteins (e.g., mutant PSTPIP1 [41], [93]); and 3) a coiled-coil domain responsible for homotrimerization of pyrin [41]. While the C-terminal region of mouse pyrin is uncharacterized, the human counterpart contains a SPRY domain that mediates interactions with Casp1 and pro-IL-1β and that is the site of mutations associated with Mediterranean fever [94]. It has been reported that the binding of mutant PSTPIP1 to the B-box activates pyrin by causing a conformational change that releases the pyrin domain from sequestration, thus promoting the recruitment of the ASC-Casp1 complex [41]. Similarly, it is tempting to speculate that pyrin would act as a sensor for an endogenous danger signal inducible by PTX in vivo (but not ex vivo), which would mimic the effect of mutant PSTPIP1 by binding to the B-box.
In conclusion, this study provides fundamental insights into how the immune system recognizes PTX. By analogy, it helps to appreciate how microbial agents may influence the course of autoimmune diseases, such MS, which is worsened by common infections. By identifying PTX as a pyrin activator as well as an inducer of lymphocyte crawling, this study provides a new convenient model for investigating the mechanisms that control the activation of the pyrin inflammasome and the trafficking of lymphocytes at the blood-brain interface. This study also provides the first physiological demonstration of a role of wild-type pyrin in vivo, as opposed to the mutated forms found in patients suffering from Mediterranean fever. Finally, it identifies a pyrin-dependent innate immune response that could potentially be targeted for the treatment of autoimmune diseases.
Materials and Methods
Ethics statement
All mice experiments were approved by the Laval University Animal Protection Committee (permit #2012140) and were conducted in compliance with the guidelines of the Canadian Council on Animal Care.
Mice
The mice used in this study are detailed in Table S1. They were acclimated to our facility for at least 1 week, housed under specific pathogen-free conditions and tested at 8–10 weeks of age.
Toxin injection
Mice were injected intraperitoneally with PTX (20 µg/kg), PTX-A (5.3 µg/kg), PTX-B (14.5 µg/kg), PTXmut (20 µg/kg), LPS from Escherichia coli O55:B5 (1 mg/kg) or PBS. The toxins were purchased from List Biological Laboratories, except LPS (Sigma-Aldrich). In most experiments, mice were given a single injection and killed 3 or 6 h later. In indicated experiments, they were given 2 injections, 2 days apart, and killed 24 h after the last injection.
Ganciclovir treatment
Mice were injected intraperitoneally twice daily for 6 days with GCV (Hoffmann-La Roche, 50 mg/kg, diluted in saline). Control mice were treated identically, except that GCV was substituted by saline.
Bone marrow transplantation
Recipient mice were exposed to 10 gray of radiation using a 137caesium source (Gammacell 40 Exactor) and injected via a tail vein with 107 bone marrow cells freshly collected from donor mice. The cells were harvested by flushing femurs with DPBS containing 2% FBS using a syringe with a 25-gauge needle, filtered through a 40-µm nylon mesh, centrifuged and resuspended in DPBS. The mice were treated with antibiotics (Septra; 100 µg/ml trimethoprim and 500 µg/ml sulfamethoxazole in drinking water) for 3 days before and 3 weeks after transplantation. Chimeric mice were injected with PTX 2 months after transplantation.
EAE induction by active immunization
Mice were subcutaneously injected into both flanks with a total of 200 µl of emulsion containing 300 µg of MOG peptide 35–55 (Feldan) dissolved in saline and mixed with an equal volume of complete Freund's adjuvant containing 500 µg of killed Mycobacterium tuberculosis H37 RA (Difco Laboratories). The mice were also injected intraperitoneally with 20 µg/kg of PTX immediately and 2 days after immunization.
EAE induction by adoptive transfer
Mice were intraperitoneally injected with 20×106 lymphoid cells. These were isolated from abdominal lymph nodes and spleens of mice killed 8 days after active EAE induction, and then cultured for 2 days in DMEM supplemented with MOG peptide 35–55 (15 µg/ml), murine IL-12 (5 ng/ml), murine IL-23 (20 ng/ml), FBS (10%), 1% modified Eagle's medium nonessential amino acids (Wisent), penicillin (100 U/ml), streptomycin (100 µg/ml) and amphotericin B (250 ng/ml).
Evaluation of EAE symptoms
Mice were weighed and scored daily as follows: 0, no visual sign of disease; 0.5, partial tail paralysis; 1, complete tail paralysis; 1.5, weakness in one hind limb; 2, weakness in both hind limbs; 2.5, partial hind limb paralysis; 3, complete hind limb paralysis; 3.5, partial forelimb paralysis; 4, complete forelimb paralysis; 5, dead or killed for humane reasons.
Cell culture
Primary cultures of abdominal stromal cells were prepared from peritoneal wall biopsies and mesentery of adult mice. The tissues were washed in PBS and digested in 0.25% trypsin-EDTA solution for 50 min at 37°C with gentle agitation. The mixture was filtered on a 70-µm cell strainer and centrifuged at 1,500 rpm for 5 min. Cells were cultured in DMEM supplemented with 15% heat-inactivated FBS, penicillin (100 U/ml), streptomycin (100 µg/ml) and amphotericin B (250 ng/ml) until confluence. Cells were subcultured in DMEM containing 10% FBS and used at passage 3. For experimentation, 1.5×105 cells were seeded in 6-well plates, starved for 24 h and incubated for 3 h with 100 µg/ml PTX, 10 ng/ml recombinant mouse IL-1β (BioLegend) or PBS as control.
Ex vivo stimulation of leukocytes
Peritoneal leukocytes were harvested by flushing the peritoneum of naive mice with 5 ml D-PBS. Cells were pooled in DMEM supplemented with 2% FBS, transferred in 24-well plates at a density of 2×106 cells per well in 500 µl containing or not 500 ng/ml PTX, PTXmut or heat-inactivated PTX (heated at 80°C for 30 min). After a 6 h-incubation at 37°C, the supernatant was collected, and the cells were washed with PBS and then lyzed with Cell Lysis Buffer 2 (R&D Systems).
RNA isolation and qRT-PCR
Total RNA was isolated from tissue or cells by homogenization in TRI-reagent (Sigma-Aldrich) followed by purification using the GenElute Mammalian Total RNA Miniprep Kit (Sigma-Aldrich). First strand cDNA was generated from 1 (cells) or 5 (tissues) µg of total RNA using Superscript III (Invitrogen) with random hexamers and 20-mer oligo-dT primers, then purified using the GenElute PCR Clean-Up Kit (Sigma-Aldrich). The product (20 ng) was analyzed using the LightCycler 480 system with the SYBR Green I Master mix according to the manufacturer's instructions (Applied Biosystems). The primers were as follows: IL-1β, 5′-TCAAATCTCGCAGCAGCACATC-3′ and 5′ - CCAGCAGGTTATCATCATCATCCC-3′; IL-6, 5′-GTCCTTCCTACCCCAATTTCCAA-3′ and 5′ - GAATGTCCACAAACTGATATGCTTAGG-3′; pyrin, 5′-CCCTACTGGATGAGATGATTGAAGAAC-3′ and 5′-TCCAACAGCTCAGAGGCAGACAT-3′. The PCR conditions consisted of 45 cycles of denaturation (10 s at 95°C), annealing (10 s at 60°C), elongation (14 s at 72°C) and reading (5 s at 74°C). The number of mRNA copies was determined using the second derivative method [95].
ELISA
Blood samples were obtained by intracardiac puncture in EDTA tubes and centrifuged for 20 min at 2000×g to collect plasma. Peritoneal fluid was collected by washing the peritoneal cavity with 2 ml of DPBS, and then centrifuged for 3 min at 1000×g to remove peritoneal cells. Samples were analyzed using the mouse IL-1β Quantikine ELISA kit or IL-6 Duoset ELISA kit (R&D Systems).
Western blotting
Peritoneal fluid and cell samples were homogenized in extraction buffer (20 mM Tris-HCl at pH 7.5, 150 mM NaCl, 1% Triton X-100, 1 mM ethylenediaminetetraacetic acid, 1 mM ethylene glycol tetraacetic acid, 2.5 mM pyrophosphate, 1 mM β-glycerophosphate, 1× protease inhibitor cocktail [Sigma]). Immunoblot analysis was carried out with the Criterion system (Bio-Rad). Accordingly, proteins were resolved in 4–20% Tris-HCl Criterion precasted gels (Bio-Rad), transferred to polyvinylidene difluoride membranes (Applied Biosystems), placed in blocking buffer (PBS, 0.1% Tween 20 and 0.4% I-Block [Applied Biosystems]), and then incubated for 1 h with antibodies to IL-1β (1∶5000, National Cancer Institute), Casp1 (1∶1000, Imgenex), TLR4 (1∶1000, Imgenex), Myd88 (1∶1000, Imgenex) or pyrin (1∶10000, Dr. Jae Jin Chae), followed by 45 min in appropriate secondary horseradish peroxidase (HRP)-linked antibodies (Cell Signaling Technology). Signal visualization was enhanced by chemiluminescence using a phototope-HRP detection kit (Cell Signaling Technology). In cell lysates, to control for protein loading, immunoblots were stripped with Restore Western blot stripping buffer (Pierce) and blotted for β-actin using monoclonal anti-β-actin antibody (1∶5000, Sigma). Data were normalized to β-actin for peritoneal cells and equal amounts of protein were loaded for peritoneal fluid (20 µg). Band densities were quantified with UN-SCAN-IT software 5.3, measuring the gray scale intensity, subtracting single region from the background value, checking for saturation and with a linear optical density calculation.
Flow cytometry
Peritoneal cells were blocked for 10 min with 5 µg/ml anti-CD16/CD32 antibody (clone 2.4G2, BD Biosciences) and stained for 30 min on ice with the following antibodies (1 µl each per 106 cells): rat anti-F4/80-FITC (clone BM8, eBioscience), rat anti-CD45-V500 (clone 30-F11, BD Biosciences), rat anti-CD11b-V450 (clone M1/70, BD Biosciences), rat anti-Ly6G-PerCP-Cy5.5 (clone 1A8, BD Biosciences), rat anti-CD3-PE (clone 17A2, BD Biosciences), rat-anti-CD19-PE-Cy7 (clone 1D3, BD Biosciences). Cells were analyzed with a BD FACSCanto II flow cytometer and FlowJo software (Tree Star).
In situ hybridization
Tissue sections were analyzed by in situ hybridization as described previously [96]. Combined in situ hybridization and immunohistochemistry was performed according to a modified protocol [97].
Immunostaining
Immunostaining was performed as described previously [53] using the following primary antibodies: rat anti-CD3 (clone 17A2, BD Biosciences, 1∶500), rat anti-Ly6G (clone 1A8, BD Biosciences, 1∶5000) and rabbit anti-S100A4 (Abcam, 1∶200).
Stereology
Cells were counted using the optical fractionator method as described previously [53]. The counting parameters were as follows: distance between counting frames, 400×400 µm; counting frame size, 120×120 µm; dissector height, 10 µm; guard zone thickness, ≥2 µm.
Microscopy
Micrographs were taken using a Retiga EX monochrome camera mounted on a Nikon E800 microscope. Images were adjusted for contrast, brightness and sharpness using Photoshop 12 (Adobe Systems).
Statistics
Data are expressed as mean ± standard error and were analyzed using non-parametric tests, because they were discontinuous, were not normally distributed or the variances were not equal. Means were compared using the Wilcoxon or Kruskal-Wallis test. Correlation between qRT-PCR results was evaluated using the Spearman's test. EAE incidence curves were constructed using the Kaplan-Meier method and compared using the Wilcoxon test. All these analyses were performed using JMP 10 (SAS Institute) with a significance level of 5%.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. AscherioA, MungerKL, LunemannJD (2012) The initiation and prevention of multiple sclerosis. Nat Rev Neurol 8 : 602–612.
2. KochMW, MetzLM, AgrawalSM, YongVW (2013) Environmental factors and their regulation of immunity in multiple sclerosis. J Neurol Sciol 324 : 10–612.
3. MarrieRA, WolfsonC, SturkenboomMC, GoutO, HeinzlefO, et al. (2000) Multiple sclerosis and antecedent infections: a case-control study.sclerosis. NeurologySciol 54 : 2307–2310.
4. HernanMA, ZhangSM, LipworthL, OlekMJ, AscherioA (2001) Multiple sclerosis and age at infection with common viruses. study.sclerosis. Epidemiologyol 12 : 301–610.
5. GoldacreMJ, WottonCJ, SeagroattV, YeatesD (2004) Multiple sclerosis after infectious mononucleosis: record linkage study.osis. J Epidemiol Community Health 58 : 1032–1050.
6. ThackerEL, MirzaeiF, AscherioA (2006) Infectious mononucleosis and risk for multiple sclerosis: a meta-analysis.is. Ann Neurol 59 : 499–503.
7. NielsenTR, RostgaardK, NielsenNM, Koch-HenriksenN, HaahrS, et al. (2007) Multiple sclerosis after infectious mononucleosis.erosis: a meta-analysis.is. Arch Neurol 64 : 72–5503.
8. AscherioA, MungerKL (2007) Environmental risk factors for multiple sclerosis. Part I: the role of infection. Ann Neuroll 61 : 288–993.
9. ZaadstraBM, ChorusAM, van BuurenS, KalsbeekH, van NoortJM (2008) Selective association of multiple sclerosis with infectious mononucleosis.ection. Mult Sclerl 14 : 307–133.
10. RamagopalanSV, ValdarW, DymentDA, DeLucaGC, YeeIM, et al. (2009) Association of infectious mononucleosis with multiple sclerosis. A population-based study. Neuroepidemiology 32 : 257–623.
11. HandelAE, WilliamsonAJ, DisantoG, HandunnetthiL, GiovannoniG, et al. (2010) An updated meta-analysis of risk of multiple sclerosis following infectious mononucleosis. PLoS One 5: e12496.
12. SibleyWA, BamfordCR, ClarkK (1985) Clinical viral infections and multiple sclerosiserosis following infectious mononucleosis. Lancet 1 : 1313–1353.
13. AndersenO, LygnerPE, BergstromT, AnderssonM, VahlneA (1993) Viral infections trigger multiple sclerosis relapses: a prospective seroepidemiological study. J Neurol 240 : 417–223.
14. PanitchHS (1994) Influence of infection on exacerbations of multiple sclerosisective seroepidemiological study. Ann Neurol 36 SupplS25–823.
15. EdwardsS, ZvartauM, ClarkeH, IrvingW, BlumhardtLD (1998) Clinical relapses and disease activity on magnetic resonance imaging associated with viral upper respiratory tract infections in multiple sclerosis. J Neurol Neurosurg Psychiatry 64 : 736–413.
16. BuljevacD, FlachHZ, HopWC, HijdraD, LamanJD, et al. (2002) Prospective study on the relationship between infections and multiple sclerosis exacerbations.er respiratory tract infections in multiple sclerosis. Brain 125 : 952–603.
17. TremlettH, van der MeiIA, PittasF, BlizzardL, PaleyG, et al. (2008) Monthly ambient sunlight, infections and relapse rates in multiple sclerosis.is exacerbations.er respiratory tract infections in multiple sclerosis. Neuroepidemiology 31 : 271–903.
18. MunzC, LunemannJD, GettsMT, MillerSD (2009) Antiviral immune responses: triggers of or triggered by autoimmunity? Nat Rev Immunolgy 9 : 246–583.
19. KakalachevaK, MunzC, LunemannJD (2011) Viral triggers of multiple sclerosis.of or triggered by autoimmunity? Biochim Biophys Acta 1812 : 132–403.
20. GovermanJ, WoodsA, LarsonL, WeinerLP, HoodL, et al. (1993) Transgenic mice that express a myelin basic protein-specific T cell receptor develop spontaneous autoimmunity. Cell 72 : 551–603.
21. YokoteH, MiyakeS, CroxfordJL, OkiS, MizusawaH, et al. (2008) NKT cell-dependent amelioration of a mouse model of multiple sclerosis by altering gut flora.ous autoimmunity. Am J Pathol 173 : 1714–1723.
22. LeeYK, MenezesJS, UmesakiY, MazmanianSK (2011) Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis.immunity. Proc Natl Acad Sci U S A 108 Suppl 14615–4622.
23. BererK, MuesM, KoutrolosM, RasbiZA, BozikiM, et al. (2011) Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination.lomyelitis.immunity. Nature 479 : 538–412.
24. StromnesIM, GovermanJM (2006) Active induction of experimental allergic encephalomyelitis.gger autoimmune demyelination.lomyelitis.immunity. Nat Protoc 1 : 1810–1892.
25. StromnesIM, GovermanJM (2006) Passive induction of experimental allergic encephalomyelitis.ger autoimmune demyelination.lomyelitis.immunity. Nat Protoc 1 : 1952–1960.
26. CarbonettiNH (2010) Pertussis toxin and adenylate cyclase toxin: key virulence factors of Bordetella pertussis and cell biology tools. Future Microbiol 5 : 455–690.
27. PlautRD, CarbonettiNHM (2008) Retrograde transport of pertussis toxin in the mammalian cell.tors of Bordetella pertussis and cell biology tools. Cell Microbiolol 10 : 1130–1190.
28. BokochGM, KatadaT, NorthupJK, HewlettEL, GilmanAG (1983) Identification of the predominant substrate for ADP-ribosylation by islet activating protein.d cell biology tools. J Biol Chemiolol 258 : 2072–2050.
29. CodinaJ, HildebrandtJ, IyengarR, BirnbaumerL, SekuraRD, et al. (1983) Pertussis toxin substrate, the putative Ni component of adenylyl cyclases, is an alpha beta heterodimer regulated by guanine nucleotide and magnesium. Proc Natl Acad Sci U S A 80 : 4276–4280.
30. MangmoolS, KuroseH (2011) G(i/o) protein-dependent and -independent actions of Pertussis Toxin (PTX).is an alpha beta heterodimer regulated by guanine nucleotide and magnesium. Toxins (Basel) Sci U S A 3 : 884–990.
31. KerfootSM, LongEM, HickeyMJ, AndoneguiG, LapointeBM, et al. (2004) TLR4 contributes to disease-inducing mechanisms resulting in central nervous system autoimmune diseaser regulated by guanine nucleotide and magnesium. J Immunol 173 : 7070–7070.
32. RichardJF, RoyM, Audoy-RemusJ, TremblayP, VallieresL (2011) Crawling phagocytes recruited in the brain vasculature after pertussis toxin exposure through IL6, ICAM1, and ITGαM. guanine nucleotide and magnesium. Brain Pathol 21 : 661–671.
33. RoyM, RichardJF, DumasA, VallieresL, P, VallieresL (2012) CXCL1 can be regulated by IL-6 and promotes granulocyte adhesion to brain capillaries during bacterial toxin exposure and encephalomyelitis.magnesium. J Neuroinflammation 9 : 18.
34. AkiraS, UematsuS, TakeuchiO (2006) Pathogen recognition and innate immunity. Cell 124 : 783–801.
35. TakeuchiO, AkiraS (2010) Pattern recognition receptors and inflammation. Cell 140 : 805–201.
36. SchroderK, TschoppJ (2010) The inflammasomes. Cell 140 : 821–321.
37. RathinamVA, VanajaSK, FitzgeraldKA (2012) Regulation of inflammasome signaling. Nat Immunol 13 : 333–221.
38. LatzE, XiaoTS, StutzA, KA (2013) Activation and regulation of the inflammasomes. Nat Rev Immunol 13 : 397–411.
39. RichardsN, SchanerP, DiazA, StuckeyJ, SheldenE, et al. (2001) Interaction between pyrin and the apoptotic speck protein (ASC) modulates ASC-induced apoptosis. J Biol Chemunol 276 : 39320–39329.
40. YuJW, WuJ, ZhangZ, DattaP, IbrahimiI, et al. (2006) Cryopyrin and pyrin activate caspase-1, but not NF-kappaB, via ASC oligomerization.ed apoptosis. Cell Death Differ 13 : 236–499.
41. YuJW, Fernandes-AlnemriT, DattaP, WuJ, JulianaC, et al. (2007) Pyrin activates the ASC pyroptosome in response to engagement by autoinflammatory PSTPIP1 mutants. Mol Cellth Differ 28 : 214–279.
42. GavrilinMA, MitraS, SeshadriS, NateriJ, BerheF, et al. (2009) Pyrin critical to macrophage IL-1beta response to Francisella challenge.lammatory PSTPIP1 mutants. J Immunolh Differ 182 : 7982–7999.
43. GavrilinMA, AbdelazizDH, MostafaM, AbdulrahmanBA, GrandhiJ, et al. (2012) Activation of the pyrin inflammasome by intracellular Burkholderia cenocepacia.ry PSTPIP1 mutants. J Immunolh Differ 188 : 3469–3477.
44. YuJW, FariasA, HwangI, Fernandes-AlnemriT, AlnemriES (2013) Ribotoxic stress through p38 mitogen-activated protein kinase activates in vitro the human pyrin inflammasome. J Biol ChemDiffer 288(16): 11378–83.
45. ChaeJJ, KomarowHD, ChengJ, WoodG, RabenN, et al. (2003) Targeted disruption of pyrin, the FMF protein, causes heightened sensitivity to endotoxin and a defect in macrophage apoptosis. Mol CellhemDiffer 11 : 591–604.
46. ChaeJJ, WoodG, MastersSL, RichardK, ParkG, et al. (2006) The B30.2 domain of pyrin, the familial Mediterranean fever protein, interacts directly with caspase-1 to modulate IL-1beta production. Proc Natl Acad Sci U S A 103 : 9982–9974.
47. PapinS, CueninS, AgostiniL, MartinonF, WernerS, et al. (2007) The SPRY domain of Pyrin, mutated in familial Mediterranean fever patients, interacts with inflammasome components and inhibits proIL-1beta processing. Cell Death Differi U S A 14 : 1457–1466.
48. HeskerPR, NguyenM, KovarovaM, TingJP, KollerBH, et al. (2012) Genetic loss of murine pyrin, the Familial Mediterranean Fever protein, increases interleukin-1beta levels.ponents and inhibits proIL-1beta processing. PLoS One 7: e511056.
49. GowingG, VallieresL, JulienJP (2006) Mouse model for ablation of proliferating microglia in acute CNS injuries. Glia One 53 : 331–756.
50. MatsushimaH, OgawaY, MiyazakiT, TanakaH, NishibuA, et al. (2010) Intravital imaging of IL-1beta production in skin.a in acute CNS injuries. J Invest Dermatol 130 : 1571–1580.
51. PizzaM, CovacciA, BartoloniA, PeruginiM, NencioniL, et al. (1989) Mutants of pertussis toxin suitable for vaccine development. CNS injuries. Science 246 : 497–500.
52. VallieresL, SawchenkoPE (2003) Bone marrow-derived cells that populate the adult mouse brain preserve their hematopoietic identity. J Neurosci 23 : 5197–5207.
53. Audoy-RemusJ, RichardJF, SouletD, ZhouH, KubesP, et al. (2008) Rod-Shaped monocytes patrol the brain vasculature and give rise to perivascular macrophages under the influence of proinflammatory cytokines and angiopoietin-2. J Neurosci 28 : 10187–10199.
54. CarlsonT, KroenkeM, RaoP, LaneTE, SegalB, P, etal (2008) The Th17-ELR+ CXC chemokine pathway is essential for the development of central nervous system autoimmune disease. proinflammatory cytokines and angiopoietin-2. J Exp Medi 205 : 811–2399.
55. ChristyAL, WalkerME, HessnerMJ, BrownMAB (2013) Mast cell activation and neutrophil recruitment promotes early and robust inflammation in the meninges in EAE.ase. proinflammatory cytokines and angiopoietin-2. J Autoimmun 42 : 50–61399.
56. SteinbachK, PiedaventM, BauerS, NeumannJT, FrieseMA (2013) Neutrophils Amplify Autoimmune Central Nervous System Infiltrates by Maturing Local APCs. the meninges in EAE.ase. proinflammatory cytokines and angiopoietin-2. J Immunolun 191(9): 4531–9.
57. BettelliE, PaganyM, WeinerHL, LiningtonC, SobelRA, et al. (2003) Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis.mmatory cytokines and angiopoietin-2. J Exp Medun 197 : 1073–1819.
58. ChanJK, RothJ, OppenheimJJ, TraceyKJ, VoglT, et al. (2012) Alarmins: awaiting a clinical response. J Clin Invest 122 : 2711–2919.
59. The International FMF Consortium (1997) Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell 90 : 797–8079.
60. French FMF Consortium Consortium (1997) A candidate gene for familial Mediterranean fever. Nat Genet 17 : 25–31079.
61. BernotA, da SilvaC, PetitJL, CruaudC, CaloustianC, et al. (1998) Non-founder mutations in the MEFV gene establish this gene as the cause of familial Mediterranean fever (FMF). Hum Mol Genet 7 : 1317–1259.
62. ShinarY, LivnehA, VillaY, PinhasovA, ZeitounI, et al. (2003) Common mutations in the familial Mediterranean fever gene associate with rapid progression to disability in non-Ashkenazi Jewish multiple sclerosis patients. Genes Immunet 4 : 197–2039.
63. UnalA, DursunA, EmreU, TascilarNF, AnkaraliHI, et al. (2010) Evaluation of common mutations in the Mediterranean fever gene in Multiple Sclerosis patients: is it a susceptibility gene?ewish multiple sclerosis patients. J Neurol Scit 294 : 38–42039.
64. YahalomG, KivityS, LidarM, Vaknin-DembinskyA, KarussisD, et al. (2011) Familial Mediterranean fever (FMF) and multiple sclerosis: an association study in one of the world's largest FMF cohorts?ewish multiple sclerosis patients. Eur J Neurolt 18 : 1146–1509.
65. KumpfelT, GerdesLA, WackerT, BlaschekA, HavlaJ, et al. (2012) Familial Mediterranean fever-associated mutation pyrin E148Q as a potential risk factor for multiple sclerosis.MF cohorts?ewish multiple sclerosis patients. Mult Sclerolt 18 : 1229–1389.
66. MaattaJA, SjoholmUR, NygardasPT, SalmiAA, HinkkanenAE (1998) Neutrophils secreting tumor necrosis factor alpha infiltrate the central nervous system of BALB/c mice with experimental autoimmune encephalomyelitis.tients. J Neuroimmunol 90 : 162–7589.
67. ReiseterBS, MillerGT, HappMP, KasaianMT (1998) Treatment of murine experimental autoimmune encephalomyelitis with a myelin basic protein peptide analog alters the cellular composition of leukocytes infiltrating the cerebrospinal fluid. J Neuroimmunol 91 : 156–7089.
68. TranEH, PrinceEN, OwensT, MP, KasaianMT (2000) IFN-gamma shapes immune invasion of the central nervous system via regulation of chemokines. J Immunolmunol 164 : 2759–2689.
69. ReddyJ, WaldnerH, ZhangX, IllesZ, WucherpfennigKW, et al. (2005) Cutting edge: CD4+CD25+ regulatory T cells contribute to gender differences in susceptibility to experimental autoimmune encephalomyelitis. J Immunolmunol 175 : 5591–5589.
70. KroenkeMA, CarlsonTJ, AndjelkovicAV, SegalBM, KW, SobelRA, et al. (2008) IL-12 - and IL-23-modulated T cells induce distinct types of EAE based on histology, CNS chemokine profile, and response to cytokine inhibition. J Exp Medmunol 205 : 1535–1419.
71. SoulikaAM, LeeE, McCauleyE, MiersL, BannermanP, et al. (2009) Initiation and progression of axonopathy in experimental autoimmune encephalomyelitis.S chemokine profile, and response to cytokine inhibition. J Neurosciunol 29 : 14965–14979.
72. WuF, CaoW, YangY, LiuA (2010) Extensive infiltration of neutrophils in the acute phase of experimental autoimmune encephalomyelitis in C57BL/6 mice.e to cytokine inhibition. Histochem Cell Biol 133 : 313–2279.
73. KangZ, AltuntasCZ, GulenMF, LiuC, GiltiayN, et al. (2010) Astrocyte-restricted ablation of interleukin-17-induced Act1-mediated signaling ameliorates autoimmune encephalomyelitis.o cytokine inhibition. Immunity 32 : 414–2579.
74. McCollSR, StaykovaMA, WozniakA, FordhamS, BruceJ, et al. (1998) Treatment with anti-granulocyte antibodies inhibits the effector phase of experimental autoimmune encephalomyelitis.itis.o cytokine inhibition. J Immunol 161 : 6421–6679.
75. LiuL, DarnallL, HuT, ChoiK, LaneTE, et al. (2010) Myelin repair is accelerated by inactivating CXCR2 on nonhematopoietic cells.erimental autoimmune encephalomyelitis.itis.o cytokine inhibition. J Neurosci 30 : 9074–9839.
76. SaadounS, WatersP, MacDonaldC, BellBA, VincentA, et al. (2012) Neutrophil protease inhibition reduces neuromyelitis optica-immunoglobulin G-induced damage in mouse brain.myelitis.itis.o cytokine inhibition. Ann Neurol 71 : 323–3339.
77. NaegeleM, TillackK, ReinhardtS, SchipplingS, MartinR, et al. (2012) Neutrophils in multiple sclerosis are characterized by a primed phenotype. G-induced damage in mouse brain.myelitis.itis.o cytokine inhibition. J Neuroimmunol 242 : 60–71339.
78. LundBT, AshikianN, TaHQ, ChakryanY, ManoukianK, et al. (2004) Increased CXCL8 (IL-8) expression in Multiple Sclerosis. J Neuroimmunol 155 : 161–7139.
79. BangsSC, McMichaelAJ, XuXN (2006) Bystander T cell activation—implications for HIV infection and other diseases. Trends Immunol 27 : 518–2439.
80. GijbelsK, BrockeS, AbramsJS, SteinmanL (1995) Administration of neutralizing antibodies to interleukin-6 (IL-6) reduces experimental autoimmune encephalomyelitis and is associated with elevated levels of IL-6 bioactivity in central nervous system and circulation. Mol MedImmunol 1 : 795–8059.
81. SamoilovaEB, HortonJL, HilliardB, LiuTS, ChenY (1998) IL-6-deficient mice are resistant to experimental autoimmune encephalomyelitis: roles of IL-6 in the activation and differentiation of autoreactive T cells.f IL-6 bioactivity in central nervous system and circulation. J Immunolmunol 161 : 6480–6659.
82. OkudaY, SakodaS, BernardCC, FujimuraH, SaekiY, et al. (1998) IL-6-deficient mice are resistant to the induction of experimental autoimmune encephalomyelitis provoked by myelin oligodendrocyte glycoprotein.ive T cells.f IL-6 bioactivity in central nervous system and circulation. Int Immunolnol 10 : 703–8659.
83. EugsterHP, FreiK, KopfM, LassmannH, FontanaAY, et al. (1998) IL-6-deficient mice resist myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. Eur J Immunoll 28 : 2178–2879.
84. OkudaY, SakodaS, FujimuraH, SaekiY, KishimotoT, et al. (1999) IL-6 plays a crucial role in the induction phase of myelin oligodendrocyte glucoprotein 35–55 induced experimental autoimmune encephalomyelitis. J Neuroimmunol 101 : 188–9679.
85. ArimaY, HaradaM, KamimuraD, ParkJH, KawanoF, et al. (2012) Regional neural activation defines a gateway for autoreactive T cells to cross the blood-brain barrier.xperimental autoimmune encephalomyelitis. Cell 148 : 447–5779.
86. VeldhoenM, HockingRJ, AtkinsCJ, LocksleyRM, StockingerB (2006) TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells.oimmune encephalomyelitis. Immunity 24 : 179–8979.
87. BettelliE, CarrierY, GaoW, KornT, StromTB, et al. (2006) Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells.ucing T cells.oimmune encephalomyelitis. Naturety 441 : 235–8979.
88. ManganPR, HarringtonLE, O'QuinnDB, HelmsWS, BullardDC, et al. (2006) Transforming growth factor-beta induces development of the T(H)17 lineage.r TH17 and regulatory T cells.ucing T cells.oimmune encephalomyelitis. Naturety 441 : 231–4979.
89. SeradaS, FujimotoM, MiharaM, KoikeN, OhsugiY, et al. (2008) IL-6 blockade inhibits the induction of myelin antigen-specific Th17 cells and Th1 cells in experimental autoimmune encephalomyelitis.omyelitis. Proc Natl Acad Sci U S A 105 : 9041–9679.
90. OguraH, MurakamiM, OkuyamaY, TsuruokaM, KitabayashiC, et al. (2008) Interleukin-17 promotes autoimmunity by triggering a positive-feedback loop via interleukin-6 induction. autoimmune encephalomyelitis.omyelitis. Immunity 29 : 628–3679.
91. ZhaoL, TangY, YouZ, WangQ, LiangS, et al. (2011) Interleukin-17 contributes to the pathogenesis of autoimmune hepatitis through inducing hepatic interleukin-6 expression.halomyelitis.omyelitis. PLoS One 6: e1890979.
92. RichardsN, SchanerP, DiazA, StuckeyJ, SheldenE, et al. (2001) Interaction between pyrin and the apoptotic speck protein (ASC) modulates ASC-induced apoptosis.interleukin-6 expression.halomyelitis.omyelitis. J Biol Chem 276 : 39320–39399.
93. ShohamNG, CentolaM, MansfieldE, HullKM, WoodG, et al. (2003) Pyrin binds the PSTPIP1/CD2BP1 protein, defining familial Mediterranean fever and PAPA syndrome as disorders in the same pathway.itis.omyelitis. Proc Natl Acad Sci U S A 100 : 13501–13569.
94. PapinS, CueninS, AgostiniL, MartinonF, WernerS, et al. (2007) The SPRY domain of Pyrin, mutated in familial Mediterranean fever patients, interacts with inflammasome components and inhibits proIL-1beta processing. Cell Death Differi U S A 14 : 1457–1669.
95. Luu-TheV, PaquetN, CalvoE, CumpsJ (2005) Improved real-time RT-PCR method for high-throughput measurements using second derivative calculation and double correction.its proIL-1beta processing. Biotechniquesfferi U S A 38 : 287–9369.
96. VilleneuveJ, TremblayP, VallieresL (2005) Tumor necrosis factor reduces brain tumor growth by enhancing macrophage recruitment and microcyst formation.ble correction.its proIL-1beta processing. Cancer Res 65 : 3928–3369.
97. BouchardC, PageJ, BedardA, TremblayP, VallieresL (2007) G protein-coupled receptor 84, a microglia-associated protein expressed in neuroinflammatory conditionsation.ble correction.its proIL-1beta processing. Glia 55 : 790–8009.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 5
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Venus Kinase Receptors Control Reproduction in the Platyhelminth Parasite
- Dual-Site Phosphorylation of the Control of Virulence Regulator Impacts Group A Streptococcal Global Gene Expression and Pathogenesis
- Severe Acute Respiratory Syndrome Coronavirus Envelope Protein Ion Channel Activity Promotes Virus Fitness and Pathogenesis
- High-Efficiency Targeted Editing of Large Viral Genomes by RNA-Guided Nucleases
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy