
Dual-Site Phosphorylation of the Control of Virulence Regulator Impacts Group A Streptococcal Global Gene Expression and Pathogenesis
Group A Streptococcus (GAS) causes a variety of human diseases ranging from mild throat infections to deadly invasive infections. The capacity of GAS to cause infections at such diverse locations is dependent on its ability to precisely control the production of a broad variety of virulence factors. The control of virulence regulator (CovR) is a master regulator of GAS genes encoding virulence factors. It is known that CovR can be phosphorylated on aspartate-53 in vitro and that such phosphorylation increases its regulatory activity, but what additional factors influence CovR-mediated gene expression have not been established. Herein we show for the first time that CovR is phosphorylated in vivo and that phosphorylation of CovR on threonine-65 by the threonine/serine kinase Stk prevents aspartate-53 phosphorylation, thereby decreasing CovR regulatory activity. Further, while CovR-mediated gene repression is highly dependent on aspartate-53 phosphorylation, CovR-mediated gene activation proceeds via a phosphorylation-independent mechanism. Modifications in CovR phosphorylation sites significantly affected the expression of GAS virulence factors during infection and markedly altered the ability of GAS to cause disease in mice. These data establish that multiple inter-related pathways converge to influence CovR phosphorylation, thereby providing new insight into the complex regulatory network used by GAS during infection.
Published in the journal:
. PLoS Pathog 10(5): e32767. doi:10.1371/journal.ppat.1004088
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004088
Summary
Group A Streptococcus (GAS) causes a variety of human diseases ranging from mild throat infections to deadly invasive infections. The capacity of GAS to cause infections at such diverse locations is dependent on its ability to precisely control the production of a broad variety of virulence factors. The control of virulence regulator (CovR) is a master regulator of GAS genes encoding virulence factors. It is known that CovR can be phosphorylated on aspartate-53 in vitro and that such phosphorylation increases its regulatory activity, but what additional factors influence CovR-mediated gene expression have not been established. Herein we show for the first time that CovR is phosphorylated in vivo and that phosphorylation of CovR on threonine-65 by the threonine/serine kinase Stk prevents aspartate-53 phosphorylation, thereby decreasing CovR regulatory activity. Further, while CovR-mediated gene repression is highly dependent on aspartate-53 phosphorylation, CovR-mediated gene activation proceeds via a phosphorylation-independent mechanism. Modifications in CovR phosphorylation sites significantly affected the expression of GAS virulence factors during infection and markedly altered the ability of GAS to cause disease in mice. These data establish that multiple inter-related pathways converge to influence CovR phosphorylation, thereby providing new insight into the complex regulatory network used by GAS during infection.
Introduction
Bacteria causing infections in humans must closely modulate virulence factor production in response to different environmental challenges [1], [2], [3]. It has long been recognized that two-component gene regulatory systems (TCS) are a major mechanism by which bacteria react to external stimuli, and thus are critical to the virulence of numerous pathogenic bacteria [4], [5], [6], [7]. Although there is diversity in TCS composition [8], standard TCS consist of a membrane-embedded histidine kinase that can respond to environmental signals by either phosphorylating or dephosphorylating a cognate response regulator, usually on an aspartate residue in the N-terminal receiver domain [9]. The aspartate phosphorylation status of the response regulator alters its gene regulation effect thereby allowing the organism to remodel its expression profile [10].
The critical role of TCS is exemplified by their status as possible targets for novel antimicrobials [11], [12], [13], [14]. However, much remains to be learned about the mechanisms and effects of bacterial response regulator phosphorylation. One of the model systems for understanding how response regulators influence bacterial pathogenesis has been the control of virulence regulator (CovR, also known as CsrR) from group A Streptococcus [15], [16], [17]. GAS causes a wide range of infectious and post-infectious syndromes in humans [18]. CovR is a member of the OmpR/PhoB family of response regulators and is a key repressor of numerous GAS virulence factors such that CovR-inactivated strains are hypervirulent [15], [19], [20]. In vitro studies have shown that phosphorylation of CovR at amino acid residue aspartate-53 (D53) results in homodimerization and increased DNA-binding affinity, but in vivo phosphorylation of CovR has not been demonstrated [21], [22], [23]. covR is co-transcribed with an adjacent gene encoding the sensor kinase CovS [15]. Although it has been assumed that CovS controls the D53 phosphorylation status of CovR, numerous groups have reported that inactivation of CovS produces a distinct phenotype from inactivation of CovR suggesting that factors in addition to CovS influence CovR phosphorylation and thus activity [19], [21], [24], [25].
Recently, Agarwal et al. found that the GAS eukaryotic-like serine/threonine kinase Stk phosphorylates CovR on threonine residues in vitro although neither the exact site nor the functional consequences of such phosphorylation has been determined [26]. GAS Stk and CovR homologues are present in group B Streptococcus (GBS), and GBS Stk has been shown to phosphorylate CovR on threonine-65 (T65) [27], [28], [29]. Stk-mediated threonine phosphorylation of GBS CovR resulted in a decreased ability of CovR to be phosphorylated on D53, decreased CovR binding to DNA from the cylX promoter, and relieved CovR-mediated repression of the cyl operon which encodes the potent β-hemolysin/cytolysin [29], [30], [31]. Whether GAS Stk influences CovR-mediated gene regulation in a fashion similar to GBS is unknown.
To gain new insights into the functional consequence of GAS CovR phosphorylation on host-pathogen interaction, we performed a series of biochemical, genetic, and virulence assays. First, we used the recently developed Phos-Tag technology to demonstrate that CovR is phosphorylated in vivo. Using a combination of mass spectrometry, Phos-Tag assays, and targeted mutagenesis, we determined that Stk phosphorylates GAS CovR at T65 and that such phosphorylation negatively influences CovR phosphorylation on D53. In this way, Stk phosphorylation of CovR on T65 significantly impacts GAS global gene expression and virulence. Moreover, we discovered that, in contrast to CovR-mediated gene repression, CovR-mediated activation of gene expression is not primarily determined by CovR D53 phosphorylation status. Taken together, these data provide crucial new information about how the phosphorylation status of the receiver domain of a TCS response regulator influences host-pathogen interaction and provide a key platform for achieving a fuller understanding of the mechanisms influencing response regulator phosphorylation status in vivo.
Results
CovR is phosphorylated in vivo
Although the importance of CovR phosphorylation has been well established in GAS and GBS over the past 15 years, in vivo phosphorylation of CovR has not been demonstrated in either species [22], [27], [28], [29], [30], [32], [33], [34]. Recently, Phos-Tag gels, in which the migration of phosphorylated proteins is retarded compared to non-phosphorylated proteins, have been used to assay protein phosphorylation status in vitro and in vivo [35], [36], [37], [38]. To investigate whether Phos-Tag gels would be suitable for assessing CovR phosphorylation, we first compared in vitro CovR phosphorylation using Phos-Tag gel and native polyacrylamide gel electrophoresis (PAGE), which has been the typical method for studying CovR phosphorylation [22], [39]. Purity of the recombinant proteins used in these assays is shown in Figure S1A in Text S1. Phosphorylation of recombinant CovR by the small molecule phosphodonor acetyl phosphate induces homodimerization, which results in slower migration on a native polyacrylamide gel (Figure 1A) [22]. An alanine substitution at D53 abrogates phosphorylation at the altered site [22] and thereby homodimerization (Figure 1A). Similarly, acetyl-phosphate mediated phosphorylation of wild-type CovR, but not CovR-D53A, could be readily detected using the Phos-Tag method, as demonstrated by the appearance of a higher migrating band in the phosphorylated sample (Figure 1B). Heating of the CovR samples, which removes the heat-labile aspartate-phosphorylation, eliminated the higher running band, confirming that the Phos-tag assay was able to detect CovR aspartate-phosphorylation (Figure 1B) [22].

Next, we sought to use the Phos-Tag assay to determine whether CovR is phosphorylated in vivo. To this end, strain MGAS10870, a serotype M3 strain which has been fully sequenced and is known to contain a wild-type CovR/S TCS [40], and its isogenic CovR-inactivated derivative (10870ΔcovR) were grown to mid-exponential phase in THY medium. The cells were pelleted, lysed, and cell lysates were run on a Phos-Tag gel, transferred to a nitrocellulose membrane, and probed with a polyclonal anti-CovR antibody. The performance of the anti-CovR antibody is shown in Figure S2 in Text S1. For strain MGAS10870, we observed two distinct bands reacting with the anti-CovR antibody which were at the same location as recombinant CovR and recombinant CovR phosphorylated with acetyl phosphate (Figure 1C). These data are consistent with partial CovR phosphorylation in strain MGAS10870 during growth in a standard laboratory medium (THY medium). Heating of the MGAS10870 cell extract led to the disappearance of the majority of the slower migrating band whereas treating of the sample with calf intestinal phosphatase (CIP) completely removed the slower migrating band (Figure 1C). Conversely, no bands were observed in the lysate of strain 10870ΔcovR (Figure 1C). Next we assayed the phosphorylation status of CovR grown in THY medium supplemented with 15 mM MgCl2 as Mg2+ is thought to increase CovR D53 phosphorylation [41]. Following growth in a high Mg2+ medium, only the slower migrating band was detected by the anti-CovR antibody suggesting that CovR phosphorylation is complete under the high Mg2+ condition (Figure 1D). Given that GAS CovR and GBS CovR are highly similar (84% identity, 92% similarity at the amino acid level), we also assayed the cell lysate from the GBS strain SGBS001 and found that our anti-CovR antibody did react with the GBS cell lysate and that GBS CovR appears to be partially phosphorylated during growth in THY (Figure 1E). Phosphorylation of GBS CovR was removed by heat or CIP-treatment. We conclude that the Phos-Tag assays can be used to determine GAS and GBS CovR phosphorylation status in vitro and in vivo and that during growth in a standard laboratory medium, the majority of GAS CovR phosphorylation is heat-labile, consistent with aspartate phosphorylation.
Stk phosphorylates CovR on T65 in vitro
The fact that some, but not all, CovR phosphorylation disappeared upon heating suggested that CovR may be phosphorylated at heat-stable sites, such as threonine, in addition to being phosphorylated at the heat-labile aspartate site [35]. It was recently shown that Stk phosphorylates CovR in vitro but the phosphorylation site was not identified [26]. Thus, we first sought to determine the amino acid residue in GAS CovR that is the target for Stk mediated phosphorylation. To this end, we overexpressed and purified the kinase domain of Stk (StkKD) from the serotype M3 strain MGAS10870 (Figure S1A in Text S1). StkKD encompasses the N-terminal 315 amino acids and has been shown previously to be sufficient to phosphorylate CovR on threonine residues in vitro [26]. CovR was incubated with StkKD, subjected to chymotrypsin or trypsin digestion, and then analyzed by LC/MS/MS (see Materials and Methods). No specific phosphorylated peptide was detected in the trypsin digest. However, in the chymotrypsin digest, the CovR-derived peptide EVTRRLQTEKTTY was predicted to be phosphorylated at position 3, which corresponds to T65 (Figure 2A). In order to validate the site of phosphorylation we then instructed the mass spectrometer to specifically acquire MS/MS of only this phosphorylated peptide in addition to acquiring MS/MS/MS of specific fragments. Fragmentation of the peptide EVT65RRLQTEKTTY showed fragments that lost phosphoric acid (m/z 804, marked with blue arrow in Figure 1A) as well as a specific y102+ fragment at m/z 648.61, which was predicted to be RRLQTEKTTY. This fragment was not phosphorylated therefore suggesting that the phosphorylated amino acid residue was T65. Further fragmentation of this y102+ fragment proved that this fragment was indeed RRLQTEKTTY (Figure 2B). Thus, in vitro, StkKD phosphorylates CovR on T65.

Phos-tag gel assays confirm phosphorylation of CovR T65 by Stk in vitro
We next sought to confirm our mass spectroscopy finding that T65 is the site of CovR phosphorylation by Stk and to investigate the relationship between CovR D53 and T65 phosphorylation. To this end, we generated a recombinant CovR variant with alanine at the T65 phosphorylation site (CovR-T65A). We performed circular dichroism (CD) spectroscopy on CovR-T65A and the previously generated CovR-D53A (Figure S1A in Text S1) to ensure that the mutations did not result in aberrantly folded proteins. The resulting CD spectra of all recombinant CovR variants were identical to the spectrum of the wild-type CovR protein indicating that our introduced changes had not altered the overall secondary protein structure (Figure S1B in Text S1).
Given that CovR threonine phosphorylation is not expected to induce protein dimerization, native PAGE is not suitable for analysis of CovR threonine phosphorylation status. Thus, we used Phos-tag PAGE to assay for CovR T65 phosphorylation by incubating CovR with StkKD and then performing standard Western blot using the anti-CovR antibody. This approach eliminates the strong signal from highly phosphorylated (and therefore appearing as several bands) StkKD, which overwhelmed the weaker CovR-P signal when using anti-threonine antibodies (data not shown). As depicted in Figure 3A, the Phos-tag analysis demonstrated wild-type CovR phosphorylation following incubation with StkKD. Although Jin et al. found GAS StkKD to be an Mn2+ dependent threonine kinase [42], we did not observe any difference in StkKD-CovR phosphorylation reactions containing 10 mM Mg2+ or Mn2+ ions, respectively, in the kinase buffer (Figure 3A). Therefore, we used 10 mM MgCl2 throughout the following experiments. Consistent with our mass spectrometry data, incubation of CovR-T65A with StkKD resulted in no detectable phosphorylation using the Phos-tag assay (Figure 3A). In contrast, StkKD was able to phosphorylate CovR with an alanine substituted for threonine at position 73 (CovR-T73A, data not shown), further supporting the specific role of T65 in Stk-dependent CovR phosphorylation.

Interplay between acetyl-phosphate and Stk mediated phosphorylation
Having established that Stk phosphorylates CovR on T65, we sought to investigate whether CovR D53 and T65 phosphorylation events influence each other. To this end, we generated the recombinant CovR variant T65E (Figure S1A in Text S1). Introduction of a negative charge at this position mimics constitutive phosphorylation of T65 (i.e. a phospho-mimetic mutation) [29]. Similar to the CovR-D53A and -T65A variants, we observed no difference in CD spectra for the CovR-T65E protein compared to wild-type indicating normal protein folding (Figure S1B in Text S1). Next, we determined whether the CovR variants could be phosphorylated in vitro by either acetyl-phosphate or StkKD. By Phos-Tag analysis, CovR-T65A could be phosphorylated by acetyl phosphate similar to wild-type CovR (Figure 3B). However, analogous to the CovR-D53A protein, the T65E substitution in CovR completely impeded acetyl-phosphate mediated phosphorylation (Figure 3B). Consequently, only a phosphor-mimetic (T65E), not a neutral (T65A) mutation at T65 interferes with phosphorylation at CovR D53.
StkKD was unable to phosphorylate either the CovR-T65A or -T65E variants (Figure 3C). Moreover, StkKD readily phosphorylated CovR-D53A to the point where we observed reproducibly higher amounts of phosphorylation following incubation with StkKD for the CovR-D53A protein compared to wild-type CovR (Figure 3C). Hence, in vitro, the D53 and T65 CovR phosphorylation sites influence each other.
Creation and characterization of GAS strains with altered CovR phosphorylation sites
To better understand the functional consequences of CovR phosphorylation, we next created the following GAS strains from the serotype M3 strain MGAS10870: a CovS-knockout strain (10870ΔcovS), a strain with an alanine at CovR position 53 (CovR-D53A), a strain with an alanine at CovR position 65 (CovR-T65A), and a strain with glutamate at CovR position 65 (CovR-T65E) (Figure 4A). We previously had created the CovR knockout strain 10870ΔcovR [43]. The growth curves of the strains in a standard laboratory medium (THY) were indistinguishable (Figure 4B), whereas the phenotypes of the various strains on sheep blood-agar plates were quite distinct (Figure 4A). Compared to wild-type, the 10870ΔcovR, CovR-D53A, and CovR-T65E strains had a larger colony phenotype indicating increased production of the GAS hyaluronic acid capsule which is known to be negatively regulated by CovR [15]. Conversely, strains 10870ΔcovS and CovR-T65A had a phenotype similar to strain MGAS10870. Consistent with these observations, strains 10870ΔcovR, CovR-D53A, and CovR-T65E produced significantly more capsule during growth in THY compared to strains MGAS10870, 10870ΔcovS, and CovR-T65A (Figure 4C). In many GAS strains, CovS positively influences the production of the cysteine proteinase, streptococcal pyrogenic exotoxin B (SpeB) [44]. Similarly, strain 10870ΔcovS had significantly decreased SpeB production compared to wild-type as measured by casein hydrolysis (Figure 4D). No significant difference in SpeB production was observed between the CovR variant strains and strain MGAS10870 (Figure 4D).

Genome-wide characterization of the effects of CovR phosphorylation on GAS gene expression
To determine the genome-wide effects of alterations in CovR phosphorylation status on GAS gene expression, four replicates of each strain were grown to mid-exponential phase (Figure 4B), and transcriptome analyses were performed using RNA-Seq. A minimum of 2.3 million paired reads were resolved per strain for a minimum average base coverage of 174 indicating excellent sequencing depth. Gene transcripts were detected for 1609 genes or approximately 83% of the 1951 genes in strain MGAS10870. Principal components analysis, which creates a global view of the variance in the data set, showed that strains 10870ΔcovR, CovR-D53A, and CovR-T65E were the most distant from wild-type (Figure S3 in Text S1). Strain 10870ΔcovS was closest to wild-type whereas strain CovR-T65A had a phenotype intermediate between wild-type and strain 10870ΔcovR.
Gene transcript levels were considered significantly different between the wild-type and a derivative strain if the mean transcript level had at least a 1.5-fold change and the multiple-testing adjusted P value was ≤0.05. Compared to wild-type, there were significant differences in transcript levels for 200 genes in strain 10870ΔcovR, or some 10% of the genome (Table S1). Known CovR-influenced genes whose transcript levels were higher in strain 10870ΔcovR included hasA, the initial gene in the hyaluronic acid capsule-encoding operon [45], prtS, which encodes an IL-8 degrading enzyme [46], and grab, which encodes the protein G-related α2-macroglobulin-binding protein [47] (Figure 5A). Genes with decreased transcript level in the CovR knockout strain included the dipeptide permease encoding operon (dppA-E) [48] and gene spyM3_1493 which encodes a platelet acetyltransferase degrading enzyme known as esterase or Sse [49].

Consistent with D53 phosphorylation being critical to CovR function, the transcriptome of strain CovR-D53A was quite similar to that of the CovR-inactivated strain. Pairwise comparison of log2 transcript level ratios of strains CovR-D53A and 10870ΔcovR relative to the wild-type strain MGAS10870 showed a Pearson correlation r2 value of 0.65 (P<0.001). The transcript levels of genes encoding virulence factors well-established to be repressed by CovR such as hasA, prtS, and ska, which encodes the plasminogen-activating streptokinase [50], were increased relative to wild-type in strain CovR-D53A (Figure 5A). However, the transcript levels of the dipeptide permease operon and the esterase-encoding gene spyM3_1493, although decreased in the CovR knockout strain, were not significantly altered compared to wild-type in the CovR-D53A strain (Figure 5A).
We previously found that changing T65 to glutamate (T65E) prevented phosphorylation of CovR at D53 by the small molecule phosphodonor acetyl phosphate in vitro. Thus we hypothesized that the CovR-T65E strain would have a transcriptome similar to the CovR-D53A strain. Indeed, among all the strains studied in this experiment, the Pearson correlation of log2 transcript levels ratios relative to the wild-type strain were the highest for strain CovR-D53A and CovR-T65E (r2 = 0.86) (Figure 5B). Genes whose transcript levels were significantly increased in strain CovR-T65E relative to wild-type included the key virulence factor-encoding genes hasA, mac-1 (also known as ideS), which encodes an immunoglobulin degrading enzyme [51], prtS, and sagA, which encodes the cytolysin streptolysin S [52] (Figure 5A). Similar to strain CovR-D53A, we observed no difference in the transcript levels of the CovR-activated genes dppA and spyM3_1493 between strain CovR-T65E and strain MGAS10870.
The 10870ΔcovR, CovR-D53A and CovR-T65E strains each had approximately 200 genes with altered gene transcript levels relative to strain MGAS10870. In contrast, the replacement of CovR T65 with an alanine resulted in a strain with only 111 genes whose transcript levels were significantly different compared to wild-type (Figure 5C). The transcript levels of approximately half of the virulence factor encoding genes affected in the CovR-knockout strain were also affected in the CovR-T65A strain including ska, mac-1, and prtS. Genes whose transcript levels were affected in the CovR-knockout and CovR-D53A strains but not in strain CovR-T65A included sagA and mf, which encodes a DNA degrading enzyme [53]. As we had observed for strains CovR-D53A and CovR-T65E, there was no significant transcript level difference compared to wild-type for the CovR-activated genes dppA and spyM3_1493 in strain CovR-T65A.
In contrast to the profound transcriptome effects of altering the CovR phosphorylation sites D53and T65, inactivating CovS had a relatively modest impact on GAS gene transcription during growth in THY (Figure 5C). Only 39 genes had significantly different transcript levels between the CovS-knockout and wild-type strain, which was the lowest number of differentially transcribed genes observed for any of the strains studied. Also, the effect of CovS inactivation led a distinct pattern of differential gene expression compared to the strains in which CovR had been altered. Specifically, in the CovR-isoallelic strains, all significant transcript level differences in CovR-repressed genes involved an increase in transcript levels relative to wild-type (Figure 5A). Conversely, CovS inactivation could result in either an increase or decrease in transcript level for CovR-repressed genes compared to wild-type, an effect that has been previously described for a CovS-inactivated serotype M1 strain [25]. For example, compared to strain MGAS10870, mac-1, prtS, and ska transcript levels were all significantly increased in strain 10870ΔcovS whereas ropB, which encodes an transcription factor activating speB expression [54], and grab transcript levels were decreased (Figure 5A).
CovR-T65A, but not CovR-T65E or CovR-D53A, retains the ability to repress gene expression in response to Mg2+
Adding Mg2+ to standard laboratory medium is known to increase the repression of a subset of CovR-regulated genes, presumably by increasing CovS-mediated CovR D53 phosphorylation (Figure 1C) [41]. Thus, we next sought to specifically analyze the response of CovR-regulated genes to Mg2+ in our isogenic and isoallelic strains using quantitative real-time PCR (qRT-PCR). In accordance with previous data, in the wild-type strain MGAS10870, we observed significantly higher gene transcript levels during growth in a low Mg2+ medium compared to a high Mg2+ medium for hasA, prtS, mac-1, spyM3_0132, and spyM3_0105 (Figure 6A) [41], [55]. This effect did not appear to be due to altered CovR levels as there was no significant difference in covR gene transcript levels between the two media (Figure 6A). Similarly, we observed no significant differences in transcript levels between the two media for sagA or dppA (Figure 6A). Consistent with the idea that CovS phosphorylation on CovR D53 mediates the response to Mg2+, we saw no difference in hasA, prtS, or spyM3_0105 gene transcript levels in either strain 10870ΔcovR, 10870ΔcovS, CovR-D53A, or CovR-T65E between growth in low and high Mg2+ concentration (Figure 6B–D). Conversely, for strains MGAS10870 and CovR-T65A, the gene transcript levels of hasA, prtS, and spy_0105 were significantly decreased in the high Mg2+ medium consistent with the hypothesis that the CovR-T65A strain can still be phosphorylated on D53 in vivo. We next examined the CovR-activated genes dppA and spyM3_1493. There was no difference in dppA transcript level in response to altered Mg2+ concentration for any of the studied strains (Figure 6E) whereas there was a significant decrease in the transcript level of spyM3_1493 in strains MGAS10870 and CovR-T65A (Figure 6F) under high Mg2+ condition. Thus, of the strains tested here, a Mg2+ response for CovR-regulated genes was only observed for strains MGAS10870 and CovR-T65A.

Responsiveness to Mg2+ is independent of DNA binding affinity of CovR-D53∼P
The finding that some, but not all CovR-regulated genes, have altered gene transcript levels in the presence of increased Mg2+ concentration has previously been noted [41], but the explanation for this observation is not known. One possibility that has been postulated is that Mg2+-responsive promoters are bound by CovR phosphorylated at D53 (CovR-D53∼P) with lower affinity compared to Mg2+ non-responsive promoters [41]. Consequently, Mg2+-responsive genes would only be fully repressed by CovR under conditions that increase CovR phosphorylation above that observed in a standard laboratory medium (compare Figure 1C and 1D for different CovR phosphorylation levels under different growth conditions). To test this hypothesis, we measured the DNA binding properties of wild-type CovR-D53∼P for the Mg2+ non-responsive promoters covR and dppA in comparison to the Mg2+ responsive promoters prtS and hasA. Unexpectedly, we observed that CovR-D53∼P bound to DNA from both groups with similar affinity (Figure S4 in Text S1). For all promoters tested, CovR-D53∼P mediated shifts began at CovR concentration of 0.1–0.2 µM to form a low molecular weight complex; at ∼1 µM the higher molecular weight complex became more prominent. Hence, in vitro DNA affinity alone does not seem to be the decisive factor for the different response of CovR regulated promoters towards Mg2+ ions. However, these data do not rule out differential CovR-DNA affinity in vivo given the complex nature of CovR-DNA interaction in the bacterial cell [16].
Recombinant CovR-D53A and CovR-T65E retain the ability to bind promoter DNA from CovR-regulated genes
We hypothesize that the primary functional effect of CovR T65 phosphorylation is to prevent phosphorylation at D53, and a previous study found that recombinant CovR-D53A bound to DNA from the has promoter with the same affinity as unphosphorylated wild-type CovR [22]. However, in group B Streptococcus, recombinant CovR-T65E was unable to bind to DNA from the promoter of CovR-regulated gene cylX [29]. Thus, we sought to determine the DNA binding characteristics of GAS CovR-T65E compared to wild type CovR and CovR-D53A. To this end, ∼300 bp DNA fragments that encompass the complete promoter regions of the CovR-regulated genes hasA, prtS, and sagA were amplified from MGAS10780 genomic DNA [33], [46], [56]. Both CovR-D53A and CovR-T65E bound to all of the tested promoters (Figure 7 and Figure S5 in Text S1). The binding affinity of CovR-D53A and CovR-T65E was similar to that of unphosphorylated wild-type CovR, with initial shifts starting at 0.25–0.5 µM protein concentration (formation of a low molecular weight complex) and a full shift at ∼1.5 µM protein concentration (Figure 7). The specificity of CovR DNA binding was ascertained using a promoter that is not CovR-regulated, adcR (Figure S5 in Text S1).
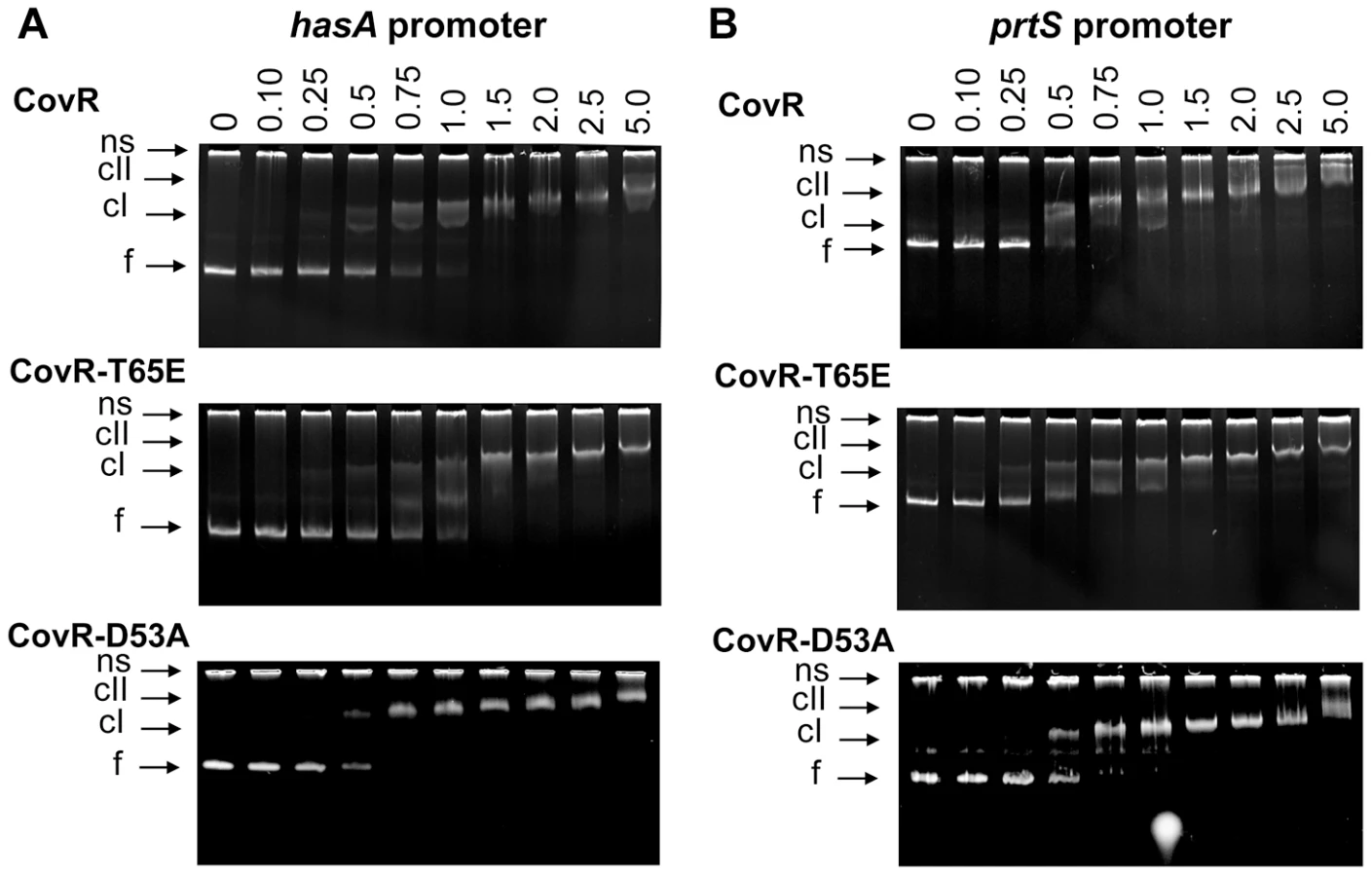
Phosphorylation of CovR significantly affects GAS virulence and gene expression during infection
To test whether CovR phosphorylation affects GAS virulence, outbred CD-1 mice were challenged intraperitoneally with the various CovS - or CovR-inactivated and CovR isoallelic strains and followed for near-mortality over 7 days. As expected, there was a significant survival difference amongst the mice challenged with the 6 strains (P<0.001 by Mantel-Cox log rank test, Figure 8A). Specifically, strain MGAS10870 was less virulent compared to the other five strains (P<0.001 after accounting for multiple comparisons). There was no significant difference in survival among mice infected with strains 10870ΔcovR, 10870ΔcovS, or CovR-T65A (P = 1.0). However, the survival time was significantly shorter for mice infected with strains CovR-D53A or CovR-T65E compared to strains 10870ΔcovR, 10870ΔcovS, or CovR-T65A (P<0.01 for all comparisons). No significant difference in survival was observed between mice infected with strain CovR-D53A or CovR-T65E (P = 0.19).

We sought to gain insight into the survival differences noted in the animal challenge experiment by determining the gene expression profile of bacteria during infection. To this end, we euthanized 4 animals per infecting strain 24 hours after infection, isolated GAS RNA from the blood of infected animals, and analyzed select gene transcript levels. As expected, the transcript levels of hasA, sagA, and prtS genes were significantly higher in the CovR-inactivated and CovR-isoallelic strains compared to wild-type (Figure 8B, data not shown). The transcript levels of ska were significantly lower in the wild-type strain compared to the other strains, but ska transcript levels were also lower in strain 10870ΔcovS and CovR-T65A compared to strains 10870ΔcovR, CovR-D53A, and CovR-T65E (Figure 8C). Similarly to what we had observed during growth in THY, inactivation of CovS significantly decreased speB transcript level and significantly increased spyM3_1493 transcript level compared to wild-type and the other tested strains (Figure 8D, 8E). Moreover, we observed decreased transcript level of spyM3_1493 and dppA specifically in strain 10870ΔcovR compared to the other tested strains (Figure 8E, 8F).
CovR amounts in vivo are lower in the CovR-T65A isoalleic strain compared to wild type
Our in vitro experiments showed that mutation of CovR T65 to alanine impedes Stk-mediated phosphorylation, while having no impact on phosphorylation of D53 by acetyl-phosphate. Hence, we predicted that the concentration of CovR-D53∼P in the CovR-T65A strain would be equal or even higher than in the parental wild type strain leading to pronounced CovR-mediated gene repression and decreased virulence. However, our expression and animal data did not confirm this hypothesis as the CovR-T65A strain showed increased virulence compared to strain MGAS10870. As CovR is known to repress its own expression [57], we compared the CovR transcript and protein levels in wild type strain MGAS10780, the CovR-deletion strain, and the CovR-T65A isoallelic strain using qRT-PCR and Western blot analysis, respectively. As expected no CovR signal was detected in strain10870ΔcovR (Figure 9A). However, the amount of CovR was significantly higher in the wild type compared to the CovR-T65A strain under both high and low Mg2+ growth conditions (Figure 9A). Similarly, the level of covR transcript was significantly higher in strain MGAS10870 compared to the CovR-T65A isoallelic strain (Figure 9B). Finally, during infection, we also observed significantly decreased covR transcript levels in the CovR-T65A strain compared to strain MGAS10870 (Figure 9B). Consequently, at least part of the characteristics of the CovR-T65A strain is likely a result of decreased covR expression due to increased CovR self-repression and thus lower CovR levels compared to wild-type.

Discussion
Until recently, studies of how protein phosphorylation participates in prokaryotic signal transduction have mainly focused on linear TCS pathways that involve phosphorylation of aspartate in response regulators by their cognate histidine kinase [9]. Increasingly, however, it is being recognized that the phosphorylation status of response regulators can be influenced by a multitude of factors, including phosphorylation on non-aspartate residues, clearly placing response regulators into the wider gene regulation network [28], [58], [59]. Understanding the mechanisms influencing the phosphorylation status of bacterial regulator proteins and determining how such phosphorylation ultimately influences bacterial gene expression is critical to a more complete understanding of bacterial pathogenesis. Herein we demonstrate that the key GAS response regulator CovR is phosphorylated in vivo and that CovR phosphorylation at both aspartate and threonine residues profoundly influences GAS global gene expression and virulence.
The recognition of CovR in 1998 as a negative regulator of GAS hyaluronic acid capsule production was accompanied by the observation that CovR is a OmpR/PhoB family member, and as such, contains a highly conserved aspartate phosphorylation site [15]. It was also determined at that time that the covR gene is located immediately upstream of covS and that the two genes are co-transcribed, although mono-cistronic covR transcripts have also been reported [15], [60]. This combination of observations has led to the supposition that CovS controls CovR aspartate phosphorylation status analogous to the situation observed for the well-studied EnvZ/OmpR TCS in Escherichia coli, although CovS has never been directly shown to phosphorylate CovR in vitro or in vivo [16]. Over the past 15 years, numerous investigations have demonstrated the critical role of CovR D53 phosphorylation in CovR-mediated gene repression, mainly relying on in vitro binding assays and transcriptional reporter systems [22], [32], [56], [57]. However, up to now, demonstration of CovR phosphorylation in vivo has been lacking. Our finding that a Phos-Tag assay can be used to measure GAS and GBS CovR phosphorylation status in vivo opens extensive possibilities into understanding the mechanisms and impact of streptococcal response regulator phosphorylation. Moreover, the fact that the CovS-inactivated and CovR-D53A strains had dramatically different transcriptomes provides additional impetus to study which factors, other than CovS, influence CovR D53 phosphorylation.
In addition to demonstrating CovR phosphorylation in vivo, another key finding of this work was the identification of T65 as the target of Stk phosphorylation in CovR using both mass spectrometry and targeted mutagenesis strategies. A previous work had established that recombinant Stk phosphorylates CovR on threonine residues but did not establish the phosphorylation site [26]. In contrast to the highly specific phosphotransfer in TCS, eukaryotic-like serine/threonine kinases, such as Stk, are well known to have multiple targets, including cell-wall division regulating proteins that are critical to cellular homeostasis [61], [62], [63]. Thus, genetic inactivation of Stk or its cognate phosphatase, Stp, as has previously been done in serotype M1 GAS strains, is unlikely to generate information specifically relevant to threonine phosphorylation of CovR [42], [64]. For example, in serotype M1 GAS strains, inactivation of both Stk and its cognate phosphatase, Stp, resulted in decreased virulence, probably as a result of their pleiotropic effects on GAS physiology [26], [64]. Thus, once we established the site of CovR-threonine phosphorylation, we used a combination of deletion (ΔcovR, ΔcovS), phosphorylation silencing (CovR-D53A and CovR-T65A), and phosphorylation mimicking (CovR-T65E) mutations to comprehensively assess the functional consequences of CovR phosphorylation. Our data indicate that the combination of competing CovR D53 and T65 phosphorylation enables the integration of thus far unknown signals sensed by Stk into the regulatory network in order to fine-tune CovR/S-mediated gene regulation during GAS host-pathogen interaction.
One surprising finding from this study was that the CovR-D53A and CovR-T65E strains were more virulent compared to strain 10870ΔcovR. A likely explanation for this finding can be gleaned from our transcriptome data. Whereas CovR-mediated repression of gene expression strongly depended on phosphorylation of D53 (which is abolished in strains CovR-D53A and CovR-T65E), it had minimal effect on CovR-mediated gene activation. A possible target to explain the virulence effects of this differential gene regulation is spyM3_1493, which encodes a platelet-activating factor acetylhydrolase recently shown to be critical for GAS pathogenesis [65]. Review of previously published microarray data from serotype M3 GAS confirms that spyM3_1493 transcript levels were lower in CovR-inactivated strains compared to the wild-type [41], [43]. Similar findings of lower transcript levels in the CovR-inactivated, but not the CovR-D53A and CovR-T65E, strains were also observed for the dpp operon, which encodes proteins involved in amino acid uptake [48]. Apparently, merely the presence of CovR, not its phosphorylation status, is crucial for the regulation of these genes. The mechanism of CovR-mediate gene activation is unknown as neither CovR nor CovR-D53∼P was sufficient to activate transcription from the dppA promoter in vitro despite the fact that CovR specifically binds to the dppA promoter [48]. Thus, whereas nearly all previous GAS CovR related work has focused on CovR-mediated gene repression, our data provide new impetus to better elucidate the mechanism underlying CovR gene activation. Moreover, these data may provide an explanation for why the majority of naturally occurring CovR variation identified in strains causing invasive infection involves alterations in N-terminal amino acid residues, rather than truncations of the CovR protein as are commonly observed for CovS [43], [66].
T65 has also previously been identified as the site for Stk-mediated phosphorylation of CovR in GBS [29]. However, despite significant parallels, such as the site of phosphorylation and negative interplay between the two phosphorylation sites [28], [29], [30], there are some striking differences between GAS and GBS in terms of the effect of CovR T65 phosphorylation. For example, GBS CovR-T65E was completely unable to bind to DNA from the CovR-regulated cylX promoter [29]. In contrast, we found that recombinant GAS CovR-T65E and CovR-D53A bound DNA from several CovR-regulated promoters at concentrations equivalent to unphosphorylated wild type CovR, which is consistent with our transcript data showing that the CovR-T65E and CovR-D53 strains maintained CovR-mediated gene activation profiles. Thus, in GAS, it appears that Stk-mediated phosphorylation of CovR T65 reduces, but does not completely abrogate CovR-DNA binding as was observed in GBS. Given that GAS CovR D53 phosphorylation increases interaction with RNA polymerase in addition to enhancing CovR-DNA interaction [22], GAS CovR T65 phosphorylation likely has additional consequences besides affecting CovR DNA binding affinity. Another key difference between the CovR system in GAS and GBS is that GAS CovR represses its own transcription whereas GBS CovR is self-activating [30], [57]. There is significant variance in the covR promoter regions between GAS and GBS, including differences at previously identified key GAS CovR binding sites [57]. Abolishing CovR threonine phosphorylation in GBS via the T65A substitution increased covR transcript level whereas we observed decreased covR transcript and decreased CovR expression in GAS CovR-T65A [30]. Interestingly, in GBS the CovR-T65A variant was also hypervirulent in mice compared to wild-type and hypovirulent compared to the CovR-D53A and CovR-T65E strains, which was the same phenotypic hierarchy we observed in GAS [30].
These and other data on CovR-Stk interaction in GAS and GBS raise the important question of how T65 phosphorylation hampers D53 phosphorylation and vice versa [26], [42]. The best studied example of dual-site phosphorylation in a prokaryotic pathogen concerns the histidine-containing phosphocarrier (HPr) protein, which can be phosphorylated on histidine-15 as well as serine-46 [67]. The various phosphorylation states of HPr regulate its interaction with other proteins as exemplified by the fact that HPr-S46-P, but not HPr-H15∼P can interact with the regulatory protein catabolite control protein A (CcpA) [68]. Conversely, HPr-His15∼P, but not HPr-S46-P serves as component of the phosphorelay in the phosphoenol-phosphotransferase system (PTS) regulating PTS-sugar transport [67]. In contrast to HPr, it does not appear that the phosphorylated forms of CovR are active in distinct functional systems but instead serve to influence the final effect of CovR on gene expression. Rather than obstructing specific protein-protein interactions (e.g. CovR-CovS), it is likely that CovR T65 phosphorylation induces an allosteric change in the CovR N-terminal receiver domain that serves to inhibit D53 phosphorylation by interfering with the general activation mechanism for OmpR/PhoB family members [69]. Such a change would explain why even the small molecule phosphodonor acetyl phosphate is unable to phosphorylate CovR-T65E. Similarly, although the CovR homolog OmpR is not known to be regulated by serine/threonine phosphorylation, fluorescence-labeling of OmpR cysteine-67, which corresponds to T65 in CovR, also impaired OmpR aspartate-phosphorylation and vice versa [70]. A crystal structure of the N-terminal domain of CovR is not available, but modeling of CovR on the structure of the related response regulator PhoP in complex with the phosphomimic BeF3− (PBD code 2PL1) shows that D53 and T65 are ∼9 Å apart meaning that interference due to direct electrostatic repulsion can be excluded [71]. We are currently engaged in generating crystal structures of the N-terminal domain of CovR and its various phosphorylated forms in an attempt to better understand the molecular consequences of CovR dual-site phosphorylation.
In summary, we have employed a combination of biochemical, genetic, and virulence assays to generate new insights into how phosphorylation of CovR mediated by distinct pathways affects GAS pathogenesis. Further studies of how bacteria modulate the phosphorylation status of regulatory proteins may generate new opportunities to prevent or treat serious bacterial infections.
Materials and Methods
Ethics statement
This study was carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by MD Anderson Animal Care and Use Committee (Protocol Number: 07-09-09131). All efforts were made to minimize suffering.
Bacterial strains and culture media
The strains and plasmids used in this work are presented in Table 1, and primers used for strain creation are listed in Table S2. Strains were grown in a nutrient-rich standard laboratory medium (Todd-Hewitt broth with 0.2% yeast extract (THY)) at 37°C with 5% CO2 with 15 mM MgCl2 added when noted to create a high Mg2+ condition [41]. THY without supplemental MgCl2 has previously been determined to have a Mg2+ concentration of ∼1 mM [72]. When appropriate, spectinomycin and chloramphenicol were added at 150 µg/mL and 10 µg/mL, respectively. Strain MGAS10870 is a fully-sequenced, invasive, serotype M3 strain that contains a wild-type covRS operon [40]. Strains 10870ΔcovR and 10870ΔspeB have been previously described [43], [73]. Strain 10870ΔcovS was created by replacing the covS gene in strain MGAS10870 with a spectinomycin resistance cassette via insertional inactivation as described [17], [25]. Derivatives of strain MGAS10870 that differed only by the presence of a single amino acid replacement were created using the chloramphenicol-resistant, temperature-sensitive plasmid pJL1055 (gift of D. Kasper) as described [74]. For each of the CovR strains derived from strain MGAS10870, sequencing of the entire covRS operon, as well as the mga and emm genes, was performed to check for the presence of spurious mutations that could have developed during the strain construction process (none were found). Strain SGBS001 is a serotype V group B Streptococcus strain isolated from human blood in Houston.

Protein overexpression and purification
Recombinant CovR was isolated from plasmid pTXB1-covR using the IMPACT Protein Purification System (New England Biolabs), which allows for recovery of soluble CovR isoforms lacking any non-native residues (e.g. no His-tag) [39]. To generate CovR variants, single nucleotide exchanges were introduced into pTXB1-covR via Quick-change mutagenesis (Stratagene) using the respective primer pairs (see Table S2). Wild type and variant CovR proteins were overexpressed in E. coli BL21/pLysS at 18°C overnight and purified to ≥95% homogeneity as described previously [39]. All CovR proteins were extensively buffer exchanged to 50 mM CAPS pH 10.0, 100 mM NaCl. Purified CovR protein was used to immunize rabbits to generate anti-CovR antibody (Covance, Denver, PA). The specificity of the anti-CovR antibody for CovR is shown in Figure S2.
The isolated Stk kinase domain (StkKD) has been shown previously to be sufficient to phosphorylate CovR [26]. Thus, recombinant StkKD was generated by amplifying DNA encoding the Stk kinase domain (amino acids 1–315) from MGAS10870. The resulting PCR product was cloned into pET15b (Novagen) via NdeI and XhoI. The N-terminal His-tagged StkKD was overexpressed in E. coli BL21/pLysS. Cells were induced with 1 mM IPTG at an OD ∼0.6, incubated for five hours at 20°C and harvested by centrifugation. After lysing the cells in buffer containing 20 mM Tris/HCl pH 8.0, 200 mM NaCl, 20 mM imidazole, and 10% glycerol, StkKD protein was purified over a Ni-NTA column and eluted with 200 mM imidazole. The protein was concentrated to ∼10 mg/ml and stored at −20°C in 20 mM Tris/HCl pH 8.0, 150 mM NaCl, 40% glycerol. The resultant StkKD protein was ∼% pure (Figure S1A).
In vitro phosphorylation of CovR and detection of the phosphorylation status
Wild type and variant CovR proteins were phosphorylated on D53 for 2 h at 37°C in phosphorylation buffer containing 50 mM Tris/HCl pH 8.0, 10 mM MgCl2, 3 mM DTT, and 32 µM acetyl-phosphate (Sigma), as previously described [56]. Threonine phosphorylation of CovR was performed by incubating wild type or variant CovR proteins for 30 min at 37°C with a three-fold excess of StkKD in phosphorylation buffer containing 100 mM Tris/HCl pH 7.5, 10 mM MgCl2 or MnCl2, 1 mM DTT, and 10 mM ATP. To detect phosphorylated CovR species we used Phos-tag SDS polyacrylamide (PAA) gels which contain a phosphate-binding reagent that specifically retards the migration of phosphorylated proteins, thereby allowing for resolution between phosphorylated and non-phosphorylated protein species [35]. The phosphorylation samples were separated on 12% Phos-tag SDS-PAA gels containing 100 µM Phos-tag solution (Wako Pure Chemical Industries Ltd, Richmond, VA) and 200 µM MnCl2 for 70 min at 180 V. The gel was washed in transfer buffer (SDS running buffer containing 20% MeOH and 1 mM EDTA) for 15 min. The proteins were then transferred onto a nitrocellulose membrane (0.45 µm, Bio-Rad) using a Trans-blot SD semi-dry electrophoretic transfer cell (Bio-Rad) for 15 min at 15 V. After blocking the membrane overnight in 20 mM Tris/HCl pH 7.5, 150 mM NaCl, 0.05% Tween 20, and 5% dry milk, it was incubated for 1 h with affinity purified anti-CovR antibody at a 1∶5,000 dilution with blocking solution. The secondary antibody, goat anti-rabbit IgG (Pierce), was diluted 1∶10,000. The blot was developed using the SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific).
Detection of CovR phosphorylation in vivo
GAS and GBS strains were grown in 100 ml of THY or THY supplemented with 15 mM MgCl2 to OD = 0.5 and harvested by centrifugation. Cell lysates were prepared by the rapid preparation method previously described taking care to always keep the lysates chilled to minimize spontaneous dephosphorylation [36]. A total protein amount of 70 µg as determined by Bradford assay (Bio-Rad) was loaded on the gel for each sample. As phosphorylation controls, wild type cell lysates were either boiled at 100°C for 1 min, which specifically removes the heat labile aspartate phosphorylation, or incubated with 40 units calf intestine phosphatase (CIP, New England Biolabs) at 37°C for 10 min, which completely removes any phosphorylation. Samples were electrophoresed for 80 min at 150 V on a 10% Bis-Tris buffered Zn2+-Phostag SDS-PAGE. This allows the separation of unphosphorylated and phosphorylated proteins under neutral pH [38]. Subsequently, CovR species were detected by standard Western blotting using anti-CovR antibodies.
Circular dichroism spectroscopy
Wild type and mutant CovR proteins were diluted to a concentration of 0.4 mg/ml in 10 mM potassium phosphate, pH 7.5, 10 mM NaF prior to measurements. Far-UV (200–260 nm) spectra were recorded on a Jasco J-810 spectropolarimeter at 37°C and a scan speed of 20 nm/min as described [43].
Mass spectrometry
15 µg of recombinant CovR was phosphorylated using StkKD as described above. The entire reaction was run on a 12% SDS polyacrylamide gel. The band corresponding to CovR was excised and 50 pmol of in vitro phosphorylated CovR was diluted into 45 µl of 0.1 M ammonium bicarbonate, reduced with 2 µl 0.1 M DTT and alkylated with 2 µl 0.2 M iodoacetamide. The protein was digested with 1 µg of chymotrypsin (Roche, Indianapolis, IN) at 25°C or 1 µg of trypsin at 37°C overnight. 10 µl of the peptide mixture was analyzed by automated microcapillary liquid chromatography-tandem mass spectrometry (LC-MS-MS). The application of a 1.8 kV distal voltage electro-sprayed the eluted peptides directly into the LTQ Orbitrap XL ion trap mass spectrometer equipped with a nanoLC electrospray ionization source (ThermoFinningan). Full masses (MS) spectra were recorded on the peptides over a 400–2000 m/z range at 60,000 resolution in the Orbitrap, followed by five tandem mass (MS/MS) events sequentially generated in a data-dependent manner on the first, second, third, fourth and fifth most intense ions selected from the full MS spectrum (at 35% collision energy). Mass spectrometer scan functions and HPLC solvent gradients were controlled by the Xcalibur data system (ThermoFinnigan). MS/MS spectra were extracted from the RAW file with ReAdW.exe (http://sourceforge.net/projects/sashimi). The MS/MS data were searched with Inspect [75] against the MGAS315 Streptococcus pyogenes database (NC_004070) with optional modifications: +15.9994 on methionine, +57.0214 on cysteine, and +79.9663 on threonine, serine, and tyrosine, respectively. Only peptides with at least a P-value of 0.01 were analyzed further. Data from keratins and trypsin were excluded from further analysis. Peptides identified by the software to be phosphorylated were manually verified. In addition, further verification was performed by analyzing putative phosphorylated peptides in targeted MS/MS and MS/MS/MS modes.
Hyaluronic acid capsule and cysteine protease activity assays
The amount of cell-associated capsular polysaccharide was done as previously described using GAS grown to late-exponential phase (OD600 = 0.9) [15]. Functional SpeB protease activity was determined using casein hydrolysis as described [76].
Transcript level analysis
RNA was purified from various GAS strains grown to mid-exponential phase using an RNeasy mini kit (Qiagen). 1 µg of RNA per sample was converted to cDNA using a High Capacity Reverse Transcription Kit (Applied Biosystems). TaqMan real-time qRT-PCR (primers and probes listed in Table S2) was performed on an Applied Biosystems Step-One Plus System as described [77]. All samples were done at least in duplicate on two separate occasions and analyzed in duplicate. To compare gene transcript levels between the wild-type and the various derivative strains, a two-sample t-test (unequal variance) was applied with a P value of <0.05 and a mean transcript level of at least 1.5-fold change being considered statistically significant.
For RNA-Seq analysis, strains were grown in quadruplicate to mid-exponential phase and RNA was isolated as for TaqMan qRT-PCR. First and second strand cDNA synthesis was performed on 500 ng of RNA using the Ovation Prokaryotic RNA-Seq System (NuGEN). The resulting cDNA was fragmented to 200 bp (mean fragment size) with the S220 Focused-ultrasonicator (Covaris) and used to make barcoded sequencing libraries on the SPRI-TE Nucleic Acid Extractor (Beckman-Coulter). No size selection was employed. Libraries were quantitated by qPCR (KAPA Systems), multiplexed and 8 samples per lane were sequenced on the HiSeq2000 using 76 bp paired-end sequencing. The raw reads in FASTQ format were aligned to the reference genome, Streptococcus pyogenes MGAS315 (GI:21905618), using Mosaik alignment software (version: 1.1.0021, http://code.google.com/p/mosaik-aligner/) with the following alignment parameters: 8 percent maximal percentage read length allowed to be errors and 50 percent minimal percentage of the read length aligned. Duplicate fragments with a lower alignment quality were discarded. Next, the overlaps between aligned reads and annotated genes were counted using HTSeq software (http://www-huber.embl.de/users/anders/HTSeq/doc/overview.html). If the number of overlapped read of any given gene was less than one per million total mapped read for all samples, this gene was excluded from further analysis. 330 (17%) of total 1951 genes were removed due to low expression. The gene counts were normalized using the scaling factor method [78]. A negative binomial generalized linear model was fitted to each gene. Then a likelihood-ratio test was applied to examine if there was a difference in the gene transcript levels among the six strains [78]. The Benjamini-Hochberg method was used to control false discovery rate (FDR) [79]. Next, pair-wise comparisons using the likelihood ratio test were performed to compare the gene transcript levels between pairs of strains with the Holm's method used to calculate adjusted P-values to correct for multiple testing. Transcript levels were considered significantly different only if the mean transcript level difference was ≥1.5-fold and the final, adjusted P value was less than 0.05.
Electrophoretic shift assays
The ∼300 bp encompassing promoter regions of selected CovR regulated genes were amplified by PCR from MGAS10870 genomic DNA using the respective primer pairs listed in Table S2. The purified PCR products (0.4 µg) were incubated with indicated amounts of various CovR isoforms at 37°C for 15 min in TBE-buffer (89 mM Tris, 89 mM borate, 1 mM EDTA, 5% glycerol, and 10 µg/ml polydI:dC (Sigma)) as described [43]. Samples were then separated on a 6% TBE-PAA gel for 70 min at 120 V and stained with ethidium bromide.
Mouse infection studies/in vivo gene expression analysis
20 female outbred CD-1 Swiss mice per strain (Harlan-Sprague-Dawley) were injected intraperitoneally with 1.0×107 GAS CFU and monitored for near-mortality. Differences in survival were calculated using a Kaplan-Meier survival analysis with Bonferroni's correction for multiple comparisons. For in vivo gene expression assays, four mice per strain were euthanized 24 hours after infection and blood was isolated via cardiac puncture. Total RNA was isolated from blood using the QIAamp RNA Blood Mini Kit (Qiagen) with DNA contamination being eliminated using Turbo DNAfree (Ambion). Bacterial RNA was preferentially isolated from the total blood RNA using the MICROBEnrich Kit (Invitrogen) according to the manufacturer's instruction. cDNA was created from the RNA as previously described. RNA samples without reverse transcriptase were included as negative controls to ensure that no contaminating DNA was present in any of the samples. TaqMan real-time qRT-PCR was performed in triplicate on an Applied Biosystems Step-One Plus system as previously described [77].
Accession numbers
RNA-Seq data have been deposited at the short-read archive under accession # PRJNA238945.
Supporting Information
Zdroje
1. RecinosDA, SekedatMD, HernandezA, CohenTS, SakhtahH, et al. (2012) Redundant phenazine operons in Pseudomonas aeruginosa exhibit environment-dependent expression and differential roles in pathogenicity. Proc Natl Acad Sci U S A 109 : 19420–19425.
2. BohmeK, SteinmannR, KortmannJ, SeekircherS, HerovenAK, et al. (2012) Concerted actions of a thermo-labile regulator and a unique intergenic RNA thermosensor control Yersinia virulence. PLoS Pathog 8: e1002518.
3. MandlikA, LivnyJ, RobinsWP, RitchieJM, MekalanosJJ, et al. (2011) RNA-Seq-based monitoring of infection-linked changes in Vibrio cholerae gene expression. Cell Host Microbe 10 : 165–174.
4. BalasubramanianD, SchneperL, KumariH, MatheeK (2013) A dynamic and intricate regulatory network determines Pseudomonas aeruginosa virulence. Nucleic Acids Res 41 : 1–20.
5. CaiW, WannemuehlerY, Dell'annaG, NicholsonB, BarbieriNL, et al. (2013) A novel two-component signaling system facilitates uropathogenic Escherichia coli's ability to exploit abundant host metabolites. PLoS Pathog 9: e1003428.
6. LyonGJ, MayvilleP, MuirTW, NovickRP (2000) Rational design of a global inhibitor of the virulence response in Staphylococcus aureus, based in part on localization of the site of inhibition to the receptor-histidine kinase, AgrC. Proc Natl Acad Sci U S A 97 : 13330–13335.
7. ZahrtTC, DereticV (2001) Mycobacterium tuberculosis signal transduction system required for persistent infections. Proc Natl Acad Sci U S A 98 : 12706–12711.
8. GaoR, StockAM (2009) Biological insights from structures of two-component proteins. Annu Rev Microbiol 63 : 133–154.
9. StockAM, RobinsonVL, GoudreauPN (2000) Two-component signal transduction. Annu Rev Biochem 69 : 183–215.
10. KobirA, ShiL, BoskovicA, GrangeasseC, FranjevicD, et al. (2011) Protein phosphorylation in bacterial signal transduction. Biochim Biophys Acta 1810 : 989–994.
11. RaskoDA, MoreiraCG, Li deR, ReadingNC, RitchieJM, et al. (2008) Targeting QseC signaling and virulence for antibiotic development. Science 321 : 1078–1080.
12. GotohY, DoiA, FurutaE, DubracS, IshizakiY, et al. (2010) Novel antibacterial compounds specifically targeting the essential WalR response regulator. J Antibiot (Tokyo) 63 : 127–134.
13. GotohY, EguchiY, WatanabeT, OkamotoS, DoiA, et al. (2010) Two-component signal transduction as potential drug targets in pathogenic bacteria. Curr Opin Microbiol 13 : 232–239.
14. TangYT, GaoR, HavranekJJ, GroismanEA, StockAM, et al. (2012) Inhibition of bacterial virulence: drug-like molecules targeting the Salmonella enterica PhoP response regulator. Chem Biol Drug Des 79 : 1007–1017.
15. LevinJC, WesselsMR (1998) Identification of csrR/csrS, a genetic locus that regulates hyaluronic acid capsule synthesis in group A Streptococcus. Mol Microbiol 30 : 209–219.
16. ChurchwardG (2007) The two faces of Janus: virulence gene regulation by CovR/S in group A streptococci. Mol Microbiol 64 : 34–41.
17. FederleMJ, McIverKS, ScottJR (1999) A response regulator that represses transcription of several virulence operons in the group A Streptococcus. J Bacteriol 181 : 3649–3657.
18. CarapetisJR, SteerAC, MulhollandEK, WeberM (2005) The global burden of group A streptococcal diseases. Lancet Infect Dis 5 : 685–694.
19. EnglebergNC, HeathA, MillerA, RiveraC, DiRitaVJ (2001) Spontaneous mutations in the CsrRS two-component regulatory system of Streptococcus pyogenes result in enhanced virulence in a murine model of skin and soft tissue infection. J Infect Dis 183 : 1043–1054.
20. GrahamMR, SmootLM, MigliaccioCA, VirtanevaK, SturdevantDE, et al. (2002) Virulence control in group A Streptococcus by a two-component gene regulatory system: global expression profiling and in vivo infection modeling. Proc Natl Acad Sci USA 99 : 13855–13860.
21. DaltonTL, ScottJR (2004) CovS inactivates CovR and is required for growth under conditions of general stress in Streptococcus pyogenes. J Bacteriol 186 : 3928–3937.
22. GusaAA, GaoJ, StringerV, ChurchwardG, ScottJR (2006) Phosphorylation of the group A streptococcal CovR response regulator causes dimerization and promoter-specific recruitment by RNA polymerase. J Bacteriol 188 : 4620–4626.
23. BarbieriCM, WuT, StockAM (2013) Comprehensive analysis of OmpR phosphorylation, dimerization, and DNA binding supports a canonical model for activation. J Mol Biol 425 : 1612–1626.
24. DaltonTL, HobbRI, ScottJR (2006) Analysis of the role of CovR and CovS in the dissemination of Streptococcus pyogenes in invasive skin disease. Microb Pathog 40 : 221–227.
25. TrevinoJ, PerezN, Ramirez-PenaE, LiuZ, ShelburneSA3rd, et al. (2009) CovS simultaneously activates and inhibits the CovR-mediated repression of distinct subsets of group A Streptococcus virulence factor-encoding genes. Infect Immun 77 : 3141–3149.
26. AgarwalS, PancholiP, PancholiV (2011) Role of serine/threonine phosphatase (SP-STP) in Streptococcus pyogenes physiology and virulence. J Biol Chem 286 : 41368–41380.
27. RajagopalL, ClancyA, RubensCE (2003) A eukaryotic type serine/threonine kinase and phosphatase in Streptococcus agalactiae reversibly phosphorylate an inorganic pyrophosphatase and affect growth, cell segregation, and virulence. J Biol Chem 278 : 14429–14441.
28. RajagopalL, VoA, SilvestroniA, RubensCE (2006) Regulation of cytotoxin expression by converging eukaryotic-type and two-component signalling mechanisms in Streptococcus agalactiae. Mol Microbiol 62 : 941–957.
29. LinWJ, WalthersD, ConnellyJE, BurnsideK, JewellKA, et al. (2009) Threonine phosphorylation prevents promoter DNA binding of the Group B Streptococcus response regulator CovR. Mol Microbiol 71 : 1477–1495.
30. LemboA, GurneyMA, BurnsideK, BanerjeeA, de los ReyesM, et al. (2010) Regulation of CovR expression in Group B Streptococcus impacts blood-brain barrier penetration. Mol Microbiol 77 : 431–443.
31. WhidbeyC, HarrellMI, BurnsideK, NgoL, BecraftAK, et al. (2013) A hemolytic pigment of Group B Streptococcus allows bacterial penetration of human placenta. J Exp Med 210 : 1265–1281.
32. MillerAA, EnglebergNC, DiRitaVJ (2001) Repression of virulence genes by phosphorylation-dependent oligomerization of CsrR at target promoters in Streptococcus pyogenes. Mol Microbiol 40 : 976–990.
33. FederleMJ, ScottJR (2002) Identification of binding sites for the group A streptococcal global regulator CovR. Mol Microbiol 43 : 1161–1172.
34. JiangSM, IshmaelN, HotoppJD, PulitiM, TissiL, et al. (2008) Variation in the group B Streptococcus CsrRS regulon and effects on pathogenicity. J Bacteriol 190 : 1956–1965.
35. BarbieriCM, StockAM (2008) Universally applicable methods for monitoring response regulator aspartate phosphorylation both in vitro and in vivo using Phos-tag-based reagents. Anal Biochem 376 : 73–82.
36. WayneKJ, LiS, KazmierczakKM, TsuiHC, WinklerME (2012) Involvement of WalK (VicK) phosphatase activity in setting WalR (VicR) response regulator phosphorylation level and limiting cross-talk in Streptococcus pneumoniae D39 cells. Mol Microbiol 86 : 645–660.
37. BoulangerA, ChenQ, HintonDM, StibitzS (2013) In vivo phosphorylation dynamics of the Bordetella pertussis virulence-controlling response regulator BvgA. Mol Microbiol 88 : 156–172.
38. KinoshitaE, Kinoshita-KikutaE (2011) Improved Phos-tag SDS-PAGE under neutral pH conditions for advanced protein phosphorylation profiling. Proteomics 11 : 319–323.
39. ShelburneSA, OlsenRJ, SuberB, SahasrabhojaneP, SumbyP, et al. (2010) A combination of independent transcriptional regulators shapes bacterial virulence gene expression during infection. PLoS Pathog 6: e1000817.
40. BeresSB, CarrollRK, SheaPR, SitkiewiczI, Martinez-GutierrezJC, et al. (2010) Molecular complexity of successive bacterial epidemics deconvoluted by comparative pathogenomics. Proc Natl Acad Sci U S A 107 : 4371–4376.
41. GryllosI, GrifantiniR, ColapricoA, JiangS, DeforceE, et al. (2007) Mg(2+) signalling defines the group A streptococcal CsrRS (CovRS) regulon. Mol Microbiol 65 : 671–683.
42. JinH, PancholiV (2006) Identification and biochemical characterization of a eukaryotic-type serine/threonine kinase and its cognate phosphatase in Streptococcus pyogenes: their biological functions and substrate identification. J Mol Biol 357 : 1351–1372.
43. HorstmannN, SahasrabhojaneP, SuberB, KumaraswamiM, OlsenRJ, et al. (2011) Distinct single amino acid replacements in the control of virulence regulator protein differentially impact streptococcal pathogenesis. PLoS Pathog 7: e1002311.
44. KansalRG, DattaV, AzizRK, AbdeltawabNF, RoweS, et al. (2010) Dissection of the molecular basis for hypervirulence of an in vivo-selected phenotype of the widely disseminated M1T1 strain of group A Streptococcus bacteria. J Infect Dis 201 : 855–865.
45. AshbaughCD, AlbertiS, WesselsMR (1998) Molecular analysis of the capsule gene region of group A Streptococcus: the hasAB genes are sufficient for capsule expression. J Bacteriol 180 : 4955–4959.
46. SumbyP, ZhangS, WhitneyAR, FalugiF, GrandiG, et al. (2008) A chemokine-degrading extracellular protease made by group A Streptococcus alters pathogenesis by enhancing evasion of the innate immune response. Infect Immun 76 : 978–985.
47. RasmussenM, MullerHP, BjorckL (1999) Protein GRAB of Streptococcus pyogenes regulates proteolysis at the bacterial surface by binding alpha2-macroglobulin. J Biol Chem 274 : 15336–15344.
48. GusaAA, FroehlichBJ, DesaiD, StringerV, ScottJR (2007) CovR activation of the dipeptide permease promoter (PdppA) in Group A Streptococcus. J Bacteriol 189 : 1407–1416.
49. LiuM, ZhuH, ZhangJ, LeiB (2007) Active and passive immunizations with the streptococcal esterase Sse protect mice against subcutaneous infection with group A streptococci. Infect Immun 75 : 3651–3657.
50. KapurV, KanjilalS, HamrickMR, LiLL, WhittamTS, et al. (1995) Molecular population genetic analysis of the streptokinase gene of Streptococcus pyogenes: mosaic alleles generated by recombination. Mol Microbiol 16 : 509–519.
51. von Pawel-RammingenU, JohanssonBP, BjorckL (2002) IdeS, a novel streptococcal cysteine proteinase with unique specificity for immunoglobulin G. EMBO J 21 : 1607–1615.
52. NizetV, BeallB, BastDJ, DattaV, KilburnL, et al. (2000) Genetic locus for streptolysin S production by group A Streptococcus. Infect Immun 68 : 4245–4254.
53. SriskandanS, UnnikrishnanM, KrauszT, CohenJ (2000) Mitogenic factor (MF) is the major DNase of serotype M89 Streptococcus pyogenes. Microbiology 146(Pt 11): 2785–2792.
54. LyonWR, GibsonCM, CaparonMG (1998) A role for trigger factor and an Rgg-like regulator in the transcription, secretion and processing of the cysteine proteinase of Streptococcus pyogenes. EMBO J 17 : 6263–6275.
55. Tran-WinklerHJ, LoveJF, GryllosI, WesselsMR (2011) Signal transduction through CsrRS confers an invasive phenotype in group A Streptococcus. PLoS Pathog 7: e1002361.
56. GaoJ, GusaAA, ScottJR, ChurchwardG (2005) Binding of the global response regulator protein CovR to the sag promoter of Streptococcus pyogenes reveals a new mode of CovR-DNA interaction. J Biol Chem 280 : 38948–38956.
57. GusaAA, ScottJR (2005) The CovR response regulator of group A Streptococcus (GAS) acts directly to repress its own promoter. Mol Microbiol 56 : 1195–1207.
58. ChaoJD, PapavinasasundaramKG, ZhengX, Chavez-SteenbockA, WangX, et al. (2010) Convergence of Ser/Thr and two-component signaling to coordinate expression of the dormancy regulon in Mycobacterium tuberculosis. J Biol Chem 285 : 29239–29246.
59. XuH, CaimanoMJ, LinT, HeM, RadolfJD, et al. (2010) Role of acetyl-phosphate in activation of the Rrp2-RpoN-RpoS pathway in Borrelia burgdorferi. PLoS Pathog 6: e1001104.
60. Chiang-NiC, TsouCC, LinYS, ChuangWJ, LinMT, et al. (2008) The transcriptional terminator sequences downstream of the covR gene terminate covR/S operon transcription to generate covR monocistronic transcripts in Streptococcus pyogenes. Gene 427 : 99–103.
61. CousinC, DerouicheA, ShiL, PagotY, PoncetS, et al. (2013) Protein-serine/threonine/tyrosine kinases in bacterial signaling and regulation. FEMS Microbiol Lett 346 : 11–19.
62. PancholiV, BoelG, JinH (2010) Streptococcus pyogenes Ser/Thr kinase-regulated cell wall hydrolase is a cell division plane-recognizing and chain-forming virulence factor. J Biol Chem 285 : 30861–30874.
63. BeilharzK, NovakovaL, FaddaD, BrannyP, MassiddaO, et al. (2012) Control of cell division in Streptococcus pneumoniae by the conserved Ser/Thr protein kinase StkP. Proc Natl Acad Sci U S A 109: E905–913.
64. BugryshevaJ, FroehlichBJ, FreibergJA, ScottJR (2011) Serine/threonine protein kinase Stk is required for virulence, stress response, and penicillin tolerance in Streptococcus pyogenes. Infect Immun 79 : 4201–4209.
65. LiuM, ZhuH, LiJ, GarciaCC, FengW, et al. (2012) Group A Streptococcus secreted esterase hydrolyzes platelet-activating factor to impede neutrophil recruitment and facilitate innate immune evasion. PLoS Pathog 8: e1002624.
66. IkebeT, AtoM, MatsumuraT, HasegawaH, SataT, et al. (2010) Highly frequent mutations in negative regulators of multiple virulence genes in group A streptococcal toxic shock syndrome isolates. PLoS Pathog 6: e1000832.
67. DeutscherJ, FranckeC, PostmaPW (2006) How phosphotransferase system-related protein phosphorylation regulates carbohydrate metabolism in bacteria. Microbiol Mol Biol Rev 70 : 939–1031.
68. SchumacherMA, AllenGS, DielM, SeidelG, HillenW, et al. (2004) Structural basis for allosteric control of the transcription regulator CcpA by the phosphoprotein HPr-Ser46-P. Cell 118 : 731–741.
69. GaoR, MackTR, StockAM (2007) Bacterial response regulators: versatile regulatory strategies from common domains. Trends Biochem Sci 32 : 225–234.
70. AmesSK, FrankemaN, KenneyLJ (1999) C-terminal DNA binding stimulates N-terminal phosphorylation of the outer membrane protein regulator OmpR from Escherichia coli. Proc Natl Acad Sci U S A 96 : 11792–11797.
71. BachhawatP, StockAM (2007) Crystal structures of the receiver domain of the response regulator PhoP from Escherichia coli in the absence and presence of the phosphoryl analog beryllofluoride. J Bacteriol 189 : 5987–5995.
72. GryllosI, LevinJC, WesselsMR (2003) The CsrR/CsrS two-component system of group A Streptococcus responds to environmental Mg2+. Proc Natl Acad Sci USA 100 : 4227–4232.
73. OlsenRJ, LauciricaDR, WatkinsME, FeskeML, Garcia-BustillosJR, et al. (2012) Polymorphisms in regulator of protease B (RopB) alter disease phenotype and strain virulence of serotype M3 group A Streptococcus. J Infect Dis 205 : 1719–1729.
74. CarrollRK, IiiSA, OlsenRJ, SuberB, SahasrabhojaneP, et al. (2011) Naturally occurring single amino acid replacements in a regulatory protein alter streptococcal gene expression and virulence in mice. J Clin Invest 121 : 1956–68.
75. TannerS, ShuH, FrankA, WangLC, ZandiE, et al. (2005) InsPecT: identification of posttranslationally modified peptides from tandem mass spectra. Anal Chem 77 : 4626–4639.
76. LukomskiS, SreevatsanS, AmbergC, ReichardtW, WoischnikM, et al. (1997) Inactivation of Streptococcus pyogenes extracellular cysteine protease significantly decreases mouse lethality of serotype M3 and M49 strains. J Clin Invest 99 : 2574–2580.
77. VirtanevaK, PorcellaSF, GrahamMR, IrelandRM, JohnsonCA, et al. (2005) Longitudinal analysis of the group A Streptococcus transcriptome in experimental pharyngitis in cynomolgus macaques. Proc Natl Acad Sci USA 102 : 9014–9019.
78. AndersS, HuberW (2010) Differential expression analysis for sequence count data. Genome Biol 11: R106.
79. ReinerA, YekutieliD, BenjaminiY (2003) Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics 19 : 368–375.
80. LiJ, KasperDL, AusubelFM, RosnerB, MichelJL (1997) Inactivation of the alpha C protein antigen gene, bca, by a novel shuttle/suicide vector results in attenuation of virulence and immunity in group B Streptococcus. Proc Natl Acad Sci U S A 94 : 13251–13256.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 5
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Venus Kinase Receptors Control Reproduction in the Platyhelminth Parasite
- Dual-Site Phosphorylation of the Control of Virulence Regulator Impacts Group A Streptococcal Global Gene Expression and Pathogenesis
- Severe Acute Respiratory Syndrome Coronavirus Envelope Protein Ion Channel Activity Promotes Virus Fitness and Pathogenesis
- High-Efficiency Targeted Editing of Large Viral Genomes by RNA-Guided Nucleases
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy