
Large Scale RNAi Reveals the Requirement of Nuclear Envelope Breakdown for Nuclear Import of Human Papillomaviruses
Certain human papillomaviruses (HPV) are the etiological cause of cervical cancers and other epithelial tumors. Recent advances in the development of anti-HPV vaccines and their increasing deployment provide hope for a significant decrease of these cancers in the future. However, many details of the transmission of HPV between infected and uninfected individuals are still not well understood. In particular, the multistep process of entry into host cells is only partially characterized. Here, we used a systematic RNAi silencing approach to identify host cell proteins required during HPV16 infection, the most prevalent cancer-causing HPV. Our results indicated that HPV entry requires cells to divide. Cell division is important, since it removes the barrier between the nucleoplasm and the cytosol, so that the virus can access the nucleus for transcription and replication. Our study provides new mechanistic insights into the entry process of an important pathogen, and explains why it can only infect the stem cells or transiently amplifying cells of human skin or mucosa. This work also highlights a novel nuclear import strategy for DNA viruses, which typically use the nuclear import machinery to access the nucleoplasm for infection, Thus, HPV entry displays another unique aspect uncommon to most other viruses.
Published in the journal:
. PLoS Pathog 10(5): e32767. doi:10.1371/journal.ppat.1004162
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004162
Summary
Certain human papillomaviruses (HPV) are the etiological cause of cervical cancers and other epithelial tumors. Recent advances in the development of anti-HPV vaccines and their increasing deployment provide hope for a significant decrease of these cancers in the future. However, many details of the transmission of HPV between infected and uninfected individuals are still not well understood. In particular, the multistep process of entry into host cells is only partially characterized. Here, we used a systematic RNAi silencing approach to identify host cell proteins required during HPV16 infection, the most prevalent cancer-causing HPV. Our results indicated that HPV entry requires cells to divide. Cell division is important, since it removes the barrier between the nucleoplasm and the cytosol, so that the virus can access the nucleus for transcription and replication. Our study provides new mechanistic insights into the entry process of an important pathogen, and explains why it can only infect the stem cells or transiently amplifying cells of human skin or mucosa. This work also highlights a novel nuclear import strategy for DNA viruses, which typically use the nuclear import machinery to access the nucleoplasm for infection, Thus, HPV entry displays another unique aspect uncommon to most other viruses.
Introduction
Papillomaviruses are small, nonenveloped DNA viruses that cause mostly benign lesions [1]. Several human papillomavirus (HPV) types are, however, etiological agents for anogenital cancers, and tumors of the head and neck [1]. Among them, HPV16 is the most prevalent, and the best studied [2]. No specific drug treatment against HPV-related cancers exists, but the increasing deployment of anti-HPV vaccines will likely help to reduce the incidence rate of the vaccine types [3]. The spherical HPV particles have an icosahedral (T = 7) architecture, and a diameter of 50–55 nm. The capsid is formed by 72 homo-pentamers of the major structural protein, L1, and contains in addition 12–72 copies of the minor structural protein, L2 [4]. The 8kB DNA genome is circular and double-stranded. During initial infection, HPVs enter basal stem cells or transiently amplifying cells in squamous epithelia [1]. Replication and assembly of new viruses occur in the nucleus, when the host cells differentiate into spinous and granular keratinocytes [1].
Since authentic viruses are difficult to be propagated in sufficient amount and purity, most of what is known about early interactions between HPV and host cells has been learned using virus-like particles (VLPs) and pseudoviruses (PsV). These surrogate particles have the protein composition and architecture of the authentic HPV, but do not carry the viral genome[5], [6]. Instead, the PsV contain a pseudogenome that, when delivered to cells, expresses a reporter protein [6], [7].
Binding of VLPs and PsV of HPVs to tissue culture cells depends on heparan sulfate proteoglycans (HSPG) [8]–[11]. Highly sulfated domains of the HS induce the first of several conformational changes in the incoming virion [12]–[14]. The next change is caused by cyclophilin B, a peptidylprolyl isomerase [15], [16]: The N-terminal peptide of L2 buried within the capsid is exposed so that it can be cleaved by furin [14], [15], [17], [18]. The alterations in the capsid lead to a loss of affinity to HSPGs and transfer to a secondary receptor [8], [14], [15], [19], [20], such as alpha6-integrin, annexin A2 heterotetramer, growth factor receptors, and the tetraspanin CD151 [21]–[28]. However, the exact functions of these receptors in PV entry is disputed [29].
HPV entry into host cells requires endocytosis. Compared with other animal viruses, endocytic uptake is slow and asynchronous (t1/2 = 4–12 h)[8], [30], [31]. Reporter gene expression of the pseudogenome is not detected until 24–48 h postinfection (p.i.) [31]. The endocytic mechanism used by high-risk HPV16, 18, and 31 is ligand-induced, clathrin-, caveolin-, lipid raft-, and dynamin-independent, and critically dependent on actin dynamics [27], [31], [32]. The viruses are internalized in small vesicles, transported to late endosomes or endolysosomes, and the Golgi complex [31], [33], [34].
The subsequent steps are poorly understood. It is known, however, that a late step involving penetration into the cytosol or initial uncoating of the capsid requires exposure of the virus to low pH [31]. Also, the major capsid protein L1 dissociates from a subviral complex composed of the viral DNA (vDNA) and L2 probably within endosomal compartments [33], [35]. In transit to the nucleus, the subviral complex is routed to the Golgi complex [33], [34]. After penetration into the cytosol [36], the vDNA moves complexed with L2 into the nucleus and eventually localizes to ND10 nuclear domains [37], [38]. How nuclear import of this subviral complex occurs is not known. Of note, entry of HPV16 requires progression of cells through the cell cycle into early mitosis [39]. However, it is unclear which entry step is influenced by cell cycle progression.
In this study, we used a large-scale RNA interference (RNAi) screen to identify host cell factors and processes involved in HPV entry. Such screens have emerged as a powerful tool to analyze pathogen-host interactions [40]. By focusing our follow-up studies on a cluster of genes that suppresses HPV16 infectivity, we uncovered a crucial role for mitosis in HPV16 nuclear entry. Mitosis was important, since nuclear import of the HPV16 vDNA genome requires changes in nuclear envelope (NE) permeability facilitated by nuclear envelope breakdown (NEB). These changes allowed association of the subviral complex with host cell chromatin aligned on the metaphase plate.
Results
RNAi screening
To identify cellular factors and processes involved in HPV16 entry, we performed an automated, high-content, high-throughput RNAi screen in human epithelial cells (HeLa) (Figure 1A). For infection, PsV were used that contained a pseudogenome encoding the green fluorescent protein (GFP) under the control of the cytomegalovirus (CMV) early promoter (HPV16-GFP). Expression of GFP allowed identification and quantification of cells in which the pseudogenome had been successfully delivered to the cytosol, uncoated, imported into the nucleus, transcribed, and translated.

The number of cells and the fraction of GFP-expressing cells were determined 36 h p.i. using automated microscopy (Figure S1). The multiplicity of HPV16 was kept low so that only about 10% of the cells in control wells were positive. This allowed us to not only identify genes required for infection, but also those that act as enhancer genes, i.e. proteins that increased the fraction of infected cells when depleted. Moreover, using a low amount of siRNA (5 nM), we aimed to minimize off-target effects and reduce the number of false-positive hits. To increase reliability, we further used a fully automated computational pipeline for high-content image analysis [41]. Several iterations of supervised machine learning were applied to identify virus infection, mitosis, apoptosis, and technical phenotypes.
The RNAi screen was carried out in two steps. Three non-overlapping siRNAs against each gene were used in a primary screen, and three additional siRNAs in a secondary follow-up screen. In the primary screen, 7,000 human genes selected for the potential to be inhibited by small compounds (the druggable genome library, Qiagen) were screened. The library encompasses a well-studied and well-annotated cross-section of genes. Most of the genes have known functions, and many inhibitors and other reagents are readily available for follow-up studies. The primary screen, which was performed three times, led to the identification of 315 genes in which the median of the three siRNAs reproducibly reduced or enhanced infection by at least 3 robust z-scores (Table S1, all). To provide a resource for future studies of HPV-host cell interaction, the full set of images and analysis results is available at http://www.infectome.org.
The follow-up screen was directed against the genes identified as hits in the primary screen. It made use of three additional siRNAs per gene from another vendor (Ambion) except for a few genes against which no additional siRNAs were available (Table S1, red). The secondary screen was also repeated three times. It confirmed 162 (56%) of the tested 291 hits from the primary screen, when the same criteria as the primary screen were applied.
The final hit list represented a broad range of cell functions (Table S1). Among these functions, several have been previously implicated in HPV16 infection: e.g. phosphoinositide 3-kinase (PI3K) signaling (represented by PI3K subunits PIK3CA, PIK3CB), sodium/proton exchange (SLC9A6, SLC9A8, SCNN1B), minus-end directed microtubular transport (motor complex dynein (DYNC1H1, DYNC1L1I2); microtubule-regulating kinases (MARK1, MASTL)), tyrosine kinase signaling (LMTK2, LCK), dephosphorylation (phosphatases (INPP5B, PtPN4, PPP2R5C, ACP5)), signaling by the small G-protein Ras (HRAS, RASA4), and the matrix metallopeptidases (MMP12) [21], [31], [42]–[44]. Other hits, such as metabolic and catabolic proteins, or of the biosynthetic machinery were expected for any virus. However, the majority of hits had no prior connection to HPV16 infection.
To extract information regarding critical processes in HPV entry, the list was subjected to rigorous bioinformatic analysis. An algorithm that combined function - and interaction-based information was used [45]. When assigned to functional annotation clusters representing sets of proteins that share common annotations within public databases [DAVID; [46]], the hits were enriched within 32 clusters and several highly connected, functional annotation networks (Table S2). The function-based information was combined next with data on protein interactions among individual host factors [STRING; [47]].
The result of the combined analysis is shown for 117 host factors in Figure 1B (compare Figure S2, in which the hits were subdivided by their effect, i.e. reducing or enhancing infection upon knockdown). Automatic annotation was used to visualize the interaction networks (gray to black lines) both within and between functional annotation clusters (dashed boxes). A high number of cellular regulators appeared to be hits, ranging from receptor molecules, kinases, transporters, and ion channels. Since the regulation of the cellular processes leading to entry, such as endocytosis and intracellular trafficking are poorly characterized with regard to signal transduction and the influence of ion flux regulation, these findings are likely to open new avenues in HPV and cell biological research.
To us the most interesting was a cluster of mitosis regulators such as AURKB, ANAPC and INCENP. It showed many interconnections within and outside the cluster (Figure 1B). The high abundance of genes and multiple interconnections among them highlighted the importance of this cluster. Interestingly, most of these genes enhanced infection upon knockdown (Figure S2A). In contrast, genes in a cluster known to regulate nucleoplasm architecture such as HDAC11, CCNE2, and POLI reduced infection upon depletion (Table S2).
Infection is completed during mitosis
To follow up on the involvement of the mitotic and cell cycle regulators in HPV16 entry, we first characterized the temporal relationship between mitosis and HPV infection. For this, we followed the GFP expression from HPV16-GFP in relation to chromosome condensation, segregation, and decondensation in HeLa cells that stably express the histone 2B tagged with mCherry (H2B-mCherry [48]). It was clear from this analysis that GFP expression only occurred if mitosis had taken place as monitored by chromosome segregation (Figure 2A first row, Video S1). In fact, the GFP signal was first detected on average 4.5 h after chromosome decondensation in the newly formed nuclei (Figure 2B). Given that expression and functional chromophore formation of the GFP in vitro require 0.5 and 3–3.5 h, respectively [49], the time course suggested that the establishment of HPV infection correlated with completion of mitosis. That mitosis defined the onset of viral gene expression was further supported by the observation that the onset of GFP expression did not change with the number of pseudogenomes delivered into cells (Figure 2B).

Entry into early mitosis is required for infection
Next, we aimed to relate our finding that mitosis defined the onset of viral gene expression to previous findings that indicated the requirement for cell cycle progression into early mitosis for infection [39]. To corroborate the cell cycle requirements for infection in HeLa and HaCaT cells, we used inhibitors known to block at three specific stages. Progression in the synthesis (S-)phase was blocked by aphidicolin, gap2-phase to mitosis (G2/M) transition by purvalanol A or CGP74514A, and chromosome segregation/cytokinesis during mitosis by monastrol (Figure 2C). As expected, the inhibitors caused significant changes in the fraction of cells in G1-, S-, and G2/M-phase (Figure S3).
Arresting cells in S-phase by aphidicolin blocked infection efficiently (Figure 2D) and reversibly (Figure 2E). In line with our observations in fixed cells, GFP expression could not be detected in live cells arrested in S-phase (Figure 2A middle row, Video S2). This effect was not limited to transformed cells, as infection of primary human keratinocytes (PHK) was blocked by aphidicolin (Figure 2F). S-phase arrest by aphidicolin also blocked infection of HPV6 and HPV18 PsV, and of raft-derived HPV16 (Figure 2G, H). Together, this suggested that cell cycle progression through S-phase was required for infection of various HPV types independent of whether PsV or raft-derived virions were used for infection of transformed or primary cells. Similar to S-phase arrest, inhibition of the G2/M transition by purvalanol A and CPG74514A prevented infection (Figure 2D). In contrast, if mitosis was interrupted at the stage of chromosome segregation by monastrol treatment, GFP expression did occur (Figure 2D). Similarly, GFP expression occurred in live cells treated with monastrol (Figure 2A last row, Video S3). Monastrol prevents monopolar spindle formation after NEB by inhibiting the kinesin KIF11 [50], [51]. This was also obvious in the live HeLa cells, which featured condensation, erratic alignment, and decondensation of chromosomes consistent with mitotic progression into prometaphase/metaphase followed by a block in chromosome segregation and cytokinesis. Thus, our results corroborated that early steps in mitosis were important for HPV16 entry, but later steps such as spindle segregation and cytokinesis were not. The early stages of mitosis are complex and highly regulated. They involve changes in cytoskeletal organization, vesicular transport, permeability of the nuclear envelope induced by NEB, and transcriptional regulation [52]. As hypothesized previously [39], any of these changes may affect HPV16 entry.
Nuclear import of HPV16 is blocked in interphase cells
Earlier studies indicated that HPV16 particles bind to all cells in a population [19], [31], [53], i.e. irrespective of their cell cycle phase. The key question therefore was which step in the entry program of the virus is inhibited in interphase cells. To study this question, we followed the fate of the PsV in aphidicolin-treated, S-phase-arrested cells and analyzed the efficacy of virus endocytosis, intracellular trafficking of viruses to the perinuclear area, viral uncoating, and nuclear import of vDNA.
First, the endocytic uptake of fluorescently labeled particles into cells was analyzed in S-phase arrested cells by flow cytometry. Internalization was unaffected (Figure S4A). When a monoclonal antibody against L1 (L1-7) that detects partially uncoated particles but not mature virions [27], [54] was used to localize the virus, a perinuclear accumulation was observed in both control and S-phase-arrested cells (Figure 3A). This indicated that intracellular trafficking and partial uncoating of the endocytosed viruses occurred as efficiently as in unperturbed cells. Neither the number and the intensity of fluorescent spots, nor their 3D distance from the nuclear border significantly differed in cells arrested in S-phase (Figure 3B–D). An inhibitor exchange experiment indicated moreover that the incoming viruses passed the acid-dependent step in interphase cells as well. In this experiment, infection for 24 h in the presence or absence of aphidicolin was followed by NH4Cl treatment for 48 h. Infection in the aphidicolin-treated cells was normal compared to the control (Figure S4B). Since NH4Cl prevents low pH-activation of HPV16 in endosomes [31], we concluded that the aphidicolin-induced block occurred after low pH activation of the virus in endosomes or the trans-Golgi-network (TGN).
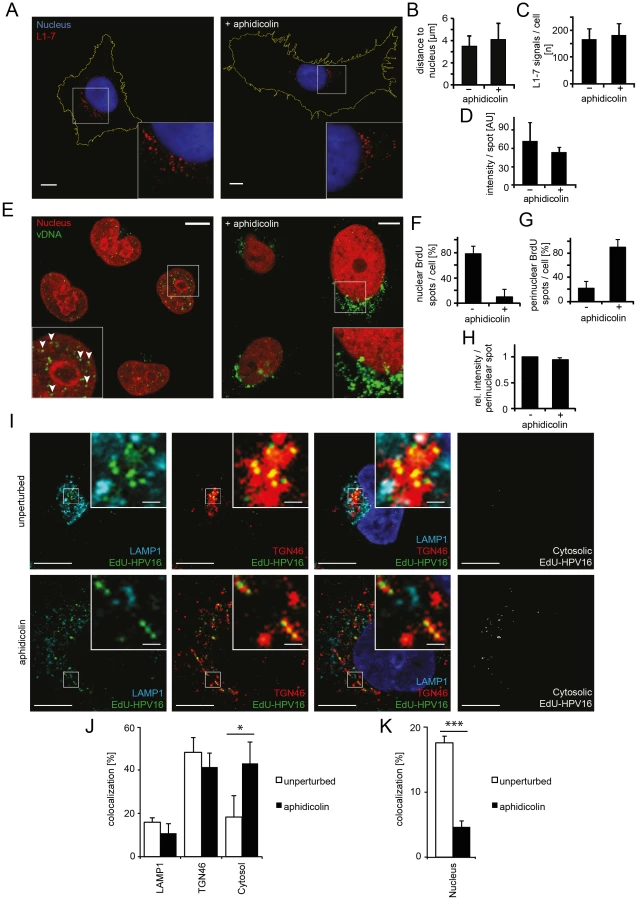
Finally, the exposure (uncoating) and intracellular localization of vDNA were analyzed. Cells were infected with HPV16 that contained DNA labeled with 5-bromo-2′-deoxy-uridine (BrdU), a nucleoside analog that allows immunodetection after partial uncoating of the DNA [38]. At 24 h p.i., the vDNA was readily detected in the nuclei of infected cells with BrdU-specific antibodies (Figure 3E, left). In cells arrested in S-phase, the vDNA was detectable to a similar extent as in untreated cells (Figure 3E, right, 3H) suggesting that the vDNA was accessible to antibodies. However, the signal was located in the perinuclear area instead of within the nucleoplasm (Figure 3E–G). To more thoroughly analyze the intracellular localization of the viral DNA in aphidicolin-treated cells, PsV containing 5-ethynyl-2′-deoxyuridine (EdU)-labeled vDNA (EdU-HPV16) were used for infection, which allowed a more sensitive detection. Irrespective of whether cells were arrested in S-phase or not, the vDNA localized to the same extent to LAMP1-, and TGN46-positive structures (Figure 3I, J). Since LAMP1 and TGN46 are markers for late endosomes and the TGN, respectively, the results supported that vesicular trafficking of the virus was unperturbed. The nuclear localization of EdU-labeled vDNA was clearly reduced in aphidicolin-treated cells (Figure 3K), as observed for the BrdU-labeled vDNA. Instead, the proportion of virus that did not localize to LAMP1 - and TGN46-positive structures, or the nucleus, increased significantly (Figure 3J). It is likely that this vDNA signal reflected the localization of the subviral complex to the cytosol after penetration, as our previous results indicated no perturbation in HPV internalization, vesicular trafficking and uncoating.
Our observations, thus, indicated that whereas endocytic uptake of the virus, intracellular transport, acid-activation, and uncoating were normal in interphase cells, the vDNA failed to be imported into the nucleus from the cytosol. Taken together, these data showed that mitosis was required for infection, because it allowed nuclear import of the viral genome.
HPV16 does not require nuclear pore complexes for nuclear import
Viruses that replicate in the nucleus generally exploit the nuclear import machinery of the cell and nuclear pore complexes in the nuclear envelope [55]–[57]. S-phase arrest typically does not impair nuclear import (and export) through nuclear pore complexes, or of viruses that replicate in the nucleus [58], [59]. However, to rule out a perturbation in nucleo-cytoplasmic shuttling, we infected aphidicolin-treated cells with Herpes Simplex Virus type 1 (HSV-1) that requires functional nuclear pore complexes for import of its DNA genome and subsequent infection [60]. A recombinant HSV-1 that expresses GFP under the control of the same promoter as in the HPV16 PsV reporter constructs was used [7], [61]. It allowed us to monitor effects on gene expression in addition to nuclear import of HSV-1 vDNA.
We found that cells were infected by HSV-1 to the same extent whether the cells were arrested in S-phase or not (Figure S5A, B, Video S4). Therefore, neither the nuclear pore machinery nor the subsequent gene expression appeared to be affected by this treatment. Also, the nuclear import of the importin beta binding domain fused to GFP (IBB-GFP) occurred to a similar extent in unperturbed and S-phase arrested cells (Figure S5C, D) indicating that karyopherin-mediated transport was unaffected.
Since the nucleo-cytoplasmic shuttling appeared not to be impaired under our experimental conditions, it was unlikely that the block in HPV16 vDNA import upon S-phase arrest was caused by perturbation of the nuclear pore complexes. To substantiate this, we impaired nuclear import by depleting NUP153 by RNAi. NUP153 is a nuclear pore protein essential for nuclear pore basket formation, nuclear pore complex anchoring and import of a variety of nuclear proteins [62]. While NUP153 knockdown clearly reduced HSV-1 infection, HPV16 infection was largely unaffected (Figure S5E). Taken together, the data indicated that HPV16 did not require nucleo-cytoplasmic transport via nuclear pore complexes for import of the vDNA.
Transient loss of NE barrier function facilitates HPV16 infection
So how does the vDNA of HPV16 enter the nucleoplasm? One possibility is that it relies on NEB, which removes the nucleo-cytoplasmic barrier during mitosis. The NE is disassembled during G2/M transition, and reassembles during telophase after segregation of the chromosomes prior to cytokinesis [52]. It is thought that the provirus of certain gamma retroviruses uses this time window to access the nucleoplasm [63].
If HPV16 uses the same strategy as the gamma retroviruses, one would expect that damage to the integrity of the NE would allow the vDNA resident in the cytoplasm to gain access to the nucleus. To induce perforation of the nuclear membranes without mitosis, we made use of parvovirus H1, a small DNA virus known to enter the nucleus by inducing holes in the NE [64]. These hole are large enough to allow sudden entry of antibodies into the nucleus and, at least in some cells, chromatin leakage into the cytoplasm. To confirm that H1 induced NE perforation in our cells, we monitored the fluorescence signal of IBB-GFP. In unperturbed cells, IBB-GFP was found to be intranuclear during interphase but dispersed in the cytosol during mitosis as a result of NEB, as expected (Figure 4B, Video S5, [65]). The IBB-GFP signal persisted in the nucleus in aphidicolin-treated cells because neither mitosis nor NEB occurred (Figure 4C, Video S6). In contrast when incubated with H1, 20±13% of the aphidicolin-treated cells exhibited translocation of the IBB-GFP signal to the cytosol consistent with the expected extent of NE perforation (Figure 4D, E, Video S7, [64], [65]).

When S-phase-arrested cells were infected with HPV16, and the NE was perforated by superinfection with H1 (Figure 4A), a small but readily detectable number of cells (1±0.5%) was infected suggesting that H1-induced perforation of the NE was sufficient for productive entry of vDNA into the nucleus (Fig 4F, G, Video S8). Without H1 there was no detectable infection (Figure 4G, Video S2). These findings were consistent with the idea that vDNA/L2 complexes cannot enter the nucleus as long as the NE is intact. Therefore, it is likely that mitosis is necessary for infection, because it causes a transient loss of the barrier function provided by the NE. The small number of infected cells after the transient NE perforation indicated that although sufficient, NE perforation is not highly efficient to allow nuclear import. This is likely due to the short, temporary state of NE perforation combined with the restricted diffusion of the subviral complex previously observed for other macromolecular complexes [66].
The vDNA and L2 associate with host cell chromosomes on the metaphase plate during mitosis
If the subviral DNA/L2 complex gains access to the nucleoplasm after NEB, the question remained of how the complexes ensure incorporation into the newly forming daughter nuclei. To answer this question, we analyzed the localization of EdU-labeled vDNA in mitotic cells after infection of unperturbed cells with EdU-HPV16. During mitotic progression the vDNA spots were found to be increasingly associated with condensed host cell chromatin (Figure 5A, B). The association increased from prophase over prometaphase to metaphase, after which no further increase during ana - and telophase was observed (Figure 5A, B). This suggested that the vDNA associated with the host cell chromosomes after NEB.

To analyze the dynamics of association of the subviral complex with condensed host cell chromatin during mitosis, and the contribution of L2, we expressed L2 fused to GFP and followed their intracellular localization in live HeLa H2B-mCherry cells. As expected, L2-GFP localized to the nucleus during interphase (Figure 5C, Video S9), since it is imported by karyopherin-mediated transport [67]. Upon nuclear envelope breakdown, it was dispersed throughout the cytosol (Figure 5C, Video S9). Upon metaphase plate formation, L2-GFP associated with host cell chromatin, and remained associated with it throughout cytokinesis (Figure 5C, Video S9). These findings supported a model in which the association of the subviral complex with condensed host cell chromatin was mediated by L2. Since a visible L2-GFP association with host cell chromatin was observed almost exclusively after metaphase plate formation, i.e. mostly within 5–15 min post metaphase plate formation (Figure 5D), it is likely that the subviral complex is recruited to host cell chromatin specifically during this time window.
The timing of NE absence is rate-limiting for infection
These findings explained, why HPV16 nuclear entry was blocked until mitosis occurred during cell cycle progression. However, the question remained why RNAi-mediated silencing of certain mitosis regulators would enhance infectivity of HPV16.
To analyze whether any particular mitotic phenotype may correlate with enhanced infection, we depleted some of the mitotic regulators that enhanced HPV infection or, as controls, genes that reduced infection. We examined this in HeLa cells expressing H2B-mRFP and IBB-GFP, and determined whether defects in mitosis could be observed by live cell microscopy.
A variety of mitotic phenotypes were observed including defects in metaphase plate formation, in chromosome segregation, and in cytokinesis (Figure 6A), but entry into mitosis and NEB readily occurred (Figure 6A–C, Table S3). These phenotypes did not correlate with the effect on infection. The only correlation with infection was the duration of the time window during which the NE was absent. Prolonged NE absence always correlated with enhanced infection (Figure 6C, Table S3, Videos S10–14, compare Table S1). In fact, consistent with prolonged mitosis, RNAi of genes that enhanced infection resulted on average in a significantly increased mitotic index (≥1.5 z-scores) over those that reduced infection (Figure S6). This was supported by additional observations: First, careful analysis of live cell videos of individual monastrol-treated cells showed that the longer mitosis lasted, the earlier GFP expression from HPV16 infection could be detected, following completion of the mitosis (Figure S7A). Secondly, when examining cell divisions with and without added virus, we observed that, although HPV16 infection did not affect the number of mitoses per cell number, it did increase the percentage of prolonged mitosis events, 9 versus 27% (Figure S7B–E). Third, expression of L2-GFP induced a prolongation of mitosis (Figure S7F, compare Video S14, 15). The duration of mitosis increased with higher amounts of L2-GFP signal (Figure S7F). If higher amounts of L2-GFP were present in cells (i.e. ≥100% signal above background), these cells entered mitosis, but failed to divide, and subsequently became apoptotic, indicating that the checkpoint was not overcome (Video S16). Taken together, these observations indicated that the time of NE absence was rate-limiting for infection and nuclear import of the vDNA, and that association of L2 with host cell chromatin was able to increase the length of mitosis thus favoring nuclear import.

To test whether prolongation of mitoses enhanced the probability of nuclear import (and infection), we depleted cells of mitotic regulators from the screen such as AURKB, INCENP, and ANAPC by RNAi to induce prolonged mitosis. They were then infected for 24, 36, and 48 h to cover the time when the first cells expressed GFP to the time when the number of GFP expressing cells reached maximum [31]. Strikingly, the infectivity was strongly enhanced at 24 h p.i. in cells depleted of the mitotic regulators, whereas the effects at 48 h p.i. were minor (Figure 6D). These data were consistent with a higher probability of vDNA localization to the nucleoplasm if the time window without an intact NE was longer.
To verify our hypothesis, we depleted two additional genes by RNAi that prolong mitosis according to published work (the mitotic phosphatase PPP2R2A and the centromere protein CENPE, [68], [69]). As expected, depletion of PPP2R2A and CENPE led to prolongation of mitosis (Figure S8A, Table S3, S4). When PPP2R2A - and CENPE -depleted cells were infected with HPV16-GFP, infection was increased (Figure S8B), which supported the notion that the length of NE absence may be a kinetic bottleneck for import of the vDNA during mitosis.
Discussion
To screen for cell factors that influence entry and other early events in the HPV infection cycle, we employed a two-step strategy with a total of six siRNAs per gene. We relied on a robust, image-based infection assay, supervised classification of phenotypes and an improved bioinformatic pipeline [41], [45]. To avoid some of the pitfalls inherent in the RNAi approach [40], we focused our validation on gene clusters rather than individual hits.
The primary screen involved 7000 human genes targeted in the so-called druggable library of siRNAs (Qiagen). Of the 315 potential hits, 291 were followed up in a secondary screen with additional siRNAs, and 162 could be confirmed. Our confidence in the results was strengthened by the identification of several factors previously described to be involved in HPV16 infection. The results of the follow-up studies demonstrated that not only genes that give reduced infectivity after silencing can lead to novel insights but also those hits that show enhanced infection.
A recent genome-wide RNAi-screen for HPV16 PsV infection in HeLa-S3 cells revealed the importance of retrograde membrane trafficking to the Golgi complex and a role for the retromer as an important determinant in HPV16 entry into cells [34]. Out of the 261 and 162 hits identified by Lipovsky et al. and by us, respectively, four were common (Figure S9). This low number of overlapping genes is not unusual when RNAi screens are compared, and the overlap typically increases significantly, when clusters of genes are compared [40], [41], [70]. This was also the case for the two HPV screens when significantly enriched functional annotation clusters were compared (Figure S9). For example, both studies identified the importance of a cluster associated with the Golgi complex. A more thorough comparison was not possible, because the results of the Lipovsky screen are only partially published.
Besides the Golgi cluster, the bioinformatic analysis of our screen revealed a number of clusters with multiple hits associated with known pathways or complexes. The most prominent was a set of genes involved in the regulation of mitosis and the cell cycle. This cluster has not been observed in screens performed with other viruses by us or published by others ([40], [45], [71], unpublished data), and may be unique to papillomaviruses. Most of the genes in the cluster were inhibitory, i.e. they showed enhanced infection after silencing suggesting that they function to limit infection. Moreover, it was striking that the hit list failed to include nuclear import factors or nuclear pore components that have been seen as prominent hits in screens against other viruses including those that replicate in the nucleus [40], [45]. This suggested that, unlike these other viruses, HPV may not be using the nuclear pores and nuclear transport receptors for accessing the nucleus during the host cell entry.
Our results showed that the virus was efficiently internalized by endocytosis, routed to the perinuclear area, and partially uncoated in interphase cells. However, the viral DNA failed to enter the nucleus. Therefore no transcription of the marker protein GFP occurred. Mitosis was the key step that allowed infection to proceed. The expression of GFP from the vDNA correlated perfectly with mitosis; the GFP became detectable 4.5±0.4 h after mitosis was completed. Moreover, since late steps in mitosis such as chromosome separation and cytokinesis were not required, the critical process could be narrowed down to one of the early steps.
That the critical step was the disruption of the NE was indicated by two observations. First, co-infection experiments in interphase cells showed that transient NE perforation by parvovirus H1 allowed HPV infection. This implied that the viruses were able to reach a stage in their entry program where they were ready to enter the nucleus productively, but could only do so if the NE integrity was compromised. Secondly, analysis of some of the hits from the siRNA screen indicated that mitosis provided a kinetic bottleneck for the nuclear import of viral genomes. The average time during mitosis when the NE is disrupted is about 45–60 min in HeLa cells. After depletion of INCENP, ANAPC, and AURKB, the time window was extended 3–5 fold resulting in a doubling or more in infection. This suggested that the longer the period without a functional membrane barrier, the better the chances for the virus to be productively included when daughter nuclei were formed. We also noted that while the addition of HPV16 itself did not promote division in the cell population, it did cause a significant increase in the fraction of cells that displayed prolonged mitosis, a process that appears to depend on the amount of L2 associating with host cell chromatin. This may be a mechanism for the virus to promote vDNA import into nuclei, a difficult step in the entry program. The results were consistent with observations of Pyeon and coworkers, who showed that cell cycle progression into early mitosis is required for HPV16 PsV infection [39]. Our data allowed us to define the key step as the breakdown of the NE barrier. Since raft-derived HPV16, as well as HPV6 and 18 PsV exhibted similar requirements, it is likely that cell cycle progression into early mitosis represents a general requirement for papillomavirus infection.
We propose that after virus internalization, trafficking, conformational alterations, and penetration into the cytosol, L2/vDNA complexes wait in the perinuclear space until NEB occurs. When the nuclear components are rendered accessible, the subviral complexes associate with the condensed chromatin, and are included in newly forming daughter cell nuclei. Such a mechanism has not been observed for other viruses that replicate in the nucleus except for certain retroviruses that also depend on mitosis for provirus integration [57], [63]. Most other DNA viruses make use of the nuclear import machinery and the nuclear pore complexes.
The inclusion of the HPV16 genome into the newly forming nuclei involves the participation of the minor capsid protein L2, since it is the only viral protein that accompanies the vDNA into the nucleus [17], [35], [38]. L2 contains two functional nuclear localization signals (NLSs) [67].While these are necessary for nuclear import during virus assembly, they are hardly important for nuclear inclusion during virus entry. In fact, the N-terminal NLS is cleaved off by the cellular protein convertase furin in an earlier stage of entry [11], [17], [18]. The C-terminal NLS is involved in binding both to dynein light chains and to the vDNA [43], [72], [73]. Strikingly, L2 associated with condensed host cell chromatin on the metaphase plate, which may indicate an active recruitment of the subviral complex during mitosis to ensure enclosure of the vDNA into newly forming nuclei. It is tempting to speculate that L2 interacts specifically with cell proteins or nuclear structures that facilitate nuclear localization. Such a role has been observed for the p12 protein of murine leukemia virus and the E2 protein of HPV [74], [75]. It is interesting to note that HPV appear to encode two proteins that secure vDNA localization to daughter nuclei during cell division. Whereas E2 links the replicated, episomal vDNA to chromosomes by cellular adapter molecules during cytokinesis in suprabasal layers, L2 may fulfill a similar role during entry. The association of HPV16 vDNA with host cell chromatin and its enclosure into newly forming nuclei is efficient, since the majority of vDNA located on host chromosomes during metaphase and in the nucleus after cytokinesis.
That HPV16 entry did not induce mitosis or nuclear envelope breakdown suggested that nuclear entry of the vDNA is dependent on physiologically occurring cell divisions. This provides an explanation for the narrow cell specificity of HPV; it exclusively infects basal stem cells or transiently amplifying cells in human skin or mucosa [1]. Only these are mitotically active. One possible advantage may be that, since the transcription and replication of HPV vDNA is tightly coupled to the differentiation program of keratinocytes [76], it may be beneficial to limit viral transcription and replication during G1 phase, and thus avoid immune detection. In addition, it may be favorable to restrict productive entry to basal cells in order to provide a reservoir of persistently infected cells. The advantage may outweigh the risks that the prolonged viral entry process would trigger innate immune responses, which might lead to viral clearance during entry.
Materials and Methods
All experimental results are derived from at least three independent experiments.
Cell lines
HeLa cells were from ATCC. HaCaT cells that originated from Norbert Fusenig at the DKFZ, Heidelberg, Germany, were a gift from the Schiller lab (NIH, NCI, Bethesda, USA). HeLa H2B-mRFP/IBB-GFP and HeLa H2B-mCherry were a gift of D. Gerlich, Vienna [48]. HeLa IBB-EGFP cells were obtained after transfection of HeLa Kyoto cells with pIBB-EGFP [65] and clonal selection with 500 µg/ml G418 (PAA). Cells were maintained in DMEM (Gibco) supplemented with 10% fetal calf serum (Biochrom). Medium for stable cell lines was supplemented with 500 µg/ml G418. For HeLa H2B-mRFP/IBB-GFP cells 0.5 µg/ml puromycin was additionally added. PHK cells were from PromoCell.
Viruses
Viral preparations have been previously described for HPV16, 18 and 6 PsV using plasmids pCIneo, pRwB and p16SheLL, p18sheLL [77], [78], and p6sheLL [79], respectively, for HPV16 PsV containing BrdU-labelled DNA (BrdU-HPV16) [38], for HPV16 PsV containing EdU-labelled DNA (EdU-HPV16) [37], for fluorophore labelled HPV16 PsV [80], for recombinant HSV-1 expressing GFP under control of the CMV promoter (HSV-1-GFP) [61], and for parvovirus H1 [64]. Raft-derived HPV16 was generated in organotypic raft culture of human foreskin keratinocytes that harbor episomal HPV16 (114/B) genomes as previously described [81]. In brief, raft tissues were grown on a collagen matrix for 20 days, and subsequently subjected to dounce homogenization. The homogenate was benzonase-treated, after which the homogenate was cleared of cell debris by differential centrifugation [82].
Reagents and antibodies
Hoechst 33258 was from Invitrogen, RedDot2 was from Biotium, and all other reagents were from Sigma. The L1-7 monoclonal antibody was a gift of M. Sapp, Shreveport [54]. The TGN46 antibody (AHP1586) was from AbD Serotec. The LAMP1 (H4A3) antibody (sc-20011) was from Santa Cruz Biotechnology. AlexaFluor labeled secondary antibodies were from Invitrogen. BrdU labeling and detection kit I was from Roche. EdU Click-iT imaging kit was from Invitrogen. siRNAs were from Qiagen. The HPV16 L2-GFP expression plasmid was a gift by M. Mueller (DKFZ, Heidelberg, Germany).
RNAi screen
Procedures are detailed in the Text S1.
Infection studies
1×105 cells were treated for 16 h prior to infection with cell cycle inhibitors at 15 µM and 100 µM for aphidicolin or monastrol, respectively, or at indicated concentrations. Cells were infected with HPV16-GFP (1 viral genome equivalent (vge)/cell) or HSV-1-GFP (0.1 plaque forming units (PFU)/cell) to result in ≈20% infection for the control. For infection of PHK, cells were treated as above, but infected with HPV16-GFP (100 vge/cell). Cells were fixed 48 h p.i. or 6 h p.i. with 4% paraformaldehyde (PFA), and analyzed by flow cytometry as described [83]. In case of NH4Cl exchange for aphidicolin, cells were treated as above, but 24 h p.i., aphidicolin-containing medium was exchanged for medium with 10 mM NH4Cl/10 mM HEPES. Cells were fixed in 4% PFA at 48 h p.i. or post washout, and analyzed as above.
For infection with HPV6 and HPV18, 5×104 HeLa cells were treated for 16 h prior to infection with 3 µM aphidicolin. Cells were infected with HPV6 - and HPV18-GFP in the absence and presence of aphidicolin to result in about 20% infection for untreated control. Cells were fixed 48 h p.i. with 4% PFA, and analyzed by flow cytometry.
For infection with raft-derived HPV16, 5×104 HaCaT cells were seeded 48 h prior to infection. Cells were treated with 0.2 µM aphidicolin as above. Cells were infected in the presence or absence of aphidicolin with 5 vge/cell. Total RNA was extracted from cells with the Qiagen RNeasy Mini Kit at 48 h p.i.. Infection was measured by amplification of the viral splice transcript E1∧E4 normalized to the endogenous cellular control mRNA TATA-Box binding protein (TBP) in a one-step reverse transcription quantitative PCR using the Qiagen Quantitect Probe RT-PCR Kit. Primers and Probes for HPV16 E1∧E4 and TBP were used as described [82].
For infection studies after RNAi, 2000 or 1500 HeLa cells were seeded into 96-well optical-bottom plates for HSV-1 or HPV16 infections, respectively, and cells were reverse transfected with 5 nM of indicated siRNAs. 48 h after transfection, cells were infected with HSV-1-GFP or HPV16-GFP as above, and infection was scored at 6 h or 36 hpi, respectively, by automated microscopy and computational image analysis. Cell numbers and raw infection indices for each well were determined using a MATLAB-based infection scoring procedure described previously [84]. The raw infection indices were normalized to AllStarNegative siRNA-transfected control cells to result in relative infection percentages.
For the analysis of HPV16 infection kinetics upon knockdown of mitotic regulators, 2000 HeLa Kyoto cells were seeded into optical 96-well plates and reverse transfected with 5 nM siRNAs. 48 h after transfection, cells were infected with HPV16-GFP, and infection was scored at 24 h, 36 h and 48 hpi as above.
For video microscopy of infected cells, HeLa H2B-mCherry cells were plated on optical 96-well plates (Nunc), or 8-well Labtek chambers (Nunc), and pretreated with aphidicolin for 6-16 h for synchronization. The medium was exchanged 30 min prior to infection to medium containing solvent, monastrol or aphidicolin. Cells were infected with HPV16-GFP (30 vge/cell), or HSV-1-GFP (0.1 PFU/cell).
NE perforation
1.5×104 HeLa IBB-EGFP or H2B-mCherry were plated on 8-well Labtek chambers and treated with aphidicolin for 16 h. HeLa IBB-EGFP cells were infected with parvovirus H1 (200 PFU/cell) in the presence of aphidicolin. Alternatively, HeLa IBB-EGFP or H2B-mCherry cells were infected with or with HPV16 PsV expressing dsRed or GFP (50 vge/cell), respectively, followed by infection with parvovirus H1 (200 PFU/cell) at 12 h post HPV16 addition. Subsequently, time-lapse microscopy was started.
Video microscopy
Time-lapse images were acquired on an ImageXpress Micro (Molecular Devices) under environmental control with a CoolSnap HQ camera (Photometrics) for Videos S1–4. Alternatively, a Zeiss Axiovert Z.1 automated live cell microscope, equipped with a spinning-disk-head (Yokogawa), and a CoolSnap HQ camera was employed for image acquisition. If not otherwise stated, images from infection studies were acquired every 10 min for 48 h (HPV16) or 24 h (HSV-1, H1), with a 10× or 20× objective. For phenotyping of RNAi-mediated perturbation of mitosis and L2-GFP localization during mitosis, images were acquired every 5 min for 24 h total with a 20× objective.
Immunofluorescence analysis
To visualize vDNA using BrdU-HPV16, 3×104 HeLa H2B-mCherry cells were seeded on coverslips, and treated with aphidicolin 16 h prior to infection. Cells were infected with BrdU-HPV16 (500–1000 vge/cell) in the presence or absence of aphidicolin. Cells were fixed 20–24 h p.i. in 70% ethanol containing 15 mM glycine. For detection, the BrdU Labeling and Detection Kit I (Roche) was used according to the manufacturer's instructions, except that anti-mouse AlexaFluor488 was used as secondary antibody. In case of EdU-HPV16, HeLa Kyoto cells were used, cells were fixed in 4% PFA, and for detection the Click-iT EdU imaging kit (Invitrogen) was used. After the EdU-Click-iT reaction, cells were immunostained with TGN46 and LAMP1 antibodies.
To analyze virus trafficking and partial uncoating, 3×104 HeLa cells were seeded on coverslips, treated for 16 h with aphidicolin, and infected with HPV16 PsV (50–100 vge/cell) in the presence or absence of aphidicolin. Cells were fixed 16 hpi in 4% PFA, and immunostained with anti-L1 antibody (L1-7), Atto-488 phalloidin, and RedDot2.
Cells were imaged using a Zeiss LSM 510 (BrdU-HPV16) or LSM 780 (L1-7, TGN46, LAMP1 stainings; EdU-HPV16) confocal microscope. For presentation, images were adjusted and assembled using Adobe Photoshop CS6.
HPV16 L2-GFP expression
5000 HeLa H2B-mCherry cells were seeded into optical 96-well plates (Nunc) and transfected with 125 ng HPV16 L2-GFP expression plasmid using Lipofectamine2000 (Invitrogen). About 30–32 h post transfection, video microscopy was started.
Mitotic phenotyping after RNAi
2000 HeLa H2B-mRFP/IBB-GFP cells were seeded into optical 96-well plates (Nunc) and reverse transfected with 5–10 nM siRNA oligos (Qiagen, Table S4) using Lipofectamine RNAiMax (Invitrogen). About 48 h after transfection, video microscopy was started.
Image analysis
For image analysis of mitosis length, we used NIH ImageJ 1.44j. Mitosis start and end frames were defined by visual detection of chromosome condensation and decondensation. GFP onset was defined as 10% signal over background. vDNA positive nuclei were quantified by thresholding of BrdU signals, with BrdU positivity ≥10× the signal of uninfected cells. For quantification of L1-7 and BrdU signals, images were analyzed using the Imaris (v7.2 and v7.6.4, respectively) Cell module. For segmentation and distance measurements, cell outlines were defined by F-actin and nuclear signals. L1-7 spot locations were segmented by thresholding, whereas signal intensity measurements occurred in arbitrary units compared to control. For quantification of nuclear import of IBB-GFP, Hoechst-stained nuclei were segmented by CellProfiler (v 2.0, [85], [86]). IBB-GFP mean fluorescence intensities (fluorescence intensity/nuclear area) were measured as arbitrary units. For mitotic timing of L2-GFP expressing cells, dispersal of L2-GFP marked the onset of mitosis with NEB and chromosome decondensation during telophase the end of mitosis. L2-GFP expression levels were classified according to fluorescence intensity in interphase cells (mean intensity of nuclear L2-GFP at 25 min before NEB). Fluorescence intensities ≥20% above background (mock-transfected cells) were considered as L2-GFP expression. L2-GFP chromosomal association was assessed visually by accumulating signal on chromosomes. Colocalization of vDNA of EdU-HPV16 with host chromatin or LAMP1 and TGN46 was quantified by using the Imaris Colocalization module (v7.6.4). Cytosolic EdU-HPV16 was considered as virus that did not localize either to TGN46 or LAMP1. For mitotic phenotyping, time-lapse recordings were visually inspected and mitotic events (i.e. cytosolic dispersal followed by nuclear accumulation of IBB-GFP signal) were assigned to one or more of the following phenotypes: metaphase plate formation defect, chromosomal segregation defect, cytokinesis defect, without phenotype. For mitotic timing, dispersal of IBB-GFP marked the onset of mitosis with NEB and nuclear accumulation of IBB-GFP during telophase the end of mitosis [68].
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. DoorbarJ, QuintW, BanksL, BravoIG, StolerM, et al. (2012) The biology and life-cycle of human papillomaviruses. Vaccine 30 Suppl 5F55–70.
2. BoschFX, de SanjoseS (2003) Chapter 1: Human papillomavirus and cervical cancer—burden and assessment of causality. J Natl Cancer Inst Monogr (31): 3–13.
3. SchillerJT, CastellsagueX, GarlandSM (2012) A review of clinical trials of human papillomavirus prophylactic vaccines. Vaccine 30 Suppl 5F123–138.
4. BuckCB, ChengN, ThompsonCD, LowyDR, StevenAC, et al. (2008) Arrangement of L2 within the Papillomavirus capsid. J Virol 82 : 5190–5197.
5. BuckCB, PastranaDV, LowyDR, SchillerJT (2004) Efficient intracellular assembly of papillomaviral vectors. J Virol 78 : 751–757.
6. BuckCB, ThompsonCD, PangYY, LowyDR, SchillerJT (2005) Maturation of papillomavirus capsids. J Virol 79 : 2839–2846.
7. Buck CB, Thompson CD (2007) Production of papillomavirus-based gene transfer vectors. Curr Protoc Cell Biol Chapter 26: Unit 26 21.
8. GiroglouT, FlorinL, SchaferF, StreeckRE, SappM (2001) Human papillomavirus infection requires cell surface heparan sulfate. J Virol 75 : 1565–1570.
9. JohnsonKM, KinesRC, RobertsJN, LowyDR, SchillerJT, et al. (2009) Role of heparan sulfate in attachment to and infection of the murine female genital tract by human papillomavirus. J Virol 83 : 2067–2074.
10. JoyceJG, TungJS, PrzysieckiCT, CookJC, LehmanED, et al. (1999) The L1 major capsid protein of human papillomavirus type 11 recombinant virus-like particles interacts with heparin and cell-surface glycosaminoglycans on human keratinocytes. J Biol Chem 274 : 5810–5822.
11. KinesRC, ThompsonCD, LowyDR, SchillerJT, DayPM (2009) The initial steps leading to papillomavirus infection occur on the basement membrane prior to cell surface binding. Proc Natl Acad Sci U S A 106 : 20458–20463.
12. CerqueiraC, LiuY, KuhlingL, ChaiW, HafeziW, et al. (2013) Heparin increases the infectivity of Human Papillomavirus type 16 independent of cell surface proteoglycans and induces L1 epitope exposure. Cell Microbiol 15 : 1818–1836.
13. RichardsKF, Bienkowska-HabaM, DasguptaJ, ChenXS, SappM (2013) Multiple heparan sulfate binding site engagements are required for the infectious entry of human papillomavirus type 16. J Virol 87 : 11426–11437.
14. SelinkaHC, FlorinL, PatelHD, FreitagK, SchmidtkeM, et al. (2007) Inhibition of transfer to secondary receptors by heparan sulfate-binding drug or antibody induces noninfectious uptake of human papillomavirus. J Virol 81 : 10970–10980.
15. DayPM, GambhiraR, RodenRB, LowyDR, SchillerJT (2008) Mechanisms of human papillomavirus type 16 neutralization by l2 cross-neutralizing and l1 type-specific antibodies. J Virol 82 : 4638–4646.
16. Bienkowska-HabaM, PatelHD, SappM (2009) Target cell cyclophilins facilitate human papillomavirus type 16 infection. PLoS Pathog 5: e1000524.
17. RichardsRM, LowyDR, SchillerJT, DayPM (2006) Cleavage of the papillomavirus minor capsid protein, L2, at a furin consensus site is necessary for infection. Proc Natl Acad Sci U S A 103 : 1522–1527.
18. DayPM, LowyDR, SchillerJT (2008) Heparan sulfate-independent cell binding and infection with furin-precleaved papillomavirus capsids. J Virol 82 : 12565–12568.
19. SelinkaHC, GiroglouT, NowakT, ChristensenND, SappM (2003) Further evidence that papillomavirus capsids exist in two distinct conformations. J Virol 77 : 12961–12967.
20. DayPM, ThompsonCD, BuckCB, PangYY, LowyDR, et al. (2007) Neutralization of human papillomavirus with monoclonal antibodies reveals different mechanisms of inhibition. J Virol 81 : 8784–8792.
21. SurviladzeZ, DziduszkoA, OzbunMA (2012) Essential roles for soluble virion-associated heparan sulfonated proteoglycans and growth factors in human papillomavirus infections. PLoS Pathog 8: e1002519.
22. WoodhamAW, Da SilvaDM, SkeateJG, RaffAB, AmbrosoMR, et al. (2012) The S100A10 subunit of the annexin A2 heterotetramer facilitates L2-mediated human papillomavirus infection. PLoS One 7: e43519.
23. EvanderM, FrazerIH, PayneE, QiYM, HengstK, et al. (1997) Identification of the alpha6 integrin as a candidate receptor for papillomaviruses. J Virol 71 : 2449–2456.
24. McMillanNA, PayneE, FrazerIH, EvanderM (1999) Expression of the alpha6 integrin confers papillomavirus binding upon receptor-negative B-cells. Virology 261 : 271–279.
25. YoonCS, KimKD, ParkSN, CheongSW (2001) alpha(6) Integrin is the main receptor of human papillomavirus type 16 VLP. Biochem Biophys Res Commun 283 : 668–673.
26. DziduszkoA, OzbunMA (2013) Annexin A2 and S100A10 regulate human papillomavirus type 16 entry and intracellular trafficking in human keratinocytes. J Virol 87 : 7502–7515.
27. SpodenG, FreitagK, HusmannM, BollerK, SappM, et al. (2008) Clathrin - and caveolin-independent entry of human papillomavirus type 16—involvement of tetraspanin-enriched microdomains (TEMs). PLoS One 3: e3313.
28. SchefferKD, GawlitzaA, SpodenGA, ZhangXA, LambertC, et al. (2013) Tetraspanin CD151 mediates papillomavirus type 16 endocytosis. J Virol 87 : 3435–3446.
29. FlorinL, SappM, SpodenGA (2012) Host-cell factors involved in papillomavirus entry. Med Microbiol Immunol 201 : 437–448.
30. DayPM, LowyDR, SchillerJT (2003) Papillomaviruses infect cells via a clathrin-dependent pathway. Virology 307 : 1–11.
31. SchelhaasM, ShahB, HolzerM, BlattmannP, KuhlingL, et al. (2012) Entry of human papillomavirus type 16 by actin-dependent, clathrin - and lipid raft-independent endocytosis. PLoS Pathog 8: e1002657.
32. SpodenG, KuhlingL, CordesN, FrenzelB, SappM, et al. (2013) Human papillomavirus types 16, 18, and 31 share similar endocytic requirements for entry. J Virol 87 : 7765–7773.
33. DayPM, ThompsonCD, SchowalterRM, LowyDR, SchillerJT (2013) Identification of a role for the trans-Golgi network in human papillomavirus 16 pseudovirus infection. J Virol 87 : 3862–3870.
34. LipovskyA, PopaA, PimientaG, WylerM, BhanA, et al. (2013) Genome-wide siRNA screen identifies the retromer as a cellular entry factor for human papillomavirus. Proc Natl Acad Sci U S A 110 : 7452–7457.
35. Bienkowska-HabaM, WilliamsC, KimSM, GarceaRL, SappM (2012) Cyclophilins Facilitate Dissociation of the HPV16 Capsid Protein L1 from the L2/DNA Complex Following Virus Entry. J Virol 86 : 9875–87.
36. KamperN, DayPM, NowakT, SelinkaHC, FlorinL, et al. (2006) A membrane-destabilizing peptide in capsid protein L2 is required for egress of papillomavirus genomes from endosomes. J Virol 80 : 759–768.
37. IshiiY, TanakaK, KondoK, TakeuchiT, MoriS, et al. (2010) Inhibition of nuclear entry of HPV16 pseudovirus-packaged DNA by an anti-HPV16 L2 neutralizing antibody. Virology 406 : 181–188.
38. DayPM, BakerCC, LowyDR, SchillerJT (2004) Establishment of papillomavirus infection is enhanced by promyelocytic leukemia protein (PML) expression. Proc Natl Acad Sci U S A 101 : 14252–14257.
39. PyeonD, PearceSM, LankSM, AhlquistP, LambertPF (2009) Establishment of human papillomavirus infection requires cell cycle progression. PLoS Pathog 5: e1000318.
40. CherryS (2009) What have RNAi screens taught us about viral-host interactions? Curr Opin Microbiol 12 : 446–452.
41. SnijderB, SacherR, RamoP, LiberaliP, MenchK, et al. (2012) Single-cell analysis of population context advances RNAi screening at multiple levels. Mol Syst Biol 8 : 579.
42. FothergillT, McMillanNA (2006) Papillomavirus virus-like particles activate the PI3-kinase pathway via alpha-6 beta-4 integrin upon binding. Virology 352 : 319–328.
43. SchneiderMA, SpodenGA, FlorinL, LambertC (2011) Identification of the dynein light chains required for human papillomavirus infection. Cell Microbiol 13 : 32–46.
44. SurviladzeZ, SterkRT, DeHaroSA, OzbunMA (2013) Cellular entry of human papillomavirus type 16 involves activation of the phosphatidylinositol 3-kinase/Akt/mTOR pathway and inhibition of autophagy. J Virol 87 : 2508–2517.
45. MercerJ, SnijderB, SacherR, BurkardC, BleckCK, et al. (2012) RNAi screening reveals proteasome - and Cullin3-dependent stages in vaccinia virus infection. Cell Rep 2 : 1036–1047.
46. Huang daW, ShermanBT, LempickiRA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4 : 44–57.
47. SzklarczykD, FranceschiniA, KuhnM, SimonovicM, RothA, et al. (2011) The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res 39: D561–568.
48. SteigemannP, WurzenbergerC, SchmitzMH, HeldM, GuizettiJ, et al. (2009) Aurora B-mediated abscission checkpoint protects against tetraploidization. Cell 136 : 473–484.
49. HeimR, PrasherDC, TsienRY (1994) Wavelength mutations and posttranslational autoxidation of green fluorescent protein. Proc Natl Acad Sci U S A 91 : 12501–12504.
50. MayerTU, KapoorTM, HaggartySJ, KingRW, SchreiberSL, et al. (1999) Small molecule inhibitor of mitotic spindle bipolarity identified in a phenotype-based screen. Science 286 : 971–974.
51. KapoorTM, MayerTU, CoughlinML, MitchisonTJ (2000) Probing spindle assembly mechanisms with monastrol, a small molecule inhibitor of the mitotic kinesin, Eg5. J Cell Biol 150 : 975–988.
52. GuttingerS, LaurellE, KutayU (2009) Orchestrating nuclear envelope disassembly and reassembly during mitosis. Nat Rev Mol Cell Biol 10 : 178–191.
53. DrobniP, MistryN, McMillanN, EvanderM (2003) Carboxy-fluorescein diacetate, succinimidyl ester labeled papillomavirus virus-like particles fluoresce after internalization and interact with heparan sulfate for binding and entry. Virology 310 : 163–172.
54. SappM, KrausU, VolpersC, SnijdersPJ, WalboomersJM, et al. (1994) Analysis of type-restricted and cross-reactive epitopes on virus-like particles of human papillomavirus type 33 and in infected tissues using monoclonal antibodies to the major capsid protein. J Gen Virol 75 (Pt 12): 3375–3383.
55. WhittakerGR, KannM, HeleniusA (2000) Viral entry into the nucleus. Annu Rev Cell Dev Biol 16 : 627–651.
56. GreberUF, FassatiA (2003) Nuclear import of viral DNA genomes. Traffic 4 : 136–143.
57. CohenS, AuS, PanteN (2011) How viruses access the nucleus. Biochim Biophys Acta 1813 : 1634–1645.
58. KwantMM, van der VlietPC (1980) Differential effect of aphidicolin on adenovirus DNA synthesis and cellular DNA synthesis. Nucleic Acids Res 8 : 3993–4007.
59. SpadariS, FocherF, SalaF, CiarrocchiG, KochG, et al. (1985) Control of cell division by aphidicolin without adverse effects upon resting cells. Arzneimittelforschung 35 : 1108–1116.
60. OjalaPM, SodeikB, EbersoldMW, KutayU, HeleniusA (2000) Herpes simplex virus type 1 entry into host cells: reconstitution of capsid binding and uncoating at the nuclear pore complex in vitro. Mol Cell Biol 20 : 4922–4931.
61. HafeziW, LorentzenEU, EingBR, MullerM, KingNJ, et al. (2012) Entry of herpes simplex virus type 1 (HSV-1) into the distal axons of trigeminal neurons favors the onset of nonproductive, silent infection. PLoS Pathog 8: e1002679.
62. WaltherTC, FornerodM, PickersgillH, GoldbergM, AllenTD, et al. (2001) The nucleoporin Nup153 is required for nuclear pore basket formation, nuclear pore complex anchoring and import of a subset of nuclear proteins. EMBO J 20 : 5703–5714.
63. SuzukiY, CraigieR (2007) The road to chromatin - nuclear entry of retroviruses. Nat Rev Microbiol 5 : 187–196.
64. PorwalM, CohenS, SnoussiK, Popa-WagnerR, AndersonF, et al. (2013) Parvoviruses cause nuclear envelope breakdown by activating key enzymes of mitosis. PLoS Pathog 9: e1003671.
65. HeldM, SchmitzMH, FischerB, WalterT, NeumannB, et al. (2010) CellCognition: time-resolved phenotype annotation in high-throughput live cell imaging. Nat Methods 7 : 747–754.
66. SeksekO, BiwersiJ, VerkmanAS (1997) Translational diffusion of macromolecule-sized solutes in cytoplasm and nucleus. J Cell Biol 138 : 131–142.
67. DarshanMS, LucchiJ, HardingE, MoroianuJ (2004) The l2 minor capsid protein of human papillomavirus type 16 interacts with a network of nuclear import receptors. J Virol 78 : 12179–12188.
68. SchmitzMH, HeldM, JanssensV, HutchinsJR, HudeczO, et al. (2010) Live-cell imaging RNAi screen identifies PP2A-B55alpha and importin-beta1 as key mitotic exit regulators in human cells. Nat Cell Biol 12 : 886–893.
69. TanudjiM, ShoemakerJ, L'ItalienL, RussellL, ChinG, et al. (2004) Gene silencing of CENP-E by small interfering RNA in HeLa cells leads to missegregation of chromosomes after a mitotic delay. Mol Biol Cell 15 : 3771–3781.
70. BushmanFD, MalaniN, FernandesJ, D'OrsoI, CagneyG, et al. (2009) Host cell factors in HIV replication: meta-analysis of genome-wide studies. PLoS Pathog 5: e1000437.
71. PandaD, CherryS (2012) Cell-based genomic screening: elucidating virus-host interactions. Curr Opin Virol 2 : 784–792.
72. FlorinL, BeckerKA, LambertC, NowakT, SappC, et al. (2006) Identification of a dynein interacting domain in the papillomavirus minor capsid protein l2. J Virol 80 : 6691–6696.
73. BordeauxJ, ForteS, HardingE, DarshanMS, KlucevsekK, et al. (2006) The l2 minor capsid protein of low-risk human papillomavirus type 11 interacts with host nuclear import receptors and viral DNA. J Virol 80 : 8259–8262.
74. ElisE, EhrlichM, Prizan-RavidA, Laham-KaramN, BacharachE (2012) p12 tethers the murine leukemia virus pre-integration complex to mitotic chromosomes. PLoS Pathog 8: e1003103.
75. McBrideAA (2013) The papillomavirus E2 proteins. Virology 445 : 57–79.
76. BarksdaleSK, BakerCC (1993) Differentiation-specific expression from the bovine papillomavirus type 1 P2443 and late promoters. J Virol 67 : 5605–5616.
77. PastranaDV, BuckCB, PangYY, ThompsonCD, CastlePE, et al. (2004) Reactivity of human sera in a sensitive, high-throughput pseudovirus-based papillomavirus neutralization assay for HPV16 and HPV18. Virology 321 : 205–216.
78. RobertsJN, BuckCB, ThompsonCD, KinesR, BernardoM, et al. (2007) Genital transmission of HPV in a mouse model is potentiated by nonoxynol-9 and inhibited by carrageenan. Nat Med 13 : 857–861.
79. PastranaDV, GambhiraR, BuckCB, PangYY, ThompsonCD, et al. (2005) Cross-neutralization of cutaneous and mucosal Papillomavirus types with anti-sera to the amino terminus of L2. Virology 337 : 365–372.
80. SchelhaasM, EwersH, RajamakiML, DayPM, SchillerJT, et al. (2008) Human papillomavirus type 16 entry: retrograde cell surface transport along actin-rich protrusions. PLoS Pathog 4: e1000148.
81. McLaughlin-DrubinME, MeyersC (2005) Propagation of infectious, high-risk HPV in organotypic “raft” culture. Methods Mol Med 119 : 171–186.
82. ConwayMJ, CruzL, AlamS, ChristensenND, MeyersC (2011) Differentiation-dependent interpentameric disulfide bond stabilizes native human papillomavirus type 16. PLoS One 6: e22427.
83. CerqueiraC, LiuY, KuhlingL, ChaiW, HafeziW, et al. (2013) Heparin increases the infectivity of Human Papillomavirus Type 16 independent of cell surface proteoglycans and induces L1 epitope exposure. Cell Microbiol 15 : 1818–36.
84. EngelS, HegerT, ManciniR, HerzogF, KartenbeckJ, et al. (2011) Role of endosomes in simian virus 40 entry and infection. J Virol 85 : 4198–4211.
85. JonesTR, KangIH, WheelerDB, LindquistRA, PapalloA, et al. (2008) CellProfiler Analyst: data exploration and analysis software for complex image-based screens. BMC Bioinformatics 9 : 482.
86. CarpenterAE, JonesTR, LamprechtMR, ClarkeC, KangIH, et al. (2006) CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol 7: R100.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 5
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Venus Kinase Receptors Control Reproduction in the Platyhelminth Parasite
- Dual-Site Phosphorylation of the Control of Virulence Regulator Impacts Group A Streptococcal Global Gene Expression and Pathogenesis
- Severe Acute Respiratory Syndrome Coronavirus Envelope Protein Ion Channel Activity Promotes Virus Fitness and Pathogenesis
- High-Efficiency Targeted Editing of Large Viral Genomes by RNA-Guided Nucleases
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy